

Abstract
Sendai virus (SeV) C protein is a multifunctional protein that plays important roles in regulating viral genome replication and transcription, antagonizing the host interferon system, suppressing virus-induced apoptosis, and facilitating virus assembly and budding. We here report a novel role of SeV C protein, the limitation of double-stranded RNA (dsRNA) generation for maintaining the rate of protein synthesis in infected cells. It was found that the intracellular protein synthesis rate was maintained even after wild-type (wt) SeV infection, but markedly suppressed following C-knockout SeV infection. This indicates the requirement of C protein for maintaining protein synthesis after infection. In contrast to wt SeV infection, C-knockout SeV infection caused phosphorylation of both the translation initiation factor eIF2α and dsRNA-dependent protein kinase (PKR). Phosphorylation of eIF2α occurred mainly due to the action of PKR, since knockdown of PKR by small interfering RNA limited eIF2α phosphorylation. C protein, however, could inhibit neither poly(I):poly(C)-activated nor Newcastle disease virus-induced phosphorylation of PKR and eIF2α, suggesting that C protein does not target common pathways leading to PKR activation. Immunofluorescent staining experiments with a monoclonal antibody specifically recognizing dsRNA revealed generation of a large amount of dsRNA in cells infected with C-knockout SeV but not wt SeV. The dsRNA generation as well as phosphorylation of PKR and eIF2α induced by C-knockout SeV was markedly suppressed in cells constitutively expressing C protein. Taken together, these results demonstrate that the SeV C protein limits generation of dsRNA, thereby keeping PKR inactive to maintain intracellular protein synthesis.
Members of the Paramyxovirinae subfamily have evolved their accessory proteins, V and/or C, to antagonize the host interferon (IFN) system (9, 10, 41). Their IFN antagonistic mechanisms found so far are classified into two categories depending on the target intracellular process. One targets the JAK-STAT pathway for suppression of activation of IFN-stimulated genes, resulting in inhibition of upregulation of antiviral proteins such as double-stranded RNA (dsRNA)-dependent protein kinase (PKR), 2′,5′-oligoadenyl synthetase, and Mx proteins, and another targets signaling pathways leading to IFN-β gene activation for inhibition of IFN-β production induced in response to virus infection. For Sendai virus (SeV), a prototypic paramyxovirus, we and some other researchers have demonstrated that C protein employs both mechanisms, whereas V protein employs only the latter mechanism for counteracting the host IFN system (8, 14, 26, 27, 40). In addition to the IFN antagonistic abilities, a number of abilities have been found for SeV C protein, as follows: inhibition of virus genome replication and transcription (2-4, 48), suppression of virus-induced apoptosis (28), and promotion of efficient virus assembly and budding (16, 20, 21, 29, 44).
Accessory proteins V and/or C are encoded by the paramyxovirus P gene. The P gene is distinct from other viral genes in encoding multiple species of proteins (30). For SeV, mRNA as a faithful copy of the P gene directs synthesis of the P protein, whereas V protein is synthesized from the edited mRNA generated by means of the RNA editing; one additional G nucleotide is inserted at a specific position (editing site) on the P gene during de novo viral mRNA synthesis. On the other hand, the C protein is synthesized from both the P and V mRNAs, since the C open reading frame (ORF) is located upstream of the editing site in the +1 frame relative to the P ORF. The SeV C ORF produces a nested set of four proteins, C′, C, Y1, and Y2, which share a termination codon, UAA/728, but use four different ribosomal initiation sites, ACG/81, and AUGs/114, 183, and 201, respectively.
During progress of our research on the IFN antagonistic ability of SeV, we found that viral protein synthesis was hardly suppressed in IFN-pretreated cells that contain a plentiful amount of antiviral proteins. This finding indicates that SeV has the ability to directly inhibit functions of antiviral proteins or to keep them inactive, consistent with a previous finding that Had-2 cells, a mouse FM3A cell line constitutively expressing IFN-α, are susceptible to SeV but resistant to Newcastle disease virus (NDV) (1). Since this ability does not appear to be explained by the previously found IFN antagonistic abilities, we decided to investigate the mechanisms underlying this novel antagonism.
Of a variety of antiviral proteins induced by IFN, PKR is one of the most important players in the inhibition of viral protein synthesis (6). PKR is expressed in most cell types at a low abundance even in the absence of IFN and is usually present in an inactive form. Following virus infection, dsRNA generated during viral genome replication and transcription binds to PKR, leading to activation with concomitant homodimerization and autophosphorylation. Activation of PKR appears to be associated with phosphorylation on Thr446 and Thr451 in the activation loop of PKR (43). PKR is also activated by the dsRNA binding protein PACT (38). PKR activation leads to phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α) on Ser51, which prevents the recycling of eIF2 through inactivation of the guanine exchange factor eIF2B that is required for ongoing translation, leading to inhibition of translation and consequent suppression of virus replication (47). Phosphorylation of eIF2α is also mediated by other kinases, such as the endoplasmic reticulum (ER) stress sensor protein, PERK (PKR-like ER kinase) (17).
Since the IFN antagonistic abilities previously found for SeV have been attributed to accessory proteins, we speculated that the novel ability would also be attributable to either V or C protein. We thus attempted to identify the responsible viral protein using the following accessory protein knockout recombinant viruses: V(−), which does not express V protein, and 4C(−), which does not express all four C proteins. In this study, we provide evidence for the requirement of C protein for the novel ability and further demonstrate that C protein limits dsRNA arising from viral genome replication and transcription, thereby keeping PKR inactive to maintain the rate of protein synthesis in infected cells. Although numerous viruses inhibiting the function of PKR have been known, this study is the first report about viral protein that limits dsRNA generating during viral genome replication and transcription.
MATERIALS AND METHODS
Cells and viruses.
HeLa and Vero cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). BHK cells were cultured in Eagle's minimum essential medium supplemented with 10% FBS and 10% triphosphate broth. Human glioblastoma U118 cells were maintained in DMEM supplemented with 10% FBS and 1 mM sodium pyruvate (32). A U118 cell clone constitutively expressing SeV C protein (U118-C) or a control U118 cell clone (U118-vec) was isolated from G418 (Geneticin)-resistant colonies generated by transfection with a C expression vector, pEFneo-C (13), or an empty vector, pEFneo, respectively. pEFneo-C is a plasmid that can express C, Y1, and Y2, since it contains the region from AUG/114 to UAA/728 in the C ORF. The vesicular stomatitis virus (VSV) New Jersey strain was grown in BHK cells. The SeV pB strain (50), DIH4 (a gift from L. Roux, Geneva, Switzerland), and the NDV Ulster strain were propagated in embryonated eggs (12), while recombinant SeVs, the wild-type (wt) Z strain, V(−), and 4C(−) (23, 29), were grown in Vero cells in the presence of 3 μg/ml trypsin (14). Titers of all the viruses except for DIH4 were determined by an immunofluorescent infectious focus assay in Vero cells and expressed as cell infectious units per milliliter as described previously (24), and all the infections were performed at a multiplicity of infection (MOI) of 10, unless otherwise mentioned. When DIH4 was used, it was inoculated onto cells at the minimal dose able to infect all cells cultured in a dish or a well. This dose was determined by a serial dilution of a DIH4 stock, in which DIH4 infection could completely suppress the cytopathic effect (CPE) of cells that were infected with wt SeV at an MOI of 10.
Western blot analysis.
Cells were lysed in a lysis buffer (10 mM Tris-HCl, pH 7.4, 300 mM NaCl, 0.5% Triton X-100, 5% glycerol, 1 mM EDTA, 10 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM dithiothreitol) containing a protease inhibitor cocktail (Sigma-Aldrich Co., St. Louis, MO). The cell lysates (10 μg protein) were resolved by sodium dodecyl sulfate-7.5 to 15% polyacrylamide gel electrophoresis and electroblotted onto nitrocellulose membranes. Immunoblots were then probed with anti-IRF9 (sc-496; Santa Cruz Biotechnology), anti-STAT1 (sc-417; Santa Cruz Biotechnology), anti-PKR (k-17; Santa Cruz Biotechnology), anti-phospho-PKR (p-PKR) (p-Tyr446; Epitomics, CA), anti-eIF2α (Abcam), anti-phospho-eIF2α (p-eIF2α) (p-Ser51; Cell Signaling Technology), anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (clone 6C5; Ambion), anti-actin antibody (clone C4; ICN Biomedicals, OH), anti-C protein, anti-V protein, anti-SeV, anti-VSV, or anti-NDV serum and developed using the ECL-Plus Western blotting kit (Amersham Biosciences, Uppsala, Sweden). In some experiments, band intensities were quantified using the Fluorochem 8000 imaging system (Alpha Innotech Corporation). Anti-C protein serum was prepared from rabbits, into which keyhole limpet hemocyanin conjugated to a synthetic peptide, MEEAWSLAAHIVQE (the C-terminal sequence of SeV C protein), was injected six times at appropriate intervals (Operon Biotechnologies, Inc., Tokyo, Japan). Anti-V protein serum recognizing a synthetic peptide, GHRREHIIYERDC (a sequence of the V unique region), was prepared previously (26).
Radioisotope labeling.
Cells were incubated for 30 min with a medium consisting of a methionine- and cysteine-free medium (20 vol) and normal DMEM (1 vol) and then supplemented for 1 h with 20 MBq/ml of Escherichia coli hydrolysate containing [35S]methionine and [35S]cysteine (American Radiolabeled Chemicals, St. Louis, MO). Proteins (10 μg) in the cell extracts were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the dried gels were scanned using the BAS-1500 imaging system (Fujifilm, Japan).
RNA transfection.
A short interfering RNA (siRNA) was custom synthesized by Ambion, Inc. The sequence for PKR siRNA was targeted to the 3′ end sequence (PKRc, CTTCTTCATGTATGTGACA) according to Ong et al. (35). U118 cells cultured in a 24-well plate were transfected with the siRNA specific for PKR or a control siRNA specific for GAPDH (Ambion) using Lipofectamine (Invitrogen) according to the manufacturer's instructions. At 40 h later, the cells were infected with 4C(−) and harvested at 30 h postinfection (p.i.). Lipofectamine reagent was also used for transfection with poly(I):poly(C) (Amersham Biosciences).
Immunofluorescent staining.
Cells were grown in a 24-well plate and infected with SeV and/or NDV. Cells were fixed with 3% paraformaldehyde and permeabilized with 0.5% Triton X-100 dissolved in phosphate-buffered saline. The mouse monoclonal antibody J2 (Scicons, Hungary) was diluted 1:1,000, and the J2 reactive antigens were visualized using the tyramide signal amplification cyanine 3 system (Perkin Elmer) and horseradish peroxidase-conjugated goat anti-mouse immunoglobulin antibody (Amersham). Stained cell samples were examined under the phase difference microscope and fluorescence microscope (Olympus IX70 microscope system). For detection of SeV N protein, cells were fixed with cold methanol and treated with an anti-SeV N monoclonal antibody (mAb1.180) kindly provided by C. Örvell (25, 36), followed by treatment with rhodamine-labeled anti-mouse immunoglobulin G goat serum (Tago, Inc., Burlingame, CA).
Northern blot analysis.
BHK cells cultured in a 24-well plate were infected with SeV for 24 h and then lysed with a buffer containing 1% Triton X-100. From the cell lysates, intracellular nucleocapsids were immunoprecipitated with anti-SeV antiserum and protein A-Sepharose beads (Pharmacia) and then treated with Trizol reagent (Invitrogen) to purify RNA. The purified RNA, after glyoxalation, was electrophoresed in a 1% agarose gel and capillary transferred onto a GeneScreen Plus membrane (DuPont NEN Research Products). Then the membrane was hybridized with a radiolabeled riboprobe, which is complementary to the 5′ terminus of the viral genomic RNA (from nucleotides [nt] 14865 to 15281), washed according to a manufacturer's protocol (NorthenMax-Gly; Ambion), and then exposed to an X-ray film (Fujifilm, Japan). The riboprobe used for this experiment was synthesized as follows. First, the genomic RNA of SeV was reverse transcribed with a reverse transcription primer, 5′-CGGGATCC(nt 14865)GACCTGTATCCTGTGA(nt 14880)-3′. Then, the cDNA was amplified by PCR with the reverse transcription primer and another primer, 5′-GGGAATTCTAATACGACTCACTATA(nt 15384)ACCAGACAAGAGTTTA(nt 15369)-3′. The PCR product was digested with restriction enzymes BamHI and KpnI and then inserted into the cloning site of the plasmid pGEM-3zf(+). This cloned plasmid, followed by digestion with EcoRI, was used as the template for an in vitro transcription reaction with SP6 RNA polymerase (Takara, Japan) in the presence of [32P]UTP. The radiolabeled RNA was purified and used as the riboprobe.
RESULTS
Novel role of C protein in maintaining protein synthesis in infected cells.
We first checked the ability of SeV to antagonize IFN-induced antiviral proteins. HeLa cells were treated with IFN-α for 18 h for induction of a plentiful amount of antiviral proteins and then infected with wt SeV or VSV. IFN-α pretreatment resulted in striking inhibition of the VSV protein synthesis (Fig. 1A) as expected. In contrast, wt SeV counteracted IFN-α pretreatment and maintained a viral protein synthesis level similar to that in IFN-untreated cells (Fig. 1B). Since the SeV IFN antagonistic abilities found so far had been assigned to the accessory proteins, accessory protein-knockout recombinant SeVs were also examined. V(−), which does not express V protein, exhibited significant resistance to IFN-α pretreatment, although the P and C protein levels were slightly affected by IFN pretreatment (Fig. 1B). The viral protein expression level of V(−) without IFN pretreatment was higher than that of wt SeV, since knockout of the V gene augments viral gene expression (23). On the other hand, 4C(−), which does not express all four C proteins, C′, C, Y1, and Y2, was very sensitive to IFN-α pretreatment and exhibited a striking decrease in viral protein synthesis (Fig. 1B), demonstrating the requirement of C protein for maintaining the viral protein expression level in the presence of antiviral proteins induced by IFN-α.
FIG. 1.
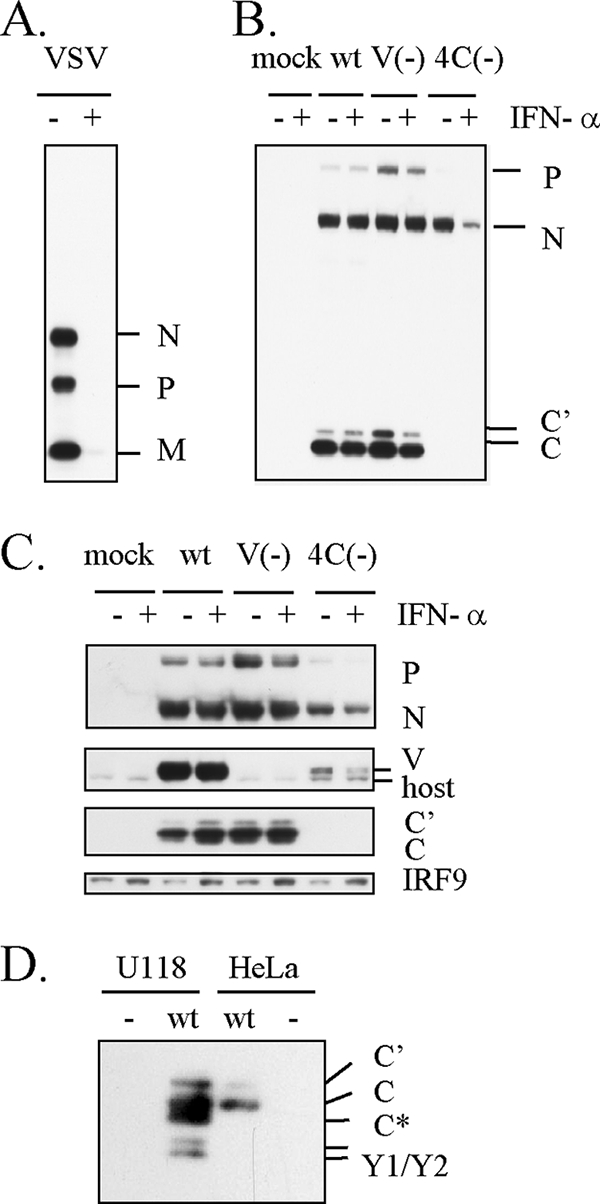
Viral protein expression levels in IFN-α-pretreated cells. HeLa (A, B) or U118 (C) cells were mock treated (−) or treated with IFN-α (1,000 IU/ml) (+) for 18 h and then mock infected or infected with VSV, wt SeV, V(−), or 4C(−). The cells were harvested at 8 h p.i. for VSV (A) and at 30 h p.i. for wt SeV, V(−), and 4C(−) (B, C). Viral proteins and IRF9 were detected by Western blot analysis with anti-VSV, anti-SeV, anti-C, anti-V serum, or anti-IRF9 antibody. In panel D, C proteins in U118 and HeLa cells infected with wt SeV were compared by Western blot analysis with anti-C serum. Positions of C′, C, Y1, and Y2 bands were determined according to their estimated molecular weights. C* migrated faster than C but slower than Y1.
In HeLa cells, 4C(−) infection induces a large amount of IFN-β, thereby upregulating IFN-stimulated gene products, including antiviral proteins, because of the lack of previously found IFN antagonistic abilities of C protein (11, 14, 26, 27). It was thus possible that IFN-β newly produced by 4C(−) infection might cause the decrease in protein synthesis. To check this, we used U118 human glioblastoma cells, which have a global defect in type I IFN genes and consequently cannot produce IFN-α and IFN-β (32). Resistance of wt SeV and V(−) to IFN-α pretreatment was also observed in U118 cells (Fig. 1C). In contrast, viral protein synthesis in 4C(−)-infected U118 cells was significantly suppressed irrespective of IFN-α pretreatment, and the suppressive effect was enhanced by IFN-α pretreatment. The 4C(−) infection caused no elevation of the basal level of IRF9, one of the IFN-stimulated gene products, confirming the lack of IFN genes of U118 cells. C protein thus seems to be involved in maintaining the viral protein synthesis level irrespective of IFN, and this novel ability does not appear to be attributable to the IFN antagonistic abilities previously found. In SeV-infected U118 cells, an extra band, designated C*, which migrated faster than C but slower than Y1, was observed (Fig. 1D). C* seems to be an N-terminally truncated product of C, because the anti-C serum used here recognizes the C-terminal sequence.
We next examined the effect of SeV infection on the host cellular protein synthesis level in U118 cells. U118 cells were infected with recombinant SeVs and pulse-labeled for 1 h at 24 h p.i. or 30 h p.i. with [S35]methionine and [S35]cysteine. As shown in Fig. 2A and B, 4C(−) showed reduced incorporation of radiolabel into host and viral proteins in contrast to wt SeV or V(−), demonstrating that C protein affected not only viral protein synthesis but also host cellular protein synthesis. Interestingly, NDV exhibited a feature distinct from wt SeV, a striking decrease in the protein synthesis rate (Fig. 2C), although NDV and SeV belong to the same subfamily, Paramyxovirinae. C protein supplied in trans by wt SeV infection could restore the protein synthesis rate of cells infected with 4C(−) (Fig. 2A) but not NDV (Fig. 2C). This suggests that C protein targets SeV-specific processes rather than common intracellular processes. For the subsequent experiments, we used primarily the U118 cell line to avoid the effect of IFN.
FIG. 2.

Protein synthesis rates of U118 cells infected with wt SeV, V(−), 4C(−), or NDV. U118 cells were mock infected (−) or infected with wt SeV, V(−), or 4C(−), or they were infected with a combination of wt SeV and 4C(−) (A, B) or with NDV or a combination of NDV and wt SeV (C). At 24 h later (B) or 30 h later (A to C), the cells were labeled for 1 h with [35S]methionine and [35S]cysteine. (A) For detection of SeV V and C proteins, samples were also analyzed by Western blotting with anti-V or anti-C serum. Protein synthesis rates were estimated by summing up densities of all bands but those shown on the top and bottom. (A, C) Percent ratios of virus-infected cells to mock-infected cells (percent protein synthesis) are also shown at the bottom. In panel B, percent protein synthesis was calculated from mean values of the protein synthesis rates from three independent experiments. Standard deviations are shown as error bars.
Phosphorylation of eIF2α and PKR in 4C(−)-infected cells.
The translation initiation factor eIF2α is an important regulator of translation. Phosphorylation of eIF2α on Ser51 leads to inhibition of translation. As shown in Fig. 3A, eIF2α was significantly phosphorylated on Ser51 after 4C(−) infection, whereas little phosphorylation was observed for wt SeV, V(−) infection, or a mixed infection of wt SeV and 4C(−). In accordance with phosphorylation of eIF2α, PKR was phosphorylated on Tyr446 only in 4C(−)-infected cells (Fig. 3A). Transfection with siRNA specific for PKR significantly decreased PKR expression levels in both mock-infected and 4C(−)-infected cells, whereas the control siRNA specific for GAPDH did not (Fig. 3B and C). Knockdown of PKR decreased the ratio of p-eIF2α to eIF2α in 4C(−)-infected cells, with an increase in the accumulation of SeV N protein (Fig. 3C), suggesting that PKR is a major factor responsible for phosphorylation of eIF2α.
FIG. 3.
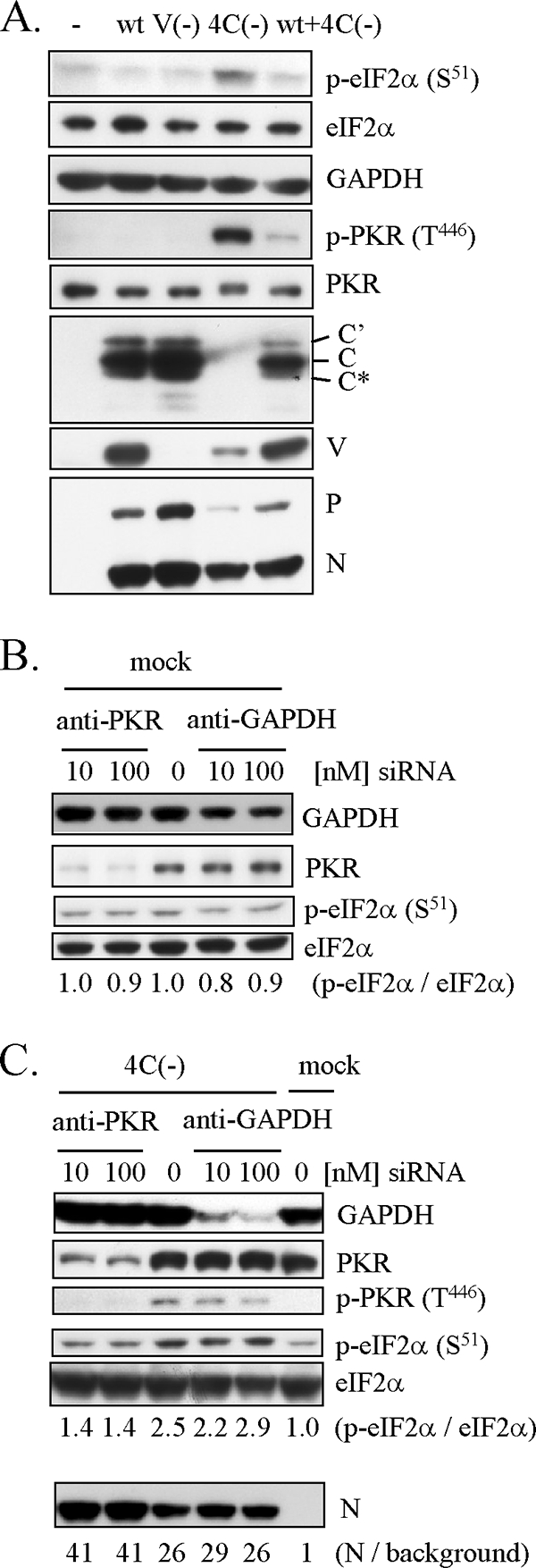
Phosphorylation of eIF2α and PKR in U118 cells infected with 4C(−). (A) U118 cells were mock infected (−) or infected with wt SeV, V(−), 4C(−), or a combination of wt SeV and 4C(−) and harvested at 30 h p.i. (B, C) U118 cells were transfected with increasing amounts of the siRNA specific for PKR or the control siRNA specific for GAPDH. At 40 h later, the cells were mock infected (B) or infected with 4C(−) (C) and harvested at 30 h p.i. (A to C) Proteins (10 μg) were analyzed by Western blotting with anti-V, anti-C, anti-SeV serum, anti-p-eIF2α (S51), anti-eIF2α, anti-p-PKR (T446), anti-PKR, or anti-GAPDH antibody. Ratios of p-eIF2α to eIF2α (B, C) and ratios of N to background (C) are also shown.
Effect of C protein on poly(I):poly(C)-activated or NDV-mediated phosphorylation of PKR and eIF2α.
To determine whether C protein could inhibit the common intracellular pathways leading to activation of PKR, we examined the effect of C protein on the poly(I):poly(C)-activated or NDV-mediated phosphorylation of PKR and eIF2α. U118 cells were preinfected with wt SeV to supply C protein and then transfected with poly(I):poly(C) (Fig. 4A) or superinfected with NDV (Fig. 4B). Transfection with poly(I):poly(C) increased the phosphorylation level of PKR and eIF2α in a dose-dependent manner. However, there was no significant difference between wt SeV-infected and mock-infected cells (Fig. 4A). Both PKR and eIF2α were phosphorylated following infection with NDV (Fig. 4B), consistent with the limitation of protein synthesis seen in NDV-infected cells (Fig. 2C), but this phosphorylation was not inhibited by SeV preinfection. In this experiment and a subsequent experiment for which results are shown in Fig. 5D, the SeV pB strain, which exhibited the same phenotype as that of the wt SeV Z strain (Fig. 3A and 4C) but caused less CPE than wt SeV, was used in order to prevent the cells from being detached from the bottom of flasks. Taken together, these results suggest that C protein does not target common pathways leading to activation of PKR but targets SeV-specific processes.
FIG. 4.
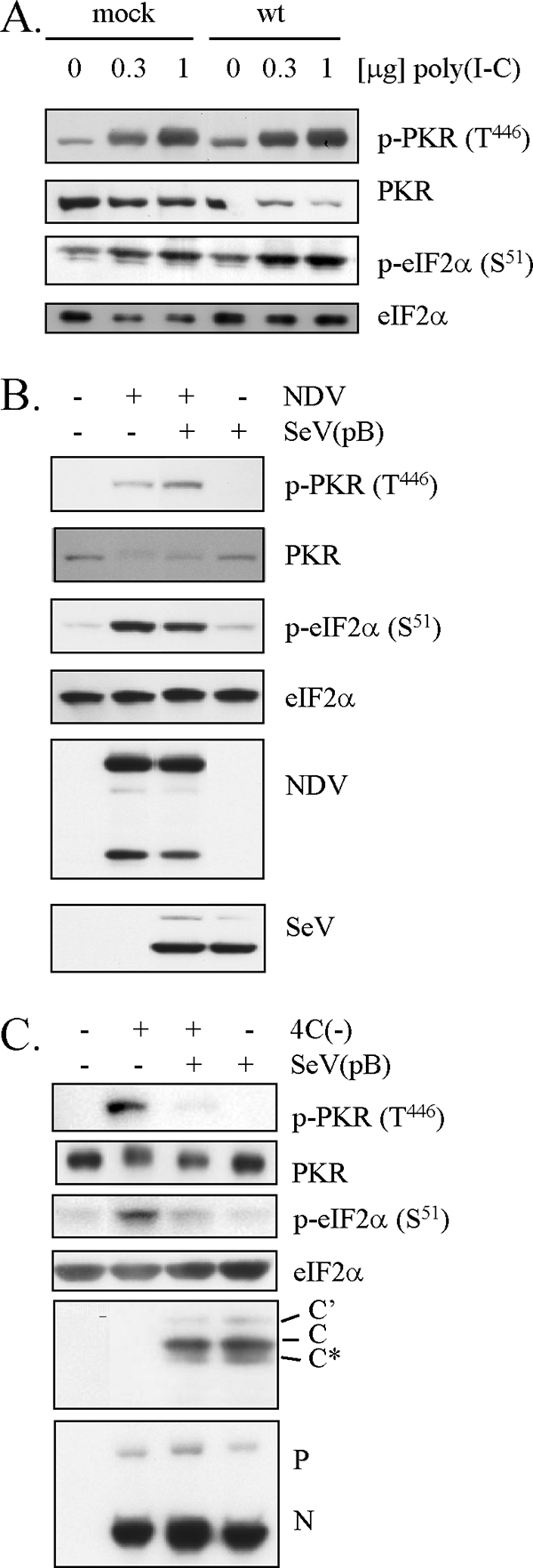
Effect of SeV infection on dsRNA-activated or NDV-mediated phosphorylation of PKR and eIF2α. (A) U118 cells were mock infected or infected with wt SeV. At 20 h later, the cells were treated with increasing amounts of poly(I):poly(C) for 5 h. (B) U118 cells were mock infected or infected with SeV pB. At 26 h later, the cells were mock superinfected or superinfected with NDV. At 22 h after superinfection, the cells were harvested. (C) U118 cells were mock infected or infected with 4C(−), SeV pB, or a combination of 4C(−) and SeV pB. At 30 h p.i., the cells were harvested. (A to C) Proteins (10 μg) were analyzed by Western blotting with anti-p-PKR (T446), anti-PKR, anti-p-eIF2α (S51), or anti-eIF2α antibody, or anti-SeV, anti-NDV, or anti-C serum.
FIG. 5.
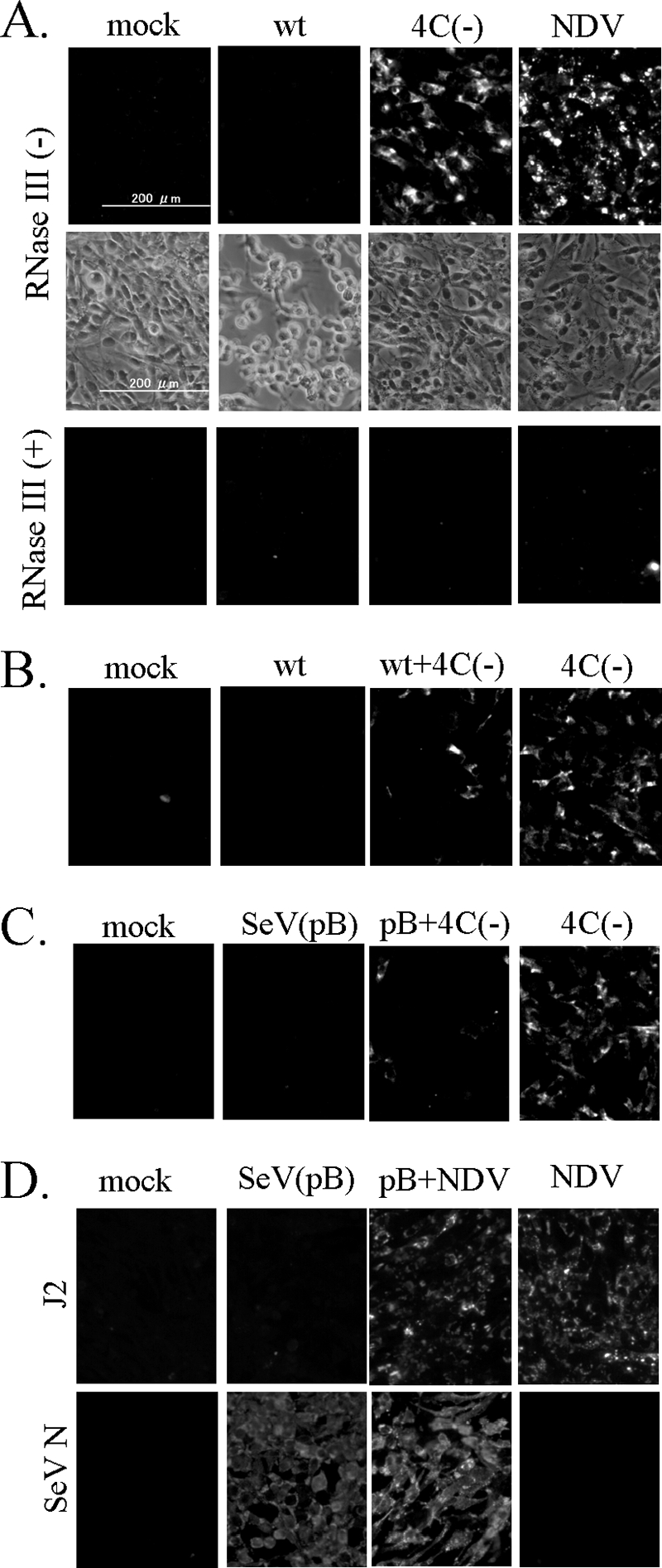
Immunofluorescent staining of SeV-infected cells with an anti-dsRNA monoclonal antibody. (A) U118 cells were mock infected or infected with wt SeV, 4C(−), or NDV. At 30 h later, the cells were fixed, permeabilized, and then mock treated (−) or treated with 5 units of RNase III (+) at 37°C for 1 h. (B) U118 cells were mock infected or infected with wt SeV, 4C(−), or a combination of wt SeV and 4C(−) for 30 h and then fixed, followed by permeabilization. (C) U118 cells were mock infected or infected with SeV pB, 4C(−), or a combination of SeV pB and 4C(−) for 30 h and then fixed, followed by permeabilization. (D) U118 cells were mock infected or infected with SeV pB. At 25 h later, the cells were mock superinfected or superinfected with NDV. The cells were fixed and permeabilized at 20 h after superinfection. (A to D) The fixed cells were processed for immunofluorescent staining with the mouse anti-dsRNA monoclonal antibody J2 as the first antibody and observed under the phase difference microscope (A) or fluorescence microscope. (D) For detection of an SeV antigen, cells were fixed with cold methanol and processed for immunofluorescent staining with an anti-SeV N monoclonal antibody (mAb1.180).
Detection of dsRNA signals in cells infected with wt SeV, 4C(−), or NDV.
PKR is constitutively expressed at a small amount and present in an inactive form. Since C protein regulates SeV genome replication and transcription (2-4, 48), we hypothesized that C protein plays a role in limiting generation of dsRNA. Thus, we decided to check intracellular dsRNA by using immunofluorescent staining with a monoclonal antibody, J2, specifically recognizing the dsRNA of more than 40 bp in length (45). The J2 monoclonal antibody seems to be suitable for detecting dsRNA-activating PKR, since full activation of PKR requires approximately 50 bp of duplexed RNA (42). As shown in Fig. 5A, no positive signals for dsRNA were seen in wt SeV-infected U118 cells with extensive CPE, as previously reported for other cell lines (49), whereas clearly positive dsRNA signals were observed for cells infected with 4C(−) or NDV. Treatment of the fixed cells with RNase III, which specifically binds to and cleaves dsRNA, eliminated the signals. Generation of dsRNA signals in 4C(−)-infected cells was prevented by wt SeV Z strain coinfection (Fig. 5B) and by SeV pB strain coinfection as well (Fig. 5C), confirming that both strains have the same phenotype. On the other hand, NDV-mediated dsRNA signals were not affected by prior infection with SeV pB (Fig. 5D). This indicates that C protein does not inhibit dsRNA arising from NDV genome replication and transcription. Thus, there was a good correlation between generation of dsRNA and phosphorylation of PKR and eIF2α.
Little ability of a copyback DI genome to generate dsRNA.
It has been reported that SeV stocks that are not contaminated with copyback defective interfering (DI) genomes have little ability to induce IFN-β production, but SeV stocks containing large amounts of copyback DI genomes strongly activate the IFN-β gene (22, 40, 46). This is thought to be due to dsRNA arising from copyback DI genome replication, although there is no direct evidence (46). Therefore, the possibility should be excluded that dsRNA signals seen in 4C(−)-infected cells were due to copyback DI genomes possibly present in 4C(−) virus stocks. Thus, DIH4, a well-studied copyback DI particle, was examined for generation of dsRNA. We first confirmed the reported ability of DIH4 to strongly activate the IFN-β gene (data not shown). Northern blot analysis showed that coinfection with the parent SeV and DIH4 prevented parent SeV genome replication with simultaneous enhancement of DIH4 genome replication (Fig. 6A). Prevention of wt SeV and 4C(−) genome replication was also indirectly shown by inhibition of their CPE (Fig. 6B). Unexpectedly, however, immunofluorescent staining experiments revealed no signals for dsRNA in cells coinfected with DIH4 (Fig. 6C), where the DIH4 genome but not the parent genome replicated well (Fig. 6A). It should be noted that C protein is supplied for DIH4 genome replication in both the cases wt SeV plus DIH4 and 4C[−] plus DIH4, since a DIH4 stock used here contains parent SeV that expresses functional C protein. These results demonstrate that DIH4 generates little dsRNA during DIH4 genome replication. Therefore, dsRNA signals seen in 4C(−)-infected cells do not seem to be due to copyback DI genomes such as DIH4.
FIG. 6.
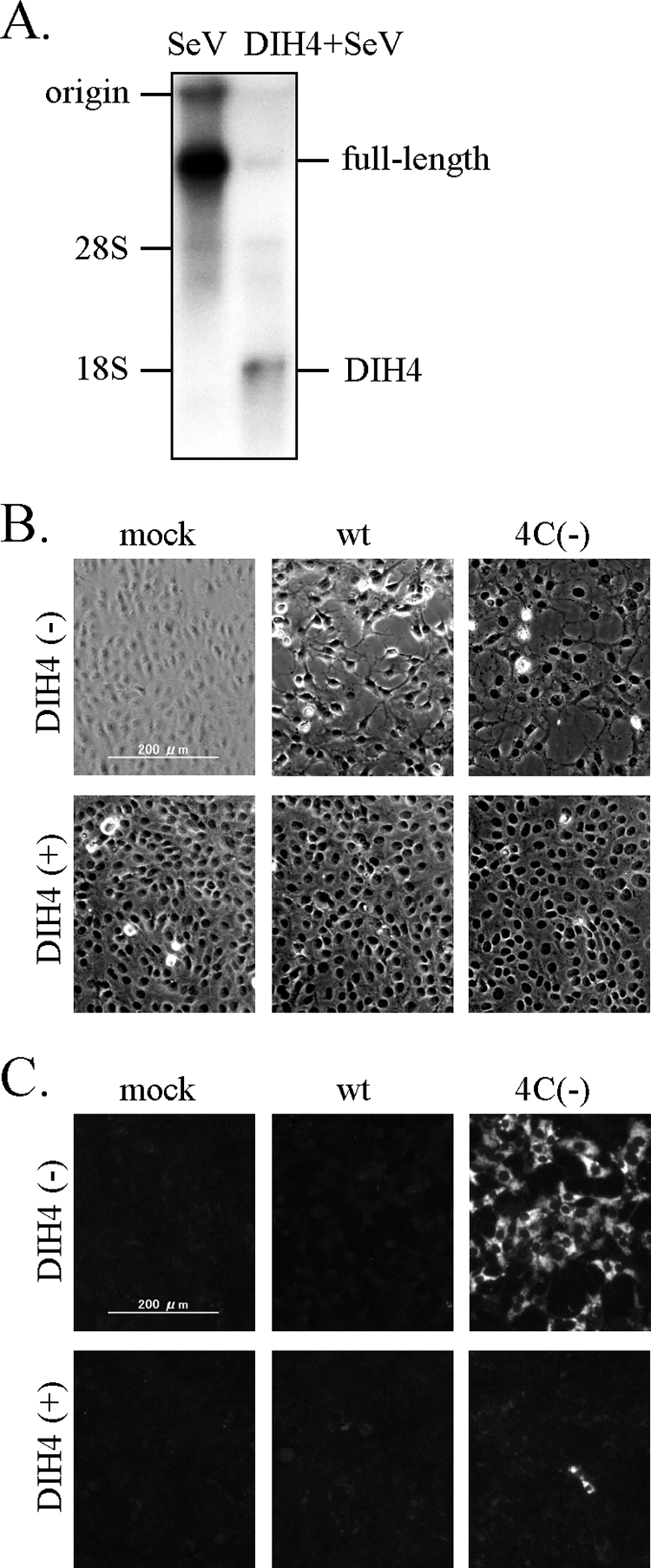
Level of dsRNA signal in cells infected with a copyback DI particle, DIH4. (A) BHK cells were singly infected with the parent SeV or a combination of the parent SeV and DIH4 for 24 h. RNAs within intracellular viral nucleocapsids were detected by Northern blot analysis with a specific riboprobe. (B and C) Vero cells were mock infected or infected with wt SeV or 4C(−), together with (+) or without (−) DIH4 for 40 h. Fixed cells were processed for immunofluorescent staining with the anti-dsRNA monoclonal antibody J2 and observed under the phase difference microscope (B) or fluorescence microscope (C).
Effect of constitutive expression of C protein on 4C(−)-mediated dsRNA generation and phosphorylation of PKR and eIF2α.
A U118 cell line that constitutively expresses SeV C protein (U118-C) was isolated from G418-resistant colonies following transfection with pEFneo-C, a C expression plasmid, into U118 cells. As a control, a U118 cell line (U118-vec) was also isolated from G418-resistant colonies following transfection with pEFneo empty vector. As shown in Fig. 7A, apparent expression of SeV C protein was observed for U118-C cells. Treatment of U118-vec cells with IFN-α upregulated STAT1α, IRF9, and PKR, which are IFN-stimulated gene products, whereas no response to IFN-α was observed for U118-C cells, indicating a blockage of the JAK-STAT pathway in U118-C cells. These data confirmed the constitutive expression of functional C protein in U118-C cells. These cell lines were infected with wt SeV or 4C(−) and examined for viral protein synthesis (Fig. 7B). Levels of SeV N protein expression at 24 h p.i. in U118-C cells were apparently lower than those in U118-vec cells for either infection, probably due to the suppressive effect of C protein on viral genome replication and transcription. However, the amounts of N protein in U118-C cells reached comparable levels by 48 or 65 h p.i. for both wt SeV and 4C(−) infections. Notably, the level of C protein expression in U118-C cells decreased at 65 h after 4C(−) infection, probably due to degradation. Thus, a comparison was made within 48 h after 4C(−) infection between U118-vec and U118-C (Fig. 7C). As shown in Fig. 7C, phosphorylation of both PKR and eIF2α was significantly suppressed in U118-C cells. We also examined dsRNA generation after 4C(−) infection (Fig. 7D). In this experiment, cells were infected at a low MOI to prevent replication of DI genomes that might be present in 4C(−) stocks, since DI genome replication requires viral proteins supplied by the parent SeV replication in the same cell. Even under these conditions, a number of U118-vec cells exhibited dsRNA signals at 48 h after 4C(−) infection (Fig. 7D), suggesting that the property of dsRNA generation is attributable to 4C(−) itself. Generation of dsRNA seen for 4C(−) infection was also suppressed in U118-C cells (Fig. 7D). Taken together, these results demonstrate that C protein plays a crucial role in limiting dsRNA generation and subsequent phosphorylation of PKR and eIF2α in SeV-infected cells.
FIG. 7.

Effect of constitutive expression of C protein on 4C(−)-mediated dsRNA generation, PKR activation, and eIF2α phosphorylation. (A) After an overnight serum starvation, U118-C or U118-vec cells were mock treated (−) or treated with IFN-α (1,000 IU/ml) (+) for 24 h. U118/pB indicates original U118 cells infected with SeV pB for 24 h. Proteins (10 μg) were analyzed by Western blotting with anti-C, anti-actin, anti-STAT1, anti-IRF9, or anti-PKR antibody. (B) U118-C or U118-vec cells were infected with wt SeV or 4C(−) at an MOI of 0.1. Cells were harvested at the indicated time points, and proteins were analyzed by Western blotting with anti-SeV, anti-C, or anti-GAPDH antibody. (C) U118-C or U118-vec cells were infected with 4C(−) at an MOI of 10. Cells were harvested at the indicated time points and proteins (10 μg) were analyzed by Western blotting with anti-p-PKR (T446), anti-PKR, anti-p-eIF2α (S51), anti-eIF2α, or anti-GAPDH antibody. (D) U118-C or U118-vec cells were infected with 4C(−) at an MOI of 0.1. Cells were fixed at 24 h p.i. and 48 h p.i. and processed for immunofluorescent staining with the anti-dsRNA monoclonal antibody J2.
DISCUSSION
A number of viruses have evolved their own strategies to inhibit function of PKR (31). Some viruses synthesize dsRNA-binding proteins (e.g., influenza A virus NS1 and reovirus σ3), which function to sequester any free dsRNA molecules. Some make RNA with secondary structures (e.g., adenovirus-associated RNAs I and II), which bind directly to PKR but do not lead to the activation of the kinase domain. However, there has been no report about viral proteins that function as a regulator of dsRNA generation. This study has thus presented the first example of such viral proteins through elucidation of the mechanism by which C protein limits PKR activation.
We have found that in the absence of C protein expression, SeV infection caused activation of PKR, phosphorylation of eIF2α, and inhibition of host and viral protein synthesis. We have used U118 cells to demonstrate that inhibition of protein synthesis is not mediated by IFN. Knockdown of PKR by siRNA transfection resulted in a decrease in the phosphorylation level of eIF2α, indicating a key role of PKR in eIF2α phosphorylation leading to translational inhibition. Although C protein had no ability to inhibit the common PKR activation pathway, we found a great difference in generation of dsRNA between wt SeV- and 4C(−)-infected cells. The 4C(−) infection generated a large amount of dsRNA, whereas the wt SeV infection did not. Not only dsRNA generation but also phosphorylation of PKR and eIF2α seen in 4C(−)-infected cells was strikingly suppressed by constitutive expression of C protein. Our data suggest that C protein limits dsRNA generation, thereby keeping PKR and eIF2α unphosphorylated and consequently maintaining protein synthesis in infected cells. The ability to limit dsRNA generation is important primarily for inhibition of not only PKR but also other dsRNA-dependent antiviral proteins, such as 2′,5′-oligoadenyl synthetase, and furthermore may partly contribute to the previously found property of wt SeV that is a weaker activator for the IFN-β gene than 4C(−) (27).
It has been reported that SeV stocks containing large amounts of DI genomes, especially copyback DI genomes, strongly induce IFN-β production, presumably due to generation of much dsRNA, whereas SeV virus stocks containing less DI barely activate the IFN-β gene (46). Therefore, we needed to exclude the possibility that generation of dsRNA seen for 4C(−) infection could be attributed to copyback DI genomes, which might be present in 4C(−) stocks. We found that a number of cells showed dsRNA signals even when cells were infected with 4C(−) stocks at a low MOI (Fig. 7D). In this case, DI genome replication is prevented, since it requires viral proteins supplied by replication of the parent SeV, 4C(−). In addition, we did not find any dsRNA signal in cells where DIH4, a copyback DI genome, replicated. These findings suggest that dsRNA generation is attributable to the property of 4C(−) itself but not contaminating DI genomes. Since the DIH4 genome strongly activates the IFN-β gene (46), this activation does not appear to be initiated with dsRNA stimulation. Alternatively, DIH4 may generate a larger amount of positive-sense trailer RNA or aberrant RNA transcripts present in a form of 5′-triphosphate single-strand RNA without long duplex forms, which bind to the RIG-I molecule, thereby activating the IFN-β gene (18, 39). However, the possibility cannot be excluded that dsRNAs with short base pairs are generated during DI genome replication, because J2 monoclonal antibody requires more than 40 bp in length for recognition of dsRNA (45).
Garcin et al. pointed out the sequence similarity of the N-terminal region of C protein to that of reovirus σ3 protein (7). However, no data have been accumulated indicating functional similarity between them. Instead, an inability of C protein to bind to dsRNA has been reported by Ostertag et al. (37). Furthermore, our present study revealed that C protein could not prevent artificial dsRNA, poly(I):poly(C), or NDV infection from activating PKR (Fig. 4A and B). Therefore, C protein seems to be totally different from σ3 protein in its functions.
How does C protein limit generation of dsRNA? Limitation of dsRNA signals has been demonstrated also for other negative-strand RNA viruses, such as influenza A virus strain PR8 and La Crosse virus, by immunofluorescent staining with mouse monoclonal antibody J2 (49). This property thus seems to be a consequence of the common strategy of viral genome replication and transcription of negative-strand RNA viruses. The most striking feature to minimize dsRNA generation is the tight coupling of genome/antigenome RNA synthesis and encapsidation (15). Full-length antigenomes and genomes of SeV are indeed found only as assembled nucleocapsids. On the other hand, leader RNA and viral mRNAs are synthesized in large quantities as naked positive-sense RNA transcripts in infected cells, but synthesis of naked negative-sense RNA is limited to the trailer RNA. Accordingly, no source of dsRNA is found in SeV-infected cells, when genome/antigenome synthesis and encapsidation are firmly coupled. However, assuming that this coupling would become unreliable, extension of the trailer RNA beyond the trailer RNA/L gene junction could occur without encapsidation, resulting in generation of naked negative-sense RNA whose 3′ sequence could anneal to the 5′ sequence of the L mRNA. Thus, we hypothesize that C protein plays a key role in the precise coupling of genome/antigenome synthesis and encapsidation. Alternatively, one could hypothesize that limitation of dsRNA generation is related to the inhibitory effect of C protein on viral genome replication and transcription. C protein affects viral RNA synthesis in a promoter-specific fashion and inhibits leader promoter-dependent positive-sense RNA synthesis but not trailer promoter-dependent negative-sense RNA synthesis (2). Indeed, it was found that the level of positive-sense RNA products, including viral mRNA and antigenome in 4C(−)-infected cells, was extremely higher than that in wt SeV-infected cells in contrast to the steady-state level of negative-sense RNA genomes (19). An increase in N-P gene read-through positive-sense RNA transcripts was also observed for 4C(−)-infected cells, suggesting possible enhancement of other read-through positive-sense RNA transcripts, including L-trailer read-through transcripts. Since the L-trailer read-through transcripts can anneal to the trailer RNA, it could become a possible source of dsRNA, as pointed out previously (46).
During progress of our research, C knockout measles virus (MeV) was found to induce phosphorylation of eIF2α and translational inhibition in infected cells (34). More recently, the P and V proteins of simian virus 5 (SV5) have been reported to play similar roles in limiting PKR activation to maintain protein synthesis (5). However, these studies did not elucidate how these proteins regulate activation of PKR. Thus, the present study gives the first example of paramyxovirus mechanisms underlying regulation of PKR activation. SeV, MeV, and SV5 belong to the same subfamily, Paramyxovirinae, but to different genera. SV5 does not express C protein, but MeV expresses C protein, which is a different protein on the amino acid sequence level (only 19% homologous to the SeV C protein) (33). Therefore, it is surprising that these different proteins have acquired similar roles during evolution. On the other hand, at least the Ulster strain of NDV does not seem to have viral proteins that limit dsRNA generation to regulate the PKR activation and phosphorylation of eIF2α (Fig. 2C, 4B, and 5A). It is thus intriguing to learn how the NDV Ulster strain has survived without acquiring antagonisms against PKR-mediated translational inhibition during evolution.
In summary, we have studied a role of SeV C protein in restricting PKR activation for maintaining protein synthesis. Our findings reveal that SeV C protein does not target the common intracellular pathway leading to PKR activation but limits dsRNA generation, probably by regulating viral genome replication and transcription.
Acknowledgments
We thank M. Mikami for her excellent technical assistance; A. Kato and Y. Nagai (Tokyo) for providing the recombinant wt SeV, 4C(−), and V(−); L. Roux (Geneva) for providing DIH4; C. Örvell for providing the anti-SeV N monoclonal antibody (mAb1.180); F. Weber (Freiburg) for advice about obtaining the mouse monoclonal antibody J2; and A. Kawai (Kyoto) for his encouragement to K.T.
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (no. 18590446 and no. 19790337), by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science, and Technology (no. 16017236), by the Yakult Foundation, by Fukui University, and by Wajinkai of Shiga University of Medical Science, Japan.
Footnotes
Published ahead of print on 6 August 2008.
REFERENCES
- 1.Aoki, K., and M. Kawakita. 1996. Differential sensitivity of two related viruses, Newcastle disease virus and Sendai virus, to interferon in mouse Had-2 cells: selective inhibition of translation of NDV mRNA. Arch. Virol. 1411847-1862. [DOI] [PubMed] [Google Scholar]
- 2.Cadd, T., D. Garcin, C. Tapparel, M. Itoh, M. Homma, L. Roux, J. Curran, and D. Kolakofsky. 1996. The Sendai paramyxovirus accessory C proteins inhibit viral genome amplification in a promoter-specific fashion. J. Virol. 705067-5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curran, J., R. Boeck, and D. Kolakofsky. 1991. The Sendai virus P gene expresses both an essential protein and an inhibitor of RNA synthesis by shuffling modules via mRNA editing. EMBO J. 103079-3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curran, J., J. B. Marq, and D. Kolakofsky. 1992. The Sendai virus nonstructural C proteins specifically inhibit viral mRNA synthesis. Virology 189647-656. [DOI] [PubMed] [Google Scholar]
- 5.Gainey, M. D., P. J. Dillon, K. M. Clark, M. J. Manuse, and G. D. Parks. 2008. Paramyxovirus-induced shutoff of host and viral protein synthesis: role of the P and V proteins in limiting PKR activation. J. Virol. 82828-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.García, M. A., J. Gil, I. Ventoso, S. Guerra, E. Domingo, C. Rivas, and M. Esteban. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 701032-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcin, D., J. Curran, M. Itoh, and D. Kolakofsky. 2001. Longer and shorter forms of Sendai virus C proteins play different roles in modulating the cellular antiviral response. J. Virol. 756800-6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcin, D., P. Latorre, and D. Kolakofsky. 1999. Sendai virus C proteins counteract the interferon-mediated induction of an antiviral state. J. Virol. 736559-6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gotoh, B., T. Komatsu, K. Takeuchi, and J. Yokoo. 2001. Paramyxovirus accessory proteins as interferon antagonists. Microbiol. Immunol. 45787-800. [DOI] [PubMed] [Google Scholar]
- 10.Gotoh, B., T. Komatsu, K. Takeuchi, and J. Yokoo. 2002. Paramyxovirus strategies for evading the interferon response. Rev. Med. Virol. 12337-357. [DOI] [PubMed] [Google Scholar]
- 11.Gotoh, B., T. Komatsu, K. Takeuchi, and J. Yokoo. 2003. The C-terminal half-fragment of the Sendai virus C protein prevents the gamma-activated factor from binding to a gamma-activated sequence site. Virology 31629-40. [DOI] [PubMed] [Google Scholar]
- 12.Gotoh, B., Y. Ohnishi, N. M. Inocencio, E. Esaki, K. Nakayama, P. J. Barr, G. Thomas, and Y. Nagai. 1992. Mammalian subtilisin-related proteinases in cleavage activation of the paramyxovirus fusion glycoprotein: superiority of furin/PACE to PC2 or PC1/PC3. J. Virol. 666391-6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gotoh, B., K. Takeuchi, T. Komatsu, and J. Yokoo. 2003. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J. Virol. 773360-3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gotoh, B., K. Takeuchi, T. Komatsu, J. Yokoo, Y. Kimura, A. Kurotani, A. Kato, and Y. Nagai. 1999. Knockout of the Sendai virus C gene eliminates the viral ability to prevent the interferon-alpha/beta-mediated responses. FEBS Lett. 459205-210. [DOI] [PubMed] [Google Scholar]
- 15.Gubbay, O., J. Curran, and D. Kolakofsky. 2001. Sendai virus genome synthesis and assembly are coupled: a possible mechanism to promote viral RNA polymerase processivity. J. Gen. Virol. 822895-2903. [DOI] [PubMed] [Google Scholar]
- 16.Hasan, M. K., A. Kato, M. Muranaka, R. Yamaguchi, Y. Sakai, I. Hatano, M. Tashiro, and Y. Nagai. 2000. Versatility of the accessory C proteins of Sendai virus: contribution to virus assembly as an additional role. J. Virol. 745619-5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He, B. 2006. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 13393-403. [DOI] [PubMed] [Google Scholar]
- 18.Hornung, V., J. Ellegast, S. Kim, K. Brzozka, A. Jung, H. Kato, H. Poeck, S. Akira, K. K. Conzelmann, M. Schlee, S. Endres, and G. Hartmann. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314994-997. [DOI] [PubMed] [Google Scholar]
- 19.Irie, T., N. Nagata, T. Yoshida, and T. Sakaguchi. 2008. Paramyxovirus Sendai virus C proteins are essential for maintenance of negative-sense RNA genome in virus particles. Virology 374495-505. [DOI] [PubMed] [Google Scholar]
- 20.Irie, T., N. Nagata, T. Yoshida, and T. Sakaguchi. 2008. Recruitment of Alix/AIP1 to the plasma membrane by Sendai virus C protein facilitates budding of virus-like particles. Virology 371108-120. [DOI] [PubMed] [Google Scholar]
- 21.Irie, T., Y. Shimazu, T. Yoshida, and T. Sakaguchi. 2007. The YLDL sequence within Sendai virus M protein is critical for budding of virus-like particles and interacts with Alix/AIP1 independently of C protein. J. Virol. 812263-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnston, M. D. 1981. The characteristics required for a Sendai virus preparation to induce high levels of interferon in human lymphoblastoid cells. J. Gen. Virol. 56175-184. [DOI] [PubMed] [Google Scholar]
- 23.Kato, A., K. Kiyotani, Y. Sakai, T. Yoshida, and Y. Nagai. 1997. The paramyxovirus, Sendai virus, V protein encodes a luxury function required for viral pathogenesis. EMBO J. 16578-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiyotani, K., S. Takao, T. Sakaguchi, and T. Yoshida. 1990. Immediate protection of mice from lethal wild-type Sendai virus (HVJ) infections by a temperature-sensitive mutant, HVJpi, possessing homologous interfering capacity. Virology 17765-74. [DOI] [PubMed] [Google Scholar]
- 25.Komada, H., S. Kusagawa, C. Örvell, M. Tsurudome, M. Nishio, H. Bando, M. Kawano, H. Matsumura, E. Norrby, and Y. Ito. 1992. Antigenic diversity of human parainfluenza virus type 1 isolates and their immunological relationship with Sendai virus revealed by using monoclonal antibodies. J. Gen. Virol. 73875-884. [DOI] [PubMed] [Google Scholar]
- 26.Komatsu, T., K. Takeuchi, and B. Gotoh. 2007. Bovine parainfluenza virus type 3 accessory proteins that suppress beta interferon production. Microbes Infect. 9954-962. [DOI] [PubMed] [Google Scholar]
- 27.Komatsu, T., K. Takeuchi, J. Yokoo, and B. Gotoh. 2004. C and V proteins of Sendai virus target signaling pathways leading to IRF-3 activation for the negative regulation of interferon-beta production. Virology 325137-148. [DOI] [PubMed] [Google Scholar]
- 28.Koyama, A. H., H. Irie, A. Kato, Y. Nagai, and A. Adachi. 2003. Virus multiplication and induction of apoptosis by Sendai virus: role of the C proteins. Microbes Infect. 5373-378. [DOI] [PubMed] [Google Scholar]
- 29.Kurotani, A., K. Kiyotani, A. Kato, T. Shioda, Y. Sakai, K. Mizumoto, T. Yoshida, and Y. Nagai. 1998. Sendai virus C proteins are categorically nonessential gene products but silencing their expression severely impairs viral replication and pathogenesis. Genes Cells 3111-124. [DOI] [PubMed] [Google Scholar]
- 30.Lamb, R. A., and G. D. Parks. 2007. Paramyxoviridae: the viruses and their replication, p. 1449-1496. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 1. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 31.Langland, J. O., J. M. Cameron, M. C. Heck, J. K. Jancovich, and B. L. Jacobs. 2006. Inhibition of PKR by RNA and DNA viruses. Virus Res. 119100-110. [DOI] [PubMed] [Google Scholar]
- 32.Miyakoshi, J., K. D. Dobler, J. Allalunis-Turner, J. D. McKean, K. Petruk, P. B. Allen, K. N. Aronyk, B. Weir, D. Huyser-Wierenga, D. Fulton, et al. 1990. Absence of IFNA and IFNB genes from human malignant glioma cell lines and lack of correlation with cellular sensitivity to interferons. Cancer Res. 50278-283. [PubMed] [Google Scholar]
- 33.Nagai, Y. 1999. Paramyxovirus replication and pathogenesis. Reverse genetics transforms understanding. Rev. Med. Virol. 983-99. [DOI] [PubMed] [Google Scholar]
- 34.Nakatsu, Y., M. Takeda, S. Ohno, R. Koga, and Y. Yanagi. 2006. Translational inhibition and increased interferon induction in cells infected with C protein-deficient measles virus. J. Virol. 8011861-11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ong, C. L., J. C. Thorpe, P. R. Gorry, S. Bannwarth, A. Jaworowski, J. L. Howard, S. Chung, S. Campbell, H. S. Christensen, G. Clerzius, A. J. Mouland, A. Gatignol, and D. F. Purcell. 2005. Low TRBP levels support an innate human immunodeficiency virus type 1 resistance in astrocytes by enhancing the PKR antiviral response. J. Virol. 7912763-12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Örvell, C., and M. Grandien. 1982. The effects of monoclonal antibodies on biologic activities of structural proteins of Sendai virus. J. Immunol. 1292779-2787. [PubMed] [Google Scholar]
- 37.Ostertag, D., T. M. Hoblitzell-Ostertag, and J. Perrault. 2007. Overproduction of double-stranded RNA in vesicular stomatitis virus-infected cells activates a constitutive cell-type-specific antiviral response. J. Virol. 81503-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel, R. C., and G. C. Sen. 1998. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 174379-4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314997-1001. [DOI] [PubMed] [Google Scholar]
- 40.Poole, E., B. He, R. A. Lamb, R. E. Randall, and S. Goodbourn. 2002. The V proteins of simian virus 5 and other paramyxoviruses inhibit induction of interferon-beta. Virology 30333-46. [DOI] [PubMed] [Google Scholar]
- 41.Randall, R. E., and S. Goodbourn. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 891-47. [DOI] [PubMed] [Google Scholar]
- 42.Robertson, H. D., and M. B. Mathews. 1996. The regulation of the protein kinase PKR by RNA. Biochimie 78909-914. [DOI] [PubMed] [Google Scholar]
- 43.Romano, P. R., M. T. Garcia-Barrio, X. Zhang, Q. Wang, D. R. Taylor, F. Zhang, C. Herring, M. B. Mathews, J. Qin, and A. G. Hinnebusch. 1998. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol. Cell. Biol. 182282-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakaguchi, T., A. Kato, F. Sugahara, Y. Shimazu, M. Inoue, K. Kiyotani, Y. Nagai, and T. Yoshida. 2005. AIP1/Alix is a binding partner of Sendai virus C protein and facilitates virus budding. J. Virol. 798933-8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schönborn, J., J. Oberstrass, E. Breyel, J. Tittgen, J. Schumacher, and N. Lukacs. 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 192993-3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strahle, L., D. Garcin, and D. Kolakofsky. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351101-111. [DOI] [PubMed] [Google Scholar]
- 47.Sudhakar, A., A. Ramachandran, S. Ghosh, S. E. Hasnain, R. J. Kaufman, and K. V. Ramaiah. 2000. Phosphorylation of serine 51 in initiation factor 2α (eIF2α) promotes complex formation between eIF2α(P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry 3912929-12938. [DOI] [PubMed] [Google Scholar]
- 48.Tapparel, C., S. Hausmann, T. Pelet, J. Curran, D. Kolakofsky, and L. Roux. 1997. Inhibition of Sendai virus genome replication due to promoter-increased selectivity: a possible role for the accessory C proteins. J. Virol. 719588-9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weber, F., V. Wagner, S. B. Rasmussen, R. Hartmann, and S. R. Paludan. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 805059-5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yokoo, J., B. Gotoh, T. Komatsu, K. Takeuchi, and T. Miyadai. 1999. Replication-incompetent Sendai virus can suppress the antiviral action of type I interferon. Arch. Virol. 1441043-1055. [DOI] [PubMed] [Google Scholar]