

Abstract
The Staudinger ligation provides a means to form an amide bond between a phosphinothioester and azide. This reaction holds promise for the ligation of peptides en route to the total chemical synthesis of proteins. (Diphenylphosphino)methanethiol is the most efficacious of known reagents for mediating the Staudinger ligation of peptides, providing high (>90%) isolated yields for equimolar couplings in which a glycine residue is at the nascent junction. Surprisingly, the yields are lower (<50%) for non-glycyl couplings due to an aza-Wittig reaction that diverts the reaction towards a phosphonamide byproduct. Here, the partitioning of the reaction towards Staudinger ligation (and away from the aza-Wittig reaction) is shown to increase with increasing electron density on phosphorous. This electron density can be tuned either by installing functional groups on the phenyl substituents of (diphenylphosphino)methanethiol or by changing the polarity of the solvent. Installing p-methoxy groups and using solvents of low polarity (such as toluene and dioxane) provide especially high (>80%) isolated yields for the ligation of two non-glycyl residues. These conditions retain the high chemoselectivity of the reaction, and do not lead to a substantial change in reaction rate. The traceless Staudinger ligation is now poised to enable the iterative ligation of peptides with little regard for their sequence as well as the synthesis of amide bonds for other purposes.
Introduction
Total chemical synthesis is beginning to provide ready access to natural proteins, as well as enable the creation of nonnatural ones.1 Many proteins have already been assembled from synthetic peptides. “Native chemical ligation”—the coupling of a peptide (or protein) containing a C-terminal thioester with another peptide containing an N-terminal cysteine residue has been especially powerful.2–4 “Expressed protein ligation” is a method by which C-terminal thioesters for native chemical ligation can be accessed with recombinant DNA techniques.5,6
A limitation of native chemical ligation is its intrinsic reliance on having a cysteine residue at the ligation junction. Cysteine is uncommon, comprising only 1.7% of all residues in proteins.7 Modern peptide synthesis is typically limited to peptides of ≤40 residues,8 whereas most proteins contain hundreds of residues. Hence, most proteins cannot be prepared by any method that allows for peptides to be coupled only at cysteine residues.1
An alternative strategy is to employ auxiliaries that act as cysteine surrogates to mediate the chemical ligation of peptide fragments.1,9,10 After the ligation, the auxiliary is removed to reveal the native amide bond. Efforts with extant auxiliaries have, however, revealed a requirement for a glycine residue to be present at the ligation junction.11,12 Although these methods are not yet sequence-independent, they do represent an advance beyond native chemical ligation, as glycine is among the most common amino acids in proteins (7.2% of all residues).7
An elegant new strategy for peptide coupling entails the decarboxylative condensation of α-ketoacids and N-alkylhydroxylamines.13 This reaction suffers, however, from the accumulation of numerous intermediates, leading to low overall rates. Modifications to the reactants or reaction conditions that overcome this intrinsic deficiency are not apparent. Moreover, many of the α-ketoacids that correspond to the proteinogenic amino acids are not readily accessible.
The traceless Staudinger ligation was designed to overcome such limitations and provide a general means to synthesize proteins from component peptides (Scheme 1).14,15 In this reaction, a peptide with a C-terminal phosphinothioester (1) is coupled with a second peptide having an N-terminal azido acid (2) through the intermediacy of an iminophosphorane (3). The iminophosphorane can form a tetrahedral intermediate (4), which collapses to give an amidophosphonium salt (5) that is hydrolyzed in aqueous solution. The final ligation product is a peptide (6) without either residual atoms14,15 or racemization.16 In nonaqueous solvents, amidophosphonium salt 5 can fragment to form peptide 6 and a thiaphosphiranium salt.17
SCHEME 1.

Putative Mechanism for the Traceless Staudinger Ligation Mediated by (Diphenylphosphino)methanethiol
(Diphenylphosphino)methanethiol (HSCH2PPh) is the most efficacious of known reagents for effecting the traceless Staudinger ligation.17 This reagent has been used in the orthogonal assembly of a protein,18 site-specific immobilization of peptides and proteins to a surface,19,20 and synthesis of glycopeptides.21 The reaction is rapid, displaying t1/2 = 7 min for the coupling of AcGlySCH2PPh2 and 2-azido-N-benzyl-acetamide (175 mM each) in a wet organic solvent.17 This value is limited by the encounter of the reactants to form the initial phosphazide intermediate. Another phosphine, o-(diphenylphosphino)benzoic acid, has been used to mediate the non-traceless Staudinger ligation for biomolecular labeling experiments in vitro22 and in vivo,23,24 and drug delivery.25 This reaction leaves a phosphine oxide in the ligation product, making it inappropriate for synthetic applications.
The putative mechanism for the traceless Staudinger ligation appears to be indifferent to amino acid substitution at the ligation sites (Scheme 1). Yet, as with auxiliary-mediated ligations,11,12 all reported traceless Staudinger ligations of peptides that proceed with a high (>90%) yield have had a glycine residue at the ligation junction.26 Reactions of more encumbered residues proceed with a lower (<50%) yield.17,27 Likewise, yields for the synthesis of glycopeptides are diminished by steric strain.21 Somehow, steric effects are being manifested during the reaction.
Herein, we overcome the limitation of having a glycine residue at the ligation junction during a traceless Staudinger ligation. To do so, we discern the basis for the inefficiency of non-glycyl couplings. We then discover that the rational tuning of the electronic structure of tetrahedral intermediate 4 (Scheme 1) by both substitution on the phenyl groups of (diphenylphosphino)methanethiol and solvent effects can enable the traceless Staudinger ligation between non-glycyl residues to proceed in high yield. This discovery expands the synthetic utility of the traceless Staudinger ligation for the synthesis of proteins as well as other molecules.
Results and Discussion
NMR-Based Assay to Monitor the Staudinger Ligation
We recently reported on a means to monitor the traceless Staudinger ligation by 13C NMR spectroscopy using an organic azide enriched with 13C at its α-carbon.17 This NMR-based assay offers the ability to observe the reaction continuously and without its perturbation, and (most importantly) reveals a product-distribution profile that is unperturbed by the vagaries of chemical purification. This NMR-based assay was used for determining all product yields herein, unless noted otherwise.
Effect of Phenyl Substitutents
Ala+Ala couplings were examined as a model ligation involving two non-glycyl residues.28 As reported previously, the coupling of alanyl phosphinothioester 10 with 13C-labeled alanyl azide 11 in DMF results in a yield of only 36% (Scheme 2).17 This moderate yield is due largely to the formation of a byproduct, phosphonamide 13, which results from the aza-Wittig reaction of the iminophosphorane.
SCHEME 2.

Reaction of Alanyl Phosphinothioester 10 with Alanyl Azide 11
The key species in the partitioning towards Staudinger ligation product 6 (Scheme 1, Path A) and aza-Wittig byproduct 9 (Path B) is tetrahedral intermediate 4. In the Staudinger ligation (Path A), an electron pair on the oxygen of tetrahedral intermediate 4 displaces a thiol to form amidophosphonium salt 5. In the alternative aza-Wittig reaction (Path B), an electron pair from that oxygen forms a covalent bond with the oxophilic phosphorus atom, resulting in oxazaphosphetane 8. Why is Path A disfavored relative to Path B in an Ala+Ala coupling? In such a non-glycyl coupling, both the thioester group of 1 (RC(O)SCH2PPh2) and the azido group of 2 (R′N3) are secondary. In tetrahedral intermediate 4, the bulky R and R′ groups of a non-glycyl coupling are brought into close proximity. The ensuing steric strain could be relieved upon formation of the O–P bond in oxazaphosphetane 8. We reasoned that tuning the electron density on the phosphorus atom could discourage the formation of this bond, and thereby alter the partitioning of tetrahedral intermediate 4. The result would be a higher yield for the coupling of non-glycyl residues.
To test our hypothesis, we synthesized alanyl phosphinothioesters 14 and 15, and determined their abilities relative to alanyl phosphinothioester 10 to undergo a traceless Staudinger ligation with an alanyl azide. The p-chloro groups in phosphinothioester 14 should decrease the electron density on phosphorus in tetrahedral intermediate 4. According to our hypothesis, this decrease in electron density should lead to greater partitioning toward the aza-Wittig reaction (Path B, Scheme 1). Conversely, the p-methoxy groups of phosphinothioester 15 should increase the electron density on phosphorus in tetrahedral intermediate 4, and thereby favor the Staudinger ligation. Phosphinothioesters 10, 14, and 15 were synthesized by a route analogous to that used to synthesize related phosphinothioesters (Scheme 3).14,29
SCHEME 3.

Synthesis of Alanyl Phosphinothioesters 10, 14, and 15
The reaction of alanyl azide 11 with each alanyl phosphinothioester—10, 14, and 15—was performed in DMF, and yields were obtained with the NMR-based assay.17 Under these conditions, alanyl phosphinothioester 10 provided the desired amide 12 product in 47% yield. In agreement with our hypothesis, the p-chloro compound 14 afforded a diminished, 34%, yield of amide 12. The p-methoxy compound 15 also followed the expected trend, conferring the amide product in 61% yield. This yield represents a substantial increase from any reported previously for a non-glycyl Staudinger ligation.17,27 The yield of amide product in this Ala+Ala coupling correlates inversely with the Hammett σp value of the phenyl substituent in the phosphinothioester (Figure 1).
FIGURE 1.

Plot of the yield of amide 12 product versus Hammett σp value for the reaction of alanyl phosphinothioesters 10, 14, and 15 with 13C-labeled alanyl azide 11 in DMF and dioxane. The lines depict a linear least-squares fit of the data.
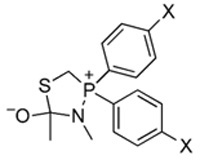
Density functional theory (DFT) calculations30 were performed with the Gaussian 98 package31 to quantify the effect of the phenyl substituents on the electron density on phosphorus in tetrahedral intermediate 4 (R = R′ = CH3). The DFT calculations indicated that this electron density increases considerably as the substituent became more electron-donating (Table 1). (By comparison, the electron density on nitrogen remains relatively constant.) Accordingly, both experiment and theory are consistent with the weakening of the O⋯P interaction in tetrahedral intermediate 4 as being responsible for the observed increase in the yield of amide product.
TABLE 1.
Effect of para Substituents on the Calculated Gas-Phase Electron Density on Phosphorus in Tetrahedral Intermediate 4
 | |||
|---|---|---|---|
| X |
|||
| Parameter | CI | H | OMe |
| σp | +0.23 | 0.00 | −0.27 |
| P, charge | +0.131 | +0.062 | +0.044 |
| N, charge | −0.342 | −0.326 | −0.322 |
Effect of Solvent Polarity
Previous efforts aimed at optimizing the Staudinger ligation showed that the reaction proceeds more rapidly in polar solvents, presumably due to favorable stabilization of a polar transition state in the rate-determining step—phosphazide formation.17 To determine the effect of solvent on the partitioning of iminophosphorane 3 between Staudinger ligation product 6 (Path A in Scheme 1) and aza-Wittig byproduct 9 (Path B), the reaction of p-methoxy phosphinothioester 15 and 13C-labeled alanyl azide 11 was performed in nine solvents of varying polarity (acetone, acetonitrile, dimethylformamide, dioxane, methanol, methylene chloride, nitromethane, tetrahydrofuran, and toluene), and the yields were obtained by using the NMR-based assay.17
The yield of amide product exhibited a strong inverse correlation with solvent polarity–polarizability (SPP) value32 (Figure 2). Thus, the highest yields (in contrast to the highest rates17) were obtained with nonpolar solvents. The yield of AcAlaAlaNHBn product was high (83%) in dioxane and nearly quantitative (99%) in toluene. These yields represent a substantial increase from those published previously for peptide ligation at two non-glycyl residues.17,26 Each of the previously described alanyl phosphinothioesters (p-chlorophenyl 14, p-methoxyphenyl 15, and phenyl 10) were reacted with alanyl azide 11 in dioxane. The yields of AcAlaAlaNHBn in dioxane again correlated well with the Hammett σp constant for the substituent, but were uniformly greater than the yields in the more polar solvent, DMF (Figure 1).
FIGURE 2.

Plot of the yield of amide 12 product versus SPP value for the reaction of alanyl phosphinothioester 15 with 13C-labeled alanyl azide 11. The lines depict a linear least-squares fit of the data.
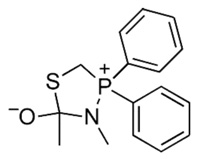
To obtain insight on the ability of non-polar solvents to provide higher yields of the desired peptide product, we performed DFT calculations on the effect of solvent on the electron density on phosphorus in tetrahedral intermediate 4 (R = R′ = CH3). The calculations indicated that this electron density increases in nonpolar solvents (Table 2), presumably due to the dispersal of charge into the phenyl groups. (Again, the electron density on nitrogen remains relatively constant.)
TABLE 2.
Effect of Solvent Polarity on the Calculated Electron Density on Phosphorus and Nitrogen in Tetrahedral Intermediate 4
 | ||
|---|---|---|
| Solvent |
||
| Parameter | H2O | THF |
| P, charge | +0.082 | +0.028 |
| N, charge | −0.348 | −0.335 |
All of the experimental and theoretical results from reactions with different phenyl substituents and in solvents of different polarity are consistent. These results indicate that increasing the electron density on phorphorus leads to a higher yield of amide product. This finding can be depicted by the dissection of tetrahedral intermediate 4 into resonance forms 4P+ (which has less charge dispersal) and 4Pδ+. To emphasize the importance of 4P+ and 4Pδ+ to the product distribution, we have refined the mechanism of the Staudinger ligation to be that in Scheme 4.
SCHEME 4.

Refined Mechanism of the Staudinger Ligation Including the Resonance Forms of Tetrahedral Intermediate 4
Coupling of Non-Glycyl Residues
Using those conditions most favorable for non-glycyl ligations, a series of model ligations were performed to determine the scope of these reactions. Dioxane was chosen as the solvent, because many starting materials and products were insoluble in the most nonpolar solvent tested, toluene. 13C-Labeled alanyl azide 11 was reacted (dioxane, 12 h) with alanyl phosphinothioester 15 and phenylalanyl phosphinothioester 25 to form AcAlaAlaNHBn (12) and AcPheAlaNHBn (26), respectively. After chromatography on silica gel, each of these couplings afforded the desired dipeptide in >80% isolated yield (Table 3). (Yields from the NMR-based assay17 were indistinguishable from those in Table 3.) These yields, which are for the coupling of equimolar reactants, represent a substantial improvement upon those reported previously for traceless Staudinger ligations at two non-glycyl amino acids,17,26,33 Unlike with (diphenylphosphino)methanethiol, increasing temperature was not found to increase yield of ligation product (data not shown).
TABLE 3.
Isolated Yields of Amide Product from the Reactions of Phosphinothioesters 15 and 25 with 13C-Labeled Alanyl Azide 11
 |
|---|
The Staudinger ligation is highly chemoselective when (diphenylphosphino)methanethiol is the coupling reagent.17,19 To determine the effect of the p-methoxy groups on chemoselectivity, the coupling of 13C-labeled alanyl azide 11 and alanyl phosphinothioester 15 was performed in the presence of glycyl amine 2717 (Scheme 5). Gratifyingly, no decrease in the AcAlaAlaNHBn 12 product was observed, indicating that this new phosphinothioester provides the high chemoselectivity of reagents reported previously.17
SCHEME 5.

Chemoselectivity of the Reaction of Alanyl Phosphinothioester 15 and 13C-Labeled Alanyl Azide 11
Finally, Staudinger ligations involving a glycyl residue are rapid.17,19 The NMR-based assay17 was used to monitor the rate at which 13C-labeled alanyl azide 11 reacted with alanyl phosphinothioester 15 (Table 3). In dioxane, this reaction was found to proceed with a second-order rate constant of k2 = 2.1 × 10−3 M−1s−1, which is similar to that reported previously for a Gly+Gly coupling mediated by (diphenylphosphino)methanethiol in DMF/D2O (6:1) (k2 = 7.7 × 10−3 M−1s−1).17
Conclusions
This work has illuminated factors critical for achieving the traceless Staudinger ligation of peptides at non-glycyl sites using novel phosphinothiols. Proper tuning of the electron density on phosphorous enables non-glycyl amino acid residues to be coupled rapidly and in high (>80%) yield, providing a benchmark for the sequence-independent ligation of peptides at amino acid residues. These findings enhance the utility of the Staudinger ligation for the iterative ligation of peptides in the convergent synthesis of proteins.
Experimental Procedures
[2-13C]-(2S)-2-Azido-N-benzyl-1-propionamide (11)
13C-Labeled alanyl azide 11 was synthesized as described previously.17
ClCH2P(O)(C6H4-p-Cl)2 (16), ClCH2P(O)(C6H5)2 (17)
Phosphine oxides 1629 and 1714 were prepared according to reports published previously. Spectral data. Spectral data were as reported previously.14,29
ClCH2P(O)(C6H4-p-OMe)2 (18)
Chloromethylphosphonic dichloride (20 g, 120 mmol) was dissolved in anyhdrous THF (120 mL). A solution of 4-methoxyphenylmagnesium bromide (0.5 M) in THF (480 mL, 240 mmol) was added dropwise over 1 h. The resulting mixture was stirred at reflux for 24 h. The reaction was then quenched by the addition of water (20 mL), and the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2, and the resulting solution was washed once with water (50 mL) and once with brine (50 mL). The organic layer was dried over anhydrous MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The residue was purified by automated chromatography (3% v/v MeOH in CH2Cl2). Phosphine oxide 18 was isolated as a white solid in 63% yield. Spectral data. 1H NMR (CDCl3, 400 MHz) δ 7.77–7.72 (m, 4H), 7.04–7.00 (m, 4H), 4.00 (d, J = 7.0 Hz, 2H), 3.88 (s, 6H) ppm; 13C NMR (CDCl3, 125 MHz) δ 162.9, 133.5, 133.4, 121.5, 120.37, 114.4, 114.2, 55.4, 38.1 (d, J = 73.2 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ 27.38 ppm; MS (ESI) m/z 310.0530 (MNa+ [C15H16ClO3PNa+] = 310.0526).
AcSCH2P(O)(C6H4-p-Cl)2 (19) and AcSCH2P(O)(C6H5)2 (20)
Phosphine oxides 1929 and 2014 were synthesized from phosphine oxides 16 and 17 according to reports published previously. Spectral data. Spectral data were as reported previously.14,29
AcSCH2P(O)(C6H4-p-OMe)2 (21)
Phosphine oxide 18 (5.8 g, 18.2 mmol) was dissolved in DMF (50 mL). Potassium thioacetate (2.5 g, 21.8 mmol) was then added, and the reaction mixture was stirred under Ar(g) for 18 h. The solvent was then removed under reduced pressure. The resulting oil was purified by automated chromatography (3% v/v MeOH in CH2Cl2). Phosphine oxide 21 was isolated as a clear, colorless oil in 87 % yield. Spectral data. 1H NMR (CDCl3, 400 MHz) δ 7.70–7.65 (m, 4H), 6.99–6.97 (m, 4H), 3.85 (s, 6H), 3.70 (d, J = 8.7 Hz, 2H), 2.28 (s, 3H) ppm; 13C NMR (CDCl3, 125 MHz) δ 162.9, 133.2, 123.4, 123.3, 114.4, 55.6, 30.4, 28.0 (d, J = 70.6 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ 29.19 ppm; MS (ESI) m/z 373.0627 (MNa+ [C17H19O4PSNa+] = 373.0634).
AcSCH2P(C6H4-p-Cl)2 (22) and AcSCH2P(C6H5)2 (23)
Phosphines 2229 and 2314 were synthesized from phosphine oxides 19 and 20 according to reports published previously. Spectral data Spectral data were as reported previously.14,29
AcSCH2P(C6H4-p-OMe)2 (24)
Phosphine oxide 21 (1.06 g, 2.95 mmol) was dissolved in anhydrous chloroform (10 mL). Trichlorosilane (8 mL, 79 mmol) was added, and the resulting solution was stirred under Ar(g) for 72 h. The solvent was then removed under reduced pressure. (CAUTION: Excess trichlorosilane in the removed solvent was quenched by the slow addition of saturated sodium bicarbonate in a well-ventilated hood.) The residue was purified by flash chromatography (silica gel, 3% v/v MeOH in CH2Cl2). Phosphine 24 was isolated as a white solid in 92% yield. Spectral data. 1H NMR (CDCl3, 400 MHz) δ 7.38–7.36 (m, 4H), 6.93–6.89 (m, 4H), 3.85 (s, 6 H), 3.47 (bs, 2H), 2.32 (s, 3H) ppm; 13C NMR (CDCl3, 125 MHz) δ 194.4, 160.5, 134.2, 134.0, 127.9, 127.8, 114.2, 114.2, 55.2, 30.3, 26.4 (d, J = 20.4 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ –19.71 ppm; MS (ESI) m/z 357.0679 (MNa+ [C17H19O3PSNa+] = 357.0685).
AcAlaSCH2P(C6H4-p-Cl)2 (14)
NaOH (44.8 mg, 1.12 mmol) was added to a solution of phosphine 22 (384 mg, 1.12 mmol) dissolved in degassed MeOH (12 mL). The resulting mixture was allowed to stir under Ar(g) for 1.5 h. The solvent was then removed under reduced pressure. The residue was dissolved in CH2Cl2, and this solution was washed with 2 N HCl and brine. The organic layer was dried over MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The resulting residue was used below without further purification. In a separate round-bottom flask, N-acetylalanine (157 mg, 1.2 mmol) was dissolved in anhydrous DMF (8 mL). HOBT (148 mg, 1.1 mmol) was added to the resulting solution, followed by DCC (138 mg, 1.1 mmol). Once precipitate (DCU) was observed, freshly deprotected phosphinothiol from above was added (330 mg, 1.09 mmol). The reaction mixture was allowed to stir under Ar(g) for 4 h. The solvent was then removed under reduced pressure. The residue was purified by automated chromatography (50% v/v ethyl acetate in hexanes). Phosphinothioester 14 was isolated as a white solid in 93% yield.Spectral data 1H NMR (CDCl3, 400 MHz) δ 7.36–7.32 (m, 8H), 5.28 (bt, 1H), 4.70 (bt, 1H), 3.46 (d, J = 6.3 Hz, 2H), 2.03 (s, 3H), 1.62 (m, 1H), 1.32 (d, J = 6.3 Hz, 3H) ppm; 13C NMR (CDCl3, 125 MHz) δ 199.6, 169.6, 135.8, 134.1, 133.9, 129.0, 128.9, 54.9, 25.2 (d, J = 24.6 Hz), 23.12, 18.77 ppm; 31P NMR (CDCl3, 161 MHz) δ –16.94 ppm; MS (ESI) m/z 436.0059 (MNa+ [C18H18Cl2NO2PSNa+] = 436.0065).
AcAlaSCH2P(C6H4)2 (10)
NaOH (148 mg, 3.71 mmol) was added to a solution of phosphine 23 (1.02 g, 3.71 mmol) dissolved in degassed MeOH (35 mL). The resulting mixture was allowed to stir under Ar(g) for 1.5 h. The solvent was then removed under reduced pressure. The residue was dissolved in CH2Cl2, and this solution was washed with 2 N HCl and brine. The organic layer was dried over MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The resulting residue was used without further purification below. In a separate round bottom flask, N-acetylalanine (557 mg, 4.25 mmol) was dissolved in anhydrous DMF (40 mL). HOBT (527 mg, 3.90 mmol) was added to the resulting solution, followed by DCC (805 mg, 3.90 mmol). Once precipitate (DCU) was observed, freshly deprotected phosphinothiol from above was added (900 mg, 3.87 mmol). The reaction mixture was allowed to stir under Ar(g) for 4 h. The solvent was then removed under reduced pressure. The residue was purified by flash chromatography (silica gel, 70% v/v ethyl acetate in hexanes). Phosphinothioester 10 was isolated as a white solid in 83% yield. Spectral data. 1H NMR (CDCl3, 400 MHz) δ 7.44–7.37 (m, 10H), 5.89 (bt, 1H), 4.71 (bt, 1H), 3.53 (s, 2H), 2.02 (s, 3H), 1.64 (m, 1H), 1.32 (d, J = 5.8 Hz, 3H) ppm; 13C NMR (CDCl3, 125 MHz) δ 199.8, 169.7, 136.5, 132.7, 132.6, 132.6, 129.1, 128.5, 128.5, 54.8, 25.3 (d, J = 24.3 Hz), 22.99, 18.66 ppm; 31P NMR (CDCl3, 161 MHz) δ –17.97 ppm; MS (ESI) m/z 368.0853 (MNa+ [C18H20NO2PSNa+] = 368.0845).
AcAlaSCH2P(C6H4-p-OMe)2 (15)
NaOH (330 mg, 8.26 mmol) was added to a solution of phosphine 24 (2.76 g, 8.26 mmol) dissolved in degassed MeOH (80 mL). The resulting mixture was allowed to stir under Ar(g) for 1.5 h. The solvent was then removed under reduced pressure. The residue was dissolved in CH2Cl2, and this solution was washed with 2 N HCl and brine. The organic layer was dried over MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The resulting residue was used below without further purification. In a separate round-bottom flask, N-acetylalanine (1.13 g, 8.70 mmol) was dissolved in anhydrous DMF (40 mL). HOBT (1.05 g, 7.80 mmol) was added to the resulting solution, followed by DCC (984 mg, 7.80 mmol). Once precipitate (DCU) was observed, freshly deprotected phosphinothiol from above was added (2.30 g, 7.86 mmol). The reaction mixture was allowed to stir under Ar(g) for 4 h. The solvent was then removed under reduced pressure. The residue was purified by automated chromatography (70% v/v ethyl acetate in hexanes). Phosphinothioester 15 was isolated as a white solid in 94% yield. Spectral data. 1H NMR (CDCl3, 400 MHz) δ 7.39–7.35 (m, 4H), 6.92–6.90 (m, 4H), 5.96 (bt, 1H), 5.70 (bt, 1H), 3.83 (s, 6H), 3.45 (m, 1H), 2.02 (s, 3H), 1.32 (d, J = 6.4 Hz, 2H), 1.15 (d, J = 6.7 Hz, 3H) ppm; 13C NMR (CDCl3, 125 MHz, numbers in parentheses indicate doubling due to rotational isomerism) δ 200.0, 170.2, 160.5, 134.26, 127.6, 126.6, 125.9, 117.5, 114.2, 114.2, 111.0, 55.2 (55.0), 42.2, 26.0 (d, J = 23.2 Hz), 23.30 (23.04), 18.76 ppm; 31P NMR (CDCl3, 161 MHz) δ –19.02 ppm; MS (ESI) m/z 428.1065 (MNa+ [C20H24NO4PSNa+] = 428.1056).
AcPheSCH2P(C6H4-p-OMe)2 (25)
NaOH (180 mg, 4.49 mmol) was added to a solution of phosphine 24 (1.50 g, 4.49 mmol) dissolved in degassed MeOH (45 mL). The resulting mixture was allowed to stir under Ar(g) for 1.5 h. The solvent was then removed under reduced pressure. The residue was dissolved in CH2Cl2, and this solution was washed with 2 N HCl and brine. The organic layer was dried over MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The resulting residue was used below without further purification. In a separate round-bottom flask, N-acetylphenylalanine (920 mg, 4.44 mmol) was dissolved in anhydrous DMF (20 mL). HOBT (546 mg, 4.04 mmol) was added to the resulting solution, followed by DIC (510 mg, 4.04 mmol). After stirring for 20 min, freshly deprotected phosphinothiol (1.18 g, 4.04 mmol) from above was added. The reaction mixture was allowed to stir under Ar(g) for 4 h. The solvent was then removed under reduced pressure. The residue was purified by flash chromatography (silica gel, 50% v/v ethyl acetate in hexanes). Phosphinoester 28 was isolated as an off-white, crusty solid in 90% yield. Spectral data. 1H NMR (CDCl3, 400 MHz) δ 7.37–7.33 (m, 4H), 7.26–7.25 (m, 4H), 7.09–7.07 (m, 2H), 6.91–6.89 (m, 3H), 5.71 (m, 1H), 4.96 (m, 1H), 3.81 (s, 6H), 3.50–3.34 (m, 2H), 3.12–2.96 (m, 2H), 1.93 (s, 3H) ppm; 13C NMR (CDCl3, 125 MHz) δ 199.2, 169.9, 160.8, 135.6, 134.4, 129.5, 128.9, 127.8, 127.4, 114.5, 55.4, 38.5, 26.6 (d, J = 23.1 Hz), 23.34, 21.30 ppm; 31P NMR (CDCl3, 161 MHz) δ –18.49 ppm; MS (ESI) m/z 504.1393 (MNa+ [C26H28NO4PSNa+] = 504.1374).
Ligation of 15 and 11
Alanyl phosphinothioester 15 (85 mg, 0.21 mmol) and alanyl azide 11 (43 mg, 0.21 mmol) were added to a reaction vessel and dissolved in anhydrous dioxane (2 mL). The resulting reaction mixture was allowed to stir for 12 h. Amide 12 was isolated as a white solid in 81% yield (silica gel, 3% v/v MeOH in CH2Cl2). Spectral data. Spectra data were as reported previously.17
Ligation of 25 and 11
Alanyl phosphinothioester 25 (100 mg, 0.21 mmol) and alanyl azide 11 (43 mg, 0.21 mmol) were added to a reaction vessel and dissolved in anhydrous dioxane (2 mL). The resulting reaction mixture was allowed to stir for 12 h. Amide 26 was isolated as a white solid in 84% yield (silica gel, 3% v/v MeOH in CH2Cl2). Spectral data. 1H NMR (CDCl3/CD3OD, 1:1, 400 MHz) δ 7.35–7.17 (m, 10H), 4.62–4.59 (m, 1H), 4.39–4.34 (m, 1H), 4.34 (bs, 2H), 3.10–2.90 (m, 2H), 1.94 (s, 3H), 1.35 (d, J = 7.0 Hz, 3H) ppm; 13C NMR (CDCl3, 125 MHz) δ 172.7, 171.7, 137.7, 136.3, 128.9, 128.0, 49.7, 42.6, 38.6, 22.1, 20.9, 17.6, 16.3; MS (ESI) m/z 391.1799 (MNa+ [C21H25N3O3Na+] = 391.1794).
Computational Methods
A global minimum-energy structure for the tetrahedral intermediate 4 was determined by Monte Carlo calculations using the program Macromodel® and the Merck Molecular Force Field (MMFF®). The minimum-energy structure thus obtained was imported into Gaussian 9831 and minimized further with DFT calculations30 at the B3LYP/6-31+g(d,p) level of theory. (If a negative frequency was obtained, then the structure was optimized again at the same level of theory.) Single-point energy calculations were then performed in a variety of solvents by using a conductor-like polarizable continuum model (CPCM).34 Mulliken charges were obtained from the single-point energy calculations.
Supplementary Material
General experimental procedures, NMR spectra of compounds 10, 14, 15, 18, 21, 24, 25, and 26, and atom coordinates and absolute energies from the theoretical calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We are grateful to Dr. J. A. Hodges and L. D. Lavis for contributive discussions. This work was supported by grant GM44783 (NIH). M.B.S. was supported by an ACS Division of Organic Chemistry Fellowship, sponsored by Abbott Laboratories.
References
- 1.Nilsson BL, Soellner MB, Raines RT. Annu. Rev. Biophys. Biomolec. Struct. 2005;34:91–118. doi: 10.1146/annurev.biophys.34.040204.144700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawson PE, Kent SBH. Annu. Rev. Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 3.Kent S. J. Peptide Sci. 2003;9:574–593. doi: 10.1002/psc.475. [DOI] [PubMed] [Google Scholar]
- 4.Tam JP, Xu J, Eom KD. Biopolymers. 2001;60:194–205. doi: 10.1002/1097-0282(2001)60:3<194::AID-BIP10031>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Muir TW. Annu. Rev. Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 6.Evans TC, Jr, Xu M-Q. Chem. Rev. 2002;102:4869–4883. doi: 10.1021/cr9601369. [DOI] [PubMed] [Google Scholar]
- 7.McCaldon P, Argos P. Proteins. 1988;4:99–122. doi: 10.1002/prot.340040204. [DOI] [PubMed] [Google Scholar]
- 8.Bray BL. Nat. Rev. Drug Discov. 2003;2:587–593. doi: 10.1038/nrd1133. [DOI] [PubMed] [Google Scholar]
- 9.Canne LE, Bark SJ, Kent SBH. J. Am. Chem. Soc. 1996;118:5891–5896. [Google Scholar]
- 10.Offer J, Dawson PE. Org. Lett. 2000;2:23–26. doi: 10.1021/ol991184t. [DOI] [PubMed] [Google Scholar]
- 11.Offer J, Boddy CNC, Dawson PE. J. Am. Chem. Soc. 2002;124:4642–4646. doi: 10.1021/ja016731w. [DOI] [PubMed] [Google Scholar]
- 12.Marinzi C, Bark SJ, Offer J, Dawson PE. Bioorg. Med. Chem. 2001;9:2323–2328. doi: 10.1016/s0968-0896(01)00136-5. [DOI] [PubMed] [Google Scholar]
- 13.Bode JW, Fox RM, Baucom KD. Angew. Chem. Int. Ed. Engl. 2006;45:1248–1252. doi: 10.1002/anie.200503991. [DOI] [PubMed] [Google Scholar]
- 14.Nilsson BL, Kiessling LL, Raines RT. Org. Lett. 2001;3:9–12. doi: 10.1021/ol006739v. [DOI] [PubMed] [Google Scholar]
- 15.Nilsson BL, Kiessling LL, Raines RT. Org. Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 16.Soellner MB, Nilsson BL, Raines RT. J. Org. Chem. 2002;67:4993–4996. doi: 10.1021/jo025631l. [DOI] [PubMed] [Google Scholar]
- 17.Soellner MB, Nilsson BL, Raines RT. J. Am. Chem. Soc. 2006;128:8820–8828. doi: 10.1021/ja060484k. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson BL, Hondal RJ, Soellner MB, Raines RT. J. Am. Chem. Soc. 2003;125:5268–5269. doi: 10.1021/ja029752e. [DOI] [PubMed] [Google Scholar]
- 19.Soellner MB, Dickson KA, Nilsson BL, Raines RT. J. Am. Chem. Soc. 2003;125:11790–11791. doi: 10.1021/ja036712h. [DOI] [PubMed] [Google Scholar]
- 20.Gauchet C, Labadie GR, Poulter CD. J. Am. Chem. Soc. 2006;128:9274–9275. doi: 10.1021/ja061131o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.For examples, see: He Y, Hinklin RJ, Chang JY, Kiessling LL. Org. Lett. 2004;6:4479–4482. doi: 10.1021/ol048271s. Bianchi A, Bernardi A. Tetrahedron Lett. 2004;45:2231–2234. Bianchi A, Russo A, Bernardi A. Tetrahedron: Asymmetry. 2005;16:381–386. Liu L, Hong Z-Y, Wong C-H. ChemBioChem. 2006;2006:429–432. doi: 10.1002/cbic.200500437.
- 22.For examples, see: Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. Saxon E, Luchansky SJ, Hang HC, Yu C, Lee SC, Bertozzi CR. J. Am. Chem. Soc. 2002;124:14893–14902. doi: 10.1021/ja027748x. Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. Proc. Natl. Acad. Sci. U.S.A. 2002;99:19–24. doi: 10.1073/pnas.012583299. Ovaa H, van Swieten PF, Kessler BM, Leeuwenburgh MA, Fiebiger E, van den Nieuwendijk AMCH, Galardy V, van der Marel GA, Ploegh HL, Overkleeft HS. Angew. Chem. Int. Ed. Engl. 2003;42:3626–3629. doi: 10.1002/anie.200351314. Wang CC-Y, Seo TS, Li Z, Ruparel H, Ju J. Bioconjug. Chem. 2003;14:697–701. doi: 10.1021/bc0256392. Restituyo JA, Comstock LR, Petersen SG, Stringfellow T, Rajski SR. Org. Lett. 2003;5:4357–4360. doi: 10.1021/ol035635s. Vocadlo DJ, Hang HC, Kim E-J, Hanover JA, Bertozzi CR. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9116–9121. doi: 10.1073/pnas.1632821100. Hang HC, Yu C, Kato DL, Bertozzi CR. Proc. Natl. Acad. Sci. U.S.A. 2003;100:14846–14851. doi: 10.1073/pnas.2335201100. Luchansky SJ, Argade S, Hayes BK, Bertozzi CR. Biochemistry. 2004;43:12358–12366. doi: 10.1021/bi049274f. Hang HC, Yu C, Pratt MR, Bertozzi CR. J. Am. Chem. Soc. 2004;126:6–7. doi: 10.1021/ja037692m. Hosoya T, Hiramatsu T, Ikemoto T, Nakanishi M, Aoyama H, Hosoya A, Iwata T, Maruyama K, Endo M, Suzuki M. Org. Biomol. Chem. 2004;2:637–641. doi: 10.1039/b316221d. Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Cheng J, Jaunbergs J, Weinbaum C, Tamanoi F, Falck J, Zhao Y. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12479–12484. doi: 10.1073/pnas.0403413101. Tsao M-L, Tian F, Schultz PG. ChemBioChem. 2005;6:2147–2149. doi: 10.1002/cbic.200500314. Sprung R, Nandi A, Chen Y, Kim SC, Barma D, Falck JR, Zhao YJ. Proteome Res. 2005;4:950–957. doi: 10.1021/pr050033j. Comstock LR, Rajski SR. Nucleic Acids Res. 2005;33:1644–1652. doi: 10.1093/nar/gki306. Rose MW, Xu J, Kale TA, O'Doherty G, Barany G, Distefano MD. Peptide Sci. 2005;80:164–171. doi: 10.1002/bip.20239. Xu J, DeGraw AJ, Duckworth BP, Lenevich S, Tann C-M, Jenson EC, Gruber SJ, Barany G, Distefano MD. Chem. Biol. Drug Des. 2006;68:85–96. doi: 10.1111/j.1747-0285.2006.00420.x.
- 23.Prescher JA, Dube DH, Bertozzi CR. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 24.Dube DH, Prescher JA, Quang CN, Bertozzi CR. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4819–4824. doi: 10.1073/pnas.0506855103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azoulay M, Tuffin G, Sallem W, Florent J-C. Bioorg. Med. Chem. Lett. 2006;16:3147–3149. doi: 10.1016/j.bmcl.2006.03.073. [DOI] [PubMed] [Google Scholar]
- 26.Yoon-Sik Lee and his coworkers have reported that 14 different non-glycyl couplings mediated by (diphenylphosphino)methanethiol proceed in "quantitative yield"( Kim H, Cho JK, Aimoto S, Lee Y-S. Org. Lett. 2006;8:1149–1151. doi: 10.1021/ol0530629.). These results cannot be reproduced in our laboratory (ref 17), and are inconsistent with reports from other laboratories ( Merkx R, Rijkers DTS, Kemmink J, Liskamp RMJ. Tetrahedron Lett. 2003;44:4515–4518. He Y, Hinklin RJ, Chang J, Kiessling LL. Org. Lett. 2004;6:4479–4482. doi: 10.1021/ol048271s.).
- 27.Merkx R, Rijkers DTS, Demink J, Liskamp RMJ. Tetrahedron Lett. 2003;44:4515–4518. [Google Scholar]
- 28.We chose an Ala+Ala coupling because alanine is especially abundant in proteins (8.0% of all residues; ref 7).
- 29.Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. J. Org. Chem. 2005;70:7123–7132. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]
- 30.Koch W, Holthausen MC. A Chemist's Guide to Density Functional Theory. 2nd Ed. Germany: Wiley–VCH: Weinheim; 2001. [Google Scholar]
- 31.Frisch MJ, Trucks GW, Schlegel GW, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian 98, Revision A.9. Pittsburgh PA: Gaussian, Inc.; 1998. [Google Scholar]
- 32.Catalan J, Lopez V, Perez P, Martinvillamil R, Rodriguez JG. Liebigs Ann. 1995:241–252. [Google Scholar]
- 33.Merkx R, Rijkers DTS, Kemmink J, Liskamp RMJ. Tetrahedron Lett. 2003;44:4515–4518. [Google Scholar]
- 34.(a) Klamt A, Schüürmann G. J. Chem. Soc., Perkin Trans. 1993;2:799. [Google Scholar]; (b) Andzelm J, Kölmel C, Klamt A. J. Chem. Phys. 1995;103:9312–9320. [Google Scholar]; (c) Barone V, Cossi M. J. Phys. Chem. A. 1998;102:1995–2001. [Google Scholar]; (d) Cossi M, Rega N, Scalmani G, Barone V. J. Comput. Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental procedures, NMR spectra of compounds 10, 14, 15, 18, 21, 24, 25, and 26, and atom coordinates and absolute energies from the theoretical calculations. This material is available free of charge via the Internet at http://pubs.acs.org.