

Abstract
Described is a convergent 13-step synthesis of a pentacyclic compound which has previously been transformed into haouamine A, therefore constituting a formal total synthesis of this unique marine alkaloid.
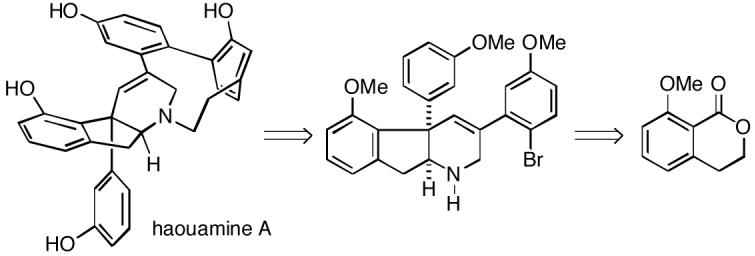
In 2003, Zubia and coworkers isolated two novel polycyclic alkaloids from the marine ascidian Aplidium haouarianum collected off the southern coast of Spain.1 The compounds were named haouamine A and B, and were assigned structures 1 and 2, respectively, based upon NMR analysis and X-ray crystallography. Interestingly, these metabolites exist in solution as a dynamic 2:1 interconverting mixture of stereoisomers generated either by nitrogen inversion or by atropisomerism of the paracyclophane system. A unique feature of these alkaloids is the 3-aza-[7]-paracyclophane moiety, which is so highly strained that the B-ring is in fact non-planar and exists in a boat-like conformation, as can be seen from the X-ray structure. The absolute configuration of these metabolites has not yet been established. Haouamine A has high and selective activity against the human colon carcinoma cell line HT-29 with an IC50 of 0.1 μg/mL. Haouamine B has only slight cytotoxic activity against mouse endothelial cells MS-1.
During the past year several research groups have begun to address the synthesis of the haouamines.2 Rawal and coworkers,2a and Grundl and Trauner2b have devised nice approaches to the indenotetrahydropyridine nucleus of the alkaloids. More recently, Wipf and Furegati have reported that the very strained aza-paracyclophane system of the haouamines cannot be constructed by standard biaryl coupling methodology.2c However, they were able to prepare a haouamine 3-aza-[7]-paracyclophane model system via a late stage aromatization of a non-benzenoid B-ring precursor.
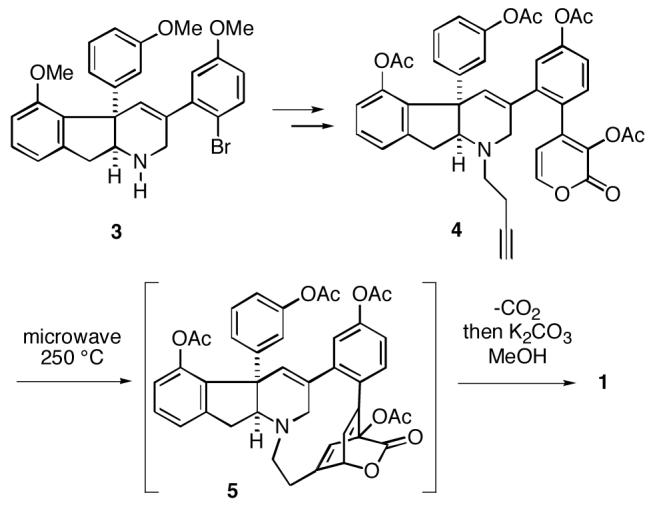
In 2006, Baran and Burns published the first total synthesis of haouamine A (1).3 In the course of this work it was also found that metal-mediated biaryl coupling technology cannot be used to generate the paracyclophane. A clever solution to this problem was to convert intermediate aryl bromide 3 to α-pyrone alkyne 4, which upon heating underwent an intramolecular Diels-Alder reaction to produce cycloadduct 5 (Scheme 1). Subsequent spontaneous loss of carbon dioxide from adduct 5 gave tetraacetyl-haouamine A (albeit in low conversion from 4) which could be transformed to the alkaloid 1.
Scheme 1.
In this communication we describe a new and efficient approach to the Baran indenotetrahydropyridine pentacyclic intermediate 3, which therefore constitutes a formal total synthesis of haouamine A. Our strategy for construction of 3 was to use an intramolecular nitrone/olefin dipolar cycloaddition to produce the requisite indene system with its attendant functionality and stereochemistry.4,5 The initial plan was then to utilize the ring closing metathesis chemistry of vinyl chlorides which we had previously developed to establish the tetrahydropyridine ring.6
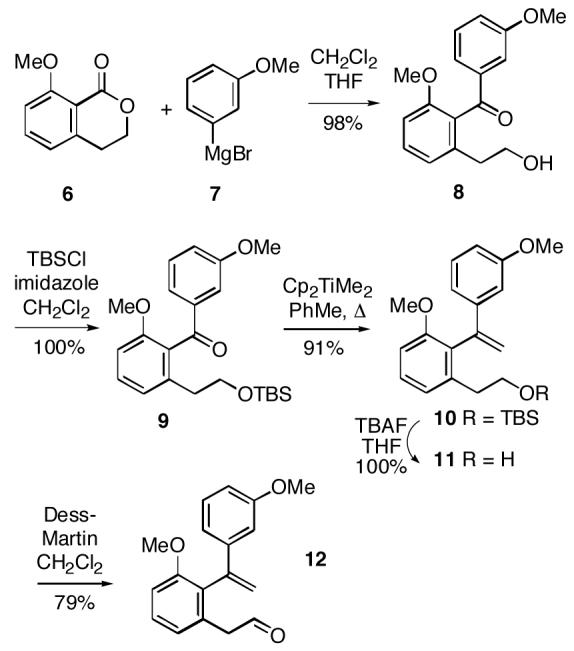
To prepare the intermediate necessary for the nitrone/olefin cycloaddition, we began with known, readily prepared lactone 6,7 which underwent addition of commercially available Grignard reagent 7 to afford keto alcohol 8 in high yield (Scheme 2). This alcohol was protected as silyl ether 9. Olefination of ketone 9 could be effected cleanly with the Tebbe-Petasis reagent8,9 to give 10, and subsequent removal of the silyl group afforded alcohol 11. Finally, Dess-Martin oxidation10 of alcohol 11 generated aldehyde 12.
Scheme 2.
With this aldehyde in hand, we began to explore the proposed intramolecular nitrone/olefin dipolar cycloaddition. Treatment of aldehyde 12 with N-benzylhydroxylamine in toluene at room temperature for one hour produced nitrone 13, as monitored by 1H NMR (Scheme 3). Subsequent heating of the solution of 13 at 115 °C for 36 hours, followed by column chromatography, produced the desired linear cycloadduct 14 in 63% isolated yield. The structure and stereochemistry of this isoxazolidine was confirmed by NMR and X-ray analysis. In addition to adduct 14, an impure chromatographic fraction was isolated which contained the bridged cycloadduct 15, but despite some effort, this compound could not be obtained in pure form for full characterization. However, heating the fraction containing 15 in toluene at 115 °C for 24 h produced the linear adduct 14, increasing the overall yield to 76%. This process undoubtedly involves a well precedented thermal retro-cycloaddition of the bridged adduct 15 to starting nitrone 13.4,5 In view of these results, the cycloaddition was conducted in toluene-d8 in an NMR tube, and was periodically monitored. It was found that bridged cycloadduct 15 is the kinetic product of the reaction, and over time is converted to the more stable linear adduct 14. For preparative purposes, it proved most convenient to heat the nitrone 13 in toluene solution in an oil bath maintained at 130 °C for 45 hours, which provided the desired adduct 14 in 73% isolated yield after chromatography.
Scheme 3.

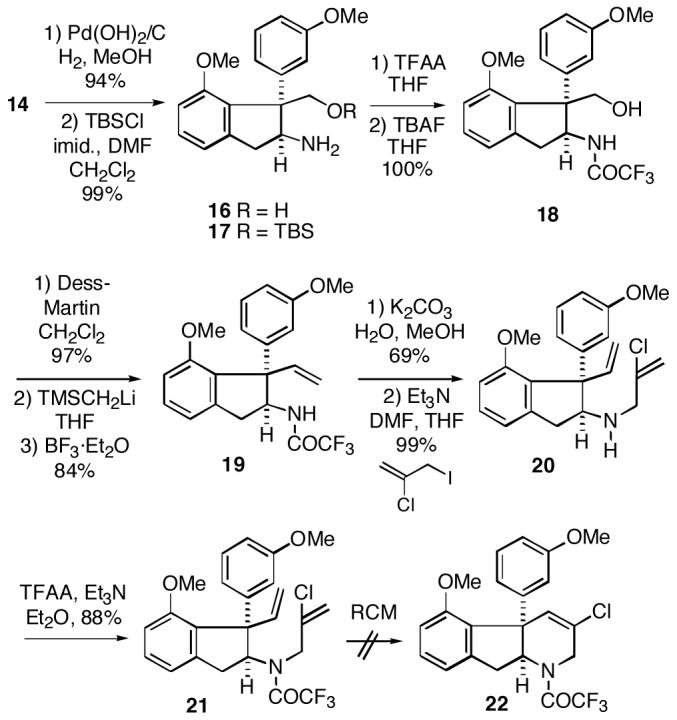
As noted above, our original intent was to construct a functionalized tetrahydropyridine ring potentially useful for the haouamines via a ring closing metathesis of a vinyl chloride.6 Towards this end, isoxazolidine 14 was first hydrogenated with Pearlman’s catalyst to afford amino alcohol 16 (Scheme 4). Protection of this alcohol as the TBS ether 17, followed by conversion of the amine to the trifluoroacetamide and removal of the silyl group led to alcohol 18. Dess-Martin oxidation10 of alcohol 18 to the corresponding aldehyde and subsequent Peterson olefination then afforded intermediate 19. All attempts to directly N-alkylate the anion derived from trifluoroacetamide 19 with 3-iodo-2-chloropropene failed.11 Therefore, it was necessary to first remove the trifluoroacetyl group to generate the corresponding free amine, which could be successfully alkylated to yield diene amine 20. Reintroduction of the TFA protecting group gave the desired metathesis substrate 21 in high overall yield. Unfortunately, all attempts to effect ring closing metathesis of 21 to produce indenotetrahydropyridine 22 failed despite screening a number of catalysts. In most cases only the starting diene 21 was recovered. We believe the problem here is steric in origin, since based upon our earlier work it appears that the non-chlorinated olefin must initially form a metal carbene species for this RCM process to occur as desired.6
Scheme 4.
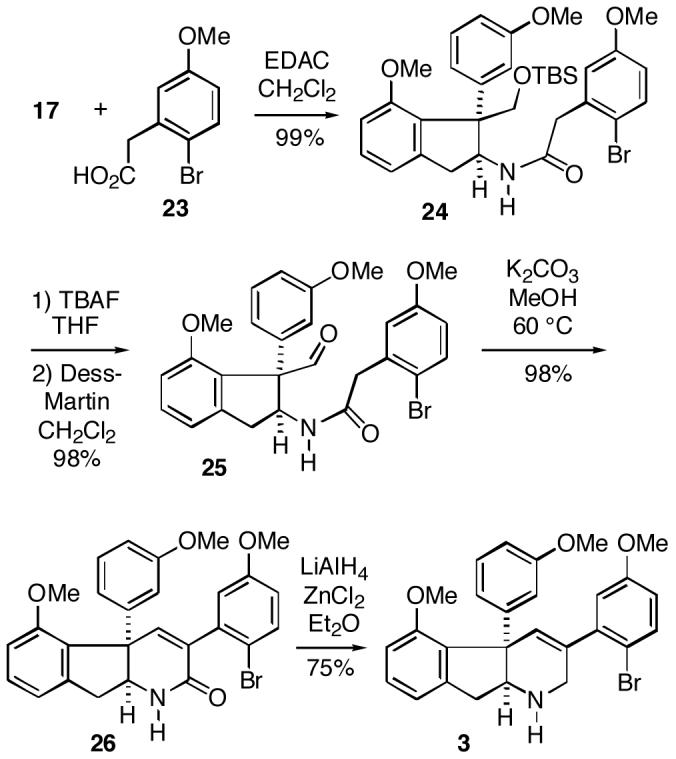
Since the metathesis strategy proved untenable, we turned to what proved to be a shorter and more convergent approach for introducing both the tetrahydropyridine and A-rings. It was possible to couple amine 17 with known phenylacetic acid 2312 leading to amide 24 (Scheme 5). Removal of the silyl protecting group of 24 with TBAF followed by Dess-Martin oxidation10 of the resulting primary alcohol afforded aldehyde 25. In a key transformation, it was found that warming aldehyde amide 25 in warm methanol in the presence of potassium carbonate effects an aldol condensation/dehydration to produce the desired pentacyclic lactam 26 in high yield. To complete the synthesis, lactam 26 was reduced with lithium aluminum hydride/zinc chloride to give the Baran pentacycle 3 in good yield.13 This compound has spectral data identical to those previously reported.3
Scheme 5.
In conclusion, we have developed a convergent synthesis of the Baran pentacycle 3 which requires 13 steps and proceeds in 34% overall yield starting from readily available bicyclic lactone 6. We are currently investigating some new strategies for efficiently converting this intermediate into haouamine A (1).
Supplementary Material
Figure 1.
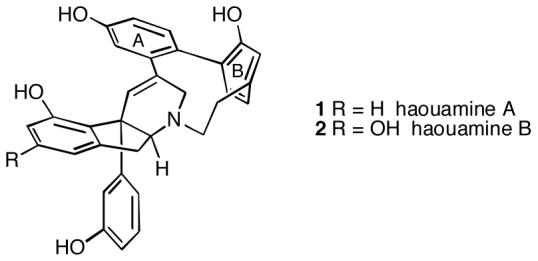
Structures of haouamines.
Acknowledgment
We are grateful to the National Institutes of Health (GM-32299 and CA-34303) for financial support of this research. We thank Dr. Hemant Yennawar (Penn State Small Molecule X-Ray Crystallographic Facility) for the crystal structure determination of cycloadduct 14.
References
- 1.Garrido L, Zubia E, Ortega MJ, Salva J. J. Org. Chem. 2003;68:293. doi: 10.1021/jo020487p. [DOI] [PubMed] [Google Scholar]
- 2(a).Smith ND, Hayashida J, Rawal VH. Org. Lett. 2005;7:4309. doi: 10.1021/ol0512740. [DOI] [PubMed] [Google Scholar]; (b) Grundl MA, Trauner D. Org. Lett. 2006;8:23. doi: 10.1021/ol052262h. [DOI] [PubMed] [Google Scholar]; (c) Wipf P, Furegati M. Org. Lett. 2006;8 doi: 10.1021/ol060455e. ASAP. [DOI] [PubMed] [Google Scholar]
- 3.Baran PS, Burns NZ. J. Am. Chem. Soc. 2006;128:3908. doi: 10.1021/ja0602997. [DOI] [PubMed] [Google Scholar]
- 4.Cf. Baldwin SW, Debenham JS. Org. Lett. 2000;2:99. doi: 10.1021/ol9911472. [DOI] [PubMed] [Google Scholar]
- 5.For a recent review of nitrone/olefin cycloadditions see:Jones RCF, Martin JN. In: Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products. Padwa A, Pearson WH, editors. Wiley; New York: 2002. Chap. 1.
- 6(a).Chao W, Weinreb SM. Org. Lett. 2003;5:2505. doi: 10.1021/ol034775z. [DOI] [PubMed] [Google Scholar]; (b) Chao W, Meketa ML, Weinreb SM. Synthesis. 2004:2058. [Google Scholar]
- 7.Rama Rao AV, Reddy DR. Synth. Commun. 1986;16:97. [Google Scholar]
- 8(a).Petasis NA, Bzowej EI. J. Am. Chem. Soc. 1990;112:6392. [Google Scholar]; (b) Payack JF, Hughes DL, Cai D, Cottrell IF, Verhoeven TR. Org. Synth. 2002;79:355. [Google Scholar]
- 9.This methylenation cannot be done by Wittig chemistry, and the Peterson reaction proceeds in poor yield. However, alkene 10 can also be prepared from ketone 9 in a yield similar to the Tebbe-Petasis reaction by addition of methyllithium, followed by dehydration with hot acetic acid.
- 10.Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277. [Google Scholar]
- 11.Nordlander JE, Catalane DB, Eberlein TH, Farkas LV, Howe RS, Stevens RM, Tripoulas NA. Tetrahedron Lett. 1978:4987. [Google Scholar]
- 12.Lebegue N, Bethegnies G, Berthelot P. Synth. Commun. 2004;34:1041. [Google Scholar]
- 13.Van der Veken P, Kertesz I, Senten K, Haemers A, Augustyns K. Tetrahedron Lett. 2003;44:6231. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.