

SUMMARY
The human gut microbiota performs functions that are not encoded in our H. sapiens genome including the processing of otherwise undigestible dietary polysaccharides. Defining the structures of proteins involved in import and degradation of specific glycans by saccharolytic bacteria complements genomic analysis of the nutrient processing capabilities of gut communities. Here we describe the atomic structure of one such protein, SusD, required for starch binding and utilization by Bacteroides thetaiotaomicron, a prominent adaptive forager of glycans in the distal human gut microbiota. The binding pocket of this unique α-helical protein contains an arc of aromatic residues that complements the natural helical structure of starch and imposes this conformation on bound maltoheptaose. Further, SusD binds cyclic oligosaccharides with higher affinity than linear forms. The structures of several SusD/oligosaccharide complexes reveal an inherent ligand recognition plasticity dominated by the three-dimensional conformation of the oligosaccharides rather than specific interactions with the composite sugars.
INTRODUCTION
Our adult human gut is home to trillions of microbes. This community (microbiota) is dominated by members of a relatively few divisions (phyla) in the domain Bacteria (the Firmicutes and Bacteroidetes), but also contains members of Archaea and Eukarya (Eckburg et al., 2005; Ley et al., 2006a,b). The distal gut microbiota is essential for digestion of a wide array of polysaccharides in our diet, as humans lack the requisite glycoside hydrolases (Sonnenburg et al., 2005; http://cazy.org). Short-chain fatty acids, derived from polysaccharide fermentation contribute as much as 10% of daily caloric intake for those on a Western-style diet (Backhed et al., 2005). Given the structural complexity and diversity of dietary glycans, a basic question is how members of the gut microbial community have evolved ways to recognize and acquire specific classes of carbohydrates among the vast and varied buffet of polysaccharides encountered in this body habitat. The answers could provide new strategies for optimizing nutrient utilization.
Bacteroides thetaiotaomicron is a prominent member of the human gut microbiota and a adept flexible forager of polysaccharides (Sonnenburg et al., 2005). Its capacity for glycan degradation is impressive: its proteome contains 246 known or predicted glycoside hydrolases and polysaccharide lyases (Xu et al., 2003) compared to 99 in our human proteome (http::/cazy.org). Most B. thetaiotaomicron genes encoding these glycoside hydrolases occur in large clusters’, termed polysaccharide utilization loci (PULs), which also contain genes for extracellular polysaccharide recognition and uptake (Bjursell et al., 2006; Xu et al., 2003). The first of these PULs to be characterized was the starch utilization system (Sus), which is required by B. thetaiotaomicron to grow on amylose, amylopectin, pullulan, and maltooligosaccharides (Anderson and Salyers, 1989a, b). The Sus system (Figure 1) contains eight genes, susRABCDEFG. SusR acts as a transcriptional activator of the locus in response to maltose (D’Elia and Salyers, 1996b). SusA, SusB, and SusG have neopullulanase, α-glycosidase, and α-amylase activity, respectively (Reeves et al., 1996; Shipman et al., 2000). SusDEFG are components of an extracellular outer membrane complex that binds to and degrades large starch molecules to maltooligosaccharides, which are imported into the periplasm via SusC, a predicted TonB-dependent β-barrel porin (Cho and Salyers, 2001; D’Elia and Salyers, 1996a; Shipman et al., 2000). SusG is the only externalized glycoside hydrolase produced by the Sus system but has a relatively low affinity for starch (Km ∼3 mM) and therefore does not contribute to starch binding to the cell surface (Shipman et al., 1999). The roles of SusE and SusF are unclear (Cho and Salyers, 2001; Shipman et al., 2000).
Figure 1. The starch utilization system (Sus) of Bacteroides thetaiotaomicron.
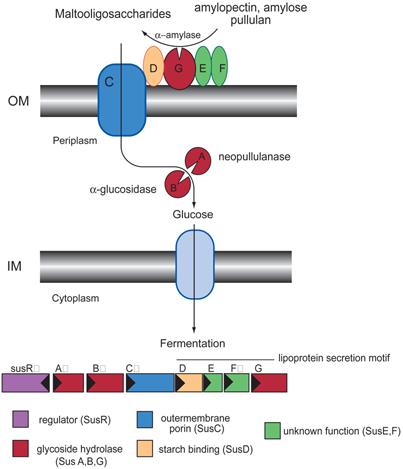
Cartoon representation of the Sus operon and its protein products (Cho and Salyers, 2001; D’Elia and Salyers, 1996a; Shipman et al., 2000; Shipman et al., 1999). The stoichiometry of the Sus complex is not known.
The lipoproteins SusDEFG are each tethered to the outer membrane surface and all five outer membrane proteins, SusCDEFG, co-purify on amylose resin with SusCD being the minimum starch-binding complex (Cho and Salyers, 2001; Shipman et al., 2000). B. thetaiotaomicron mutants containing a polar disruption in susD (ΔsusDEFG) have little to no starch binding activity, suggesting that the putative porin SusC is not sufficient for starch or oligosaccharide binding. However, a mutant containing a polar disruption in susE, (ΔsusEFG) regain 70% of the wild-type starch binding capacity (Shipman et al., 2000). Moreover, when the ΔsusEFG cells are complemented with susG, B. thetaiotaomicron can grow on starch, whereas the ΔsusDEFG mutants cannot (Cho and Giovannoni, 2003). Since SusC and SusD together promote tight binding of starch to the cell surface, it seems logical that SusD is directly involved in starch binding (Shipman et al., 1999). Interestingly, SusD does not have detectable homology to known carbohydrate-binding modules.
The B. thetaiotaomicron genome contains 101 pairs of genes related to susC-susD, with an additional 269 pairs found among four other sequenced human gut-associated Bacteroides spp. genomes sequenced to date (Xu et al., 2007). Genes encoding SusC/SusD pairs are ubiquitous components of PULs. The individual proteins may be highly divergent from one another and are often grouped with genes encoding diverse glycoside hydrolase and polysaccharide lyase activities, suggesting that each PUL has evolved to target a specific polysaccharide. Indeed, individual PULs are differentially expressed in vivo under conditions where B. thetaiotaomicron is forced to forage on various dietary and host glycans (Bjursell et al., 2006; Sonnenburg et al., 2005).
Despite the prominent representation of Sus-like systems in B. thetaiotaomicron and other sequenced human gut-associated Bacteroidetes (Xu et al., 2007), little is known about the molecular basis of carbohydrate recognition by these complexes. Therefore, we have performed structural and biochemical analyses of B. thetaiotaomicron SusD to elucidate its role in starch recognition and uptake. Our results indicate that SusD represents a novel class of polysaccharide-binding proteins, and reveal a paradigm for how a saccharolytic bacterium recognizes and acquires a specific class of glycans.
RESULTS
SusD is required for starch and maltooligosaccharide utilization
Previously, technical limitations for genetic manipulation of B. thetaiotaomicron prevented examination of the role of SusD in starch utilization in isolation from the other sus genes. We used a novel counter-selection technique to construct an in-frame deletion strain (ΔsusD) that lacked codons 2-551 of susD. Quantitative real-time PCR analysis of transcripts corresponding to the remaining genes in the susBCDEFG operon demonstrated that they are still induced in the ΔsusD strain in response to maltose (Supplemental Figure 1), confirming that only expression of SusD is eliminated.
To determine the contribution of SusD to the growth of B. thetaiotaomicron, the parent (wild-type), isogenic ΔsusD strain, and a derivative of ΔsusD containing a single, complementing copy of susD expressed from its native promoter (ΔsusD::PsusB-susD) were cultured in minimal medium containing a number of starch-like substrates as sole carbon sources. Consistent with previous findings that the Sus system is not required for growth on maltooligosaccharides containing ≤ 3 glucose units (Anderson and Salyers, 1989b), all three strains exhibited similar growth rates on glucose, maltose and maltotriose (Figure 2). However, loss of SusD abolished the ability of B. thetaiotaomicron to grow on amylopectin and pullulan as well as maltohexaose and maltoheptaose, suggesting an essential role in growth on starch molecules containing ≥ 6 glucose units. Additionally, the ΔsusD strain exhibited a reduced growth rate on maltotetraose and maltopentaose, indicating a supporting but non-essential role in utilization of shorter oligosaccharides. The loss of SusD did not affect the ability of B. thetaiotaomicron to utilize dextran, suggestingthat SusD is preferentially recognizing the α-1,4 linked sugars.
Figure 2. Growth of Bacteroides thetaiotaomicron and derivative strains on starch-like oligosaccharides and polysaccarides.
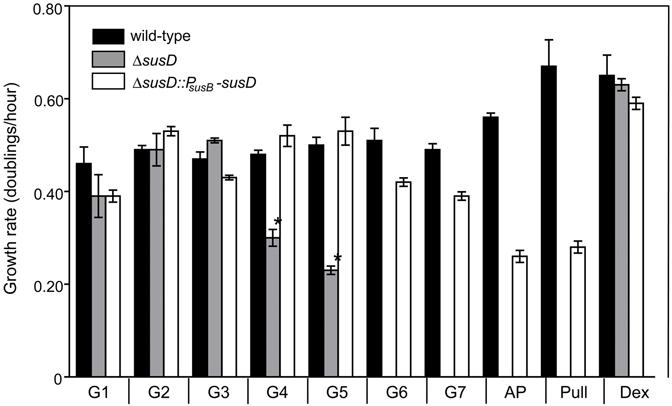
Shown are the log-phase growth rates of wild-type, ΔsusD, and complemented ΔsusD (ΔsusD::PsusB-susD) strains on glucose (G1), maltooligosaccharides of varying length (G2-G7), amylopectin (AP), and pullulan (Pull) and dextran (Dex). The ΔsusD strain is unable to grow on substrates >5 glucose units and exhibits significantly slower rates on G4 and G5 compared to wild-type on the same substrates (P<0.01, denoted by an asterisk). Complementation with a single copy of susD (expressed at ∼10% of wild-type levels; see Fig. S1) restores either full or partial growth characteristics depending on substrate size.
Overall Structure of SusD
Native SusD in B. thetaiotaomicron includes an outer membrane signal sequence that is likely removed upon translocation, and a cysteine, C25, that is lipidated and tethers the protein to the cell membrane. Therefore, only residues 26-551 of SusD were expressed in E. coli for these studies. Size exclusion chromatography of heterologously expressed SusD indicated that it is predominantly monomeric (∼60kDa) with a small fraction appearing as dimers (data not shown). This was confirmed by native PAGE analysis showing SusD in monomeric (∼80%) and dimeric (∼20%) forms (data not shown).
The apo structure of SusD was determined using multiwavelength anomalous dispersion (MAD) phasing methods and seleno-methionine substituted protein crystals. The native apo-protein structure was determined to a resolution of 1.5Å (Rwork = 19.3%, Rfree = 21.1%). The crystals belonged to the P1 space group and each asymmetric unit contained two monomeric copies of SusD. Residues 42-59 and 70-551 were defined in the electron density of both copies of the protein. It was not surprising that the first 16 residues of the protein were disordered as they likely form a flexible tether extending from the lipid anchor.
SusD has an α-helical fold comprised of 22 α-helices, three sets of 2-stranded anti-parallel β-sheets, and multiple reverse turns (Figure 3A). Eight α-helices, α1 (44-53), α4 (117-139), α5 (146-170), α6 (189-206), α7 (220-238), α8 (243-255), α17 (438-452), and α18 (457-466), pack together as four tetratricopeptide repeat (TPR) units, that form a right-handed superhelix along one side of the structure. Two additional α-helices, α19 (478-489) and α20 (495-502) also adopt a helix-loop-helix motif and pack against α17 and α7, respectively, and connect the TPR domain to the rest of the structure. SusD was not predicted to have any TPR motifs since it does not have the amino acid signature W4-L7-G8-Y11-A20-F24-A27-P32 (D’Andrea and Regan, 2003). Structural analysis using DALI (Holm and Sander, 1995) revealed the closest structural homolog of SusD is the inner membrane protein PilF from Pseudomonas aeruginosa (Kim et al., 2006). PilF is an entirely α-helical protein composed of six and a half tandem TPR repeats. Despite differences in both the lengths of each helix and overall topology of the structures, the Cα of residues 30-172 of PilF (pdb 2FI7) overlaid with an RMSD of 1.6Å to the four TPR units of SusD (Figure 3B).
Figure 3. Atomic structure of SusD.
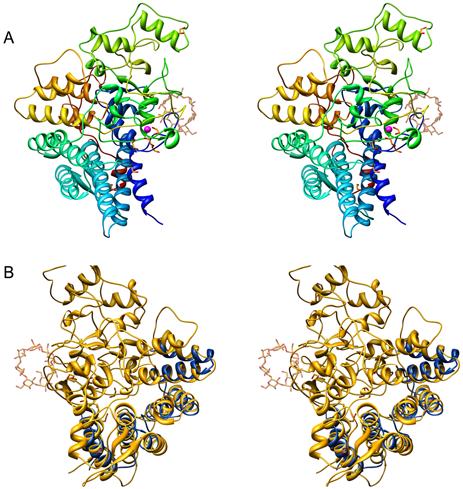
(A) Stereo ribbon diagram of apo-SusD, color-ramped from dark blue to red as the chain extends from the amino to the carboxyl end of the protein. An ordered Ca2+ ion is represented by a magenta-colored sphere while polyethylene glycol and ethylene glycol are shown as ball-and-stick figures. As a reference, a molecule of maltoheptaose from the structure of the SusD-maltoheptose complex is shown as a transparent ball-and-stick. (B) Stereo figure of SusD (yellow) highlighting residues 31-172 of PilF (blue) which contain the TPR units.
There are a number of small molecules associated with apo SusD. A single metal ion is bound in a shallow groove on the surface of SusD adjacent to α1 and the antiparallel β-sheet formed by β4 and β6. The ion was modeled as Ca2+ because it has an octahedral coordination sphere and an average metal-ligand distance of 2.4Å. The Ca2+ is coordinated by a carboxylic acid oxygen of D432, the backbone carbonyl oxygens of D430, and Q288, an ordered water, polyethylene glycol (PEG), and ethylene glycol. From the well-ordered electron density, the PEG molecule appears to be four carbon atoms in length and hydrogen bonded to the Ca2+, the side chain oxygens of D273 and D432, and the guanidyl nitrogen of R287. Several other molecules of ethylene glycol were observed in this structure, but are not found in identical positions in both copies of SusD.
C322 was oxidized to sulfenic acid in the structure of apo SusD. While unusual, sulfenic acid has been observed in a number of crystal structures for which it has no known function. C322 is surrounded by W96, W98 and W320 that create a hydrophobic patch on the surface of SusD and initiate starch binding (see below). Finally, a partially ordered molecule of morpholino-ethane sulfonic acid was observed to be bound within 4Å of W320 in chain A of the apo structure.
SusD complexed with maltoheptaose or β-cyclodextrin
The structure of SusD complexed with maltoheptaose was determined to a resolution of 2.2Å (Rwork = 18.3%, Rfree = 23.1%). C322 was not oxidized to sulfenic acid, as in the apo structure. Maltoheptaose was observed in different relaxed helical conformations in the two copies of SusD and this is likely due to the differences in crystal packing. Glc1, the non-reducing end of the maltoheptaose, was disordered in one copy of SusD (chain B) and therefore the interactions in chain A are described in detail.
Each glucose adopts the most favored 4C1 chair conformer. In both copies of SusD, Glc5 - Glc7 of maltoheptaose interact with SusD via identical ring stacking and hydrogen-bonding interactions (Figure 4A). The sugar rings of Glc5, Glc6, and Glc7 stack along the aromatic face of W98, W320, and Y296, respectively, creating an arced hydrophobic surface that complements the shape of the oligosaccharide. Maltoheptaose displays average ϕ (O5-C1-O4′-C4′), ψ C1-O4′-C4′-C5′) angles of 105.3° and -128.9°, respectively, that better reflect the natural curve and shape of double helical α-amylose (ϕ = 91.8°, ψ = -153.2°; ϕ = 85.7°, ψ = -145.3°; ϕ = 91.8°, ψ = -151.3°) rather than the single helical V-amylose (ϕ = 103.6°, ψ = 115.3°) (Gessler et al., 1999; Imberty et al., 1988). In addition to hydrophobic stacking interactions, maltoheptaose binds to SusD via both direct and solvent-mediated hydrogen bonding networks. The 2- and 3-hydroxyl groups of Glc5 are within hydrogen bonding distance of the carboxamide oxygen and nitrogen of N101, and the 6-hydroxyl of Glc5 hydrogen bonds with backbone carbonyl oxygen of W320. Both the 2- and 3-hydroxyl groups of Glc6 are hydrogen bonded to the guanidyl nitrogens of R81. The 2-hydroxyl oxygen of the reducing sugar Glc7 is within hydrogen bonding distance to backbone amines of G75 and G74. At the opposite end of maltoheptaose, Glc1 is positioned for hydrogen-bonding interactions through its 2- and 3-hydroxyl groups with the carboxylic acid side chain of D73.
Figure 4. SusD complexed with maltoheptaose.
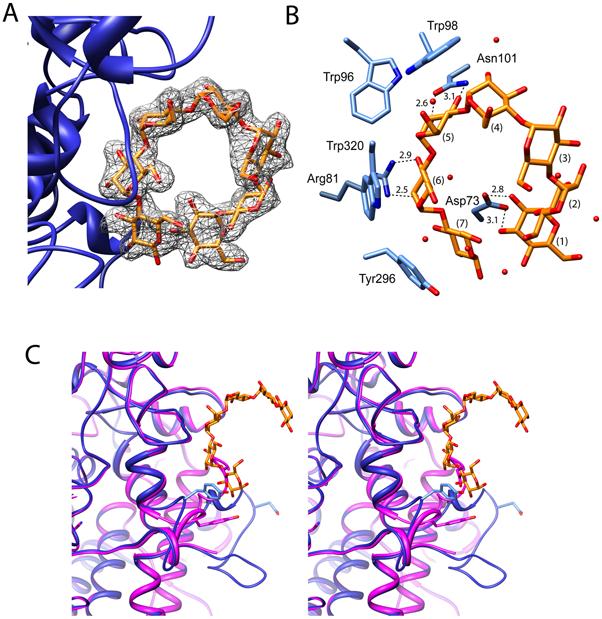
A) Shown here is the electron density of bound maltoheptaose from the corresponding omit map contoured at 3σ. B) In this panel, important hydrophobic-stacking and hydrogen-bonding interactions between the maltoheptaose and SusD are detailed. C) This panel shows a stereo diagram of SusD in the presence (blue) and absence (mauve) of bound maltoheptaose to highlight the conformational changes that occur upon oligosaccaride binding.
While the overall structures of the apo- and maltoheptaose-bound forms of SusD are nearly identical (RMS deviation of 0.44Å for 495 Cα atoms), two loops that form the sides of the binding cavity undergo a large conformational change upon polysaccharide binding (Figure 4B). For this comparison, chain B of both the apo (no MES) and bound forms of SusD were overlaid. Residues 70-77 display the greatest amount of conformational flexibility. This is at least partly due to the fact that they precede residues 60-69 that are disordered in the apo structure and only ordered in chain B of the maltoheptaose bound SusD. Upon oligosaccharide binding, residues 70-77 rotate out of the binding pocket, displacing the Cα of D70 by 18.5 Å. This rearrangement allows residues 73-76 to move proximal to the sugar, breaking the hydrogen bond between the guanidyl nitrogen of R81 and the backbone carbonyl oxygen of Q72 to form the hydrogen bond between R81 and Glc6. In turn, residues 292-297 move Y296 by 3.6Å for ring stacking interactions with Glc7.
Maltoheptaose adopts a distinctively circular shape upon binding to SusD that is akin to the expected conformation of starch. Therefore, we hypothesized that circular polysaccharides would better represent starch binding to SusD. SusD was subsequently crystallized in the presence of β-cyclodextrin and the structure (Rwork = 17.05%, Rfree = 20.6%) was determined to a resolution of 2.1Å. For the sake of clarity, the glucose moieties of β-cyclodextrin were labeled consistent with the bound maltoheptaose. We found that the overall structure and protein interactions with Glc4, Glc5, and Glc6 in β-cyclodextrin are essentially identical with those in maltoheptaose (Figure 5A). Due to the circular structure of β-cyclodextrin, Glc7 adopts a slightly different conformation compared to maltoheptaose. To compensate for this change, the phenolic side chain of Y296 shifts slightly to maintain hydrophobic stacking interactions with Glc7. In addition, both Glc7 and Glc1 are now beyond hydrogen bonding distance from residues 73-75, and make only solvent-mediated interactions with the protein. A concerted movement in residues 70-77 and 292-297 is observed in the SusD/β-cyclodextrin complex, as in the maltoheptaose complex.
Figure 5. SusD complexed with β-cyclodextrin and maltotriose.
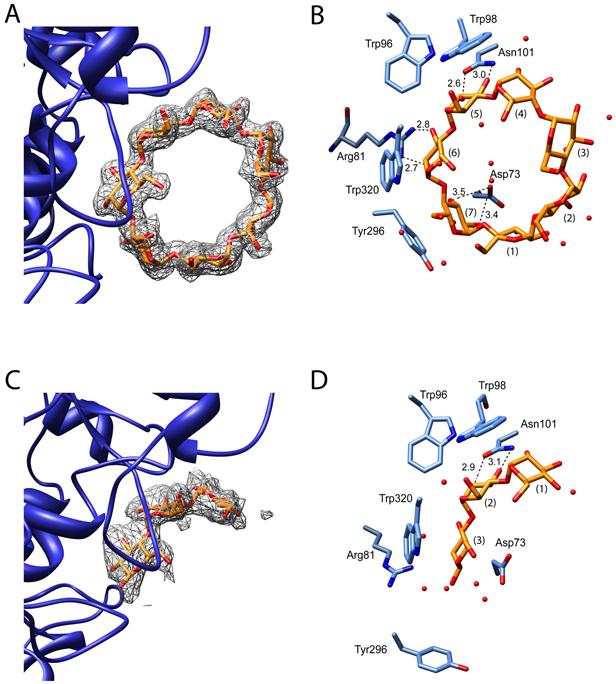
Panels (A) and (C) show the electron densities of β-cyclodextrin and maltotriose, respectively, from omit maps contoured at 3σ. Panels B and D highlight important hydrophobic-stacking and hydrogen-bonding interactions for bound β-cyclodextrin and maltotriose, respectively.
SusD complexed with α-cyclodextrin
The structure of SusD with α-cyclodextrin (G6) was also determined because we reasoned that the tighter radius of the six-glucose sugar might be a better mimic of helical starch. SusD was crystallized in the presence of α-cyclodextrin and the structure was solved to 2.1Å (Rwork=19.4%, Rfree=23.7%). Strikingly, α-cyclodextrin is wedged between two SusD molecules with each SusD binding three glucose residues (Figure 6). The Cα backbones of SusD with β-cyclodextrin and with α-cyclodextrin overlay with an RMSD of 0.36Å, with only minor shifting of the binding site residues. The two copies of SusD also interact with each other, burying ∼290Å2 on each subunit upon complex formation. It is likely that these protein-protein interactions enhance the binding of the sugar to SusD, compensating for lower affinity interactions between the protein and glucose residues with higher avidity. Including contributions from bound α-cyclodextrin, a total of ∼1100Å2 of protein is buried between the SusD subunits upon complex formation. Native PAGE analysis (see Supplemental figure S6) and size exclusion chromatography (data not shown) both suggest that SusD forms oligomers in solution. However, it is not clear whether these interactions are relevant to what is observed in this crystal structure. The banding pattern does not appear to change when α-cyclodextrin is added to the SusD sample. However, this is not entirely unexpected with the relatively weak binding affinity of α-cyclodextrin to SusD.”
Figure 6. SusD complexed with α-cyclodextrin.
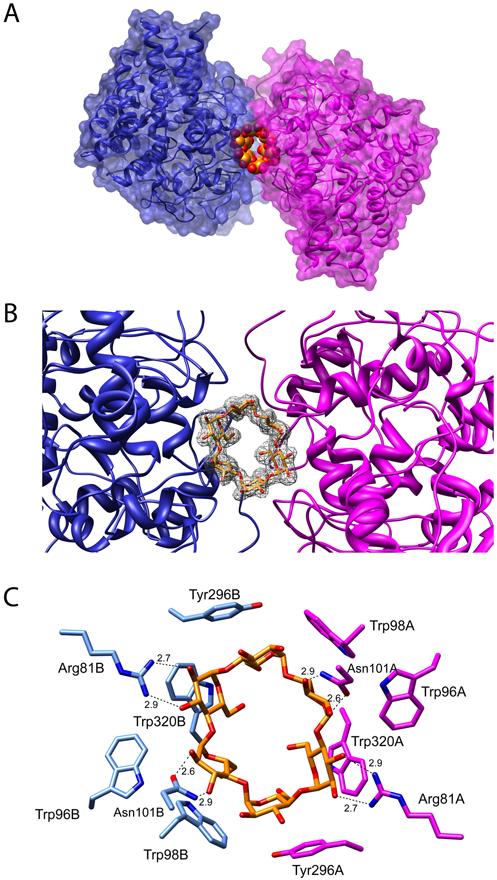
A) Ribbon and surface rendering of α-cyclodextrin complexed with two copies of SusD. B) Omit map contoured at 3σfor bound α-cyclodextrin. C) Important ring-stacking and hydrogen-bonding interactions (distances in Å) are shown for the α-cyclodextrin/SusD complex.
Each half of α-cyclodextrin binds to SusD in an equivalent manner with two-fold symmetry observed across the binding complex. In the trimeric complex of SusD with α-cyclodextrin, each glucose residue is involved in hydrophobic stacking interactions with W98, W320 or Y296 (Figure 6C). The loop defined by residues 58-72 is disordered in both subunits of this structure, as observed in the other structures except for subunit B of SusD complexed with maltoheptaose. If subunit B of SusD/maltoheptaose is superimposed on the SusD/α-cyclodextrin complex, residues 59-72 are adjacent to the starch-binding site and near the dimeric interface of SusD. Because of their proximity to the binding pocket and the inherent flexibility of this loop, it is possible that these residues either directly assist in binding linear oligosaccharides or aid in reorganizing starch-binding residues 73-77 when longer oligosaccharides bind.
SusD complexed with maltotriose
To determine which contacts define the minimal polysaccharide-binding site, apo SusD was crystallized in the presence of maltotriose and the structure was determined to a resolution of 2.3Å (Rwork = 18.2% , Rfree = 23.2%). Maltotriose binds in an identical orientation as Glc4, Glc5 and Glc6 of both maltoheptase and β-cyclodextrin (Figure 5B). The sugar rings of the bound polysaccharide only form stacking interactions with W320 and W98. There is no rearrangement of residues 70-77 or 292-297, and therefore Y296 does not participate in ligand binding. The only hydrogen bonds between maltotriose and SusD are between the 2- and 3-hydroxyls of Glc2 (Glc5 in β-cyclodextrin and maltoheptaose) and the side chain of N101, and the 6-hydroxyl of Glc2 with the backbone carbonyl oxygen of W320.
The complex of maltotriose and SusD likely represents the initial binding event between starch and SusD. The relatively rigid, hydrophobic surface created by W320 and W96 provides the initial binding surface for polysaccharide docking. As the three initial glucose residues stack along these tryptophan residues, the loops 70-77 and 290-299 open up to accommodate the larger oligosaccharides.
Isothermal titration calorimetry (ITC) suggests SusD recognizes helical starch
The SusD knockout mutants demonstrat that SusD plays a crucial role in growth of B. thetaioatomicron on starch molecules containing ≥ 6 glucose units. In addition, the crystallographic data indicate a preference for circular, or helical, oligosaccharides over linear forms. To further examine the substrate selectivity of SusD, ITC was performed with maltotriose, maltopentaose, maltoheptaose, α-cyclodextrin (G6), β-cyclodextrin (G7), and γ-cyclodextrin (G8). No interactions between SusD and maltotriose were detected via ITC and the energy associated with the binding of maltopentaose was too small to accurately determine an association constant. Of the remaining oligosaccharides tested, SusD clearly prefers the cyclic derivatives of starch over the linear forms (Figure 7). The fact that SusD binds α-cyclodextrin and γ-cyclodextrin with ∼20-fold greater affinity than maltoheptaose suggests that recognition is dominated by the helical shape of the polysaccharide rather than the stereochemistry of the composite glucose residues.
Figure 7. Isothermal titration calorimetry of the binding of various oligosaccharides to SusD.
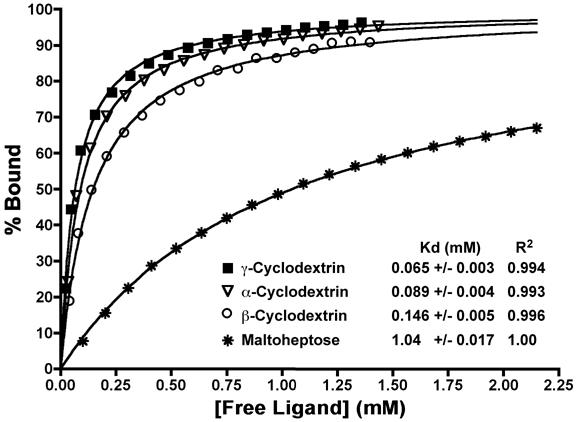
As described in the Methods, the heat of binding was converted to % of the maximum binding of the ligands to SusD and fitted to a single class of binding sites equation.
DISCUSSION
The crystal structures of SusD complexed with maltoheptaose, β-cyclodextrin, α-cyclodextrin, and maltotriose demonstrate that SusD induces marked curvature in linear oligosaccharides that closely mimics the conformations of bound cyclic polysaccharides. Further, isothermal titration calorimetry indicates that the cyclic compounds bind better than the linear forms. Together, these results suggest that SusD has a flexible starch-binding site designed to recognize the helical nature of starch rather than the stereochemistry of its composite glucose residues. This plasticity of recognition is consistent with its relatively weak binding affinity for the oligosaccharides: however, this is likely compensated by multivalent interactions between the cell and substrate.
Our studies of wild-type B. thetaioatomicron, an isogenic in-frame deletion strain (ΔsusD), and a ΔsusD strain containing a single complementing copy of susD expressed from its native promoter, demonstrate that SusD is required for the utilization of maltooligosaccharides comprised of ≥ 6 glucose units, including α1,6 branch points. SusD plays a non-essential role in growth on maltotetraose or maltopentaose, since ΔsusD is able to grow on these substrates, albeit at about half the growth rate of wild type. It is possible that these intermediate length oligosaccharides represent a transition point in B. thetaiotaomicron’s glycan utilizing capabilities, and that maltotriose and smaller sugars enter the cell via a different (non-Sus) pathway with maltotetraose and maltopentaose lying between the size limitations for these two systems. The decreased growth rate of the complemented ΔsusD::PsusB-susD strain on pullulan and amylopectin, compared to its wild-type parent, is likely due to decreased SusD expression. Quantitative RT-PCR techniques demonstrated that this strain produces ten-fold less of the susD mRNA transcript (Supplemental Figure 1) upon induction. Because the stoichiometry of the Sus complex in unknown, it is difficult to know whether the defect arises from less SusD incorporated per Sus complex or if fewer Sus complexes are present on the cell surface.
SusD has a novel α-helical fold unlike that observed for any other carbohydrate-binding modules (CBM), which are mainly comprised of β-sheets arranged in a β-barrel (Machovic and Janecek, 2006). Based on the arrangement of tetratricopeptide repeat units, the closest structural homolog to SusD is PilF, an inner membrane protein produced by P. aeruginosa,, that is involved in type IV pilus biogenesis (Kim et al., 2006). Although TPR proteins have a wide variety of functions, they tend to be involved in protein-protein interactions and assembling multiprotein complexes (D’Andrea and Regan, 2003). Previous studies suggested that PilF interacts with other members of the pilus assembly complex via the concave inner surface formed from the right-handed superhelical twist of the assembled TPR units (Kim et al., 2006). However, the concave face created by the TPR units of SusD, which comprise roughly half of the full-length PilF structure, cradles the remainder of the α-helical SusD structure in a way that the TPR domain of protein farnesyl transferase supports its catalytic domain (Kim et al., 2006). It is tempting to speculate that the TPR units of SusD form an assembly scaffold for the SusCDEFG complex since the entire Sus complex can be purified using an amylose column and SusC and SusD co-purify in the absence of SusE, SusF and SusG (Shipman et al., 2000).
As noted above, our results indicate that SusD recognizes the three-dimensional fold of the starch molecule rather than the stereochemistry of its composite glucose residues. While the crystal structures of SusD with maltoheptaose or β-cyclodextrin have nearly identical ligand-protein interactions, ITC demonstrates that cyclodextrins bind with ∼20-fold higher affinity than the linear maltoheptaose. If the binding site of SusD is designed to recognize the helical structure of α-amylose, then linear oligosaccharides would be expected to bind more weakly because of the imposition of the curved conformation. In contrast, the cyclic oligosaccharides are expected to bind better since they are already constrained to a curved conformation. The ends of bound maltoheptaose do not interact with SusD, consistent with the idea that SusD recognizes the internal helical structure of starch (‘endo’ versus ‘exo’ recognition) rather than a discrete oligosaccharide length.
The starch-binding site of SusD is dominated by tryptophan and tyrosine residues that create an arched hydrophobic surface, complementing the shape of helical oligosaccharides. In particular, W98 and W320 are juxtaposed at an angle of ∼130° to each other and their conformations are relatively unaffected by oligosaccharide binding. The sugar rings of maltotriose were found to stack along these tryptophans nearly the same as that observed with the bound maltoheptaose and β-cyclodextrin. Thus, the geometry of these two tryptophans likely aid in the initial docking of sugars based upon their overall shape. This dual tryptophan motif has also been observed in several glycoside hydrolases capable of degrading raw starch, such as glucoamylase, cyclodextrin glucosyltransferase, and barley α-amylase (Kadziola et al., 1998; Penninga et al., 1996; Robert et al., 2005; Sogaard et al., 1993; Sorimachi et al., 1997). The starch-binding site of SusD shares the most similarity with the surface starch granule-binding site of the barley α-amylase AMY1 (Robert et al., 2005). This starch granule-binding site has a Kd of ∼2mM for maltoheptaose, a value very similar to SusD and maltoheptaose. Like SusD, the starch granule site of AMY1 is dominated by two tryptophans, W278 and W279 that are situated at an angle of 159° to each other and provide a ring-stacking environment that holds adjacent glucose moieties in a helical conformation. Thus, as with SusD, the AMY1 starch-granule binding site acts as a “geometric filter”, capable of binding to helical starch polymers and thus selecting substrate by overall shape.
SusD displays some striking differences from the starch granules binding sites of glycoside hydrolyases such as AMY1. While W98 and W320 of SusD likely initiate oligosaccharide recognition, the lack of measurable binding of maltotriose by ITC demonstrates that this interaction alone is not sufficient for high affinity binding. A reorganization of the loops defined by residues 70-77, and 292-297 occurs when a longer oligosaccharide binds and brings R81 and Y296 into docking position. In addition, residues 73-75 are brought into the binding pocket so that the side chain of D73 is able to interact with Glc7 of maltoheptaose. The inherent plasticity in the SusD starch-binding pocket may allow SusD to initiate starch binding, facilitating recognition of both amylopectin and pullulan. The intermittent α1,6 linkages in these polysaccharides would likely distort the helical shape of an α1,4 glucose polymer (amylose) and therefore any protein capable of recognizing all three polysaccharides would require a plastic recognition site. As reflected in the B. thetaiotaomicron growth curves, SusD is essential for both amylopectin and pullulan utilization.
The structure of SusD with α-cyclodextrin revealed two SusD subunits binding to a single ring of sugar. While the in vivo relevance of this complex is unknown, both size exclusion chromatography and native PAGE analysis (see Supplemental Figure S6) suggest that ∼20% of heterologously expressed SusD exists as oligomers. The ability of SusD to bind oligosaccharides as either a monomer or dimer further highlights the inherent flexibility of SusD’s starch binding capacity; it is capable of monovalent interactions with flexible linear oligosaccharides as well as multivalent contacts with the more structurally static cyclic sugars.
Exactly how SusD confers the ability of the Sus complex to utilize starch is not fully understood. In previously published studies, both SusC and SusD were required, but not by themselves sufficient, for starch binding to B. thetaiotaomicron (Shipman et al., 2000). Interestingly, SusC and SusD expressed alone are more protease sensitive than when co-expressed, suggestive of SusC/SusD interactions (Shipman et al., 2000). SusC might directly participate in binding starch in conjunction with SusD or indirectly facilitate starch binding by organizing multiple copies of SusD.
Perhaps the more important question is whether SusD is involved solely in initial starch binding to the cell surface, or if it is also involved in the transit of smaller oligosaccharides to SusC for import. From the data presented here, it seems most likely that the main role of SusD is to bind starch to the surface of B. thetaiotaomicron. The plasticity of the SusD binding site increases the spectrum of recognized polysaccharides as made evident by the structural results and growth on α1,6-branched pullulan and amylopectin. This plasticity in the recognition site is the likely cause of the relatively low affinity of interaction between SusD and oligosaccharides. As exemplified in the SusD/α-cyclodextrin structure, it also seems likely that relatively weak intrinsic affinity is compensated by multivalent binding. Because of their long tethers (∼16 amino acids) to the bacterial surface, multiple SusD molecules could bind to starch either through multiple copies of SusD in a given outer membrane Sus complex or through the interactions of multiple Sus complexes with the large starch polymer. Multivalent binding can improve apparent affinity (avidity), as exemplified by studies with antibodies showing that the difference between the apparent affinity of Fab (monovalent) and intact IgG (bivalent) proteins for their ligand can be 3-4 orders of magnitude (Hornick and Karush, 1972). However, these interactions could be an artifact of crystal packing or might be indicative of interactions between SusD and the other proteins in this complex.
The binding of starch to the Sus complex could greatly enhance the catalytic efficiency of the Sus-associated α-amylase SusG by keeping the starch molecule and its products from diffusing from the complex until it is cleaved into oligosaccharides small enough for transport. The molecular weight cutoff observed in SusD binding, defined from ITC and the growth studies of the isogenic wild-type and ΔsusD strains suggests that SusD could direct these sugars into or near the SusC porin once they are smaller than 6 sugar units in length. Additional studies are necessary to better understand the interactions between SusD and the other proteins in this complex.
The Sus system is just one of 101 SusC/SusD paralog pairs found in the B. thetaiotaomicron genome. Whole genome transcriptional profiling of B. thetaiotaomicron in vitro and in the intestines of gnotobiotic mice (E. Martins and J. Gordon, unpublished results) suggests that each SusC/SusD paralog is specific for a particular glycan, with the SusD homologs binding the carbohydrate to the cell surface for initial degradation by glycoside hydrolyases and subsequent import into the periplasmic space (Bjursell et al., 2006; Sonnenburg et al., 2005). SusC/SusD paralogs are also present in soil and marine Bacteroidetes, such as Cytophaga hutchinsonii, Croceibacter atlanticus, Gramella forsetii, and Leeuwenhoekiella blandensis (Bauer et al., 2006; Cho and Giovannoni, 2003; Pinhassi et al., 2006; Xie et al., 2007). These bacteria are capable of degrading a variety of polysaccharides, including starch, cellulose, chitin, and algal material. Thus the Sus system is likely a prototype for a nutrient acquisition complex that is ubiquitous in gram-negative bacteria. Understanding the binding specificities, capacities, and transport mechanisms of Sus and Sus-like systems may not only aid the development of novel strategies for manipulating the nutrient utilization functions of the human gut microbiota, but also new ways of degrading polysaccharides for biofuels.
EXPERIMENTAL PROCEDURES
Heterologous protein expression
The susD gene (residues 26 - 551) was amplified by PCR from genomic DNA prepared from Bacteroides thetaiotaomicron ATCC 29148. The amplicon was cloned into pET28rTEV where the thrombin cleavage site of pET-28a (Novagen) has been modified to a tobacco etch virus (TEV) protease cleavage site. pET28rTEV-susD was transformed into Rosetta (DE3) pLysS (Novagen) for protein expression. Cells were grown in TB medium at 37°C with shaking (225 rpm) until they reached an O.D. of ∼0.4, at which time the temperature was adjusted to 22°C. Once the cultures reached an O.D. of ∼0.8, cells were treated with 0.2 mM ITPG to induce SusD expression, and allowed to grow 16 h at 22°C. Cells were subsequently harvested by centrifugation, frozen in liquid nitrogen, and stored at -80°C. Selenomethionine-substituted protein was produced via the methionine inhibitory pathway (Van Duyne et al., 1993), as previously described (Koropatkin et al., 2007).
Purification of native and selenomethionine-substituted SusD
Both native and selenomethionine-substituted SusD were purified using a 5 ml Hi-Trap metal affinity cartridge according to the manufacturer’s recommendations (GE Healthcare). The cell lysate was loaded onto the column in His Buffer (25 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole pH 8.0) and SusD was eluted using an imidazole (10 - 300mM) gradient. The His-tag was removed by incubation with rTEV (1:100 molar ratio relative to SusD) at room temperature for 16 h. The cleaved protein was then dialyzed against His Buffer and passed over the affinity column to remove the His-tagged rTEV and undigested SusD. Purified SusD was dialyzed against against 20 mM HEPES/100mM NaCl (pH 7.0) and concentrated to an O.D.280 of ∼22 for crystallization.
Crystallization and Data Collection
Initial crystallization conditions for apo-SusD were determined via hanging drop using the Hampton Screen (Hampton Research). Large single crystals for both SeMet SusD and the native SusD were grown at 4°C in batch plates by seeding small crystals into mother liquor that contained 4.5 mg/ml SusD, 14% poly(ethylene) glycol 8000, 50 mM NaCl, and 50mM morpholino-ethane sulfonic acid (MES). Crystals grew to dimensions of ∼0.3 × 0.3 × 0.5 mm in 1-3 weeks. All apo crystals were triclinic with unit cell dimensions of a=62.19 Å, b=68.06 Å, c=83.05 Å, α=111.1° β=93.2°, γ=109.2°. The solvent content of the crystals was approximately 51%, with two molecules in the asymmetric unit.
Crystals of SusD complexed with maltoheptaose, β-cyclodextrin or maltotriose were grown at room temperature by streak seeding the native apo crystals into batch plates containing 7.0 mg/ml SusD, 50mM sodium cacodylate (pH 6-6.5), 50-75 mM calcium acetate, 13-15% PEG 8000, and 70 mM maltotriose, 50mM maltoheptaose or 10mM β-cyclodextrin. Crystals of SusD with α-cyclodextrin were grown at room temperature by streak seeding into batch plates containing 20mg/ml SusD, 50 mM Tris-HCl (pH 8.5), 100 mM sodium acetate, 14-15% PEG 4000, and 2.5mM α-cyclodextrin.
In preparation for freezing, apo native and selenomethionine-substituted protein crystals were serially transferred to a final cryoprotectant solution containing 21% PEG 8000, 100mM NaCl, 50mM MES and 20% ethylene glycol. The crystals were flash-frozen with liquid nitrogen and diffraction maxima were collected on a 3 × 3 tiled “SBC3” CCD detector at the Structural Biology Center 19-BM beamline (Advanced Photon Source, Argonne National Laboratory, Argonne, IL). X-ray data were processed with HKL3000 and scaled with SCALEPACK (Otwinowski and Minor, 1997). Crystals of SusD complexed with maltotriose, maltoheptaose or β-cyclodextrin were serially transferred to final cryoprotectant solutions containing 17-18% PEG 8000, 60-75 mM sodium cacodylate (pH 6.5), 30 -75 mM calcium acetate, 20% ethylene glycol plus the concentration of oligosaccharide noted above. Crystals of SusD with α-cyclodextrin were serially transferred to a final cryoprotectant solution consisting of 75mM Tris-HCl pH 8.5, 100 mM sodium acetate, 20% PEG 4000, 17% ethylene glycol and 5mM α-cyclodextrin. Diffraction maxima were collected using a using an Oxford cryosystem and a Proteum R Smart 6000 CCD detector connected to a Bruker-Nonius FR591 rotating anode generator. Data were processed using the program SAINT from the Proteum2 software (Bruker AXS Inc., 2006) and merged using XPREP (SHELXTL 6.10, Bruker AXS Inc., 2000) or ProScale (Bruker AXS Inc., 2006). Data collection statistics are displayed in Supplemental Table 1.
X-ray structure determination
The structure of SusD was solved via MAD phasing from the x-ray data collected from the selenomethionine-substituted crystals. The program SOLVE (Terwilliger and Berendzen, 1999) was used to determine and refine the initial positions of the selenomethionines, and RESOLVE (Terwilliger, 2000) was then applied for solvent flattening and initial model building. Alternate cycles of manual model building in O (Jones et al., 1991) with maximum-likelihood refinement with CNS (Brunger et al., 1998) was used to build and refine the 2.0Å selenomethionine-substituted SusD (Rwork =20.4%, Rfree= 24.1%). This structure was then refined against with the native SusD x-ray data collected to 1.5Å. In all cases, the reflections used for the calculation of Rfree were selected, at random, immediately after scaling and were never used for refinement. The structures of SusD complexed with the various oligosaccharides were determined via molecular replacement using the program AMoRe (Navaza, 1994) from the CCP4 suite of programs (Collaborative computational project, 1994) with the apo SusD structure as a search model. Alternate cycles of manual model building in O and refinement using CNS were combined to complete the models. Initial coordinates and geometric constraints for the four oligosaccharides were downloaded from the HIC-Up server (xray.bmc.uu.se/hicup/). Relevant refinement statistics are presented in Supplementary Table 2.
Isothermal titration calorimetry
ITC measurements were carried out using a MicroCal VP-ITC titration calorimeter (MicroCal, Inc.). SusD was dialyzed overnight against a solution containing 20mM HEPES (pH 7.0), 100mM NaCl, and 0.5 mM CaCl2 prior to the experiment. Oligosaccharides solutions were prepared using dialysis buffer. Protein samples (0.34 to 0.48 mM) were placed in the reaction cell and the reference cell was filled with deionized water. After the temperature was equilibrated to 25°C, 19 successive 15μl injections of oligosaccharide solution (10 - 20mM) were made while stirring at 460 rpm and the resulting heat of reaction was measured. Baseline measurements were made using an identical injection regime in the absence of protein. While these measurements yielded classic saturation curves for binding, the relatively low binding constants made analysis with the MicroCal Origin software package problematic. Therefore, the raw data were analyzed by other means with emphasis on limiting the number of variables for the non-linear curve fitting analysis in the program Prism. Using a linear extrapolation from the first few injection data points, the Δheat value at zero ligand concentration was estimated and this value was used to adjust the baseline for the data set. Using the data at the higher ligand concentrations, the Δheat at equilibrium was estimated by fitting the data to an exponential equation (Y=Ymax(1-e(-kX)), where Y is the Δheat, X is the ligand concentration, and k is the exponential rate of association. Using this Ymax value and the known protein concentration, the ratio of Δheat per mole of bound ligand was determined and used to calculate the concentration of bound ligand. The concentration of free ligand was then determined by subtracting the bound ligand from total ligand concentration at each point of the curve. The resulting curves were then analyzed using the standard disassociation constant formula, Y=(Bmax-X)/(Kd+X), where Y was the concentration of bound ligand, X the concentration of free ligand, Bmax the maximum concentration of bound ligand, and Kd is the disassociation constant. As expected, Bmax always refined to the concentration of protein used for the reaction. The original ITC data are shown in Supplemental figures S2-S5.
Construction of B. thetaiotaomicron gene deletions
All bacterial strains, plasmids and primers are referenced in Supplemental Tables 3 and 4. Deletion of susD by allelic exchange was done using a novel counter-selectable system involving a modified B. thetaiotaomicron strain with a deletion of tdk (BT2275), a gene encoding thymidine kinase. A tdk deletion strain, resistant to the toxic nucleotide analog 5-fluoro-2′-deoxyuridine (FUdR) , was constructed by ligating 3.5kb fragments flanking the tdk gene into the suicide vector pKNOCK-bla-ermGb. The resulting construct was conjugated into B. thetaiotaomicron, a single-recombinant merodiploid selected on erythromycin, and plated on BHI-blood agar containing FUdR (200μg ml-1) to select for recombinants. Subsequent deletion of susD, using genomic fragments flanking the gene, was performed similarly as described for tdk, except that the Δtdk strain was used as a parent in conjunction with a different suicide vector (pExchange-tdk), that contains a cloned copy of tdk to facilitate counter-selection. Candidate susD deletions were screened by PCR and by DNA sequencing to identify isolates that had lost the gene.
Complementation of ΔsusD was accomplished by joining the susB promoter (197bp susA-susB intergenic region) to a promoterless copy of susD in the Bacteroides spp. integrative vector pNBU2-bla-ermGb (Supplemental Data) and inserting this construct into one of two tRNAser attachment sites targeted by NBU2 (Wang et al., 2000).
B. thetaiotaomicron growth on oligosaccharides
The minimal medium used for analysis of growth rates contained 100mM KH2PO4 (pH 7.2), 15mM NaCl, 8.5mM (NH4)2SO4, 4mM L-cysteine, 1.9μM hematin, 200μM L-histidine, 100nM MgCl2, 1.4nM FeSO4 · 7H2O, 50μM CaCl2, 1μg/ml vitamin K3, 5ng/ml vitamin B12, plus individual carbon sources (0.5% w/v, Sigma). Five ml cultures were inoculated with a 1:50 dilution of bacterial cells that were freshly grown in TYG medium and washed once in minimal medium lacking any added carbon source. Cultures were grown at 37°C using the NaHCO3/pyrogallol anaerobiosis method (Holdeman et al., 1977) and monitored by OD600 at 30 min intervals. Rates were calculated from ≥ 4 data points from each growth curve (OD600 range 0.2 - 0.5) that had been fit to an exponential function.
Note that suppressor mutants (5/20 cultures) were consistently observed when the ΔsusD strain was grown in medium containing maltohexaose and maltoheptaose. Analysis of four of these suppressor strains revealed that, despite lacking susD, they had regained the ability to grow on maltoheptaose and pullulan, albeit more slowly than wild-type (data not shown).
Supplementary Material
Table 1.
MAD Data Collection Statistics
| Peak | Inflection | Remote | |
|---|---|---|---|
| Wavelength (Å) | 0.97934 | 0.97951 | 0.97167 |
| Resolution (Å) | 50 - 1.76 (1.82 - 1.76) | 50 - 1.76 (1.82 - 1.76) | 50 - 1.75 (1.81 - 1.75) |
| Independent Reflections | 112867 (10400) | 112750 (10235) | 115258 (10735) |
| Completeness | 96.7 (89.0) | 96.3 (87.7) | 96.9 (90.8) |
| Redundancy | 3.8 (3.6) | 3.8 (3.6) | 3.9 (3.6) |
| Avg I/Avgσ(I) | 46.2 (15.9) | 46.7 (16.6) | 44.5 (12.4) |
| Rsym (%) | 8.5 (14.9) | 6.6 (12.7) | 6.3 (12.8) |
Table 2.
Data Collection and Refinement Statistics
| Structure | Apo | Maltotriose | Maltoheptaose | β-cyclodextrin | α-cyclodextrin |
|---|---|---|---|---|---|
| PDB Accession | 3CKC | 3CKB | 3CK9 | 3CK8 | 3CK7 |
| Resolution (Å) | 50 - 1.47 (1.52 - 1.47) | 59.5 - 2.3 (2.44 - 2.3) | 60.2 - 2.2 (2.35 - 2.2) | 60.2 - 2.1 (2.21 - 2.1) | 67.5 - 2.1 (2.21 - 2.1) |
| Unique reflections | 190539 (17881) | 47571 (5979) | 56299 (7038) | 66380 (8307) | 127524 (16027) |
| % Completeness | 95.7 (90.0) | 91.9 (73.2) | 94.6 (72.0) | 94.2 (82.7) | 93.4 (81.2) |
| Redundancy | 4.2 (3.7) | 3.5 (2.2) | 4.6 (2.3) | 4.3 (1.7) | 4.5 (1.5) |
| I/sig(I) | 54.0 (19.7) | 6.1 (2.5) | 7.3 (2.0) | 16.0 (6.6) | 6.9 (1.9) |
| Rsym (%) | 4.5 (8.4) | 7.85 (17.7) | 8.65 (19.8) | 3.3 (7.3) | 9.6 (21.3) |
| No. proteins atoms | 8048 | 8075 | 8196 | 8049 | 16002 |
| No. hetero- atoms | 1299 | 705 | 925 | 1186 | 1468 |
| Rwork(%) # reflections | 19.2 (22.8) 163414 (3298) | 18.0 (22.5) 42749 (517) | 18.5 (21.4) 50168 (629) | 16.9 (17.7) 58823 (996) | 19.4 (25.5) 114786 (1432) |
| Rfree(%) # reflections | 21.0 (24.5) 18043 (369) | 21.6 (26.2) 4791 (60) | 22.2 (26.6) 5665 (70) | 20.0 (22.0) 6638 (117) | 23.7 (26.8) 12682 (168) |
| Average B values (Å2) | |||||
| Protein Atoms | 16.09 | 7.5 | 16.2 | 15.1 | 12.0 |
| Ligand (sugar) | n/a | 18.3 | 28.2 | 29.3 | 10.5 |
| Heteroatoms | 25.73 | 11.5 | 28.9 | 24.1 | 15.8 |
| RMS deviations | |||||
| Bond length (Å) | 0.0047 | 0.0059 | 0.0064 | 0.0052 | 0.0057 |
| Bond Angles (degrees) | 1.21 | 1.16 | 1.22 | 1.18 | 1.19 |
Numbers in the parentheses represent the statistics of the highest resolution shell.
Acknowledgments
This work was supported in part by grants from the NIH (GM078800, and DK30292)
Footnotes
X-ray coordinates have been deposited in the Protein Data Bank (XXX) and will be released upon publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Anderson KL, Salyers AA. Biochemical evidence that starch breakdown by Bacteroides thetaiotaomicron involves outer membrane starch-binding sites and periplasmic starch-degrading enzymes. J Bacteriol. 1989a;171:3192–3198. doi: 10.1128/jb.171.6.3192-3198.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KL, Salyers AA. Genetic evidence that outer membrane binding of starch is required for starch utilization by Bacteroides thetaiotaomicron. J Bacteriol. 1989b;171:3199–3204. doi: 10.1128/jb.171.6.3199-3204.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- Bauer M, Kube M, Teeling H, Richter M, Lombardot T, Allers E, Wurdemann CA, Quast C, Kuhl H, Knaust F, et al. Whole genome analysis of the marine Bacteroidetes‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ Microbiol. 2006;8:2201–2213. doi: 10.1111/j.1462-2920.2006.01152.x. [DOI] [PubMed] [Google Scholar]
- Bjursell MK, Martens EC, Gordon JI. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem. 2006;281:36269–36279. doi: 10.1074/jbc.M606509200. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges N, Pannu NS, Read RJ, et al. Crystallography & NMR system (CNS): A new software system for macromolecular structure determination. Acta Cryst. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cho JC, Giovannoni SJ. Croceibacter atlanticus gen. nov., sp. nov., a novel marine bacterium in the family Flavobacteriaceae. Syst Appl Microbiol. 2003;26:76–83. doi: 10.1078/072320203322337344. [DOI] [PubMed] [Google Scholar]
- Cho KH, Salyers AA. Biochemical analysis of interactions between outer membrane proteins that contribute to starch utilization by Bacteroides thetaiotaomicron. J Bacteriol. 2001;183:7224–7230. doi: 10.1128/JB.183.24.7224-7230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative computational project, n The CCP4 suite: Programs for protein crystallography. Acta Crystallographica. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- D’Andrea LD, Regan L. TPR proteins: the versatile helix. TRENDS in Biochemical Sciences. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- D’Elia JN, Salyers AA. Contribution of a neopullulanase, a pullulanase, and an alpha-glucosidase to growth of Bacteroides thetaiotaomicron on starch. J Bacteriol. 1996a;178:7173–7179. doi: 10.1128/jb.178.24.7173-7179.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Elia JN, Salyers AA. Effect of regulatory protein levels on utilization of starch by Bacteroides thetaiotaomicron. J Bacteriol. 1996b;178:7180–7186. doi: 10.1128/jb.178.24.7180-7186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessler K, Uson I, Takaha T, Krauss N, Smith SM, Okada S, Sheldrick GM, Saenger W. V-Amylose at atomic resolution: X-ray structure of a cycloamylose with 26 glucose residues (cyclomaltohexaicosaose) Proc Natl Acad Sci U S A. 1999;96:4246–4251. doi: 10.1073/pnas.96.8.4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdeman LV, Cato ED, Moore WEC. Anaerobe Laboratory Manual. Virginia Polytechnic Institute and State University Anaerobe Laboratory; Blacksburg, Va: 1977. [Google Scholar]
- Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Hornick CL, Karush F. Antibody affinity-III. The role of multivalence. Immunochem. 1972;9:325–340. doi: 10.1016/0019-2791(72)90096-1. [DOI] [PubMed] [Google Scholar]
- Imberty A, Chanzy H, Perez S, Buleon A, Tran V. The double-helical nature of the crystalline part of A-starch. J Mol Biol. 1988;201:365–378. doi: 10.1016/0022-2836(88)90144-1. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kadziola A, Sogaard M, Svensson B, Haser R. Molecular structure of a barley alpha-amylase-inhibitor complex: implications for starch binding and catalysis. J Mol Biol. 1998;278:205–217. doi: 10.1006/jmbi.1998.1683. [DOI] [PubMed] [Google Scholar]
- Kim K, Oh J, Han D, Kim EE, Lee B, Kim Y. Crystal structure of PilF: functional implication in the type 4 pilus biogenesis in Pseudomonas aeruginosa. Biochem Biophys Res Commun. 2006;340:1028–1038. doi: 10.1016/j.bbrc.2005.12.108. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Koppenaal DW, Pakrasi HB, Smith TJ. The structure of a cyanobacterial bicarbonate transport protein, CmpA. J Biol Chem. 2007;282:2606–2614. doi: 10.1074/jbc.M610222200. [DOI] [PubMed] [Google Scholar]
- Machovic M, Janecek S. Starch-binding domains in the post-genome era. Cell Mol Life Sci. 2006;63:2710–2724. doi: 10.1007/s00018-006-6246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaza J. AMoRe: an automated package for molecular replacement. Acta Cryst. 1994;A50:157–163. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CWJ, Sweet RMRM, editors. Methods in Enzymology. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Penninga D, van der Veen BA, Knegtel RM, van Hijum SA, Rozeboom HJ, Kalk KH, Dijkstra BW, Dijkhuizen L. The raw starch binding domain of cyclodextrin glycosyltransferase from Bacillus circulans strain 251. J Biol Chem. 1996;271:32777–32784. doi: 10.1074/jbc.271.51.32777. [DOI] [PubMed] [Google Scholar]
- Pinhassi J, Bowman JP, Nedashkovskaya OI, Lekunberri I, Gomez-Consarnau L, Pedros-Alio C. Leeuwenhoekiella blandensis sp. nov., a genome-sequenced marine member of the family Flavobacteriaceae. Int J Syst Evol Microbiol. 2006;56:1489–1493. doi: 10.1099/ijs.0.64232-0. [DOI] [PubMed] [Google Scholar]
- Reeves AR, D’Elia JN, Frias J, Salyers AA. A Bacteroides thetaiotaomicron outer membrane protein that is essential for utilization of maltooligosaccharides and starch. J Bacteriol. 1996;178:823–830. doi: 10.1128/jb.178.3.823-830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, Haser R, Mori H, Svensson B, Aghajari N. Oligosaccharide binding to barley alpha-amylase 1. J Biol Chem. 2005;280:32968–32978. doi: 10.1074/jbc.M505515200. [DOI] [PubMed] [Google Scholar]
- Shipman JA, Berleman JE, Salyers AA. Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J Bacteriol. 2000;182:5365–5372. doi: 10.1128/jb.182.19.5365-5372.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman JA, Cho KH, Siegel HA, Salyers AA. Physiological characterization of SusG, an outer membrane protein essential for starch utilization by Bacteroides thetaiotaomicron. J Bacteriol. 1999;181:7206–7211. doi: 10.1128/jb.181.23.7206-7211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogaard M, Kadziola A, Haser R, Svensson B. Site-directed mutagenesis of histidine 93, aspartic acid 180, glutamic acid 205, histidine 290, and aspartic acid 291 at the active site and tryptophan 279 at the raw starch binding site in barley alpha-amylase 1. J Biol Chem. 1993;268:22480–22484. [PubMed] [Google Scholar]
- Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, Gordon JI. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–1959. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- Sorimachi K, Le Gal-Coeffet MF, Williamson G, Archer DB, Williamson MP. Solution structure of the granular starch binding domain of Aspergillus niger glucoamylase bound to beta-cyclodextrin. Structure. 1997;5:647–661. doi: 10.1016/s0969-2126(97)00220-7. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;D56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 4):849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Wang J, Shoemaker NB, Wang GR, Salyers AA. Characterization of a Bacteroides mobilizable transposon, NBU2, which carries a functional lincomycin resistance gene. J Bacteriol. 2000;182:3559–3571. doi: 10.1128/jb.182.12.3559-3571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bruce DC, Challacombe JF, Chertkov O, Detter JC, Gilna P, Han CS, Lucas S, Misra M, Myers GL, et al. Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl Environ Microbiol. 2007;73:3536–3546. doi: 10.1128/AEM.00225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- Xu J, Mahowald MA, Ley RE, Lozupone CA, Hamady M, Martens EC, Henrissat B, Coutinho PM, Minx P, Latreille P, et al. Evolution of Symbiotic Bacteria in the Distal Human Intestine. PLoS Biol. 2007;5:e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.