

Summary
Melanoma consists 4–5 % of all skin cancers, but it contributes to 71–80 % of skin cancers deaths. UV light affects cell and tissue homeostasis due to its damaging effects on DNA integrity and modification of expression of a plethora of genes. DNA repair systems protect cells from UV-induced lesions. Several animal models of melanoma have been developed (Xiphophorus, Opossum Monodelphis domestica, mouse models and human skin engrafts into other animals). This review discusses possible links between UV and genes significantly related to melanoma but does not discuss melanoma genetics. These include oncogenes, tumor suppressor genes, genes related to melanocyte-keratinocyte and melanocyte-matrix interaction, growth factors and their receptors, CRH, ACTH, α-MSH, glucocorticoids, ID1, NF-kappaB and vitamin D3.
Keywords: Ultraviolet radiation, melanocytic nevus, melanoma, animal models of melanoma, melanocyte transformation, signal transduction, receptors
INTRODUCTION
Characterization of UV radiation
Ultraviolet radiation (UVR) is divided into ultraviolet C (UVC; 200 – 280 nm), ultraviolet B (UVB; 280 – 320 nm) and ultraviolet A (UVA; 320 – 400 nm) [1]. Although UVR represents only a fraction of the solar radiation, it is responsible for the majority of its carcinogenic activity. UV photons can affect the DNA integrity, cell and tissue homeostasis, and induce mutations or affect expression of a plethora of genes including oncogenes and tumor suppressor genes. UVR depending on the dose and wavelength can also modify expression and activity of growth factors/cytokines and their receptors. It has also local and systemic immunosuppressive effects [2–4].
The UVC spectrum is highly mutagenic but does not reach the earth’s surface because it is absorbed by the stratospheric ozone layer. “Although it is generated by artificial light sources such as arc welding lamps, germicidal lamps or lasers causing irritation and damage to skin and eyes [5, 6], little is known about UVC-induced damage in humans since exposure of people to this range of UV radiation is limited. Only a small group of people are exposed to this UV spectrum range. Furthermore, UVC penetration of deeper layers of the skin is limited. On the other hand UVA and UVB wavelengths represent 95% and 5% of the UV spectrum reaching the earth’s surface, respectively, with UVA penetrating the atmospheric and stratospheric ozone, while UVB radiation is predominantly absorbed by these layers. It must also be noted that apart from natural solar light there are many artificial sources emitting UVA and UVB. UVB and UVA have also been used in many medical procedures (standard and experimental) [7–12] with comparatively high doses used; even higher than 1 minimal erythema dose (MED) (approximately 35 mJ/cm2 UVB and 60 J/cm2 UVA estimated for skin I type (skin I type is extremely pale that never tans and always burns)) [6]. The epidemiological data suggest that these therapies may lead to an increased risk of skin cancers including a risk for development of melanoma. Specifically, psoralen phototherapy with UVA irradiation (PUVA) results in increased risk of skin cancers, higher than UVB or UVA therapies alone [13].
Although UVB is about 20-fold less abundant than UVA, its energy is more efficiently absorbed by cellular molecules and is able to induce damages within cells and tissues at significantly lower doses than UVA [1, 6]. On the cellular level UVB can cause damages of DNA in direct action, since nucleic acids are among the primary chromophores for this electromagnetic energy (maximum of DNA and RNA absorption is about 260 nm). The net results of the interaction of UVB with DNA are pyrimidine (6–4) pyrimidone photoproducts (6–4PP) and cyclobutane pyrimidine dimers (CPD) between adjacent pyrimidine sites (at TT, TC CT and CC sequences, with predominance of photoproducts at T containing sites), comprising 25% and 75% of adducts, respectively. Both types of photoproducts are bulky adducts affecting spatial structure of DNA [14]. CPDs are more significant in UV-induced carcinogenesis than 6–4 PPs, since in human cells 6–4 PPs adducts can be repaired with high efficiency, while CPDs are removed slowly by transcription coupled repair (TCR) and hardly ever by genome global repair [15]. Some experimental data showed that CPDs are still present in cells recovering from UV-induced cell cycle arrest and are responsible for at least 80% of mutations induced by UVB in mammalian cells [15]. However, 6–4 PPs after absorption of UVA range can be converted into Dewar isomers (poorly repaired), thus contributing to UV-induced mutations and carcinogenesis [14]. Nevertheless others have shown an efficient repair of Dewar isomers and their precursors (6–4 PP), questioning their role in UVR induced mutagenesis [15]. If CPDs and 6–4 PPs are not repaired, they can lead to mutations in many genes. The most abundant mutations after UVB are C to T and CC to TT transitions at bipyrimidine sites or pryrimidine runs, called “UVB fingerprint mutations” [6, 16]. The tandem CC to TT transitions are the most specific mutations caused by UVR and are rarely found in internal organs [17]. UVB induces also photochemical reactions resulting in cross-linked of DNA and proteins, pyrimidine-purine adducts of unknown role [6]. UVB can also generate reactive oxygen species (ROS), but mechanism of their generation is different than after UVA irradiation [18].
UVA is generally considered to be less carcinogenic than UVB. Because of optical properties of skin and deeper penetration of long-wave radiation, these photons have nevertheless a profound impact on photoaging. Recent studies identify UVA as an important factor involved in photocarcinogenesis acting through different mechanisms than UVB [16]. Agar et al. [16] showed in human samples of squamous cell carcinoma and solar keratosis that UVA-fingerprint mutations were located mostly in basal layer of epidermis and UVB-fingerprint mutations were found in suprabasal keratinocytes, in the stratum granulosum. These findings are consistent with the depth of penetration of UVA and UVB into the skin. High-energy UVB photons are strongly attenuated by stratum corneum and in deeper layers of epidermis UV is absorbed by melanin, DNA, aminoacids, keratin, urocanic acid and other chromophores. In skin of Caucasian people, 20 – 30 % of photons at the range of 290 –320 nm reach the keratinocytes, about 10 % penetrate the epidermis reaching the upper layers of the dermis. Only 1 % of UV radiation below 300 nm can enter dermis. On the contrary most of UVA photons get into dermis, and in individuals with type I skin about 50% of these photons can penetrate into the dermis [1, 6]. Low energy UVA radiation is weakly absorbed by DNA, but can be absorbed by other cellular chromophores, and induces mainly oxidative changes in the cells. Photons at UVA range, exciting chromophores, contribute to generating of reactive oxygen species (ROS), leading indirectly to DNA damage. The main target of ROS within DNA is guanine, and guanine lesions as 8-oxo-7,8-dihydroguanine (8-oxoGua) have been considered as the most prevalent UVA-induced damage. Drobetsky et al. [19] revealed an unusual T to G transversion mutation caused by the presence of 8-oxoGua, a so-called “UVA fingerprint mutation “UV signature”. Others have also shown the prevalence of these mutations after UVA irradiation. Besaratinia et al. [20] confirmed the role of ultraviolet in the UVA range in generating G to T transversion and argued against UVA-induced CPDs. Other common DNA changes induced by UVA photons are DNA strand breaks [6]. But recent studies revealed that in human and rodent cells oxidative DNA damages are not as frequent as bipyrimidine photoproducts, and CPDs are predominant type of DNA damage resulting from UVA. In contrast to UVB photons, these photoproducts were identified at TT sites, and in UVA-irradiated cells dimers are formed by photosensitized triplet energy transfer from excited chromophores (not yet identified) to thymine [14]. Taken together these data showed that formation of UV-induced damages of DNA is complicated, and cannot be considered only as UVB-induced dimers or UVA-induced oxidative damages.
UV and DNA repair systems
There are several mechanisms and barriers that protect the body against UVR. The stratum corneum, the outermost layer of the skin, composed of dead and peeling cells, and melanin pigment attenuate penetration of UV into the skin. Antioxidative enzymes detoxify and metabolize reactive oxygen species. UVR also induces melanin synthesis and relocalization within the epidermis, which plays an important role in attenuation of UVR induced damage. DNA repair systems protect cells from UV-induced lesions. The most important DNA damage repair system involved in excision of damages caused by UVR (such as CPDs and 6–4 PP) is nucleotide excision repair (NER). This system can act in two subpathways: faster transcription-coupled repair (TCR), operates on transcribed strand of active genes and slower global genome repair (GG-NER), removes lesions within the entire genome. Oxidative damages of DNA induced by UV radiation can be repaired by the base excision repair (BER) system. BER partially overlaps with the TCR system [21]. The pivotal role of NER in preventing cells from damages induced by UV radiation can be exemplified by patients with the autosomal recessive condition xeroderma pigmentosum (XP); these patients have more than a 1000-fold increased incidents of skin cancers in comparison to general population [6]. Patients affected by this syndrome develop skin cancers mostly in sun-exposed areas, and molecular analysis of p53 mutation showed presence of UV hallmark mutations. Tumors on sun-protected areas of the skin and in internal organs occur with much lower frequency. Moreover, p53 mutations in these tumors are different (A to T and G to T transversions rather than C to T or CC to TT p53 mutations in skin) than in skin cancers, implying that the mechanisms leading to their formation are also different [6, 22].
UV and immunosuppresion
As was mentioned above, ultraviolet radiation affects function of immune system. Interestingly, UV-induced immunosuppression is not limited only to irradiated area and systemic suppression of immune system is observed [3]. The main cells affected by UV radiation are Langerhans cells and T lymphocytes. Ultraviolet radiation affects number, function and morphology of Langerhans cells making them less capable of antigen presenting. However the molecular mechanism of immunosuppression is not well known yet and is still to be clarified. Three molecular targets are proposed to be involved in initiation of suppression of immune system: DNA damage, plasma membrane damage and trans to cis isomerisation of urocanic acid. The latter affects morphology and function of Langerhans cells. Moreover, expression of immunosuppressory cytokines (such as PGE, IL-10, TGFα) or peptides (α-MSH, ACTH or β-endorphin) increases in response to exposure to UV radiation. These alternations result also in generation of regulatory antigen-specific T cells (suppressor T cells), and modulation of systemic immune system function [1, 3, 6].
EPIDEMIOLOGY OF MELANOMA
Melanoma is a neoplasm of melanocytic origin having the most rapid increase in incidence in many countries comparable to other tumors [23]. Melanoma (cutaneous and ocular) predominantly affects white populations, being 10 times less frequent in black individuals, which indicates a protective role of melanin pigment [24]. The major role of melanin is its protection against the harmful actions of solar radiation. It attenuates the penetration of ultraviolet radiation deep into the skin. Melanin can also scavenge the oxygen radical species generated by ultraviolet radiation. However, melanogenesis can induce genotoxic effects via toxic and mutagenic intermediates and melanin precursors can inhibit the lymphocytes proliferation and differentiation by inducing immunosuppresion. These photosentization properties are attributed to pheomelanin. Melanin can also increase uptake of carcinogenic environmental agents, promoting the melanoma induction. Moreover, the capacity of melanin to scavenge free radical and reactive oxygen species is an undesirable property during radio-, photo-, and chemotherapy [24, 25].
As far as age incidence is concerned, white women develop cutaneous melanoma rapidly from 15 years of age. In case of white men incidence increases slowly from 15 to 45 years of age and from 45 years it increases rapidly. In black people, an increase of melanoma rate is observed after 55 years of age [26, 27]. Every year a 3–6 % rise in new cases is observed within Caucasian population. In 1935 melanoma was diagnosed in 1 out of 1500 persons, and it is predicted to occur in 1 in 50 people by year 2010. Within other populations (Blacks, Asians, Hispanics), the incidence has been stable for over 30 years. Melanoma consists 4–5 % of all skin cancers, but it contributes to 71–80% of skin cancers deaths [26, 28].
Epidemiological studies clearly demonstrate direct role of UVR in induction of squamous cells carcinomas (SCCs) and basal cell carcinomas (BCCs) [23, 29, 30]. The relationship between sun exposure and melanoma is less evident. For example, melanomas can develop on sun-protected areas of skin and in internal organs (for example in esophagus, colon, cervix), and they can be correlated with some genetic factors indicating that their induction and progression sometimes is not related with UV [30]. Nevertheless, the majority of authors accept that main risk factors for melanoma relate to environmental exposure and genetics with epidemiologic studies linking sun exposure and sun sensitivity to melanoma development [31]. The latter is exemplified by lentigo maligna melanoma (LMM), which occurs on sites continuously exposed to sun such as the head and neck of individuals with chronically sun damaged skin, increases with advancing age and is related to cumulative sun exposure [32]. On a density basis, melanomas are significantly denser on sun-damaged skin than those on intermittently exposed skin [33]. In addition, epidemiological studies of incidence in white populations have revealed an inverse correlation with latitude and positive correlation with UV index, exemplified by prevalence of melanoma in Australia and New Zealand [28]. Migration data also provides insight into the role of UV radiation and induction of melanoma. Exposure to UV at an early age seems to have very strong effect on melanoma induction. People who spent their childhood and adolescence in countries with a high UV index, close to the equator and close to the coast have a higher risk of melanoma than individuals who have lived at higher latitude and far from coast [34]. Correlation of UV radiation, latitude and melanoma in non-white populations is disputable. While some research suggested a positive correlation between UV index and negative correlation with latitude and development of melanoma in Hispanic and black population [27], other showed no such correlation [28].
There are three distinct patterns of exposure to ultraviolet light and individual susceptibility that are related to melanoma risk: 1) intermittent sun exposure and propensity to develop multiple melanocytic nevi; 2) multiple sun burns during childhood and sun sensitivity and freckling; and 3) chronic sun exposed skin with solar lentigines (Figure 1). At the molecular level, distinct genetic alterations have been identified in melanomas at different sites and with different levels of sun exposure supporting these epidemiologic studies [32, 35, 36](See figure 1).
FIGURE 1.
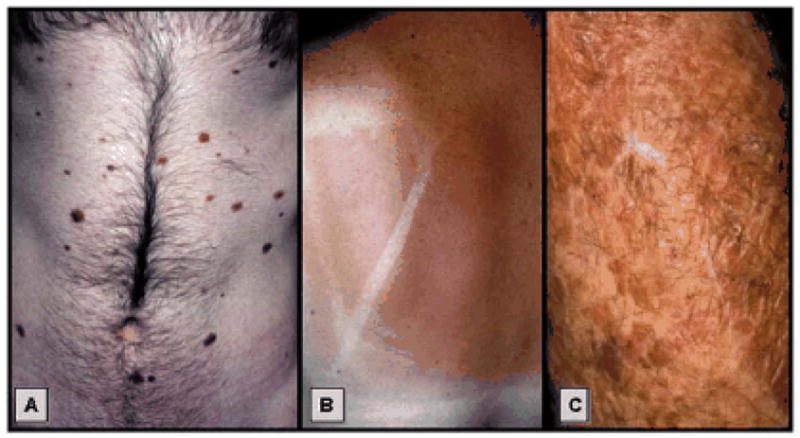
Three major setting of UV induced melanoma: A) intermittent sun exposure in nevus susceptible patient; B) multiple sun burns during childhood-aldolescence; and C) Solar damaged skin with multiple lentigines.
First, skin type, sun sensitivity, and type and amount of sun exposure all influence nevi development [29, 31]. The number of nevi, either acquired or atypical, constitutes the best predictor of individual risk of melanoma [37], and the presence of large (i.e. >5 mm) or atypical nevi (i.e. large nevi with non-uniform color and irregular borders), independent of the number of smaller nevi, is associated with an elevated risk of melanoma [36, 37]. The number and size of melanocytic nevi is related to frequency and amount of intermittent sun exposure [29, 38, 39]. Second, multiple, particularly blistering, sun burns during childhood are associated with increased number of nevi [40] and risk for melanoma [31, 39]. Three, chronically sun damaged skin in Caucasians is associated with skin wrinkling and development of numerous solar lentigos [41, 42]. This mottled skin in the elderly is the result of extensive freckling, guttate hypomelanosis, solar lentigines, and seborrhoeic keratoses. Solar lentigines are known risk factor for melanoma [39] and are the site of acquired atypical lentiginous melanocytic hyperplasias [43]. However, there exists contradictory data assessing chronic, cumulative lifetime sun exposure to melanoma risk with epidemiologic studies reporting a protective effect [31] or a negative effect [29, 44] or no correlation with risk of melanoma [34]. A protective effect of chronic exposure to UV photons was explained by the development of skin protective mechanisms during chronic and moderate-intensity sun exposure. In addition, use of sunscreens could skew the analyses, since early on they protected mostly against UVB spectrum but not against UVA, leading to increased exposure to these photons. Such unnatural overexposure to solar radiation could also be secondary to an inhibition of erythema response by some sunscreen use.
There is also a positive correlation between exposure to artificial sources of UV radiation and risk of melanoma development. For example, psoralen phototherapy with UVA irradiation (PUVA) increases the risk of melanoma development, particularly in patients with high doses and long lasting phototherapy [13].
MELANOMA EPIDEMIOLOGY MEETS HISTOLOGY
Formation of solar lentigines, (hyperpigmented, club shaped rete ridges overlying abundant solar elastotic material) are a consequence of chronic sun exposure [42]. Melanocytes are increased in density in sun damaged skin as well as within solar lentigines (See figure 2). In sun damaged skin, a high degree of variability in melanocyte density is evident, including contiguous melanocytes, atypical melanocytes, and follicular melanocytes suggestive of melanoma in situ, lentigo maligna type [45, 46]. Melanocytes in long-standing sun-exposed skin have an increased density and a confluence that is often moderate (3–6 adjacent melanocytes), but they do not exhibit pagetoid spread or nesting, histologic features diagnostic of melanoma in situ, lentigo maligna type [46]. Away from the head and neck, the diagnosis of atypical lentiginous melanocytic nevi in chronic sun-damaged skin is problematic: atypical lentiginous junctional naevi may be seen as isolated lesions and may merge with lesions that are indistinguishable from lentigo maligna (melanoma in situ) [43]. The predominant site distribution of such lesions on the trunk and limbs and the presence of a nested nevus-like pattern on biopsy differ from classical lentigo maligna, which develops mainly on the head and neck. Final diagnosis of lentigo maligna versus atypical lentiginous melanocytic nevus is often reached based on the location of lesion. Based on case studies combining dermoscopy with clinical and pathological features, atypical lentiginous junctional naevi of the elderly may evolve to lentigo maligna and in some cases to small cell (nevoid) melanomas [43].
FIGURE 2. Melanocytic hyperplasia occurring in sun damaged skin.
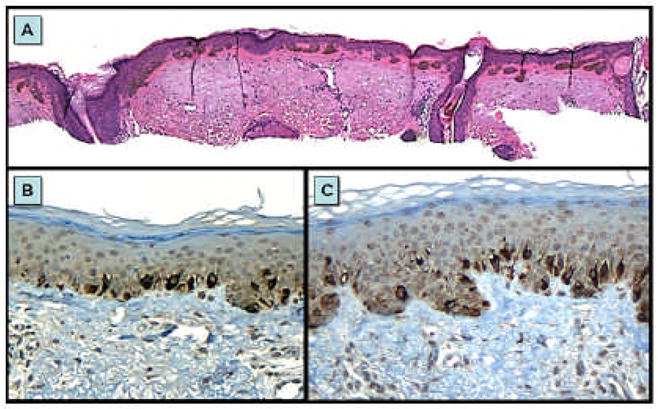
Formation of solar lentigines, (hyperpigmented, club shaped rete ridges overlying abundant solar elastotic material) after chronic sun exposure (A). Melanocytes are increased in density in sun-damaged skin (B) as well as within solar lentigines (C). Mart-1 labeling (A and C) demonstrates that both regions have an increase in melanocytes density as well as enlargement of many of the melanocytes.
Lentigo maligna (LM) is a pigmented lesion similar in appearance to solar lentigines, which occurs on sun-exposed skin, mostly the head and neck of the elderly [47]. The lesion increases in size and at some point, often many years after its onset, may become invasive melanoma (lentigo maligna melanoma (LMM)). For this reason, LM is considered a variant of melanoma in situ, characterized histologically by a proliferation of atypical melanocytes disposed as solitary cells and nests within the basal layer of the epidermis and/or adnexal epithelium. Figure 3 shows a typical lentigo maligna melanoma (LMM- invasive melanoma) arising in long standing patch of lentigo maligna (melanoma in situ) occurring in sun damaged skin of an elderly female. The early vertical growth phase of LMM is characterized by small nest of melanoma within the papillary dermis found in association with confluent nests of LM found along the dermal-epidermal junction. These patterns are highlighted by antibodies to CD117/c-kit demonstrating strong and intense expression of the both in situ and invasive components. In most melanomas, c-kit expression is restricted to epidermal melanocytes (normal, nevoid and melanoma) [48], whereas the invasive component is negative. In LMM, c-kit expression by the invasive component is consequence of activation mutations or amplification of c-kit; a molecularly distinct pathway separating this subset of melanoma from melanomas associated with intermittent sun exposure and down stream BRAF in the MAP Kinase pathway [36].
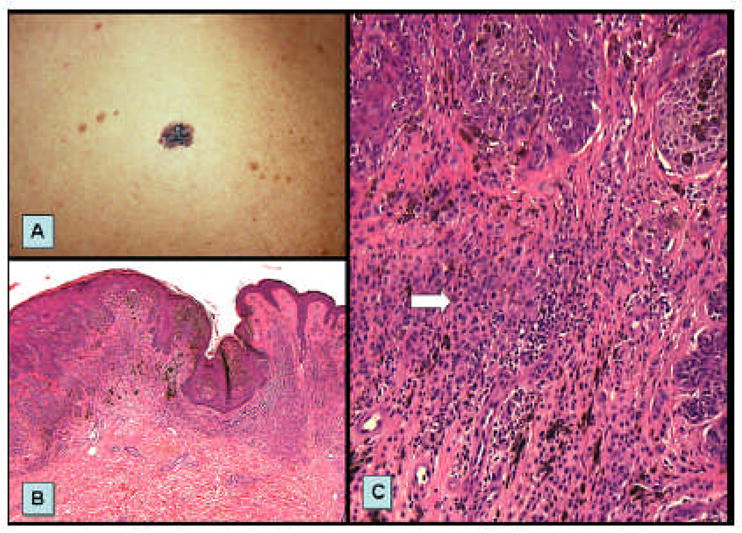
FIGURE 3. Lentigo maligna melanoma arising on sun damaged skin.
A–B. A typical lentigo maligna melanoma (LMM- invasive melanoma, dark central region) arising in long standing patch of lentigo maligna (melanoma in situ, large irregular brown patch) occurring in sun damaged skin of an elderly female. C. Early vertical growth phase LMM characterized by small nest of melanoma within the papillary dermis found in association with confluent nests of LM found along the dermal-epidermal junction. D. Antibodies to CD117/c-kit demonstrate strong and intense expression of the both in situ and invasive components.
It is a matter of debate and controversy if melanoma arises in a histological association with melanocytic nevi. One study found that 20.6 % of melanomas had a histological association with melanocytic nevi [49]. Melanomas arising in the setting or in association with melanocytic nevi are etiologically distinct from other types of melanoma [32]; they are less likely to develop in the presence of solar elastosis compared to non nevus associated melanomas. Examples of melanomas arising in association with melanocytic nevi are shown in figure 4. First example represents superficial spreading melanoma arising in the background of common melanocytic nevi. Second shows vertical growth phase melanoma arising in association with intradermal congential pattern melanocytic nevus and third shows melanoma in situ overlying cytologically banal dermal melanocytes associated with lymphocytic inflammatory host response.
FIGURE 4. Melanoma arising in association with melanocytic nevus.
A. Superificial spreading melanoma arising in the background of scattered small common melanocytic nevus cells. B. Scanning magnification shows vertical growth phase melanoma (left side) arising in association with intradermal congential pattern melanocytic nevus (right side). C. Melanoma in situ (large junctional nests of atypical melanocytes) overlying cytologically banal dermal melanocytes (arrow) associated with lymphocytic inflammatory host response.
Superficial spreading melanoma is the most common type of melanoma. Clinically it presents as irregularly shaped and variegate in color macule. Histologically it is characterized by atypical melanocytes growing in nests at the epidermal-dermal junction and pagetoid spread. Nodular melanoma presents as nodules of different level of pigmentation. Histologically it consists of atypical melanocytes growing as a nodule filling up the dermis without radial extension. Association of these types of melanoma with both melanocytic nevi and UV is still matter of debate since they arise in the skin which does not necessarily show signs of solar damage. Recently it was shown that different patterns of sun exposure were associated with melanomas arising in different anatomic locations. Superficial spreading and nodular melanomas of the head and neck were associated with chronic exposure but those arising on the trunk were more associated with only intermittent exposure [50].
ANIMAL MODELS OF MELANOMA
The role of UV radiation in melanoma development is partially supported by data obtained in studies on cultured in vitro cells and animal models. Specifically, animal models have been useful for the determination of action spectra for melanoma induction, despite their intrinsic limitations. The data obtained from animal models has suggested that the UVB spectrum plays an important role in the induction of non-melanoma skin cancers, while UVA is probably more important for melanoma induction. However, the contribution of UVB to melanoma development has also been reported, see below. On the basis of results obtained from studies on animal models of melanoma it can be assumed that induction of nevi and/or melanocytic hyperplasia (precursor of melanoma) is achieved by lower doses of ultraviolet radiation. It was found that longer time and higher doses of UV are needed for of melanoma formation [51, 52].
Xiphophorus
One of the first animal models for the studies of ultraviolet-induced skin cancers was Xiphophorus maculatus and Xiphophorus helleri, a hybrid fish model. This animal model develops spontaneous and UV-induced melanomas. UVA photons (wavelengths longer than 320 nm) contributed to melanoma development in Xiphophorus fish. The causative role in UVA-induced melanoma in this model seems to be from oxygen radical generation by melanin photosensitized reaction [25]. Unfortunately, these animals are evolutionary separated from mammals, which limits their usefulness in melanoma studies. Moreover, Xiphophorus fishes posses photoreactivation UV-induced DNA damage repair system that relies on the action of DNA photolyase. Exposure to photoreactivating light (300 – 500 nm) of complexes consisting of this enzyme and cyclobutane pyrimidine dimers returns them to their original state. Activation of this system decreased melanoma occurrence, although its presence in human cells is still a subject of debate [53].
Opossum Monodelphis domestica
The South American opossum Monodelphis domestica model is another model established for the studies of UV-induced melanomas [51]. Like the Xiphophorus model, the opossum model is susceptible to UV-induced damage, possesses a photoreactivation repair system and since it has been widely used to study UV-induced skin tumors and other lesions [4]. Similar to the Xiphophorus model, the opossum model’s usefulness in melanoma studies is limited because of different histopathology of melanocytic lesions. Melanomas in opossum originate from dermal melanocytes in contrast to human melanomas originating usually from epidermal melanocytes [4]. After UVA irradiation Monodelphis model developed non-melanoma skin cancers, melanocytic hyperplasia and malignant metastatic melanomas [51]. Furthermore, in these animals UVB radiation induced melanocytic lesions and this UV range had a higher potential to generate these alterations. Thus, while it is not clear if UVB or UVA has the strongest contribution to melanoma formation, these data support a role for UVR in the initiation of melanoma [4].
Mouse models and human xenografts
Many of experiments on UVR induction of melanoma have been performed in mouse models. Either hairless or transgenic mice have been used in most cases. In wild type mice spontaneous melanomas are rather rare, but they can be induced using DMBA, UV and other factors. In hairless mouse UVR alone usually produced non-melanoma skin cancers [54]. Using hairless mouse models UVB alone or UVB + UVA induced blue nevi, papillomas and squamous cell carcinomas, but not melanomas. DMBA can enhance UV-induced melanomagenesis. Combined treatment UV (UVB or UVA + UVB) and DMBA also induced melanomas [55]. Another studies also indicated that UVB, rather than UVA, was able to stimulate melanocyte proliferation in the hairless mouse model [56]. These results suggested that ultraviolet radiation in UVB spectrum stimulated development of pigmented lesions.
Confirmation of UVR involvement in melanoma induction/development comes also from experiments using transgenic mouse models. One of the early transgenic models is Tyr-SV40E model, with very low incidence of spontaneous melanomas, but sensitive to UV- induced melanomas. In this model melanomas were induced with UVB photons [57]. Another transgenic melanoma model is HGF/SF-transgenic mice. It was shown that single erythemal dose of UV radiation in neonatal mice was sufficient for melanoma induction. In contrast, single erythemal dose of ultraviolet in adolescent trangenic mice or chronic suberythemal UV doses in adult mice did not induce melanomas. Surprisingly, the histopathological features of these melanomas were similar to human melanoma [4, 58]. These data underline and support epidemiological data that sunburn during childhood increase risk of melanoma development and indicate UVB as causative spectrum. Experiments using INK4A/AFR deficient transgenic mice also revealed that UV radiation can stimulate nevi and melanoma development, indicating involvement of disturbances of INK4A/AFR pathway in melanoma growth after ultraviolet exposure [59, 60].
Mouse models have been extensively used in melanoma studies and provide important insight into UV-induced melanomagenesis. But it should be remembered that there are differences in UV-induced DNA damage repair in human and rodent cells. In rodent cells, CPD is repaired with much less efficiency when compared to human cells [61]. Moreover, spatial distribution of melanocytes in mouse skin is different than in humans. In adolescent or adult mouse, melanocytes presence is limited to hair follicles, with exception of ear, tail and footpads skin [62].
Hitherto human skin xenografts seem to be the more appropriate model of melanocytic lesions induction under UV exposure. Using newborn human skin xenografts into mice Atillasoy et al. [52] provided data indicating UVB spectrum as strong inducer of melanocyte proliferation and melanoma growth when combined with carcinogenic agent, DMBA. Subsequent experiments with adult skin xenografts into mice revealed that adult melanocytes are much more resistant to UV-induced alternation than neonatal melanocytes [63]. These results once again support the prevailing role of UV exposure during childhood in increased risk of melanoma development.
TUMOR SUPPRESSOR GENES AND ONCOGENES
Tumor suppressor protein p53
In human malignancies p53 tumor suppressor gene is one of the most frequently mutated gene. Its mutations are found in over 50% of different tumors. p53 is called “genomic guardian” and is involved in many fundamental processes of the cell, as cell cycle arrest, DNA repair, apoptosis, genomic integrity maintenance, cellular senescence [64]. Under the stress conditions p53 expression increases and cell cycle is arrested in check points. Failure of p53 function results in entering into S phase in spite of DNA damages, apoptosis disturbances, survival of the cell with changed DNA and generating of mutations, and consequently can lead to cancer transformation.
p53 in epidermal carcinogenesis
Unique to UV radiation mutations are found in the p53 tumor suppressor gene in precancerous and cancerous human skin lesions [16]. Mutations of p53 gene have been found in general population and XP patients in more than 90% of SCCs and approximately 50% of BCCs. Predominance of these mutations occur in bipyrimidine regions and are C to T or CC to TT transitions, i.e. marker mutations for UVB radiation. Such mutations are also found both in surrounding of BCCs and SCCs and chronically sun-exposed normal skin [17, 65]. Most of mutations caused by UVA radiation were found within generative layer of epidermis, and UVB-induced mutations were localized in upper layer of epidermis, suggesting more important role of longer wavelengths in SCCs induction.
The development of skin cancer in humans is associated with persistent changes in the basal keratinocyte stem cells predisposing to cancer. The existence and site of a melanocyte stem cell is unknown. For basal keratinocytes, chronic exposure to sun (ultraviolet) light results in low frequency point mutations in p53 gene that are fairly widely distributed in otherwise normal skin exposed to sunlight [66, 67]. These low frequency mutations are frequently found in basal cells and their progeny surrounding epithelial cancers [66–68], and appear to prime the skin or other epithelium for development of cancer. This phenomenon is known as field cancerization [68]. These low frequency mutations are not sufficient to produce cancer, but they set the stage for subsequent mutation or promoter events that do result in cancer. This is consistent with a multiple hit origin of cancer in which one mutation sets the stage for further mutations that together produce cancer; a process, which is more clearly defined in the gastrointestinal tract, in particular for colon cancer [69]. Persistent cell injury with regeneration promotes the expansion of mutant clones and development of cancer. In the skin, chronic ultraviolet light radiation (UVR) exposure promotes skin cancer by expanding the size of epidermal p53 clones [70, 71]. The size and number of epidermal p53 clones directly varies with level of sun exposure [65] and increasing age [66]. In addition, they are more numerous and larger adjacent to SCC than basal cell carcinoma and melanocytic nevi implicating them in the development of ultraviolet light related SCC [72]. Experimentally, this phenomenon has been demonstrated in mouse skin subjected to UVR [73]. Short term UVR exposures leads to formation of epidermal p53 mutant stem cell clones that increased only in cell number not area to 16+/− 6 cells, the size of the EPU in murine dorsal skin. Chronic exposure of mouse skin to UVR lead to expansion of these mutant clones to adjacent stem cell compartments/EPUs.
The human epidermis is organized into subunits, epidermal proliferative units (EPU), where each EPU is supported by one stem cell that undergoes asymmetric division producing transient amplifying cells and committed cells [74]. In the development of cancer, it is suspected that a rarely occurring set of somatic mutations in one cell is crucial for malignant transformation. The characteristics of this early, pre-transformation event is not well known. Incidental histologic findings that correlate with specific gene mutations such as focal acantholytic dyskeratosis [68] and acquisition of epidermal p53 patches or clones may well represent a pretransformational event preceding cancer [70, 73, 75, 76]. Jensen et al [77] found epidermal p53 clones either in the transient amplifying compartment only, or in both the stem cell and transiently amplifying compartment indicating that the target cells acquiring the p53 protein stabilizing mutation were either transient amplifying cells or stem cells [17]. The tendency of epidermal p53 clones to regress suggests that most arise from transient amplifying cells [78]. The size of the EPU, estimated to be about 35 basal cells in diameter in humans [79]. Mutant stem cells could be rare (quiescent and non-proliferative, reserve stem cell), populate the entire EPU with mutant daughter cells, or expand to fill other, adjacent EPU. Expansion into adjacent EPUs is suspected to be the result of replacement of stem cells by early lineage, mutant daughter cells; this is because stem cells die if they suffer DNA damage rather than repair it, thus avoiding accumulation of replication errors [71]. Some solitary basal p53 protein expressing cells found scattered along the basal layer could represent mutant melanocytes as well as melanocytes expressing wild-type p53 in G1-S arrest in response to (UV-related) DNA damage. Overcoming restriction of mutant cell clones to one EPU would represent a breach of one rate-limiting step in the pathogenesis of cancer. These mutant cells are growing exponentially and, thus, they can acquire additional errors of replication, some of which may confer a selective growth advantage.
p53 and melanoma
Similarly to association of moderate sun exposure with melanoma induction, the role of p53 mutations in melanoma is not clear. p53 mutations have not been detected in nevi and mutations of p53 in melanomas are less common than in other skin cancers [70, 75]. According to Zerp et al. [75] about 77% of mutation were C:G to T:A or CC:GG to TT:AA transitions, suggesting causative role of UVB radiation. On the other hand, Giglia-Mari and Sarasin [70] analyzed p53 mutations of melanomas and discovered high frequency of C:G to T:A mutation, but their occurrence was similar in SCCs and BCCs in general population. In contrast, A:T to G:C mutations occurred at higher rate compared to that rate found in SCCs and BCCs in general population, suggesting etiologic factors other than UVR. Nevertheless, higher percentage of defect in p53 in invasive melanoma and its metastases may suggest participation of p53 during late stages of melanomagenesis [76]. However, mutations and alteration in p53 expression even in metastatic melanomas are not as frequent as in non-melanoma skin cancers [80]. Zhang et al. [81] in his studies on activation of p53 after irradiation with UVA and UVB radiation in cultured melanomas showed that cells with wild type p53 were more sensitive to ultraviolet than cell with mutated of p53 gene, suggesting role/involvement of this protein in UV-induced apoptosis. The analysis of matched primary and metastatic melanomas for the expression of p53 did not reveal significant differences between these lesions. They suggested that changes in expression of p53 play rather minor role in direct induction/progression of melanoma and that its up-regulation by UV can represent protective response to genotoxic stress. In melanomas from XP patients, p53 mutations seem to depend on complementary group. Spatz et al. [82] revealed that p53 mutations were present in 60% of XPC melanomas and in 10% of XPV melanomas. Most of them were observed at the bipyrimidine sites and were C to T or CC to TT transitions, i.e. “UVB signature” lesions [70].
This entire picture appears to be complicated by recent data showing that p53 mutations in melanoma have a UVB signature [35] and melanomas strongly expressing p53 protein are associated with markers of high cumulative sun exposure [83]. Furthermore, p53 and CDKN2A harbor statistically higher rates of UVB signature changes (64.2% and 69.2%, respectively) than oncogenic NRAS and BRAF (15.3% and 2.4%) [35]. With respect to p53 protein expression, a surrogate marker of p53 gene mutation, a case control study from Australia [83] indicates that melanomas expressing p53 protein are distinct from non-p53 protein positive melanomas (p53 staining >10% of cell nuclei positively stained vs. <1% staining). Melanomas with high p53 nuclear labeling were positively associated with some indicators of high cumulative sun exposure: lentigo maligna melanoma subtype (OR = 3.2 vs. superficial spreading subtype), melanoma location on the head and neck (OR = 2.8 vs. back), histopathologic evidence of solar elastosis (OR = 2.1), dense freckling (OR = 6.6 vs. few freckles), and previous diagnosis of non-melanoma skin cancer (OR = 2.4) and negatively associated with high nevus density on the back (OR = 0.2 for >25 nevi vs. 0–3 nevi) and histologic evidence of a coexisting nevus (OR = 0.3). These findings support a role for p53 and UV induced mutagenesis in melanomas associated with chronic sun exposure (See figure 5).
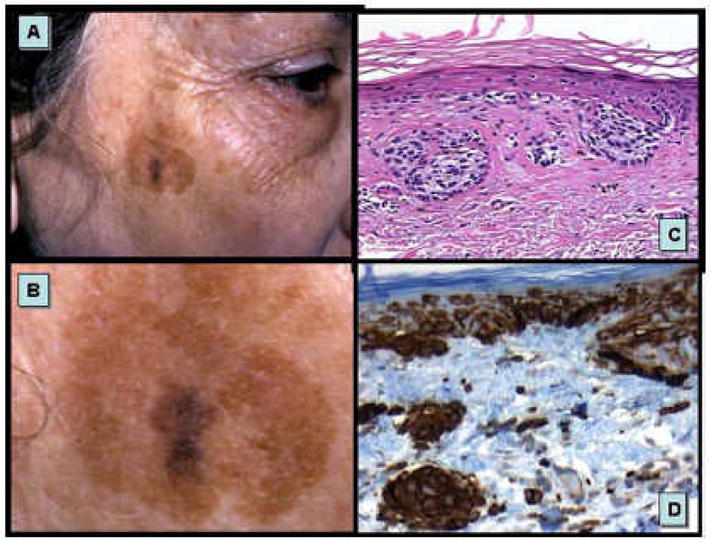
FIGURE 5.
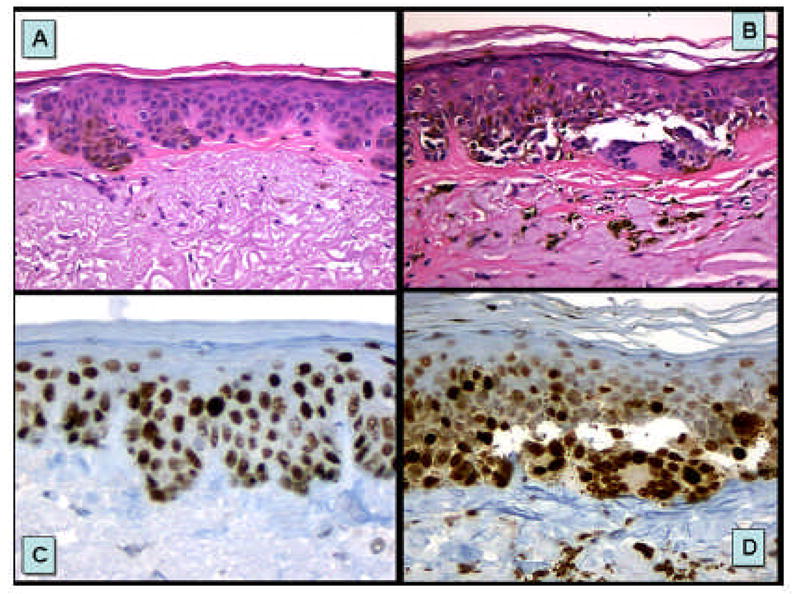
p53 mutant keratinocyte clones and mutant melanocytes occurring in sun damaged skin. Illustrated herein are mutant keratinocytes of solar lentigo (A & C) and putative p53 mutant melanocytes of melanoma in situ (B & D) arising in sun damaged skin.
Cyclin-dependent kinase inhibitor 2A (p16)
Cyclin-dependent kinase inhibitor 2A (CDKN2A; called also p16 INKa/MTS1/INK4A-ARF) is one of the best known genes involved in melanoma development. Its locus (9p21 of human chromosome) has two alternative reading frames, leading to synthesis proteins involved in control of cell proliferation, p16 INK4A and p14 AFR [76]. CDKN2A mutations have been identified in familial melanomas and its germline mutations have been identified in about 20 – 25% hereditary melanoma families [84]. Apart from germline mutations, somatic mutations have been also found in melanomas, however at lower rate (0–25%)[76]. Mutations of p16INK4A are not common in nevi, even in patients with sporadic melanoma [85], suggesting that p16INK4A protein plays a late rather than early role in the development of melanoma.
UVB fingerprints mutations were described in CDKN2A locus, encoded p16INK4A protein, both in non-melanoma cancers and in melanomas, suggesting engagement of ultraviolet radiation in inactivation p16INK4A tumor suppressor protein [35, 86]. Recent studies showed also that its mutation can contribute to melanomagenesis, apart from disturbances of control of cell proliferation, via reducing UV-induced DNA damages repair [87].
The crucial role of p16INK4A loss in UV-induced melanomas was also showed in genetically engineering mouse models with inactive INK4A/AFR gene. In these models, nevi growth was observed and melanoma development was significantly accelerated after UV exposure [59, 60].
V-raf murine sarcoma virus oncogene homolog B1 (BRAF)
BRAF is the most frequently targeted gene in melanoma. The BRAF gene, otherwise known as V-raf murine sarcoma virus oncogene homolog B1, is a member of the RAF family and is situated on chromosome 7q34. It encodes a serine-threonine protein kinase [76, 88]. RAS is a small G protein that is embedded in the plasma membrane of cells and is activated downstream of growth factor receptor tyrosine kinases, cytokines, and hormones. BRAF is a cytosolic protein and is activated following its recruitment to the plasma membrane by RAS. BRAF then phosphorylates and activates the mitogen-activated ERK (extracellular signal-regulated kinase)-activating kinase, which phosphorylates and activates ERK. ERK phosphorylates numerous cytosolic and nuclear proteins to regulate cell proliferation, differentiation, senescence, and survival [88].
More than 30 different BRAF mutations have been identified in a number of malignancies. These malignancies are thyroid, ovarian, lung, and colon cancer as well as melanoma. The highest frequency of mutations occurs in melanomas. These mutations also occur in melanocytic nevi [89]. The majority of BRAF mutations found in melanocytic nevi and melanomas substitute valine for glutamine at the amino acid at codon 600 which leads to a missense mutation at the kinase activation domain of exon 15 [90]. This mutation is otherwise known as the V600E BRAF mutation. The second most common mutation, which affects the glycine-rich P loop encoded on exon 11, is less frequent in melanomas. The BRAF mutations mimic phosphorylation of the activation site, resulting in RAS-independent activation. This in turn activates the MEK/ERK pathway leading to increased cell proliferation and inhibition of apoptosis [76, 89, 91].
BRAF mutations occur predominately in melanomas arising on intermittently sun-exposed skin rather than in melanomas arising on chronic sun-exposed or completely unexposed sites [92]. These mutations are more common in younger individuals (< 60 years old) [91]. BRAF mutations have been found in up to 70–82% of benign melanocytic nevi and 20–80% of primary melanomas [76, 88]. BRAF mutations are found in common acquired nevi (junctional, compound, and intradermal) as well as congenital and dysplastic nevi [90]. BRAF mutations are more prevalent in superficial spreading melanoma and nodular melanomas than in lentigo maligna melanomas and acral melanomas [92]. Desmoplastic melanomas lack BRAF mutations [90]. BRAF mutations are found in only about 10% of mucosal melanomas and are absent in uveal melanomas. Interestingly, BRAF mutations are identified in about 86% of Spitzoid melanomas but not in any common Spitz or atypical Spitz nevi [93]. However, the BRAF mutations are not found in Spitzoid melanomas that occur in prepubescent children [91]. Based on the prevalence of BRAF mutations in melanocytic nevi, it is likely that these mutations are an early event in melanogenesis [90]. Another supporting fact is their presence in the junctional component of the vertical growth phase of melanoma, from which the tumor is thought to arise [91].
It is still far from clear as to whether or not ultraviolet (UV) radiation causes these BRAF mutations. An argument against this association is the fact that BRAF mutations result from a single base-pair substitution of a thymidine to an adenine. In contrast to this, the typical ultraviolet ‘signature’ mutations are cytosine to thymine [90]. Melanomas in different anatomical sites and with different histopathologic subgroups may have distinct genetic pathways for melanogenesis which can not be explained by ultraviolet radiation exposure [90].
There are also arguments for the association of ultraviolet radiation and BRAF mutations. When melanocytes are exposed to ultraviolet B radiation, α-MSH binds to melanocortin receptor 1, which leads to cyclic adenosine monophosphate (cAMP) upregulation. This stimulates melanin synthesis and increases melanocyte proliferation because cAMP-dependent signaling cascade activates BRAF [93]. The fact that there is a high frequency of BRAF mutations in cutaneous melanomas as compared with uveal or mucosal melanomas suggest a link between BRAF mutations and UV exposure [90]. BRAF mutations are low in anatomic locations, which receive the lowest amount of sun exposure (mucosa) and highest sun exposure (face), whereas they are common in sites that receive intermittent UV light (trunk and extremities) [90]. In addition to bipyrimidine photoproducts UVB radiation also induces the formation of thymine-adenine dimerization photoproducts. In vitro experiments using synthetic oligonucleotides have shown that this damage induces T to A transversions. Therefore, although TA dimers are produced in 100-fold lower yield than bipyrimidine lesions, their formation could be of relevance for the predominant V600E mutation on BRAF [93].
Another option is that BRAF mutations may result from reactive oxygen species, which may be generated as a by-product of melanogenesis or pheomelanin absorption of UVA light [91]. Another theory is that since BRAF mutations have been associated with a strong inflammatory reactions in melanomas and oxidative damage also occurs in the context of inflammation (sunburn), this may be one way that BRAF mutations and UV radiation are linked [92, 93]. In support of this association melanocytic lesions often include tandem BRAF mutations, which are rarely found in non-melanocytic tumors with BRAF mutations [91]. Once BRAF mutations occur, UV exposure might also promote melanocytic tumor progression. It is possible that UV radiation provokes a stronger proliferative response in melanocytes with an activating BRAF mutation than in neighboring cells with wild-type BRAF, further promoting melanoma progression in UV-exposed sites [92, 93]. Another fact supporting the association is the fact that melanomas and their contiguous nevi, when they have one, have the same BRAF mutational status [91]. Clearly, further studies will be necessary to determine whether or not UVA or UVB contribute to the generation of BRAF mutations.
MELANOCTE-KERATINOCYTE AND MELANOCYTE-MATRIX INTERACTION
It was shown that UV radiation can act as independent inducer of melanocyte proliferation and can stimulate cell growth via modification of growth factors and their receptors. In addition, UVR can lead to melanoma development by changing the epidermal microenvironment, the nature of interactions between melanocytes with keratinocytes and matrix, and by inducting keratinocytes and fibroblasts to express melanoma-associated growth factors (for instance [2, 76, 94]). In short, UVR shifts the interactions from melanocytes with keratinocytes to melanocytes with fibroblasts and endothelium, promoting progression of melanoma.
Cadherins
Cadherins are one of the most important proteins involved in cell-to-cell adhesion. It is well documented that expression of cadherins plays a very important role in melanoma progression. In normal skin melanocytes adhere to keratinocytes via E-cadherin. During melanomagenesis switch of E-cadherin expression to N-cadherin expression takes place. N-cadherin makes adhesion between melanoma cells and fibroblast and endothelium cells possible, which can promote melanoma progression and metastasis [76, 94]. The consequence of decreased E-cadherin expression is increased β-catenin signaling promoting melanocyte malignant transformation [94]. UVR decreases E and P-cadherin expression in human melanocytes and melanoma cells. It was also found, that in keratinocytes after exposure to UVB radiation E-cadherin is cleaved, which results in disruption of cell to cell adhesion [95]. These alternations allow melanoma cells to detach from neighboring keratinocytes, promoting an invasive phenotype [94]. However, previous report did not revealed changes in E-cadherin expression in melanocytes [96]. This discrepancy could be secondary to use of different UVB doses. In the former studies the UVB doses were higher (from 20 to 90 mJ/cm2) than in latter one (20mJ/cm2). Crucial role of UVA radiation in E and N-cadherin switch is supported by in vivo studies in mouse model [97]. UVA radiation induces decrease of E-cadherin and increase of N-cadherin expression. Such results strongly support hypothesis, that UV radiation is involved in melanoma progression.
Integrins
Integrins (consist of two subunits, α and β) are another big family of adhesion molecules participating in cell adhesion to extracellular matrix. For example, αvβ3 integrin binds cells to dermal fibronectin, fibrinogen, laminins and colagens [98]. Expression of αvβ3 integrin increases melanoma cells migration, inhibits apoptosis and promotes metastasis, while its inhibition attenuates melanoma growth. It is well documented that αvβ3 integrin expression correlates with melanoma progression and invasion. The role of UV radiation in integrin expression is not fully understood. Immunochemistry staining demonstrated upregulation of α5β1 and αvβ3 integrins after UVB irradiation, but flow cytometry did not show that these alternation were significant [98]. Significant changes were found after irradiation of melanocytes with UVB in α6 integrin subunit. UVB decreases expression of this integrin, but only in the absence of laminin (important fiber of basement membrane). Laminin increases melanocytes resistance to UVB radiation. As authors suggest, that repeated exposure (sunburns) to UVB radiation could promote natural selection of resistant melanocytes, leading to melanoma development [98].
Metalloproteinases
Matrix melalloproteinases (MMPs) represent family of zinc-dependent degrading of components of extracellular matrix and basement membrane. Generally, tumor progression requires degradation of these structures. In normal tissues MMPs level is low, but in many cancers MMPs expression increases, resulting in enhanced degradation of stroma and basement membrane, thus promotion of cancer. They can be secreted by cancer cells and cells of surrounding tissue. MMPs are able to change activity of growth factors causing their cleavage [99]. Their role in melanoma progression and invasion is also well established. In melanomas, expression of MMPs can be regulated (stimulated) by UV-induced cytokines, such as TNFα and interleukins [99]. Ultraviolet radiation, changing production of the cytokins, indirectly changes MMPs synthesis, thus promotes melanoma development and invasion. UV radiation alters not only MMPs expression in melanoma cells. It was found that both UVB and UVA spectra stimulate MMPs secretion by keratinocytes and fibroblasts [100]. These data underline role of UVR in induction of cell to cell and cell to matrix adhesion disturbances and promotion of melanoma invasion.
Melanoma Inhibitory Activity
Melanoma inhibitory activity (MIA) is expressed in nevi, melanomas, but not in normal melanocytes. As was shown in vitro and in vivo, its production is correlated with melanoma progression and spreading and metastasis formation [101]. MIA plays important role in adhesion of the cells to extracellular matrix [102]. In presence of MIA interactions between cells and extracellular matrix and cell-cell adhesion are weaker. In the absence of MIA, E-cadherin is upregulated, and N-cadherin is downregulated. Moreover, MIA stimulates migration of melanoma cells [101]. MIA can bind to cell surface and directly interact with integrins causing their decreasing activity [102]. There is very little information about MIA expression and ultraviolet radiation. Marr et al. [103] showed that MIA can be produced by melanoma cells after stimulation with UV radiation. This pathway is p53-dependent. Since MIA can promote detachment of melanoma cells from basement membrane and from stroma, UV-induced MIA expression may play some role in progression of human skin melanoma.
GROWTH FACTORS AND THEIR RECEPTORS
Basic Fibroblast Growth Factor
Basic fibroblast growth factor (bFGF) is one of a few growth factors expressed in melanoma cells but not in normal melanocytes [104]. It is also expressed in keratinocytes and fibroblasts [63, 76]. bFGF is an important autocrine factor for melanoma cells and inhibition of its expression can inhibit melanoma growth as demonstrated in mouse melanoma in vivo [105]. In studies of human samples bFGF and its receptor were found, respectively, in 45% and 86% human melanomas and, respectively, in 55% and 67% of melanocytic nevi [48]. Sole overexpression of bFGF does not lead to melanoma development. Cells characterized by high level of bFGF need additional factors to become malignant [104]. One of these co-stimulants is ultraviolet radiation. Several studies showed bFGF and UV radiation have potential to induce malignant transformation of melanocytes. bFGF together with UVB radiation was able to induce in human skin pigmented lesion within 2–5 months, such as melanocyte hyperplasia and lentiginous form of melanoma [63]. When bFGF was combined with stem cell factor (SCF), endothelin 3 (ET3) and UVB melanoma occurred after 4 weeks after beginning of treatment [106]. Furthermore, studies on cells showed significant increase of bFGF at mRNA and protein level in UV-irradiated keratinocytes and fibroblasts [2, 107]. These results strongly suggest important role of the above growth factors in UVB induction of melanoma.
Hepatocyte Growth Factor
Hepatocyte growth factor (HGF) stimulates DNA synthesis in epidermal melanocytes and act as mitogen for these cells. Mainly keratinocytes and rarely melanocytes produce this factor [108]. Transgenic mouse with overexpression of HGF were created [58]. In these mice, melanocytes proliferate under stimulation with UV radiation. UV-induced HGF overexpression leads do melanoma development at neonatal age; however, only UVB but not UVA photons were able to initiate melanoma [58]. On the other hand experiments using primary fibroblasts showed high expression of HGF after irradiation with UVA (up to 8-fold compared to non-irradiated control) and UVB (up to 7-fold compared to non-irradiated control), suggesting their involvement in UVR induced stimulation of melanocytes proliferation or melanoma growth [2].
Transforming Growth Factor β
Transforming growth factor β (TGF-β) is known as an anti-mitotic factor for epidermal cells. Melanoma cells, like several other types of cancer cells, are not sensitive to inhibition of growth by TGF-β, but their precursors, melanocytes are[76, 109]. Melanoma cells and normal melanocytes are able to synthesize TGF-β, which in melanocytes is dependent on exogenous growth factors. Melanoma produce TGF-β constitutively and its expression seems to correlate with tumor progression [109]. TGF-β secreted by melanoma cells stimulated fibroblasts to produce components of intercellular matrix leading to changes in stroma favoring melanoma survival and migration. TGF-β probably can also downregulate E-cadherin expression [94]. Production of TGF-β by fibroblasts is induced by UVA and UVB radiation [2]. Increased production of TGF-β was observed in human primary keratinocytes after irradiation with UVB and UVA [110]. Thus, UVR can also affect melanoma progression through induction of TGF-β production.
Endothelin 1
Endothelin 1 (ET1) plays role in melanin synthesis by melanocytes after exposure to UV radiation. It also stimulates DNA synthesis in melanocytes. Several studies revealed effects of ET1 secreted by keratinocytes on melanocytes and melanoma cells. It was also shown that it is overexpressed in human pigmented lesions [111]. Ultraviolet radiation in UVB range results in increased secretion of ET1 by keratinocytes [2]. Upregulation of this growth factor can change significantly microenvironment of normal and transformed melanocytes. Melanoma cells show decreased expression of receptors for ET1 in comparison to melanocytes. The ability of ET1 to induce apoptosis in melanoma cells suggest its anticancerogenic potential [94]. However, UVB induces secretion of ET1 by keratinocytes, which was further able to downregulate E-cadherin [112]. Such interactions of UVB, ET1 and E-cadherin can promote melanoma progression. UVA radiation could have opposite effects on melanoma cells and microenvironment, because photons from this spectrum caused decreased of ET1 [2]. Furthermore UVA1 had no effect on ET1 expression in human dermal fibroblasts [113]. This divergent impact of UVA and UVB action on ET1 is consistent with a hypothesis on different pathways involved in UVA- and UVB-induced carcinogenesis.
Vascular Endothelial Growth Factor
Vascular endothelial growth factor (VEGF) stimulates angiogenesis under normal and pathological condition by regulation of endothelial cells proliferation. In the skin the main cells producing VEGF are keratinocytes and fibroblasts [114].
Production of VEFG can be regulated by UVR. Thus, UVR stimulates VEGF in human keratinocytes and fibroblasts [114] and in squamous cell carcinomas [115]. However, UVR has no effect on VEGF release from melanoma cells [116]. On the other hand melanocytes are one of the targets for VEGF produced by keratinocytes and fibroblasts. Normal human melanocytes constitutively produce receptors for VEGF. Moreover, expression one the receptors, VEGFR-2, is induced by UVR [117]. Thus, UVR having no effect on VEGF production in melanocytes and melanomas can change expression of the receptors for this growth factor produced locally by keratinocytes and fibroblast after UVR exposure. This can result in altered behavior of melanocytes and can promote angiogenesis potentially contributing in melanoma development and progression.
CRH, ACTH, -MSH AND GLUCOCORTICOIDS
Systemic response to stress relies on the effectors of hypothalamo-pituitary-adrenal axis [118, 119]. The noxious stimuli converge in the hypothalamus to induce production of CRH that in turn via its action on pituitary stimulates it to release ACTH. ACTH in turn stimulates production of cortisol by adrenal cortex. Glucocorticoids (cortisol) act to abrogate the inflammatory response in the tissues. Local production of glucocorticoids by the tumors can inhibit local inflammatory/immune response and contribute to the tumor progression. UVB can stimulates production of CRH, ACTH and α-MSH by human epidermal melanocytes [1, 118, 120–122]. Stimulation of CRH expression by UVB is dependent on the PKA activation [121]. Stimulation of α-MSH and ACTH expression by UVB is also dependent on the PKA activation [123]. CRH triggers the cascade of ACTH and glucocorticoid release by melanocytes [124]. UVB-induced expression of POMC is dependent on CRH [121] and CRH-induced release of cortisol is dependent on ACTH [124]. This data provide evidence of the capabilities of melanocyte derived cells to use locally neuropeptides of HPA axis and glucocorticoids [125]. Of note, skin has steroidogenic capability and melanoma cells can produce corticosteroids [124–126]. Suppression of local inflammatory response triggered by common environmental noxious stimulus (UV) is most possible physiological significance of this phenomenon. We propose that one subpopulation of melanocytes, i.e. “detector” melanocytes, would respond to environmental stimuli such as UVB with production and release of CRH, that would induce expression of POMC and release of ACTH by other melanocytes (“effector 1” melanocytes). Yet other melanocytes would respond to ACTH with the direct release of steroids (“effector 2” melanocytes). This model is presented in figure 6. Neoplastic cells derived of melanocytes are characterized by constitutive expression of CRH and POMC-derived peptides [119, 120, 127]. Since UV is a stimulus for activation of the effectors of HPA axis in melanocytes and as these effectors are apparently overexpressed by melanocyte-derived cells, it is possible that there is a link between those phenomena. The mechanism of persistent activation of HPA effectors in melanoma cells has so far not been elucidated. We might hypothesize that UV might lead to changes in the CRH promoter or a signaling system leading from UV detection to CRH promoter that in turn renders it constitutively active in the melanoma cells. Certainly similar events might involve POMC or further down enzymes engaged in the production of glucocorticoids. Direct effects of CRH, ACTH, α-MSH and glucocorticoids on melanocyte and melanoma viability and proliferation are confounding. CRH inhibits proliferation of normal and immortalized melanocytes in serum-containing medium through G2/M arrest [120]. In serum-free media CRH increases DNA synthesis and transition from G1/G0 to S phase in melanocytes. In serum-free media CRH decreased early and late apoptosis in the same cells, demonstrated by analysis with the annexin V stains and decrease of subG1 signal. Thus, CRH acts on epidermal melanocytes as a survival factor under the stress of starvation (anti-apoptotic) as well as inhibitor of growth factors induced cell proliferation [120]. In melanocyte it also acts as an antiinflamatory agent [128], which is in contrast to keratinocytes [129]. CRH and its analogs inhibit proliferation of Cloudman melanoma cells in culture and inhibit tumorogenenicity of B16 melanoma [129]. α-MSH and ACTH stimulate the proliferation of human melanocytes [130]. α-MSH reduces amount of cyclobutane pyrimidine dimers and leads to reduced apoptosis in UVB-treated normal human melanocytes [131]. ACTH and α-MSH-derived peptides increase metastatic potential of B16 melanoma cells [131]. As far as glucocorticoids are concerned, glucocorticoids inhibited proliferation of human melanoma M-5A cells [132]. M-5A cells responded with 90% growth inhibition and a 40% reduction in cell survival. Both inhibition of transition from G1/0 to S phase and from S to G2/M phase were observed. On the other hand prednisolone stimulated the proliferation of melanoma TM-1 cells [133]. Direct effects of these compounds were mostly studied in vitro (cell culture) models. In our opinion it is hard to relate these results to situation in vivo since these hormones act basically as cytokines in local inter-cellular environment. It is established that cytokines act in context [134] and exact definition of their role depends greatly on the presence of multitude of definable and indefinable factors.
FIGURE 6.
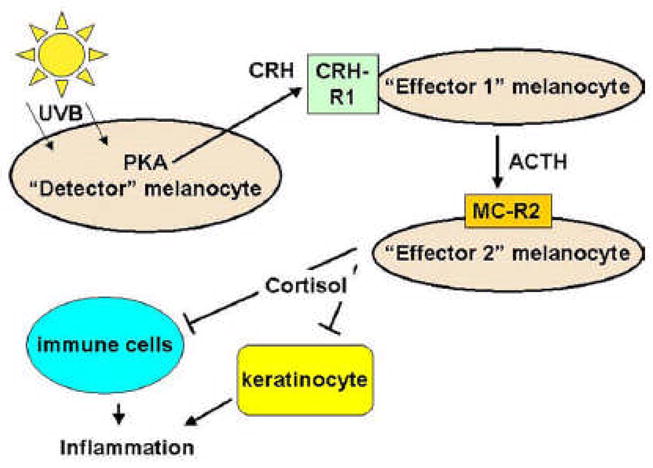
Hypothetical model of inter-melanocyte interactions through local HPA axis mediators. UVB stimulates production of CRH in “detector” melanocyte [121]. CRH stimulates “effector 1” melanocyte to produce ACTH. In turn, ACTH in stimulates steroid production by “effector 2” melanocyte [124]. Acting together, ACTH and steroids suppress local inflammatory reaction proceeding with the participation of keratinocytes [149] and immune cells [150].
INHIBITOR OF DNA BINDING 1, DOMINANT NEGATIVE HELIX-LOOP-HELIX PROTEIN (ID1)
Inhibitor of DNA binding 1, dominant negative helix-loop-helix protein (ID1) plays role in regulation of cell growth, differentiation, delayed cell senescence in mammalian cells [135]. Its upregulation seems to be important in early melanomas and can be prognostic factor for melanoma patients [136]. It has been shown that in melanoma in situ (within melanoma cells and surrounding keratinocytes) expression of ID1 correlates with loss p16/Ink4a expression. Generally ID1 expression in melanomas at late stages was not detected. ID was found only in neighboring of blood vessels. No ID1 staining was found also within nevi [135]. UV regulation of ID1 expression depends on wavelengths. In human melanocytes UVA radiation has no effect on ID1 synthesis, but in these cells after UVB exposure ID1 is found at higher level [136].
NUCLEAR FACTOR KAPPA B
Nuclear factor kappa B (NF-κB) is transcription factor which plays crucial role in the regulation of a variety of genes involved in control of cell growth and death. It is constitutively activated in many human cancers resulting in inhibition of cancer cells apoptosis. Activation of NF-κB was also found in melanocytes and melanoma cells, but in the latter level of NF- κB expression is higher than in normal melanocytes [73]. Inhibition of NF- κB activity restores susceptibility to ionizing radiation and seems to be one potential molecular target for melanoma treatment [76]. Activation of NF- κB can be induced by oxidative stress. UVA – induced oxidative stress generated increased level of NF- κB in fibroblasts [137]. Alternation of level and distribution of NF- κB was also observed in melanocytes and melanoma cells under oxidative stress conditions and after UVA and UVB exposure [138]. As was mentioned above, photons from UVA and UVB range can generate reactive oxygen species, redox alternation and oxidative stress. After irradiation of melanocytes with UVA and UVB radiation translocation of NF- κB from cytoplasm to nucleus was observed, thus carcinogenesis could be promoted by these environmental factors [138]. But some of the researchers reported that inhibition of NF- κB downregulated Fas-mediated apoptosis in melanoma cells exposed to UV radiation [139]. Thus NF- κB can induce opposite effects: in some conditions it acts as antiapoptotic factor promoting tumorogenesis and in another condition it can inhibit cancer development and induce apoptosis. Taken together, involvement of UV radiation in NF- κB - related melanoma development is still not clear, although most data show UV-induced activation of this growth factor.
VITAMIN D3
UVB wavelength of solar radiation converts 7-dehydrocholesterol to previtamin D3 in the keratinocytes, which isomerises then to vitamin D3 [140]. Vitamin D3 is hydroxylated at C25 by CYP27A1 in the liver and 25-cholecalciferol is hydroxylated at C1 by CYP27B1 in the kidney. 1,25-dihydroxycholecalciferol is a recognized inhibitor of keratinocyte proliferation and a stimulant of keratinocyte differentiation [141]. Calcipotriol, vitamin D3 derivative, is used to treat psoriasis [142]. Non-calcemic vitamin D3 analogs are currently in clinical trials for breast, colorectal, pancreatic and hepatocellular carcinoma [143–146]. All-trans-retinoic acid (ATRA) is already used to treat promyelocytic leukaemia [143]. It was recently shown that exposure to sun is actually associated with the increased survival from melanoma [147]. Exposure to sun increases levels of vitamin D3. Correlation of these two facts: inhibitory effects of vitamin D3-related compounds and increased survival of melanoma patients offers an exciting possibility for more effective melanoma treatment. It would be also of great interest to examine if melanocytes escape from the possible anti-proliferative control of 1,25-dihydroxycholecalciferol naturally produced in the epidermis and develop into neoplastic cells. Since there is a correlation between polymorphism of vitamin D receptor (VDR) and melanoma development there are possibly variants of the receptor that are less sensitive to the effects of vitamin D3. Indeed, it was recently found that certain alleles of VDR confer either protection or increased risk of cutaneous melanoma [148].
EXPERT COMMENTARY
Most data in the literature is correlational and does not really prove a direct causal relationship between UV radiation and melanoma development. UVB fingerprint mutations may be either found in genes commonly associated with melanoma development (such as best known melanoma-associated gene: p16) or not (notable example is BRAF). Melanoma cells may escape normal control of inter-cellular interactions or anti-proliferative mediators. They may use mediators such as corticosteroids to overcome the harness of immune system.
In our opinion, the ability to synthesize melanin by the melanocyte serves as a both protector of melanocyte and other skin cells DNA and possibly a cause for melanoma development. This latter pathway most probably allows for accumulation of more not yet discovered alterations in melanocyte biology that allow for development of melanoma, which is well-known for its chemo- and radioresistance.
FIVE YEAR VIEW
Looking at the exponential increase in the incidence of melanoma over the last decade, the incidence is expected to continue to increase and it is predicted that by 2010, 1 in 50 persons will have melanoma. One of the environmental factors considered to be involved in melanoma induction is ultraviolet radiation, although so far there is no direct causal data confirming the relationship between UV radiation and melanoma development. It is not clear how to prevent melanoma development (both in the induction and progression phases). The answer to this issue is very important. Epidemiological data shows that people (particularly during their childhood) who are exposed to high doses of UV radiation are at a higher risk of developing melanoma. Thus new sunscreens with higher efficiency of protection against sunburns and DNA damage and protection of children against exposure to ultraviolet radiation could result in a decrease in the morbidity rate of melanoma. The work that has been done on tumor suppressor genes, oncogenes as well as growth factors and their receptors continue to move us closer to a more effective treatment for this deadly disease. Immunotherapy trials should continue and include targeting multiple antigens that the tumor expresses as well as those that are unique to a particular patient’s tumor. Everyday we get closer to finding an effective treatment. Since it is well known that melanin can protect malignant melanocytes against chemotherapy and radiotherapy, melanogenesis inhibition could sensitize these malignant melanocytes to the killing action of therapeutic agents. We are not sure that there will be one in the next five years but researchers are working hard to find one and are hopeful that we will.
KEY ISSUES
Although both epidemiologic and experimental data does not provide direct causal relationship between UV radiation and melanoma development, it is accepted that UV radiation is an environmental risk factor for cutaneous melanomas.
The epidemiologic data shows that melanoma risk associated with UV exposure is highest in the individuals with intermittent sun exposure and sun burns during childhood.
The ability of UV radiation to induce and promote melanoma was also shown in several animal models.
Ultraviolet radiation can contribute to melanomagenesis via various pathways.
UV fingerprint mutations (especially UVB-induced mutations) are found in genes involved in melanoma development.
Ultraviolet radiation inactivates p16INK4A tumor suppressor protein thereby promoting melanoma progression.
Ultraviolet radiation decreases E and P-cadherin expression on both benign and malignant melanocytes, increasing B-catenin signaling and promotes melanocyte malignant transformation as well as disrupts cell to cell adhesion which allows melanoma cells to detach from neighboring keratinocytes thereby promoting the invasive phenotype.
Ultraviolet radiation changes cytokines such as TNF-alpha and interleukins which indirectly changes the synthesis of matrix metalloproteinases thereby promoting melanoma development and invasion.
Basic fibroblast growth factor along with ultraviolet radiation induces the formation of pigmented skin lesions including melanoma.
Malignant melanocytes secrete transforming growth factor which stimulates fibroblasts to produce components of intercellular matrix leading to changes in the stroma which favors survival and migration of these melanocytes.
Ultraviolet-B radiation can stimulate the production of CRH, ACTH and alpha-MSH by epidermal melanocytes.
Exposure to ultraviolet radiation increases the level of vitamin D3, which increases the survival of patients with melanoma. This offers a possibility of an effective treatment for melanoma.
Acknowledgments
The work was supported by National Institutes of Health grants AR047079 and AR052190 to AS.
Contributor Information
Brozyna Anna, Department of Medical Biology, Nicolaus Copernicus University, Torun, Poland, Tel: (4856)611-4776, Fax: (4856)611-4772, E-mail: abrozyna@biol.uni.torun.pl.
Zbytek Blazej, Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, 930 Madison Ave, Memphis, TN 38163, Tel: (901)448-6300, Fax: (901)448-6979, E-mail: bzbytek@utmem.edu.
Granese Jacqueline, Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, 930 Madison Ave, Memphis, TN 38163, Tel: (901)448-6300, Fax: (901)448-6979, E-mail: jackiegranese@yahoo.com.
Carlson J. Andrew, Department of Pathology & Laboratory Medicine, Albany Medical Center, 47 New Scotland Avenue, Albany, NY, Tel: (518)262-8099, Fax: (518)262-8092, E-mail: CarlsoA@mail.amc.edu.
Ross Jeffrey, Department of Pathology & Laboratory Medicine, Albany Medical Center, Albany, NY, 47 New Scotland Avenue, Albany, NY, Tel: (518)262-5461 fax: (518)262-8092, E-mail: RossJ@mail.amc.edu.
References
- 1.Slominski A, Pawelek J. Animals under the sun: Effects of UV radiation on mammalian skin. Clin Dermatol. 1998;16:503–515. doi: 10.1016/s0738-081x(98)00023-6. [DOI] [PubMed] [Google Scholar]
- 2.Brenner M, Degitz K, Besch R, Berking C. Differential expression of melanoma-associated growth factors in keratinocytes and fibroblasts by ultraviolet A and ultraviolet B radiation. Br J Dermatol. 2005;153(4):733–9. doi: 10.1111/j.1365-2133.2005.06780.x. “considerable interest” Extremely convincing experiments showing that synthesis of growth factors for melanocytes is dependent on cell type and UV wavelength. [DOI] [PubMed] [Google Scholar]
- 3.Norval M. The mechanisms and consequences of ultraviolet-induced immunosuppression. Prog Biophys Mol Biol. 2006;92(1):108–18. doi: 10.1016/j.pbiomolbio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Jhappan C, Noonan FP, Merlino G. Ultraviolet radiation and cutaneous malignant melanoma. Oncogene. 2003;22(20):3099–112. doi: 10.1038/sj.onc.1206450. [DOI] [PubMed] [Google Scholar]
- 5.Mariutti G, Matzeu M. Measurement of ultraviolet radiation emitted from welding arcs. Health Phys. 1988;54(5):529–32. doi: 10.1097/00004032-198805000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Kohen E, Santus R, Hirschberg JG. Photobiology. San Diego, CA: Academic Press Inc; 1995. [Google Scholar]
- 7.Gambichler T, Breuckmann F, Boms S, Altmeyer P, Kreuter A. Narrowband UVB phototherapy in skin conditions beyond psoriasis. J Am Acad Dermatol. 2005;52(4):660–70. doi: 10.1016/j.jaad.2004.08.047. [DOI] [PubMed] [Google Scholar]
- 8.Barbagallo J, Spann CT, Tutrone WD, Weinberg JM. Narrowband UVB phototherapy for the treatment of psoriasis: A review and update. Cutis. 2001;68(5):345–7. [PubMed] [Google Scholar]
- 9.Ledo E, Ledo A. Phototherapy, photochemotherapy, and photodynamic therapy: Unapproved uses or indications. Clin Dermatol. 2000;18(1):77–86. doi: 10.1016/s0738-081x(99)00096-6. [DOI] [PubMed] [Google Scholar]
- 10.Gasparro FP. The role of PUVA in the treatment of psoriasis. Photobiology issues related to skin cancer incidence. Am J Clin Dermatol. 2000;1(6):337–48. doi: 10.2165/00128071-200001060-00002. [DOI] [PubMed] [Google Scholar]
- 11.Breuckmann F, Gambichler T, Altmeyer P, Kreuter A. UVA/UVA1 phototherapy and puva photochemotherapy in connective tissue diseases and related disorders: A research based review. BMC Dermatol. 2004;4(1):11. doi: 10.1186/1471-5945-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheinfeld N, Deleo V. A review of studies that have utilized different combinations of psoralen and ultraviolet B phototherapy and ultraviolet A phototherapy. Dermatol Online J. 2003;9(5):7. [PubMed] [Google Scholar]
- 13.Lee E, Koo J, Berger T. UVB phototherapy and skin cancer risk: A review of the literature. Int J Dermatol. 2005;44(5):355–60. doi: 10.1111/j.1365-4632.2004.02186.x. [DOI] [PubMed] [Google Scholar]
- 14.Douki T, Reynaud-Angelin A, Cadet J, Sage E. Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation. Biochemistry. 2003;42(30):9221–6. doi: 10.1021/bi034593c. “interest” Experiments showing that dimers are the main form of damage by UVA. [DOI] [PubMed] [Google Scholar]
- 15.Courdavault S, Baudouin C, Charveron M, et al. Repair of the three main types of bipyrimidine DNA photoproducts in human keratinocytes exposed to UVB and UVA radiations. DNA Repair (Amst) 2005;4(7):836–44. doi: 10.1016/j.dnarep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Agar NS, Halliday GM, Barnetson RS, Ananthaswamy HN, Wheeler M, Jones AM. The basal layer in human squamous tumors harbors more UVA than UVB fingerprint mutations: A role for UVA in human skin carcinogenesis. Proc Natl Acad Sci U S A. 2004;101(14):4954–9. doi: 10.1073/pnas.0401141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brash DE. Roles of the transcription factor p53 in keratinocyte carcinomas. Br J Dermatol. 2006;154 Suppl 1:8–10. doi: 10.1111/j.1365-2133.2006.07230.x. [DOI] [PubMed] [Google Scholar]
- 18.Heck DE, Vetrano AM, Mariano TM, Laskin JD. UVB light stimulates production of reactive oxygen species: Unexpected role for catalase. J Biol Chem. 2003;278(25):22432–6. doi: 10.1074/jbc.C300048200. “interest” Excellent studies of ROS generation by UVB. [DOI] [PubMed] [Google Scholar]
- 19.Drobetsky EA, Turcotte J, Chateauneuf A. A role for ultraviolet a in solar mutagenesis. Proc Natl Acad Sci U S A. 1995;92(6):2350–4. doi: 10.1073/pnas.92.6.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Besaratinia A, Synold TW, Xi B, Pfeifer GP. G-to-T transversions and small tandem base deletions are the hallmark of mutations induced by ultraviolet A radiation in mammalian cells. Biochemistry. 2004;43(25):8169–77. doi: 10.1021/bi049761v. [DOI] [PubMed] [Google Scholar]
- 21.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 22.Giglia G, Dumaz N, Drougard C, Avril MF, Daya-Grosjean L, Sarasin A. p53 mutations in skin and internal tumors of xeroderma pigmentosum patients belonging to the complementation group C. Cancer Res. 1998;58(19):4402–9. [PubMed] [Google Scholar]
- 23.Carlson JA, Slominski A, Linette GP, et al. Malignant melanoma 2003: Predisposition, diagnosis, prognosis, and staging. Am J Clin Pathol. 2003;120 Suppl:S101–27. doi: 10.1309/J9M2NUM9MHYLN3DQ. “considerable interest” Summary of issues related to melanoma diagnosis. [DOI] [PubMed] [Google Scholar]
- 24.Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84(4):1155–228. doi: 10.1152/physrev.00044.2003. “considerable interest” Comprehensive review about regulation of melanogenesis in the mammalian skin. [DOI] [PubMed] [Google Scholar]
- 25.Wood SR, Berwick M, Ley RD, Walter RB, Setlow RB, Timmins GS. UV causation of melanoma in Xiphophorus is dominated by melanin photosensitized oxidant production. Proc Natl Acad Sci U S A. 2006;103(11):4111–5. doi: 10.1073/pnas.0511248103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai T, Vu C, Henson DE. Cutaneous, ocular and visceral melanoma in African Americans and Caucasians. Melanoma Res. 2005;15(3):213–7. doi: 10.1097/00008390-200506000-00012. [DOI] [PubMed] [Google Scholar]
- 27.Hu DN, Yu GP, McCormick SA, Schneider S, Finger PT. Population-based incidence of uveal melanoma in various races and ethnic groups. Am J Ophthalmol. 2005;140(4):612–7. doi: 10.1016/j.ajo.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 28.Eide MJ, Weinstock MA. Association of UV index, latitude, and melanoma incidence in nonwhite populations-US surveillance, Epidemiology, and End Results (SEER) Program, 1992 to 2001. Arch Dermatol. 2005;141(4):477–81. doi: 10.1001/archderm.141.4.477. [DOI] [PubMed] [Google Scholar]
- 29.Elwood JM, Gallagher RP. Body site distribution of cutaneous malignant melanoma in relationship to patterns of sun exposure. Int J Cancer. 1998;78(3):276–80. doi: 10.1002/(SICI)1097-0215(19981029)78:3<276::AID-IJC2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 30.Slominski A, Wortsman J, Carlson A, Matsuoka L, Balch CM, Mihm M. Malignant melanoma: An update. Arch Pathol Lab Med. 2001;125:1295–1306. doi: 10.5858/2001-125-1295-MM. [DOI] [PubMed] [Google Scholar]
- 31.Gandini S, Sera F, Cattaruzza MS, et al. Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure. Eur J Cancer. 2005;41(1):45–60. doi: 10.1016/j.ejca.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Purdue MP, From L, Armstrong BK, et al. Etiologic and other factors predicting nevus-associated cutaneous malignant melanoma. Cancer Epidemiol Biomarkers Prev. 2005;14(8):2015–22. doi: 10.1158/1055-9965.EPI-05-0097. [DOI] [PubMed] [Google Scholar]
- 33.Bulliard JL, Cox BElwood JM. Comparison of the site distribution of melanoma in new Zealand and Canada. Int J Cancer. 1997;72(2):231–5. doi: 10.1002/(sici)1097-0215(19970717)72:2<231::aid-ijc5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 34.Oliveria SA, Saraiya M, Geller AC, Heneghan MK, Jorgensen C. Sun exposure and risk of melanoma. Arch Dis Child. 2006;91(2):131–8. doi: 10.1136/adc.2005.086918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hocker T, Tsao H. Ultraviolet radiation and melanoma: A systematic review and analysis of reported sequence variants. Hum Mutat. 2007 doi: 10.1002/humu.20481. [DOI] [PubMed] [Google Scholar]
- 36.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–6. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 37.Bataille V, Bishop JA, Sasieni P, et al. Risk of cutaneous melanoma in relation to the numbers, types and sites of naevi: A case-control study. Br J Cancer. 1996;73(12):1605–11. doi: 10.1038/bjc.1996.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gefeller O, Tarantino J, Lederer P, Uter W, Pfahlberg AB. The relation between patterns of vacation sun exposure and the development of acquired melanocytic nevi in German children 6–7 years of age. Am J Epidemiol. 2007 doi: 10.1093/aje/kwm007. “interest” Recent data showing that intermittent exposure to high doses of UV radiation is an important factor involved in nevi development. [DOI] [PubMed] [Google Scholar]
- 39.Naldi L, Altieri A, Imberti GL, Gallus S, Bosetti C, La Vecchia C. Sun exposure, phenotypic characteristics, and cutaneous malignant melanoma. An analysis according to different clinico-pathological variants and anatomic locations (Italy) Cancer Causes Control. 2005;16(8):893–9. doi: 10.1007/s10552-005-2300-4. [DOI] [PubMed] [Google Scholar]
- 40.Carli P, Naldi L, Lovati S, La Vecchia C. The density of melanocytic nevi correlates with constitutional variables and history of sunburns: A prevalence study among Italian schoolchildren. Int J Cancer. 2002;101(4):375–9. doi: 10.1002/ijc.10629. [DOI] [PubMed] [Google Scholar]
- 41.Monestier S, Gaudy C, Gouvernet J, Richard MA, Grob JJ. Multiple senile lentigos of the face, a skin ageing pattern resulting from a life excess of intermittent sun exposure in dark-skinned caucasians: A case-control study. Br J Dermatol. 2006;154(3):438–44. doi: 10.1111/j.1365-2133.2005.06996.x. [DOI] [PubMed] [Google Scholar]
- 42.Bastiaens M, Hoefnagel J, Westendorp R, Vermeer BJ, Bouwes Bavinck JN. Solar lentigines are strongly related to sun exposure in contrast to ephelides. Pigment Cell Res. 2004;17(3):225–9. doi: 10.1111/j.1600-0749.2004.00131.x. [DOI] [PubMed] [Google Scholar]
- 43.Kossard S. Atypical lentiginous junctional naevi of the elderly and melanoma. Australas J Dermatol. 2002;43(2):93–101. doi: 10.1046/j.1440-0960.2002.t01-1-00568.x. [DOI] [PubMed] [Google Scholar]
- 44.Solomon CC, White E, Kristal AR, Vaughan T. Melanoma and lifetime UV radiation. Cancer Causes Control. 2004;15(9):893–902. doi: 10.1007/s10552-004-1142-9. [DOI] [PubMed] [Google Scholar]
- 45.Barlow JO, Maize J, Sr, Lang PG. The density and distribution of melanocytes adjacent to melanoma and nonmelanoma skin cancers. Dermatol Surg. 2007;33(2):199–207. doi: 10.1111/j.1524-4725.2006.33039.x. [DOI] [PubMed] [Google Scholar]
- 46.Hendi A, Brodland DG, Zitelli JA. Melanocytes in long-standing sun-exposed skin: Quantitative analysis using the ART-1 immunostain. Arch Dermatol. 2006;142(7):871–6. doi: 10.1001/archderm.142.7.871. [DOI] [PubMed] [Google Scholar]
- 47.Cohen LM. Lentigo maligna and lentigo maligna melanoma. J Am Acad Dermatol. 1995;33(6):923–36. doi: 10.1016/0190-9622(95)90282-1. quiz 937–40. [DOI] [PubMed] [Google Scholar]
- 48.Giehl KA, Nagele U, Volkenandt M, Berking C. Protein expression of melanocyte growth factors (bFGF, SCF) and their receptors (FGFR-1, c-kit) in nevi and melanoma. J Cutan Pathol. 2007;34(1):7–14. doi: 10.1111/j.1600-0560.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- 49.Massi D, Carli P, Franchi A, Santucci M. Naevus-associated melanomas: Cause or chance? Melanoma Res. 1999;9(1):85–91. doi: 10.1097/00008390-199902000-00011. [DOI] [PubMed] [Google Scholar]
- 50.Whiteman DC, Stickley M, Watt P, Hughes MC, Davis MB, Green AC. Anatomic site, sun exposure, and risk of cutaneous melanoma. J Clin Oncol. 2006;24(19):3172–7. doi: 10.1200/JCO.2006.06.1325. [DOI] [PubMed] [Google Scholar]
- 51.Ley RD, Applegate LA, Padilla RS, Stuart TD. Ultraviolet radiation-induced malignant melanoma in monodelphis domestica. Photochem Photobiol. 1989;50(1):1–5. doi: 10.1111/j.1751-1097.1989.tb04123.x. [DOI] [PubMed] [Google Scholar]
- 52.Atillasoy ES, Seykora JT, Soballe PW, et al. UVB induces atypical melanocytic lesions and melanoma in human skin. Am J Pathol. 1998;152(5):1179–86. [PMC free article] [PubMed] [Google Scholar]
- 53.Setlow RB, Grist E, Thompson K, Woodhead AD. Wavelengths effective in induction of malignant melanoma. Proc Natl Acad Sci U S A. 1993;90(14):6666–70. doi: 10.1073/pnas.90.14.6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Laat A, van der Leun JC, de Gruijl FR. Carcinogenesis induced by UVA (365-nm) radiation: The dose-time dependence of tumor formation in hairless mice. Carcinogenesis. 1997;18(5):1013–20. doi: 10.1093/carcin/18.5.1013. [DOI] [PubMed] [Google Scholar]
- 55.Husain Z, Pathak MA, Flotte T, Wick MM. Role of ultraviolet radiation in the induction of melanocytic tumors in hairless mice following 7,12-dimethylbenz(a)anthracene application and ultraviolet irradiation. Cancer Res. 1991;51(18):4964–70. [PubMed] [Google Scholar]
- 56.van Schanke A, Jongsma MJ, Bisschop R, van Venrooij GM, Rebel H, de Gruijl FR. Single UVB overexposure stimulates melanocyte proliferation in murine skin, in contrast to fractionated or UVA-1 exposure. J Invest Dermatol. 2005;124(1):241–7. doi: 10.1111/j.0022-202X.2004.23551.x. [DOI] [PubMed] [Google Scholar]
- 57.Klein-Szanto AJ, Silvers WK, Mintz B. Ultraviolet radiation-induced malignant skin melanoma in melanoma-susceptible transgenic mice. Cancer Res. 1994;54(17):4569–72. [PubMed] [Google Scholar]
- 58.Noonan FP, Dudek J, Merlino G, De Fabo EC. Animal models of melanoma: An HGF/SF transgenic mouse model may facilitate experimental access to UV initiating events. Pigment Cell Res. 2003;16(1):16–25. doi: 10.1034/j.1600-0749.2003.00014.x. “interest” Studies that imply that exposure to UV in childhood has a role in melanoma induction. [DOI] [PubMed] [Google Scholar]
- 59.van Schanke A, van Venrooij GM, Jongsma MJ, et al. Induction of nevi and skin tumors in Ink4a/Arf XPA knockout mice by neonatal, intermittent, or chronic UVB exposures. Cancer Res. 2006;66(5):2608–15. doi: 10.1158/0008-5472.CAN-05-2476. [DOI] [PubMed] [Google Scholar]
- 60.Recio JA, Noonan FP, Takayama H, et al. Ink4a/Arf deficiency promotes ultraviolet radiation-induced melanomagenesis. Cancer Res. 2002;62(22):6724–30. [PubMed] [Google Scholar]
- 61.Hanawalt PC. Revisiting the rodent repairadox. Environ Mol Mutagen. 2001;38(2–3):89–96. doi: 10.1002/em.1057. “considerable interest” Excellent review of DNA repair systems in rodent and human cells. [DOI] [PubMed] [Google Scholar]
- 62.Kunisada T, Lu SZ, Yoshida H, et al. Murine cutaneous mastocytosis and epidermal melanocytosis induced by keratinocyte expression of transgenic stem cell factor. J Exp Med. 1998;187(10):1565–73. doi: 10.1084/jem.187.10.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berking C, Takemoto R, Binder RL, et al. Photocarcinogenesis in human adult skin grafts. Carcinogenesis. 2002;23(1):181–7. doi: 10.1093/carcin/23.1.181. “interest” Excellent article identifying UVB and bFGF as co-stimulants for induction of pigmented lesions. [DOI] [PubMed] [Google Scholar]
- 64.Hussain SP, Harris CC. p53 biological network: At the crossroads of the cellular-stress response pathway and molecular carcinogenesis. J Nippon Med Sch. 2006;73(2):54–64. doi: 10.1272/jnms.73.54. [DOI] [PubMed] [Google Scholar]
- 65.Jonason AS, Kunala S, Price GJ, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci U S A. 1996;93(24):14025–9. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Backvall H, Asplund A, Gustafsson A, Sivertsson A, Lundeberg J, Ponten F, et al. Genetic tumor archeology: Microdissection and genetic heterogeneity in squamous and basal cell carcinoma. Mutat Res. 2005;571(1–2):65–79. doi: 10.1016/j.mrfmmm.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 67.Ren ZP, Ahmadian A, Ponten F. Benign clonal keratinocyte patches with p53 mutations show no genetic link to synchronous squamous cell precancer or cancer in human skin. Am J Pathol. 1997;150(5):1791–803. [PMC free article] [PubMed] [Google Scholar]
- 68.Carlson JA, Scott D, Wharton J, Sell S. Incidental histopathologic patterns: Possible evidence of ‘field cancerization’ surrounding skin tumors. Am J Dermatopathol. 2001;23(5):494–6. doi: 10.1097/00000372-200110000-00020. [DOI] [PubMed] [Google Scholar]
- 69.Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–28. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 70.Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutat. 2003;21(3):217–28. doi: 10.1002/humu.10179. [DOI] [PubMed] [Google Scholar]
- 71.Cairns J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc Natl Acad Sci U S A. 2002;99(16):10567–70. doi: 10.1073/pnas.162369899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Backvall H, Wolf O, Hermelin H, Weitzberg E, Ponten F. The density of epidermal p53 clones is higher adjacent to squamous cell carcinoma in comparison with basal cell carcinoma. Br J Dermatol. 2004;150(2):259–66. doi: 10.1111/j.1365-2133.2004.05683.x. [DOI] [PubMed] [Google Scholar]
- 73.Franco AV, Zhang XD, Van Berkel E, et al. The role of NF-kappa B in TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis of melanoma cells. J Immunol. 2001;166(9):5337–45. doi: 10.4049/jimmunol.166.9.5337. [DOI] [PubMed] [Google Scholar]
- 74.Potten CS, Booth C. Keratinocyte stem cells: A commentary. J Invest Dermatol. 2002;119(4):888–99. doi: 10.1046/j.1523-1747.2002.00020.x. [DOI] [PubMed] [Google Scholar]
- 75.Zerp SF, van Elsas A, Peltenburg LT, Schrier PI. p53 mutations in human cutaneous melanoma correlate with sun exposure but are not always involved in melanomagenesis. Br J Cancer. 1999;79(5–6):921–6. doi: 10.1038/sj.bjc.6690147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang HW, Wortsman J, Matsuoka L, Granese J, Carlson JA, Mihm M, Slominski A. Molecular spectrum of pigmented skin lesions: from nevus to melanoma. Expert Rev Dermatol. 2006;1(5):679–700. 22. “considerable interest” Comprehensive review on molecular alternations in melanoma. [Google Scholar]
- 77.Jensen UB, Lowell S, Watt FM. The spatial relationship between stem cells and their progeny in the basal layer of human epidermis: A new view based on whole-mount labelling and lineage analysis. Development. 1999;126(11):2409–18. doi: 10.1242/dev.126.11.2409. [DOI] [PubMed] [Google Scholar]
- 78.Berg RJ, van Kranen HJ, Rebel HG, et al. Early p53 alterations in mouse skin carcinogenesis by UVB radiation: Immunohistochemical detection of mutant p53 protein in clusters of preneoplastic epidermal cells. Proc Natl Acad Sci U S A. 1996;93(1):274–8. doi: 10.1073/pnas.93.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Asplund A, Guo Z, Hu X, Wassberg C, Ponten F. Mosaic pattern of maternal and paternal keratinocyte clones in normal human epidermis revealed by analysis of X-chromosome inactivation. J Invest Dermatol. 2001;117(1):128–31. doi: 10.1046/j.0022-202x.2001.01385.x. [DOI] [PubMed] [Google Scholar]
- 80.Hartmann A, Blaszyk H, Cunningham JS, et al. Overexpression and mutations of p53 in metastatic malignant melanomas. Int J Cancer. 1996;67(3):313–7. doi: 10.1002/(SICI)1097-0215(19960729)67:3<313::AID-IJC1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 81.Zhang H. p53 plays a central role in UVA and UVB induced cell damage and apoptosis in melanoma cells. Cancer Lett. 2006;244(2):229–38. doi: 10.1016/j.canlet.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 82.Spatz A, Giglia-Mari G, Benhamou S, Sarasin A. Association between DNA repair-deficiency and high level of p53 mutations in melanoma of Xeroderma pigmentosum. Cancer Res. 2001;61(6):2480–6. [PubMed] [Google Scholar]
- 83.Purdue MP, From L, Kahn HJ, et al. Etiologic factors associated with p53 immunostaining in cutaneous malignant melanoma. Int J Cancer. 2005;117(3):486–93. doi: 10.1002/ijc.21196. [DOI] [PubMed] [Google Scholar]
- 84.Carlson JA, Slominski A, Linette GP, Mihm MC., Jr Ross JS. Biomarkers in melanoma: Predisposition, screening and diagnosis. Expert Rev Mol Diagn. 2003;3(2):163–84. doi: 10.1586/14737159.3.2.163. [DOI] [PubMed] [Google Scholar]
- 85.Wang JL, Zheng BY, Li XD, et al. p16INK4A and p14ARF expression pattern by immunohistochemistry in human papillomavirus-related cervical neoplasia. Mod Pathol. 2005;18(5):629–37. doi: 10.1038/modpathol.3800308. [DOI] [PubMed] [Google Scholar]
- 86.Soufir N, Moles JP, Vilmer C, et al. p16 uvmutations in human skin epithelial tumors. Oncogene. 1999;18(39):5477–81. doi: 10.1038/sj.onc.1202915. [DOI] [PubMed] [Google Scholar]
- 87.Sarkar-Agrawal P, Vergilis I, Sharpless NE, DePinho RA, Runger TM. Impaired processing of DNA photoproducts and ultraviolet hypermutability with loss of p16INK4A or p19ARF. J Natl Cancer Inst. 2004;96(23):1790–3. doi: 10.1093/jnci/djh307. [DOI] [PubMed] [Google Scholar]
- 88.Gray-Schopfer VC, Karasarides M, Hayward R, Marais R. Tumor necrosis factor-alpha blocks apoptosis in melanoma cells when BRAF signaling is inhibited. Cancer Res. 2007;67(1):122–9. doi: 10.1158/0008-5472.CAN-06-1880. [DOI] [PubMed] [Google Scholar]
- 89.Thomas NE, Edmiston SN, Alexander A, et al. Number of nevi and early-life ambient UV exposure are associated with BRAF-mutant melanoma. Cancer Epidemiol Biomarkers Prev. 2007;16(5):991–7. doi: 10.1158/1055-9965.EPI-06-1038. [DOI] [PubMed] [Google Scholar]
- 90.Gill M, Celebi JT. B-RAF and melanocytic neoplasia. J Am Acad Dermatol. 2005;53(1):108–14. doi: 10.1016/j.jaad.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 91.Thomas NE, Berwick M, Cordeiro-Stone M. Could BRAF mutations in melanocytic lesions arise from DNA damage induced by ultraviolet radiation? J Invest Dermatol. 2006;126(8):1693–6. doi: 10.1038/sj.jid.5700458. [DOI] [PubMed] [Google Scholar]
- 92.Lang J, MacKie RM. Prevalence of exon 15 BRAF mutations in primary melanoma of the superficial spreading, nodular, acral, and lentigo maligna subtypes. J Invest Dermatol. 2005;125(3):575–9. doi: 10.1111/j.0022-202X.2005.23833.x. [DOI] [PubMed] [Google Scholar]
- 93.Papp T, Schipper H, Kumar K, Schiffmann D, Zimmermann R. Mutational analysis of the BRAF gene in human congenital and dysplastic melanocytic naevi. Melanoma Res. 2005;15(5):401–7. doi: 10.1097/00008390-200510000-00008. [DOI] [PubMed] [Google Scholar]
- 94.Herlyn M, Berking C, Li G, Satyamoorthy K. Lessons from melanocyte development for understanding the biological events in naevus and melanoma formation. Melanoma Res. 2000;10(4):303–12. doi: 10.1097/00008390-200008000-00001. “interest” Comprehensive review about interactions of pigmented cells matrix in melanoma. [DOI] [PubMed] [Google Scholar]
- 95.Hung CF, Chiang HS, Lo HM, Jian JS, Wu WB. E-cadherin and its downstream catenins are proteolytically cleaved in human HaCaT keratinocytes exposed to UVB. Exp Dermatol. 2006;15(4):315–21. doi: 10.1111/j.0906-6705.2006.00411.x. [DOI] [PubMed] [Google Scholar]
- 96.Nakazawa K, Nakazawa H, Bonnard M, Damour O, Collombel C. Ca2+ and UVB radiation have no effect on E-cadherin-mediated melanocyte-keratinocyte adhesion. Pigment Cell Res. 1995;8(5):255–62. doi: 10.1111/j.1600-0749.1995.tb00672.x. [DOI] [PubMed] [Google Scholar]
- 97.Pastila R, Leszczynski D. Ultraviolet A exposure alters adhesive properties of mouse melanoma cells. Photodermatol Photoimmunol Photomed. 2005;21(5):234–41. doi: 10.1111/j.1600-0781.2005.00166.x. [DOI] [PubMed] [Google Scholar]
- 98.Krengel S, Stark I, Geuchen C, et al. Selective down-regulation of the alpha6-integrin subunit in melanocytes by UVB light. Exp Dermatol. 2005;14(6):411–9. doi: 10.1111/j.0906-6705.2005.00295.x. [DOI] [PubMed] [Google Scholar]
- 99.Shellman YG, Makela M, Norris DA. Induction of secreted matrix metalloproteinase-9 activity in human melanoma cells by extracellular matrix proteins and cytokines. Melanoma Res. 2006;16(3):207–11. doi: 10.1097/01.cmr.0000215033.92693.73. [DOI] [PubMed] [Google Scholar]
- 100.Oh JH, Kim A, Park JM, Kim SH, Chung AS. Ultraviolet B-induced matrix metalloproteinase-1 and -3 secretions are mediated via PTEN/Akt pathway in human dermal fibroblasts. J Cell Physiol. 2006;209(3):775–85. doi: 10.1002/jcp.20754. [DOI] [PubMed] [Google Scholar]
- 101.Poser I, Tatzel J, Kuphal S, Bosserhoff AK. Functional role of MIA in melanocytes and early development of melanoma. Oncogene. 2004;23(36):6115–24. doi: 10.1038/sj.onc.1207797. [DOI] [PubMed] [Google Scholar]
- 102.Bauer R, Humphries M, Fassler R, Winklmeier A, Craig SE, Bosserhoff AK. Regulation of integrin activity by MIA. J Biol Chem. 2006;281(17):11669–77. doi: 10.1074/jbc.M511367200. [DOI] [PubMed] [Google Scholar]
- 103.Marr DG, Poser I, Shellman YG, Bosserhoff AK, Norris DA. Ultraviolet radiation induces release of MIA: A new mechanism for UVR-induced progression of melanoma. Int J Oncol. 2004;25(1):105–11. [PubMed] [Google Scholar]
- 104.Dotto GP, Moellmann G, Ghosh S, Edwards M, Halaban R. Transformation of murine melanocytes by basic fibroblast growth factor cDNA and oncogenes and selective suppression of the transformed phenotype in a reconstituted cutaneous environment. J Cell Biol. 1989;109(6 Pt 1):3115–28. doi: 10.1083/jcb.109.6.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang T, Yang M, Chen J, Watkins T, Xiuyun C. Inhibition of B16 melanoma growth in vivo by retroviral vector-mediated human ribonuclease inhibitor. Angiogenesis. 2005;8(1):73–81. doi: 10.1007/s10456-005-5714-4. [DOI] [PubMed] [Google Scholar]
- 106.Berking C, Takemoto R, Satyamoorthy K, et al. Induction of melanoma phenotypes in human skin by growth factors and ultraviolet B. Cancer Res. 2004;64(3):807–11. doi: 10.1158/0008-5472.can-03-3438. [DOI] [PubMed] [Google Scholar]
- 107.Wu CS, Yu CL, Wu CS, Lan CC, Yu HS. Narrow-band ultraviolet-B stimulates proliferation and migration of cultured melanocytes. Exp Dermatol. 2004;13(12):755–63. doi: 10.1111/j.0906-6705.2004.00221.x. [DOI] [PubMed] [Google Scholar]
- 108.Hirobe T, Osawa M, Nishikawa S. Hepatocyte growth factor controls the proliferation of cultured epidermal melanoblasts and melanocytes from newborn mice. Pigment Cell Res. 2004;17(1):51–61. doi: 10.1046/j.1600-0749.2003.00110.x. [DOI] [PubMed] [Google Scholar]
- 109.Moretti S, Pinzi C, Spallanzani A, et al. Immunohistochemical evidence of cytokine networks during progression of human melanocytic lesions. Int J Cancer. 1999;84(2):160–8. doi: 10.1002/(sici)1097-0215(19990420)84:2<160::aid-ijc12>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 110.Dissanayake NS, Mason RS. Modulation of skin cell functions by transforming growth factor-beta1 and ACTH after ultraviolet irradiation. J Endocrinol. 1998;159(1):153–63. doi: 10.1677/joe.0.1590153. [DOI] [PubMed] [Google Scholar]
- 111.Kadono S, Manaka I, Kawashima M, Kobayashi T, Imokawa G. The role of the epidermal endothelin cascade in the hyperpigmentation mechanism of lentigo senilis. J Invest Dermatol. 2001;116(4):571–7. doi: 10.1046/j.1523-1747.2001.01296.x. [DOI] [PubMed] [Google Scholar]
- 112.Jamal S, Schneider RJ. UV-induction of keratinocyte endothelin-1 downregulates E-cadherin in melanocytes and melanoma cells. J Clin Invest. 2002;110(4):443–52. doi: 10.1172/JCI13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Trompezinski S, Pernet I, Mayoux C, Schmitt D, Viac J. Transforming growth factor-beta1 and ultraviolet A1 radiation increase production of vascular endothelial growth factor but not endothelin-1 in human dermal fibroblasts. Br J Dermatol. 2000;143(3):539–45. doi: 10.1111/j.1365-2133.2000.03707.x. [DOI] [PubMed] [Google Scholar]
- 114.Yano K, Kadoya K, Kajiya K, Hong YK, Detmar M. Ultraviolet B irradiation of human skin induces an angiogenic switch that is mediated by upregulation of vascular endothelial growth factor and by downregulation of thrombospondin-1. Br J Dermatol. 2005;152(1):115–21. doi: 10.1111/j.1365-2133.2005.06368.x. [DOI] [PubMed] [Google Scholar]
- 115.Kosmadaki MG, Yaar M, Arble BL, Gilchrest BA. UV induces VEGF through a TNF-alpha independent pathway. Faseb J. 2003;17(3):446–8. doi: 10.1096/fj.02-0379fje. [DOI] [PubMed] [Google Scholar]
- 116.Shellman YG, Park YL, Marr DG, et al. Release of vascular endothelial growth factor from a human melanoma cell line, WM35, is induced by hypoxia but not ultraviolet radiation and is potentiated by activated Ras mutation. J Invest Dermatol. 2003;121(4):910–7. doi: 10.1046/j.1523-1747.2003.12511.x. [DOI] [PubMed] [Google Scholar]
- 117.Kim EJ, Park HY, Yaar M, Gilchrest BA. Modulation of vascular endothelial growth factor receptors in melanocytes. Exp Dermatol. 2005;14(8):625–33. doi: 10.1111/j.0906-6705.2005.00345.x. [DOI] [PubMed] [Google Scholar]
- 118.Slominski A, Wortsman J. Neuroendocrinology of the skin. Endocr Rev. 2000;21(5):457–87. doi: 10.1210/edrv.21.5.0410. “considerable interest” Comprehensive review about neuroendocrine interactions in the skin. [DOI] [PubMed] [Google Scholar]
- 119.Slominski A, Wortsman J, Pisarchik A, et al. Cutaneous expression of corticotropin-releasing hormone (CRH), urocortin, and CRH receptors. FASEB J. 2001;15(10):1678–93. doi: 10.1096/fj.00-0850rev. “considerable interest” Comprehensive review about cutaneous CRH/POMC system. [DOI] [PubMed] [Google Scholar]
- 120.Slominski A, Zbytek B, Pisarchik A, Slominski RM, Zmijewski MA, Wortsman J. CRH functions as a growth factor/cytokine in the skin. J Cell Physiol. 2006;206(3):780–91. doi: 10.1002/jcp.20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zbytek B, Wortsman J, Slominski A. Characterization of a ultraviolet B-induced corticotropin-releasing hormone-proopiomelanocortin system in human melanocytes. Mol Endocrinol. 2006;20(10):2539–47. doi: 10.1210/me.2006-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chakraborty AK, Funasaka Y, Slominski A, et al. UV light and MSH receptors. Ann N Y Acad Sci. 1999;885:100–16. doi: 10.1111/j.1749-6632.1999.tb08668.x. [DOI] [PubMed] [Google Scholar]
- 123.Im S, Moro O, Peng F, et al. Activation of the cyclic AMP pathway by alpha-melanotropin mediates the response of human melanocytes to ultraviolet B radiation. Cancer Res. 1998;58(1):47–54. [PubMed] [Google Scholar]
- 124.Slominski A, Zbytek B, Szczesniewski A. CRH stimulation of corticosteroids production in melanocytes is mediated by ACTH. Am J Physiol Endocrinol Metab. 2005;288(4):E701–6. doi: 10.1152/ajpendo.00519.2004. [DOI] [PubMed] [Google Scholar]
- 125.Slominski A, Wortsman J, Tuckey RC, Paus R. Differential expression of HPA axis homolog in the skin. Mol Cell Endocrinol. 2007:265–266. 143–9. doi: 10.1016/j.mce.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Slominski A, Zbytek B, Szczesniewski A, Wortsman J. Cultured human dermal fibroblasts do produce cortisol. J Invest Dermatol. 2006;126(5):1177–8. doi: 10.1038/sj.jid.5700204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Slominski A, Wortsman J, Luger T, Paus R, Solomon S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol Rev. 2000;80(3):979–1020. doi: 10.1152/physrev.2000.80.3.979. “considerable interest” Comprehensive review about CRH/POMC system in the skin. [DOI] [PubMed] [Google Scholar]
- 128.Zbytek B, Pfeffer LM, Slominski AT. CRH inhibits NF-kappaB signaling in human melanocytes. Peptides. 2006;27(12):3276–83. doi: 10.1016/j.peptides.2006.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zbytek B, Slominski AT. CRH mediates inflammation induced by lipopolysaccharide in human adult epidermal keratinocytes. J Invest Dermatol. 2007;127(3):730–2. doi: 10.1038/sj.jid.5700607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Abdel-Malek Z, Swope VB, Suzuki I, et al. Mitogenic and melanogenic stimulation of normal human melanocytes by melanotropic peptides. Proc Natl Acad Sci U S A. 1995;92(5):1789–93. doi: 10.1073/pnas.92.5.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lunec J, Pieron C, Thody AJ. MSH receptor expression and the relationship to melanogenesis and metastatic activity in B16 melanoma. Melanoma Res. 1992;2(1):5–12. doi: 10.1097/00008390-199205000-00002. [DOI] [PubMed] [Google Scholar]
- 132.Osman AM, Jansen PW, Smets LA, Benckhuijsen C. Glucocorticoid receptors and cell cycle progression in human melanoma cell lines. J Cell Physiol. 1985;125(2):306–12. doi: 10.1002/jcp.1041250220. [DOI] [PubMed] [Google Scholar]
- 133.Kimura M, Nishihira T, Kasai M, Sato H. Differentiative and proliferative effects of (But)2cAMP, n-butyric acid and prednisolone on the malignant melanoma cell line (TM-1) in vitro and in vivo. Tohoku J Exp Med. 1980;131(1):29–35. doi: 10.1620/tjem.131.29. [DOI] [PubMed] [Google Scholar]
- 134.Nathan C, Sporn M. Cytokines in context. J Cell Biol. 1991;113(5):981–6. doi: 10.1083/jcb.113.5.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Polsky D, Young AZ, Busam KJ, Alani RM. The transcriptional repressor of p16/Ink4a, Id1, is up-regulated in early melanomas. Cancer Res. 2001;61(16):6008–11. [PubMed] [Google Scholar]
- 136.Zhang H, Rosdahl I. Ultraviolet A and B differently induce intracellular protein expression in human skin melanocytes-a speculation of separate pathways in initiation of melanoma. Carcinogenesis. 2003;24(12):1929–34. doi: 10.1093/carcin/bgg171. [DOI] [PubMed] [Google Scholar]
- 137.Vile GF, Tanew-Ilitschew A, Tyrrell RM. Activation of NF-kappa B in human skin fibroblasts by the oxidative stress generated by UVA radiation. Photochem Photobiol. 1995;62(3):463–8. doi: 10.1111/j.1751-1097.1995.tb02369.x. [DOI] [PubMed] [Google Scholar]
- 138.Larsson P, Ollinger K, Rosdahl I. Ultraviolet (UV)A- and UVB-induced redox alterations and activation of nuclear factor-kappab in human melanocytes-protective effects of alpha-tocopherol. Br J Dermatol. 2006;155(2):292–300. doi: 10.1111/j.1365-2133.2006.07347.x. [DOI] [PubMed] [Google Scholar]
- 139.Ivanov VN, Ronai Z. p38 protects human melanoma cells from UV-induced apoptosis through down-regulation of NF-kappaB activity and Fas expression. Oncogene. 2000;19(26):3003–12. doi: 10.1038/sj.onc.1203602. [DOI] [PubMed] [Google Scholar]
- 140.Holick MF, MacLaughlin JA, Clark MB, et al. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science. 1980;210(4466):203–5. doi: 10.1126/science.6251551. [DOI] [PubMed] [Google Scholar]
- 141.Bikle DD. Vitamin D regulated keratinocyte differentiation. J Cell Biochem. 2004;92(3):436–44. doi: 10.1002/jcb.20095. [DOI] [PubMed] [Google Scholar]
- 142.Mason J, Mason AR, Cork MJ. Topical preparations for the treatment of psoriasis: A systematic review. Br J Dermatol. 2002;146(3):351–64. doi: 10.1046/j.1365-2133.2000.04713.x. [DOI] [PubMed] [Google Scholar]
- 143.Sirulnik LA, Stone RM. Acute promyelocytic leukemia: Current strategies for the treatment of newly diagnosed disease. Clin Adv Hematol Oncol. 2005;3(5):391–7. 429. [PubMed] [Google Scholar]
- 144.James SY, Mercer E, Brady M, Binderup L, Colston KW. EB1089, a synthetic analogue of vitamin D, induces apoptosis in breast cancer cells in vivo and in vitro. Br J Pharmacol. 1998;125(5):953–62. doi: 10.1038/sj.bjp.0702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Evans TR, Colston KW, Lofts FJ, et al. A phase II trial of the vitamin D analogue seocalcitol (EB1089) in patients with inoperable pancreatic cancer. Br J Cancer. 2002;86(5):680–5. doi: 10.1038/sj.bjc.6600162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dalhoff K, Dancey J, Astrup L, et al. A phase II study of the vitamin D analogue seocalcitol in patients with inoperable hepatocellular carcinoma. Br J Cancer. 2003;89(2):252–7. doi: 10.1038/sj.bjc.6601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Berwick M, Armstrong BK, Ben-Porat L, et al. Sun exposure and mortality from melanoma. J Natl Cancer Inst. 2005;97(3):195–9. doi: 10.1093/jnci/dji019. [DOI] [PubMed] [Google Scholar]
- 148.Li C, Liu Z, Zhang Z, et al. Genetic variants of the vitamin D receptor gene alter risk of cutaneous melanoma. J Invest Dermatol. 2007;127(2):276–80. doi: 10.1038/sj.jid.5700544. [DOI] [PubMed] [Google Scholar]
- 149.Barker JN, Mitra RS, Griffiths CE, Dixit VM, Nickoloff BJ. Keratinocytes as initiators of inflammation. Lancet. 1991;337(8735):211–4. doi: 10.1016/0140-6736(91)92168-2. [DOI] [PubMed] [Google Scholar]
- 150.Karalis KP, Venihaki M, Zhao J, van Vlerken LE, Chandras C. NF-kappaB participates in the corticotropin-releasing, hormone-induced regulation of the pituitary proopiomelanocortin gene. J Biol Chem. 2004;279(12):10837–40. doi: 10.1074/jbc.M313063200. [DOI] [PubMed] [Google Scholar]