

Abstract
DesA3 (Rv3229c) from Mycobacterium tuberculosis is a membrane-bound stearoyl coenzyme A Δ9 desaturase that reacts with the oxidoreductase Rv3230c to produce oleic acid. This work provides evidence for a mechanism used by mycobacteria to regulate this essential enzyme activity. DesA3 expressed as a fusion with either a C-terminal His6 or c-myc tag had consistently higher activity and stability than native DesA3 having the native C-terminal sequence of LAA, which apparently serves as a binding determinant for a mycobacterial protease/degradation system directed at DesA3. Fusion of only the last 12 residues of native DesA3 to the C terminus of green fluorescent protein (GFP) was sufficient to make GFP unstable. Furthermore, the comparable C-terminal sequence from the Mycobacterium smegmatis DesA3 homolog Msmeg_1886 also conferred instability to the GFP fusion. Systematic examination revealed that residues with charged side chains, large nonpolar side chains, or no side chain at the last two positions were most important for stabilizing the construct, while lesser effects were observed at the third-from-last position. Using these rules, a combinational substitution of the last three residues of DesA3 showed that either DKD or LEA gave the best enhancement of stability for the modified GFP in M. smegmatis. Moreover, upon mutagenesis of LAA at the C terminus in native DesA3 to either of these tripeptides, the modified enzyme had enhanced catalytic activity and stability. Since many proteases are conserved within bacterial families, it is reasonable that M. tuberculosis will use a similar C-terminal degradation system to posttranslationally regulate the activity of DesA3 and other proteins. Application of these rules to the M. tuberculosis genome revealed that ∼10% the proteins encoded by essential genes may be susceptible to C-terminal proteolysis. Among these, an annotation is known for less than half, underscoring a general lack of understanding of proteins that have only temporal existence in a cell.
Mycobacterium tuberculosis is a human pathogen that causes tuberculosis, one of the world's deadliest diseases (10). Current estimates are that up to one-third of the world's population may be infected with M. tuberculosis; over two million people die from tuberculosis-related diseases each year (5, 10, 52). Tuberculosis has persisted due to the lack of compliance with drug treatment regimes, the appearance of multiple-drug-resistant strains, and the AIDS epidemic (5, 36, 52). Consequently, increasing our understanding of the origins of pathogenicity of M. tuberculosis is a high priority of tuberculosis research.
DesA3 (encoded by gene rv3229c) is a membrane-bound stearoyl coenzyme A (CoA) desaturase that works together with oxidoreductase Rv3230c (encoded by gene rv3230c) to produce oleic acid, an essential constituent of mycobacterial membrane phospholipids and triglycerides (6, 22, 32, 36, 45). Due to the physiological importance of oleic acid to bacteria, DesA3 is among the ∼200 genes required for pathogen survival inside the granuloma enclosing a dormant tuberculosis infection (37). DesA3 is a target of the second-line antituberculosis drug isoxyl, which has been used with isoniazid in multiple-drug therapy (35, 36).
Mycobacterium smegmatis, a nonpathogenic bacterium that grows relatively faster than pathogenic mycobacteria and that transforms efficiently, has been widely used as a host for the expression of target genes from pathogenic mycobacteria (41). Thus, many genes from pathogenic mycobacteria yield folded proteins and active enzymes when expressed in M. smegmatis, whereas the same genes expressed in Escherichia coli yield neither folded proteins nor active enzymes (13, 43, 53). Currently, several different types of vectors have been developed for constitutive or inducible expression in M. smegmatis in order to access this biological capability (3, 11, 18, 34, 42).
Our previous studies showed that M. tuberculosis DesA3 expressed in M. smegmatis as a fusion with either a C-terminal His6 tag or a myc tag had higher catalytic activity and stability than DesA3 expressed with the natural C-terminal sequence (6). In this study, we have further investigated the role of the C-terminal sequence in this apparent increase in activity by the use of enhanced folding green fluorescent protein (GFP) fused to either DesA3 or peptides corresponding to the C-terminal sequence. The results indicate that native DesA3 is rapidly degraded by a mycobacterial protein degradation system that specifically targets the residues LAA at the C terminus.
MATERIALS AND METHODS
DNA and plasmid reagents.
Table 1 lists the bacterial strains and plasmids used in this work. The tetracycline-inducible mycobacterial expression vector pUV15TetORs (11) was obtained from Sabine Ehrt (Department of Microbiology and Immunology, Weill Medical College, Cornell University) and modified for this study. In brief, the vector pUV15TetORs (11) was digested using PacI and EcoRV, and the digested vector was ligated to the annealed multiple-cloning-site linker primers containing the compatible cohesive ends (L1, TAACAGCTGGCGATCGCCAGTACTGGATCCCCTGCAGGCTTAAGATTTAAATGAT; L2, ATCATTTAAATCTTAAGCCTGCAGGGGATCCAGTACTGGCGATCGCCAGCTGTTAAT) to create pTET. The vector pGFP was created to study the expression of GFP with a modified C terminus. The GFP variant with enhanced folding efficiency and fluorescence yield (39) was amplified from pUV15TetORs and cloned into pTET by using the PacI and AsiSI sites introduced in the multicloning site. A control vector, pGFPStop, that contained a stop codon after GFP was made in the same way. The linear maps of pTET and pGFP are shown in Fig. 1.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| Escherichia coli strain E. cloni 10G | F−mcrA Δ(mrr-hsdRMS-mcrBC) endA1 recA1 φ80dlacZΔM15 ΔlacX74 araD139 Δ(ara leu)7697 galU galK rpsL nupG λ−tonA | Lucigen |
| E. coli Rosetta 2 | F−ompT hsdSB(rB− mB−) gal dcm pRARE2 (Camr and tRNA genes argU, argW, argX, ileX, glyT, leuW, proL, metT, thrT, tyrU, and thrU) | Novagen |
| Mycobacterium smegmatis mc2155 | Mycobacterial expression host with high transformation efficiency | ATCC 700084 |
| Plasmids | ||
| pQE80 | Commercially available Escherichia coli expression vector | Qiagen |
| pVV16 | Shuttle vector and mycobacterial expression vector controlled by constitutive Hsp60 promoter | 23 |
| pMYC | Derived from PVV16 to express target genes with a C-terminal myc tag | This work |
| pQE80-Rv3230c | rv3230c in pQE80 | 6 |
| pVV16-DesA3 | desA3 in pVV16 | 6 |
| pVV16-DesA3-His6 | DesA3-His6 in pVV16 | This work |
| pMYC-DesA3-myc | DesA3-myc expressed from pMYC | This work |
| pMYC-DesA3-DKD | DesA3 with the last three amino acid changed from LAA to DKD expressed from pMYC | This work |
| pMYC-DesA3-LEA | DesA3 with the last three amino acid changed from LAA to LEA expressed from pMYC | This work |
| pUV15TetORs | Cloning shuttle vector and mycobacterial expression vector controlled by tetracycline-inducible promoter | 11 |
| pTet | Modified from pUV15TetORs to replace the original GFP gene with a useful multicloning site for further cloning work | This work |
| PGFPStop | Enhanced GFP gene with a stop codon in the multicloning site of pTet. | This work |
| Pgfp | Enhanced GFP gene without a stop codon in the multicloning site of pTet. This vector was used to make the constructs with variations in DesA3 C terminus described below. | This work |
| pGFP-DesA3-LAA | Expression of the GFP with a C-terminal fusion to native DesA3-LAA | This work |
| pGFP-DesA3-LAK | Expression of the GFP with a C-terminal fusion to native DesA3-LAK | This work |
| pGFP-DesA3-LAD | Expression of the GFP with a C-terminal fusion to native DesA3-LAD | This work |
| pGFP-DesA3-LKA | Expression of the GFP with a C-terminal fusion to native DesA3-LKA | This work |
| pGFP-DesA3-LDA | Expression of the GFP with a C-terminal fusion to native DesA3-LDA | This work |
| pGFP-DesA3-KAA | Expression of the GFP with a C-terminal fusion to native DesA3-KAA | This work |
| pGFP-DesA3-DAA | Expression of the GFP with a C-terminal fusion to native DesA3-DAA | This work |
FIG. 1.

Vector maps. (A) pTET derived from pUV15TetORs (11). This vector contains a tetracycline-inducible promoter (tetO), a multiple cloning site (MCS), transcription terminators (T), a constitutively expressed tetracycline repressor cassette (puv15-tetR), a hygromycin resistance cassette (hygR), a mycobacterial replication origin (MYC ori), and an E. coli replication origin (ColE1 ori). (B) pGFP obtained by insertion of GFP with no stop codon.
Total genomic DNA of Mycobacterium tuberculosis H37Rv was obtained from the TB Research Materials Facility at Colorado State University (provided by J. Belisle, Director, NIH NIAD NO1AI75320). This material was used to clone the genes Rv3229c and Rv3230c by PCR. Pfu DNA polymerase was from Stratagene (La Jolla, CA). Oligonucleotide primers were obtained from Integrated DNA Technologies, Inc. (Coralville, IA). The pSMART-HC Amp CloneSmart blunt cloning kit (Lucigen, Middleton, WI) was used for initial cloning and sequencing in E. coli.
The expression vector pQE80 (Qiagen, Valencia, CA) was used to express Rv3230c in E. coli (Table 1). The shuttle/expression vector pVV16 (23) was used to express DesA3 either with or without a C-terminal His6 tag under the control of the transcriptional and translational signals of the Hsp60 promoter in M. smegmatis (ATCC 700084). pMYC (modified from pVV16) was used to express DesA3 with a C-terminal myc tag or with two variations of the C-terminal amino acid sequence. Big Dye DNA sequencing (Applied Biosystems, Foster City, CA) was performed in the University of Wisconsin Biotechnology Center to verify the coding sequence of all expression plasmids.
Cloning of DesA3.
The untagged rv3229c gene (encoding DesA3) and the rv3229c gene modified to encode C-terminally His6- or myc-tagged DesA3 were amplified using M. tuberculosis total genomic DNA, and the primers are shown in Table S1 in the supplemental material. The PCR contained 10% dimethyl sulfoxide and consisted of 30 cycles of melting, annealing, and extending at temperatures of 94°C, 55°C, and 72°C, respectively. The resulting DNA fragments were purified by gel electrophoresis and extracted using a QIAquick gel extraction kit (Qiagen). The PCR product was ligated into pSMART-HCAmp for sequencing. A plasmid with the correct sequence was digested with NdeI and HindIII and ligated into the similarly digested pVV16 or pMYC to create expression vectors pVV16-DesA3, pVV16-DesA3-His6, or pMYC-DesA3-myc. E. cloni 10G transformants (Lucigen) containing these plasmids were cultured on either Luria-Bertani broth or agar medium containing 50 μg/ml of kanamycin.
The desA3 gene was amplified to mutate the C-terminal LAA residues to either DKD or LEA by using the primers shown in Table S1 in the supplemental material. The PCR and cloning steps were the same as those used for the cloning of DesA3 into pMYC described above. These two expression vectors were named pMYC-DesA3-DKD and pMYC-DesA3-LEA, respectively.
Cloning of GFP fused to DesA3.
The desA3 gene or variants with modifications at the 3′ end were amplified using the forward and reverse primers shown in Table S1 in the supplemental material. The PCR steps were the same as those used for the cloning of DesA3 described above. After a pSMART-HCAmp plasmid with the correct sequence was identified, it was digested with AsiSI and SwaI, and the insert was ligated into the similarly digested pGFP. The individual E. coli transformants containing these plasmids were cultured on either Luria-Bertani broth or agar medium containing 200 μg/ml of hygromycin. These vectors were used to express GFP with a C-terminal fusion to either native DesA3 or to variants containing single Lys or Asp substitutions in each of the last three amino acids of the native enzyme, LAA.
Cloning of GFP fused to DesA3 C-terminal peptides.
Primer pairs encoding several variations of the codons for the last 12 residues from DesA3 or from Msmeg_1886, the homologous desaturase from M. smegmatis, are listed in Table S2 in the supplemental material. Vector pGFP was digested using AsiSI and SwaI, and the annealed primer pairs, containing the compatible cohesive ends, were ligated to the digested vector. The individual E. coli transformants containing these plasmids were cultured on either Luria-Bertani broth or agar medium containing 200 μg/ml of hygromycin. The presence of the DNA sequence encoding the desired variation was confirmed by sequencing in the reverse direction. These vectors were used to express GFP with a C-terminal fusion to the different variations of the last 12 amino acids from DesA3 or to the native C terminus of Msmeg_1886.
Assay of DesA3.
Methods for expressing DesA3 in M. smegmatis, expressing Rv3230c in E. coli, preparing cell extracts containing these two proteins, and assaying DesA3 activity were as previously reported (6).
Expression of GFP and quantification of fluorescence.
M. smegmatis was transformed by electroporation, plated onto selective Middlebrook 7H10 agar enriched with Middlebrook oleic acid-albumin-dextrose-catalase (Becton Dickinson, Spark, MD) containing 50 μg/ml hygromycin, and incubated at 37°C. After 4 days, colonies from each transformation were used to inoculate 500 μl of Middlebrook 7H9 broth (Becton Dickinson, Spark, MD) supplemented with 0.2% glycerol, 0.05% Tween 80 and 50 μg/ml hygromycin. The liquid cultures were incubated for ∼24 h on a shaker (250 rpm) at 37°C. The culture was then diluted 100-fold into 5 ml of fresh liquid medium and grown as before for ∼15 h to late logarithmic growth phase. At that time, the culture was induced by addition of anhydrotetracycline to a final concentration of 200 ng/ml for ∼6 h with shaking. During this time, fluorescence obtained from the pGFP control plasmid reached a maximum value. After 6 h, the optical density was measured, and the cells were harvested by centrifugation and washed once in phosphate-buffered saline containing 0.05% Tween 20 (Sigma-Aldrich, St. Louis, MO). The cells were resuspended in the wash buffer to an optical density at 600 nm of 2 units per 0.1 ml and reincubated at 37°C with shaking. Following this shift back to noninducing conditions, 0.1-ml samples of the cell culture were transferred into black 384-well plates at various time intervals, and fluorescence was measured using a fluorescence microplate reader (Molecular Devices, Sunnyvale, CA) with excitation at 485 nm and detection of emission at 515 nm.
Infrared fluorescence Western blotting.
Infrared fluorescence Western blot analysis was conducted to confirm the gradual degradation of some GFP variants. Cells used to correlate GFP fluorescence with the presence of a polypeptide were harvested at various time intervals and immediately boiled in denaturing sample buffer. Proteins were separated by denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis in a 4% to 20% polyacrylamide gel and transferred to nitrocellulose membranes. The blotted proteins on the nitrocellulose membrane were blocked in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) and incubated with primary antibody to the GFP N terminus, followed by infrared-labeled goat anti-rabbit IRDye 800 secondary antibody (LI-COR Biosciences). Signals were detected and quantified using the Odyssey infrared imaging system (LI-COR Biosciences).
Other analyses.
The alignment of the M. tuberculosis DesA3 gene cluster with the genome sequences of other mycobacteria was obtained from The Institute for Genomic Research website (http://www.jcvi.org/).
RESULTS
Influence of the C terminus on DesA3 activity.
Stearoyl-CoA Δ9 desaturase activity could be reconstituted by combining fractions containing DesA3-His expressed in M. smegmatis with fractions containing Rv3230c expressed in E. coli (6). Preliminary comparisons showed that fractions containing native DesA3 prepared by the same methods had considerably lower catalytic activity. To further investigate the origin of this difference, the activity of the following three constructs was studied: DesA3, DesA3-His (containing a C-terminal His6 tag), and DesA3-myc (containing a C-terminal myc tag). Figure 2 shows that native DesA3 had much lower activity, while DesA3-His and DesA3-myc showed similar enhanced activity.
FIG. 2.

Effect of adding different C-terminal tags to DesA3 on stearoyl-CoA Δ9 desaturase activity. In this figure, T, S, and P correspond to the total, supernatant, and pellet fractions of lysates prepared from M. smegmatis expressing the different DesA3 constructs, respectively. The positions of migration of the substrate (18:0), the product (18:1), and a side product separated by thin-layer chromatography are indicated. Lanes: L1 to L3, native DesA3; L4 to L6, DesA3-His (containing a C-terminal His6 tag); L7 to L9, DesA3-myc (containing a C-terminal myc tag).
Stability of GFP-DesA3 fusions in M. smegmatis.
GFP has often been used as a reporter for the expression and stability of expressed fusion proteins (1, 4, 8, 39). Figure 3 shows that GFP, with LYK at the C terminus (construct 3A), was expressed and matured to an active form in M. smegmatis and that the induced fluorescence was stable over an ∼8-h period after removal of the inducer. Fusion of native DesA3 (∼48 kDa) to GFP gave LAA at the C terminus (construct 3B), and expression of this fusion protein gave only a low level of fluorescence. In contrast, fusion of variants of DesA3 containing a single lysine or aspartic acid substitution in the different positions of the LAA sequence (constructs 3C to 3H) gave increased GFP fluorescence except for the variant with residues KAA at the C terminus (construct 3G). For example, the GFP-DesA3-LAD fusion protein (construct 3D) gave ∼45% of the fluorescence observed from the unmodified GFP control. Upon consideration that the fusion protein would be an integral membrane protein of ∼75 kDa as opposed to the soluble GFP control (∼25 kDa), some differences in the overall level of fluorescence were expected. However, the different patterns of stability were not. The levels of fluorescence and stability observed for the other constructs are shown in Fig. 3.
FIG. 3.

Fluorescence of GFP expressed in M. smegmatis and influence of fusing DesA3 to its C terminus. The different protein constructs examined in this experiment are shown in the inset, and the three-residue sequences shown to the right of the constructs represent the C terminus. For example, in construct 3B, the C terminus of native DesA3 is LAA. The fluorescence of GFP was measured 0, 1, 2, and 4 h after the removal of the anhydrotetracycline used to induce expression.
Previous studies of the E. coli C-terminally targeted protein degradation systems ClpXP and Tsp showed that small and uncharged residues (Ala, Cys, Ser, Thr, and Val) in the last three positions of a protein sequence enhanced the degradation (26, 27). Since the GFP-DesA3 fusion had these small residues at the C terminus, it seemed likely that M. smegmatis might also have a C-terminally directed protein degradation system. This possibility was supported by the increased fluorescence observed when the C-terminal residue was changed.
GFP-peptide fusions.
In order to more simply address the role of the C terminus in the stability of GFP expressed in M. smegmatis, GFP variants with only 12 residues added to the C terminus were also studied. The 12 residues came from the C terminus of native DesA3 or mutated versions of this peptide sequence. Like the results obtained with fusions of the complete integral membrane DesA3, Fig. 4 shows that expression of the GFP in construct 4B (containing LAA at the C terminus) in M. smegmatis gave essentially no fluorescence (less than 10% of GFP control [construct 4A]). This loss of fluorescence indicated that the 12 residues from the C terminus of DesA3 were sufficient to stimulate a protein degradation process. In contrast, expression of the GFP in construct 4C, in which only the C-terminal Ala residue was changed to Asp, gave ∼60% of the fluorescence of the control GFP, further supporting the presence of a C-terminus-specific protein degradation system. Cells expressing the GFP in construct 4C (addition of 12 residues ending in LAD to GFP only) also had somewhat higher fluorescence than cells expressing the GFP in construct 3D (a GFP-DesA3 fusion ending in LAD) (Fig. 3), possibly because the former was better expressed and better folded than the latter.
FIG. 4.

Changes in the intensity and stability of fluorescence when GFP was expressed in M. smegmatis with a C-terminal fusion to a 12-residue sequence from DesA3 (constructs 4B and 4C) or the corresponding sequence of the DesA3 homolog from M. smegmatis, Msmeg_1886 (constructs 4D and 4E). The fluorescence of GFP was measured 0, 1, 2, 4, 6, and 8 h after removal of anhydrotetracycline used to induce expression. A change of the C-terminal Ala residue to Asp increased the level of GFP fluorescence.
M. smegmatis also contains an integral membrane desaturase homolog of DesA3, called Msmeg_1886. When the 12 residues from the C terminus of Msmeg_1888 were fused to the GFP in construct 4D, stable fluorescence was not observed, while expression of the GFP in construct 4E, with the C-terminal Ala mutated to Asp, gave stable fluorescence. Thus, M. smegmatis also degraded GFP when fused to the C-terminal residues of its own desaturase, suggesting that the process of C-terminal targeted degradation may be conserved for the membrane stearoyl-CoA desaturases in mycobacteria.
Amino acid specificity.
To further investigate the preference for C-terminal sequences in this mycobacterial degradation process, the last 5 residues of the 12-residue C-terminal fusion from DesA3 were systematically varied. Each of the last three residue positions was modified individually, while the fourth and fifth positions from the C terminus were modified in combination. M. smegmatis cells expressing each of the different fusion proteins were measured for GFP fluorescence, and the results are shown in Fig. 5.
FIG. 5.

Residue specificity of the mycobacterial C-terminally directed degradation/proteolysis system. The fluorescence intensity was monitored over an 8-h period for the GFP-mtX12 variants with each of the last three tail positions modified individually or with the fourth and fifth positions modified together. The last 5 amino acids of the native DesA3 are DDLAA. The amino acid modified at each position is labeled as X. In order to analyze all these variants simultaneously in a time scale that would not be influenced by the observed degradation, a single experiment instead of triplicate experiments was performed. However, similar results were obtained in two other independent experiments (data not shown).
At position 1 (defined here as the C-terminal residue) (Fig. 5A), GFP variants with charged side chains (Lys, Arg, Asp, Glu, and His), a large nonpolar side chain (Phe), or no side chain (Gly) all exhibited ∼50% of the fluorescence observed for the GFP control; notably, all variants exhibited substantially higher fluorescence than GFP with DDLAA at the C terminus. Most of these variants gave GFP fluorescence that was stable during the time of the experiment, although the Gly and Phe substitutions gave a weak time-dependent loss of fluorescence. At position 2 (the penultimate amino acid residue) (Fig. 5B), GFP variants with the set of investigated residue changes all exhibited higher fluorescence than the GFP with DDLAA at the C terminus. However, these had markedly different levels of fluorescence and stabilities. Among these, the Glu variant exhibited a level of fluorescence that was equivalent to that of the GFP positive control and was also stable over the time of the experiment. In contrast, the Phe variant initially exhibited ∼50% of the fluorescence of the GFP control but lost ∼75% of the fluorescence during the time of the incubation. The remainder of the variants exhibited ∼50% of the fluorescence of the GFP control and exhibited only minor changes in fluorescence over time. At position 3 (third residue back from the C terminus) (Fig. 5C), only the Gly variant exhibited substantial stable fluorescence. The variants with Asp, Glu, and His exhibited only weak fluorescence, and these were unstable during the time of the experiment. Furthermore, variants with basic residues (Arg and Lys) and a large nonpolar residue (Phe) were comparable to that of GFP with DDLAA at the C terminus in that they did not yield fluorescence.
In the fourth and fifth positions (Fig. 5D), variants with the substitution of AA, KK, or RR for DD did not stabilize the fluorescence, suggesting that these two positions do not strongly influence the protein degradation process in M. smegmatis.
Infrared fluorescence Western blotting.
In order to distinguish among the alternative possibilities for the origin of decreased GFP fluorescence (e.g., differential protein expression, incorrect protein folding, or protein degradation), quantitative Western blotting using a near-infrared fluorescence detection system was used. The initial fluorescence detected for the GFP control and several labile GFP constructs with modified C-terminal sequences correlated well with the relative protein amount detected by Western blot analysis, suggesting that neither protein expression nor protein folding was the reason for less fluorescence for some variants. Furthermore, Fig. 6 shows that, compared to the GFP control, which had stable fluorescence and detected protein levels, the variants that showed the gradual fluorescence decrease over time also exhibited a correlated decrease in protein detected by Western blotting. This result implicates protein degradation in the loss of fluorescence after the cells were switched to the noninducing conditions.
FIG. 6.

Infrared fluorescence Western blot analysis of selected GFP variants. (A) Infrared fluorescence Western blots. Several unstable protein constructs shown in Fig. 5 were examined in this experiment and labeled by their last three amino acids. Cells were harvested 0, 2, 4, and 8 h after removal of the inducer and immediately boiled in denaturing sample buffer. The arrow indicates the position of GFP or its C-terminal variants. (B) Integrated fluorescence intensities of target bands identified in panel A.
Combinational substitution.
The results shown in Fig. 4 and 5 show that most of the GFP fusions gave only ∼50% of the fluorescence of the GFP control, although substitution of Glu alone into the penultimate position gave an ∼2-fold increase in fluorescence compared to that of the GFP control. Figure 7 shows that combined substitution of DKD for LAA at the C terminus also gave fluorescence comparable to that of the GFP control, which was again nearly double that obtained from any of the single-substitution variants except the previously mentioned Glu at the second position. These results suggest that the last three positions of the C terminus may cooperatively determine the specificity of the degradation system.
FIG. 7.

Combinational substitution of residues into the C terminus of GFP-mtX12 variants. The fluorescence intensity was monitored over an 8-h period for the GFP-mtX12 variants with each of the last three tail positions modified individually or together. The GFP variant end with DKD (construct 7F) has fluorescence intensity comparable to that of the GFP positive control.
Catalytic activity of DesA3 C-terminal variants.
Fig. 8 shows results of catalytic assays of DesA3. Preparation of the cell-free lysates and membrane fractions takes several hours, so there is substantial opportunity for degradative processes to occur. The activity observed from DesA3 with a C-terminal myc tag (Fig. 8, lane L1) was considerably higher than that observed from native DesA3 (Fig. 8, lane L2). Since the GFP fusion studies showed that substitution of either DKD or LEA for the last three amino acids, LAA, could yield high, stable fluorescence, these changes were made to the last three residues of DesA3. Lanes L3 and L4 of Fig. 8 show that these changes gave stearoyl-CoA Δ9 desaturase activity comparable to that of the DesA3-myc lysate (Fig. 8, lane L1), while the activity of the native DesA3 with a C terminus ending in LAA (Fig. 8, lane L2) was nearly completely lost in the time of preparation. Thus, the improved fluorescence intensity and stability given by changes made to the GPF fusion proteins corresponded to improved activity of M. tuberculosis DesA3 expressed in M. smegmatis, presumably by protection of the expressed protein from degradation.
FIG. 8.
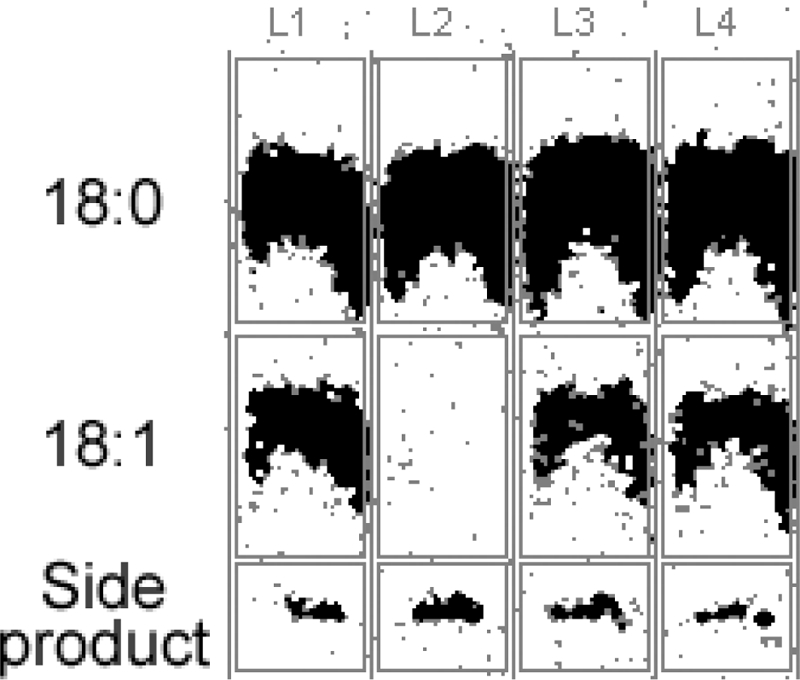
Stearoyl-CoA Δ9 desaturase activity assays for DesA3 variants expressed in M. smegmatis. Lanes: L1, the total lysate expressing DesA3-myc (containing a C-terminal myc tag); L2, native DesA3-LAA; L3, DesA3-DKD; L4, DesA3-LEA. The positions of migration of the substrate (18:0), product (18:1), and a side product in the thin-layer chromatography separation are indicated.
DISCUSSION
Protein degradation systems.
Within living cells, proteolysis is a ubiquitous and often selective process. In yeast and higher eukaryotes, some proteins are marked for degradation by modification with ubiquitin and targeted to the proteasome (7, 20, 24), and other specific routes for protein processing have been identified. Interestingly, proteolytic processing is also used to control mammalian desaturase activity (25, 31), although the proteolysis complexes used and the recognition sequences are distinct. This common proteolytic, posttranslational approach to maintaining control of stearoyl-CoA desaturase activity serves to emphasize the importance of this key branch point in fatty acid metabolism (9).
In E. coli and other bacteria, several types of C-terminally targeted protein degradation complexes have been identified, including the ClpXP, ClpAP, FtsH (HflB), and Tsp (Prc) systems, as major contributors to protein degradation in E. coli (15, 17, 26). Among these, the Clp proteases are cytoplasmic, FtsH is a membrane protease, and Tsp is a periplasmic protease, ensuring that tagged proteins can be degraded in all cellular compartments. These systems also have different substrate recognition properties and different contributions to proteolytic processing.
ClpXP, ClpAP, and Tsp are C-terminally targeted systems with preferences for small and uncharged residues (Ala, Cys, Ser, Thr, Val) in the last three sequence positions of the protein targeted for degradation (15, 26), with a few ClpAP substrates identified to alternatively have an N-terminal degradation signal (47). The ClpXP and ClpAP complexes consist of a hexameric Clp/Hsp100 family ATPase, either ClpA or ClpX, and ClpP (14, 16, 21, 28, 30, 33, 46, 48-51). ClpP is a serine peptidase whose active site faces an internal chamber. In both ClpXP and ClpAP, the ATPase mediates substrate recognition and catalyzes energy-dependent protein unfolding. The unfolded substrate then translocates through a channel in the hexameric ClpX or ClpA structure into the ClpP chamber for degradation. Both of these systems recognize proteins having a C-terminal SsrA tag (AANDENYALAA), which is appended to nascent polypeptide chains that have stalled on bacterial ribosomes by a cotranslational process (15). Although the DesA3 C terminus ends with LAA, the adjacent sequence (AKSVTEPDD) is not otherwise closely related to the sequence of the SsrA tag.
The ClpXP system is similar to ClpAP in that it recognizes the α-carboxyl group and the C-terminal LAA residues of the SsrA tag as binding determinants. However, ClpXP uses the SspB adaptor protein bound to a secondary site of the SsrA tag to enhance recognition of the tagged protein (12, 19). SspB binding to the SsrA tag inhibits the ClpAP system, pointing to structural differences between these two protease systems. About half of the known natural ClpX substrates are similar to SsrA-tagged proteins in that they have nonpolar side chains at the penultimate and C-terminal residues (30). Moreover, in some sequences, a C-terminal LAA tripeptide is sufficient to allow ClpXP-dependent degradation, while substitution of Asp for either of the last two Ala residues completely blocked substrate recognition by ClpX (12).
FtsH is the only essential protease in E. coli (17). It is a membrane-anchored protein with its active site in the cytoplasm (44). FtsH can recognize SsrA tagged proteins and substrates with a C-terminal nonpolar sequence located either in the cytoplasm or the membrane (17). It has been suggested that the major role of FtsH is to degrade abnormal membrane proteins that have their C terminus in the cytoplasm, regardless of whether they are SsrA tagged or not.
The periplasmic endoprotease Tsp cleaves at discrete sites throughout the polypeptide chain. This reaction also depends upon the identity of the substrate's C-terminal sequence and requires the presence of a free α-carboxyl group (26, 40). Tsp recognizes a relatively apolar C-terminal sequence (e.g., WVAAA or LAA), while proteins having a polar C-terminal sequence (e.g., RSEYE) are not cleaved. The identity of the C-terminal residue of this sequence is the most important determinant of the rate of reaction, and small, uncharged residues (Ala, Cys, Ser, Thr, Val) are preferred for proteolytic processing. Furthermore, nonpolar residues are preferred at the second and third positions, but larger and more-hydrophobic side chains are also acceptable at these positions.
Mycobacterial protein degradation system.
The presence of a Clp-like protease system was originally proposed for M. smegmatis based on biochemical studies (29), and more recently, the completion of the M. smegmatis genome sequence (The Institute for Genomic Research website [http://www.jcvi.org/]) has given further support to this proposal. Thus, Msmeg_4671 is annotated as the ClpX homolog, while Msmeg_4672 and Msmeg_4673 are annotated as the subunits of a dimeric ClpP homolog. In the M. tuberculosis genome, these comparable genes are rv2457c, rv2460c, and rv2461c, respectively. M. smegmatis contains the chaperone-like ATPase homologs ClpB (Msmeg_0732) and ClpC (Msmeg_6091), while the comparable genes in M. tuberculosis are rv0384c and rv3596c. M. smegmatis contains an additional ClpX isoform (Msmeg_2792), and its homolog in M. tuberculosis is rv2667. Moreover, M. smegmatis has an FtsH homolog, Msmeg_6105, and the comparable gene in M. tuberculosis is rv3610c. No apparent Tsp homologs have been identified, which may reflect the differences between the extracellular structure of mycobacteria and that of gram-negative bacteria such as E. coli.
Considering the membrane localization of DesA3, FtsH might reasonably be involved in the control of DesA3 stability. However, the degradation of DesA3 might also be controlled by either of the cytoplasmic Clp systems, as suggested by the facile degradation of soluble GFP fusions shown here.
Specificity of M. smegmatis protein degradation.
By using a set of GFP variants in which each of the last five residues of the polypeptide chain was modified, we found that the last three residues had the most influence on the stability of the expressed fusion protein. No significant improvements in the stabilities of the expressed fusion proteins were observed for the variants with combinational modifications for the fourth and fifth positions. Overall, this specificity is consistent with either or both Clp-like and FtsH protein degradation systems.
For expression in M. smegmatis, charged side chains (Lys, Arg, Asp, Glu, and His), a large nonpolar side chain (Phe), or no side chain (Gly) at the C terminus stabilized the GFP fusion proteins (position 1 [Fig. 5A]). These classes of residues were also stabilizing at the penultimate amino acid residue (position 2 [Fig. 5B]) but with the notable exception that the Glu variant had enhanced fluorescence and stability comparable to those of the GFP control. Placement of Phe at the penultimate position yielded a unique, progressive loss of fluorescence after the inducer was removed. At the third position from the C terminus (Fig. 5C), only the Gly variant exhibited and maintained fluorescence. Other variants at the third position with either acidic or basic side chains (Asp, Glu, His, Arg, and Lys) or Phe exhibited weak fluorescence, and these were not stable during the time course of the experiment.
Compared with the E. coli ClpXP and Tsp systems, the mycobacterial system(s) apparently differs in the relative contributions of the last three residues for substrate recognition. Thus, the results shown in Fig. 5 suggest that position 2 may contribute more to specificity for the mycobacterial degradation system, while position 1 appears to be dominant in ClpXP and Tsp. The increased stability observed for the GFP variant with Glu was not replicated with Asp at position 2 (Fig. 5B), which suggests a combination of both charge and size effects in the recognition process in mycobacteria.
The results shown in Fig. 5 and 7 suggested that a combination of changes from LAA to either LEA or DKD at the C terminus would promote stability of the GFP fusion protein, and this was indeed true. This result confirms that each of the last three positions of the C terminus makes some contributions to the recognition specificity for the mycobacterial degradation. Moreover, the stability of the GFP control (which ends with LYK), DesA3-myc (which ends with EDL), and DesA3-His can also be explained by the residue preferences and the combinational effects reported here.
Mycobacterium smegmatis as an expression host.
M. smegmatis is a nonpathogenic mycobacterium that grows faster than pathogenic mycobacteria and transforms efficiently (41). It has been often used as an alternative to E. coli for heterologous expression, possibly due to the better folding and posttranslational modification of mycobacterial proteins in a phylogenetically related expression host (13, 41, 43, 53). This work has made use of a tetracycline-inducible vector system that yields reproducible, titratable expression dependent on the concentration of tetracycline added to the culture medium. Our results suggest that the stability of other recombinant proteins expressed in M. smegmatis might be predicted and possibly improved by consideration of the C-terminal sequence.
Mycobacterial processing of DesA3.
DesA3 is an essential enzyme in fatty acid biosynthesis (22, 32, 36, 45). Many experiments suggest that DesA3 activity is tightly regulated under laboratory conditions that mimic the different stages of M. tuberculosis infection (2, 38). Since both DesA3 and the M. smegmatis homolog Msmeg_1886 have the requisite small residues at their C termini, a protease degradation mechanism is feasible for controlling the activity of this essential enzyme by posttranslational processing. In conjunction with the corresponding gene knockouts, appending short amino acid sequences to the C terminus of a recombinant protein may allow investigation of the role of protein stability on cellular physiology.
The rapid posttranslational degradation observed for DesA3 and for GFP tagged to the native C terminus of DesA3 provides new evidence for one mechanism used to control expression of an essential gene in M. smegmatis. Since most proteases are conserved among bacteria from the same family, the substrate selectivity rules identified here may be useful to predict protein stability or pathways of proteolytic degradation in M. tuberculosis and in other pathogenic mycobacteria. For comparison, the AKSVTEPDDLAA sequence at the end of DesA3 is exactly duplicated in the homologous protein from Mycobacterium bovis and is replaced by the more divergent SVARRTGGELAA sequence in the Mycobacterium avium protein, which nevertheless retains the critical LAA terminus. It is not presently known how control of the C-terminal degradation of DesA3 might contribute to the persistence of pathogenic M. tuberculosis in stationary phase.
Other mycobacterial degradation targets.
A search of the M. tuberculosis H37Rv genome for proteins whose last two residues consisted of any combination of the residues Ala, Cys, Ile, Leu, Ser, Thr, and Val and whose third-to-last residue was not Asp, Gly, Glu, or His revealed 549 examples out of the complete genome of 3,918 coding sequences (14%). This search may identify a subset of proteins and enzymes that have a short lifetime in the cell due to proteolytic processing. Of these 549 examples, 370 are annotated as hypothetical (67%). This may underscore a general lack of understanding of proteins that have only temporal existence in a cell. Furthermore, only 47 unique patterns of C-terminal peptides were identified out of 784 possible combinations of the specified residues (6%). This suggests that the identity of the residues at the C terminus is not statistically distributed but might instead represent some bias, such as a functional specialization.
It is also possible to use this search method to evaluate the ∼200 genes essential for survival of M. tuberculosis, which revealed 21 examples. In addition to DesA3, nine other enzymes and 11 hypothetical proteins were identified. Among these, there are a number of annotated proteins and enzymes, such as membrane transporters, biosynthetic enzymes, and other proteins, whose cellular level might reasonably need to be regulated. It is reasonable that DesA3 is among these, given the level of regulation known for the mammalian homolog. The results of this search are shown in Table 2.
TABLE 2.
Representative proteins previously identified as being essential for survival of M. tuberculosis H37Rv that also contain a C-terminal sequence that may destabilize or stabilize the protein for proteolytic processinga
| Proteinb | Annotation |
|---|---|
| Potentially susceptiblec | |
| Rv1013 | Possible polyketide synthase |
| Rv1411c | Probable lipoprotein |
| Rv1589 | Biotin synthase bioB |
| Rv1653 | Glutamate N-acetyltransferase |
| Rv2335 | Probable serine acetyltransferase |
| Rv3061c | Acyl-CoA dehydrogenase |
| Rv3229c | DesA3 |
| Rv3544c | Acyl-CoA dehydrogenase |
| Rv3758c | Probable ABC transporter |
| Rv3810 | Cell surface protein precursor |
| Potentially stabilizedd | |
| Rv0099 | Ligase most similar to long chain fatty acid-CoA ligase |
| Rv0470c | Cyclopropane mycolic acid synthase |
| Rv1028c | Probable sensor protein |
| Rv1185c | fadD21, acyl-CoA synthase |
| Rv1236 | Membrane protein probably involved in sugar transport |
| Rv1237 | Sugar (maltose) transporter |
| Rv1244 | Unknown lipoprotein |
| Rv1304 | ATP synthase A chain |
| Rv1569 | 8-Amino-7-oxononanoate synthase |
| Rv1640c | Similarity to lysyl-tRNA synthetase |
| Rv1821 | Translocase secA |
| Rv2048c | Polyketide synthase |
| Rv2072c | CobL methyl transferase |
| Rv2231 | CobC aminotransferase |
| Rv2937 | Daunorubicin resistance transmembrane protein |
| Rv2942 | mmpL1 conserved membrane protein |
| Rv3246c | mtrA response regulator |
| Rv3270 | Cation transport ATPase |
| Rv3499c | Mce protein family |
| Rv3560c | Acyl-CoA dehydrogenase |
| Rv3873 | PPE family |
The list of proteins identified here was derived from a listing of all genes associated with survival of Mycobacterium tuberculosis H37Rv (37).
Corresponding to the gene identified from the sequenced genome of Mycobacterium tuberculosis H37Rv.
Proteins that contain any combination of the residues Ala, Cys, Ile, Leu, Ser, Thr, and Val in the last two positions of the primary sequence and do not contain the stabilizing residue Asp, Gly, Glu, or His in the third-to-last position. These proteins are predicted to be susceptible to C-terminally directed proteolytic processing. The hypothetical proteins identified are Rv0636, Rv1211, Rv2004c, Rv3087, Rv3277, Rv3335c, Rv3523, Rv3616c, Rv3651, Rv3864, and Rv3910.
Proteins that contain any combination of the residues Arg, Asp, Gly, Glu, His, Lys, Phe, Pro, Trp, and Tyr in the last two positions of the primary sequence. These proteins are predicted to be stabilized against C-terminally directed proteolytic processing. The hypothetical proteins identified are Rv0100, Rv0176, Rv0204c, Rv0218, Rv0566c, Rv0687, Rv1128c, Rv1144, Rv1183, Rv1184c, Rv1405c, Rv1422, Rv1560, Rv1710, Rv1930c, Rv2038c, Rv2277c, Rv2696c, Rv2808, Rv3168, Rv3178, Rv3371, Rv3542c, Rv3614c, Rv3631, Rv3723, Rv3805c, Rv3868, Rv3876, Rv3877, and Rv3882c.
The proteins and enzymes whose stability would be enhanced by a combination of the residues Arg, Asp, Glu, Gly, His, Lys, Phe, Pro, Trp, and Tyr at the last two positions of the C terminus can also be predicted. Among the 100 possible combinations of these ten residues into dipeptides, 90 unique patterns were identified, 1,045 different proteins were identified, and 686 of these proteins were annotated as hypothetical. This result suggests at least a quarter of the M. tuberculosis H37Rv genome may be stabilized against C-terminal proteolysis. Furthermore, from the ∼200 genes essential for survival in M. tuberculosis H37Rv, 32 unique C-terminal dipeptide patterns were identified that encode 52 proteins predicted to be stable. Among these, a number of enzymes and other conserved proteins are observed, along with 31 that are annotated as hypothetical (60%). These search results are also shown in Table 2. Experimentation beyond the scope of this project will be required to evaluate the possible implications of this listing.
Conclusions.
This work has given insight into the properties of C-terminally directed proteolysis of proteins in mycobacteria. The proteolytic regulation of DesA3 was a notable finding. By use of the amino acid selectivity rules developed from this work, a listing of other potentially destabilized and stabilized proteins in the mycobacterial genome has been generated. To our knowledge, this type of broader analysis of a bacterial genome has not been previously undertaken. With understanding of the specificity of C-terminally directed proteolysis, it should be possible to perform similar predictive studies in other microbial species, both pathogenic and beneficial. This type of broader cross-correlative work may provide new insights into cellular processing of unknown proteins whose existence may be transitory or may exhibit unique longevity.
Supplementary Material
Acknowledgments
We thank Sabine Ehrt (Department of Microbiology and Immunology, Weill Medical College of Cornell University) for providing the tetracycline-inducible expression system and the vector adapted for these studies that contained the GFP gene.
This work was supported by National Institutes of Health grant R01 GM-50853 to B.G.F. National Institutes of Health Protein Structure Initiative Grant GM074901 (John L. Markley, principal investigator; G. N. Phillips, Jr., and B.G.F., coinvestigators) funded the bioinformatics work of G.E.W. and C.A.B.
Footnotes
Published ahead of print on 22 August 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Andersen, J. B., C. Sternberg, L. K. Poulsen, S. P. Bjorn, M. Givskov, and S. Molin. 1998. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl. Environ. Microbiol. 642240-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Betts, J. C., P. T. Lukey, L. C. Robb, R. A. McAdam, and K. Duncan. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43717-731. [DOI] [PubMed] [Google Scholar]
- 3.Blokpoel, M. C., H. N. Murphy, R. O'Toole, S. Wiles, E. S. Runn, G. R. Stewart, D. B. Young, and B. D. Robertson. 2005. Tetracycline-inducible gene regulation in mycobacteria. Nucleic Acids Res. 33e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blokpoel, M. C., R. O'Toole, M. J. Smeulders, and H. D. Williams. 2003. Development and application of unstable GFP variants to kinetic studies of mycobacterial gene expression. J. Microbiol. Methods 54203-211. [DOI] [PubMed] [Google Scholar]
- 5.Bloom, B. R., and C. J. Murray. 1992. Tuberculosis: commentary on a reemergent killer. Science 2571055-1064. [DOI] [PubMed] [Google Scholar]
- 6.Chang, Y., and B. G. Fox. 2006. Identification of Rv3230c as the NADPH oxidoreductase of a two-protein DesA3 acyl-CoA desaturase in Mycobacterium tuberculosis H37Rv. Biochemistry 4513476-13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciechanover, A. 1994. The ubiquitin-proteasome proteolytic pathway. Cell 7913-21. [DOI] [PubMed] [Google Scholar]
- 8.Cowley, S. C., and Y. Av-Gay. 2001. Monitoring promoter activity and protein localization in Mycobacterium spp. using green fluorescent protein. Gene 264225-231. [DOI] [PubMed] [Google Scholar]
- 9.Dobrzyn, A., and J. M. Ntambi. 2005. The role of stearoyl-CoA desaturase in the control of metabolism. Prostaglandins Leukot. Essent. Fatty Acids 7335-41. [DOI] [PubMed] [Google Scholar]
- 10.Dye, C., S. Scheele, P. Dolin, V. Pathania, and M. C. Raviglione. 1999. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA 282677-686. [DOI] [PubMed] [Google Scholar]
- 11.Ehrt, S., X. V. Guo, C. M. Hickey, M. Ryou, M. Monteleone, L. W. Riley, and D. Schnappinger. 2005. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 33e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flynn, J. M., I. Levchenko, M. Seidel, S. H. Wickner, R. T. Sauer, and T. A. Baker. 2001. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc. Natl. Acad. Sci. USA 9810584-10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garbe, T., D. Harris, M. Vordermeier, R. Lathigra, J. Ivanyi, and D. Young. 1993. Expression of the Mycobacterium tuberculosis 19-kilodalton antigen in Mycobacterium smegmatis: immunological analysis and evidence of glycosylation. Infect. Immun. 61260-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottesman, S., W. P. Clark, V. de Crecy-Lagard, and M. R. Maurizi. 1993. ClpX, an alternative subunit for the ATP-dependent Clp protease of Escherichia coli. Sequence and in vivo activities. J. Biol. Chem. 26822618-22626. [PubMed] [Google Scholar]
- 15.Gottesman, S., E. Roche, Y. Zhou, and R. T. Sauer. 1998. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 121338-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimaud, R., M. Kessel, F. Beuron, A. C. Steven, and M. R. Maurizi. 1998. Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. J. Biol. Chem. 27312476-12481. [DOI] [PubMed] [Google Scholar]
- 17.Herman, C., D. Thevenet, P. Bouloc, G. C. Walker, and R. D'Ari. 1998. Degradation of carboxy-terminal-tagged cytoplasmic proteins by the Escherichia coli protease HflB (FtsH). Genes Dev. 121348-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernandez-Abanto, S. M., S. C. Woolwine, S. K. Jain, and W. R. Bishai. 2006. Tetracycline-inducible gene expression in mycobacteria within an animal host using modified Streptomyces tcp830 regulatory elements. Arch. Microbiol. 186459-464. [DOI] [PubMed] [Google Scholar]
- 19.Hersch, G. L., T. A. Baker, and R. T. Sauer. 2004. SspB delivery of substrates for ClpXP proteolysis probed by the design of improved degradation tags. Proc. Natl. Acad. Sci. USA 10112136-12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hochstrasser, M. 1995. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 7215-223. [DOI] [PubMed] [Google Scholar]
- 21.Hoskins, J. R., S. K. Singh, M. R. Maurizi, and S. Wickner. 2000. Protein binding and unfolding by the chaperone ClpA and degradation by the protease ClpAP. Proc. Natl. Acad. Sci. USA 978892-8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hung, J. G., and R. W. Walker. 1970. Unsaturated fatty acids of Mycobacteria. Lipids 5720-722. [DOI] [PubMed] [Google Scholar]
- 23.Jackson, M., D. C. Crick, and P. J. Brennan. 2000. Phosphatidylinositol is an essential phospholipid of mycobacteria. J. Biol. Chem. 27530092-30099. [DOI] [PubMed] [Google Scholar]
- 24.Jentsch, S., and S. Schlenker. 1995. Selective protein degradation: a journey's end within the proteasome. Cell 82881-884. [DOI] [PubMed] [Google Scholar]
- 25.Kato, H., K. Sakaki, and K. Mihara. 2006. Ubiquitin-proteasome-dependent degradation of mammalian ER stearoyl-CoA desaturase. J. Cell Sci. 1192342-2353. [DOI] [PubMed] [Google Scholar]
- 26.Keiler, K. C., and R. T. Sauer. 1996. Sequence determinants of C-terminal substrate recognition by the Tsp protease. J. Biol. Chem. 2712589-2593. [DOI] [PubMed] [Google Scholar]
- 27.Keiler, K. C., P. R. Waller, and R. T. Sauer. 1996. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271990-993. [DOI] [PubMed] [Google Scholar]
- 28.Kim, Y. I., R. E. Burton, B. M. Burton, R. T. Sauer, and T. A. Baker. 2000. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol. Cell 5639-648. [DOI] [PubMed] [Google Scholar]
- 29.Knipfer, N., A. Seth, S. G. Roudiak, and T. E. Shrader. 1999. Species variation in ATP-dependent protein degradation: protease profiles differ between mycobacteria and protease functions differ between Mycobacterium smegmatis and Escherichia coli. Gene 23195-104. [DOI] [PubMed] [Google Scholar]
- 30.Levchenko, I., C. K. Smith, N. P. Walsh, R. T. Sauer, and T. A. Baker. 1997. PDZ-like domains mediate binding specificity in the Clp/Hsp100 family of chaperones and protease regulatory subunits. Cell 91939-947. [DOI] [PubMed] [Google Scholar]
- 31.Mziaut, H., G. Korza, and J. Ozols. 2000. The N terminus of microsomal delta 9 stearoyl-CoA desaturase contains the sequence determinant for its rapid degradation. Proc. Natl. Acad. Sci. USA 978883-8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okuyama, H., T. Kankura, and S. Nojima. 1967. Positional distribution of fatty acids in phospholipids from Mycobacteria. J. Biochem. 61732-737. [DOI] [PubMed] [Google Scholar]
- 33.Ortega, J., S. K. Singh, T. Ishikawa, M. R. Maurizi, and A. C. Steven. 2000. Visualization of substrate binding and translocation by the ATP-dependent protease, ClpXP. Mol. Cell 61515-1521. [DOI] [PubMed] [Google Scholar]
- 34.Parish, T., E. Mahenthiralingam, P. Draper, E. O. Davis, and M. J. Colston. 1997. Regulation of the inducible acetamidase gene of Mycobacterium smegmatis. Microbiology 143(Pt 7)2267-2276. [DOI] [PubMed] [Google Scholar]
- 35.Phetsuksiri, B., A. R. Baulard, A. M. Cooper, D. E. Minnikin, J. D. Douglas, G. S. Besra, and P. J. Brennan. 1999. Antimycobacterial activities of isoxyl and new derivatives through the inhibition of mycolic acid synthesis. Antimicrob. Agents Chemother. 431042-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phetsuksiri, B., M. Jackson, H. Scherman, M. McNeil, G. S. Besra, A. R. Baulard, R. A. Slayden, A. E. DeBarber, C. E. Barry III, M. S. Baird, D. C. Crick, and P. J. Brennan. 2003. Unique mechanism of action of the thiourea drug isoxyl on Mycobacterium tuberculosis. J. Biol. Chem. 27853123-53130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sassetti, C. M., and E. J. Rubin. 2003. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 10012989-12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnappinger, D., S. Ehrt, M. I. Voskuil, Y. Liu, J. A. Mangan, I. M. Monahan, G. Dolganov, B. Efron, P. D. Butcher, C. Nathan, and G. K. Schoolnik. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198693-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scholz, O., A. Thiel, W. Hillen, and M. Niederweis. 2000. Quantitative analysis of gene expression with an improved green fluorescent protein. Eur. J. Biochem. 2671565-1570. [DOI] [PubMed] [Google Scholar]
- 40.Silber, K. R., K. C. Keiler, and R. T. Sauer. 1992. Tsp: a tail-specific protease that selectively degrades proteins with nonpolar C termini. Proc. Natl. Acad. Sci. USA 89295-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snapper, S. B., R. E. Melton, S. Mustafa, T. Kieser, and W. R. Jacobs, Jr. 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 41911-1919. [DOI] [PubMed] [Google Scholar]
- 42.Stover, C. K., V. F. de la Cruz, G. P. Bansal, M. S. Hanson, T. R. Fuerst, W. R. Jacobs, Jr., and B. R. Bloom. 1992. Use of recombinant BCG as a vaccine delivery vehicle. Adv. Exp. Med. Biol. 327175-182. [DOI] [PubMed] [Google Scholar]
- 43.Thangaraj, H. S., F. I. Lamb, E. O. Davis, P. J. Jenner, L. H. Jeyakumar, and M. J. Colston. 1990. Identification, sequencing, and expression of Mycobacterium leprae superoxide dismutase, a major antigen. Infect. Immun. 581937-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomoyasu, T., K. Yamanaka, K. Murata, T. Suzaki, P. Bouloc, A. Kato, H. Niki, S. Hiraga, and T. Ogura. 1993. Topology and subcellular localization of FtsH protein in Escherichia coli. J. Bacteriol. 1751352-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker, R. W., H. Barakat, and J. G. Hung. 1970. The positional distribution of fatty acids in the phospholipids and triglycerides of Mycobacterium smegmatis and M. bovis BCG. Lipids 5684-691. [DOI] [PubMed] [Google Scholar]
- 46.Wang, J., J. A. Hartling, and J. M. Flanagan. 1997. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell 91447-456. [DOI] [PubMed] [Google Scholar]
- 47.Wang, L., M. Elliott, and T. Elliott. 1999. Conditional stability of the HemA protein (glutamyl-tRNA reductase) regulates heme biosynthesis in Salmonella typhimurium. J. Bacteriol. 1811211-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wawrzynow, A., D. Wojtkowiak, J. Marszalek, B. Banecki, M. Jonsen, B. Graves, C. Georgopoulos, and M. Zylicz. 1995. The ClpX heat-shock protein of Escherichia coli, the ATP-dependent substrate specificity component of the ClpP-ClpX protease, is a novel molecular chaperone. EMBO. J. 141867-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weber-Ban, E. U., B. G. Reid, A. D. Miranker, and A. L. Horwich. 1999. Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature 40190-93. [DOI] [PubMed] [Google Scholar]
- 50.Wickner, S., S. Gottesman, D. Skowyra, J. Hoskins, K. McKenney, and M. R. Maurizi. 1994. A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc. Natl. Acad. Sci. USA 9112218-12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wojtkowiak, D., C. Georgopoulos, and M. Zylicz. 1993. Isolation and characterization of ClpX, a new ATP-dependent specificity component of the Clp protease of Escherichia coli. J. Biol. Chem. 26822609-22617. [PubMed] [Google Scholar]
- 52.Young, D. B., and S. T. Cole. 1993. Leprosy, tuberculosis, and the new genetics. J. Bacteriol. 1751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang, Y., R. Lathigra, T. Garbe, D. Catty, and D. Young. 1991. Genetic analysis of superoxide dismutase, the 23 kilodalton antigen of Mycobacterium tuberculosis. Mol. Microbiol. 5381-391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.