

Abstract
Mass spectrometry analysis was used to target three different aspects of the viral infection process: the expression kinetics of viral proteins, changes in the expression levels of cellular proteins, and the changes in cellular metabolites in response to viral infection. The combination of these methods represents a new, more comprehensive approach to the study of viral infection revealing the complexity of these events within the infected cell. The proteins associated with measles virus (MV) infection of human HeLa cells were measured using a label-free approach. On the other hand, the regulation of cellular and Rock House Virus (FHV) proteins in response to FHV infection of Drosophila cells were monitored using stable isotope labeling. Three complementary techniques were used to monitor changes in viral protein expression in the cell and host protein expression. A total of 1500 host proteins were identified and quantified, of which over 200 proteins were either up- or down-regulated in response to viral infection, such as the up regulation of the Drosophila apoptotic croquemort protein, and the down regulation of proteins that inhibited cell death. These analyses also demonstrated the up-regulation of viral proteins functioning in replication, inhibition of RNA interference, viral assembly, and RNA encapsidation. Over 1000 unique metabolites were also observed with significant changes in over 30, such as the down-regulated cellular phospholipids possibly reflecting the initial events in cell death and viral release. Overall, the cellular transformation that occurs upon viral infection is a process involving hundreds of proteins and metabolites, many of which are structurally and functionally uncharacterized.
Keywords: virus, protein regulation, viral infection, metabolites, isotope labeling, mass spectrometry
Introduction
A key component in understanding biological events is to quantitatively monitor molecular concentration changes within the cell. Direct measurement of the cellular response to perturbation provides a means to identify the species that participate in cellular regulation and further allows for the determination of the molecule’s functional activity. The course of viral infection involves host cell regulation, but there is no comprehensive method to characterize these changes. Current approaches for monitoring the progress of viral infection and the cellular response, include assays based on cell cultures, antibody-antigen reactions, and nucleic acid analysis.1 Assays based on cell cultures such as the plaque assay and variations thereof remain the most commonly used.2 Other assays such as those based on a specific interaction of viral antigen with antibodies are often problematic due to the nature of affinity recognition, and are specific to a particular molecule, limiting the use for large-scale proteomics.3 Gene expression in infected cells at the mRNA level can be measured using PCR and northern blot,4 and microarrays allow simultaneous analysis of thousands of gene expression products in a single experiment, and have been applied in virology.5–7 However, these experiments do not necessarily reflect changes at the protein level in the infected cells, because changes in mRNA and protein expression levels are not directly correlated.8–10
Differential protein quantification for proteomics is typically measured by two dimensional PAGE, followed by mass spectrometry identification of the differentially expressed proteins. An alternative approach to quantify regulatory changes is stable isotope labeling combined with nanoLC and mass spectrometry.11,12 Generally, two sets of samples are differentially labeled with a stable isotope-one sample with the light form, the other with the heavy form of the label. Depending on the type of the chemical labeling reagent used, samples are differentially labeled before, during, or after enzymatic digestion, combined then subjected to a separation/enrichment technique followed by analysis by mass spectrometry for quantitative comparison. Changes in protein expression levels are quantified by determining the ratio of the peak intensities of the light and heavy forms of the generated peptides. The pairwise comparison of the peak intensities of peptides labeled with the heavy and light form of the label serves as the basis for quantitative protein analysis. It should be noted that the accuracy of the quantitative measurement depends on when the label was incorporated. Highest level of accuracy can be achieved when the label is incorporated early in the process due to the reduction of the sample losses and variability.
Stable isotope labeling has become an indispensable tool in protein quantitation. Two of the most widely used methods in which isotopes can be introduced in a proteome pool are metabolic isotope labeling and chemical isotope labeling.13,14 In metabolic isotope labeling, a stable isotope, typically either 15N or 13C, is introduced into the cell culture during cell growth or protein synthesis. Amino acids from isolated proteins are either globally or selectively labeled. While metabolic isotope labeling is simple, it is limited to protein samples derived from cell cultures. In chemical isotope labeling, chemical modification of functional groups on the peptide side chains or specific structural features on the protein allow the incorporation of stable isotopes before, during, or after proteolysis. Among the most commonly used labeling approaches are isotope coded affinity tag (ICAT),15,16–18O labeling,17–21 lysine specific labeling,22,23 and labeling of the N- or C- termini of the peptides by acylation24 or esterification.25
While stable isotope labeling has been widely used in comparative proteomics, label-free approach is becoming a viable and an attractive alternative.26–28 The method is relatively simple as it utilizes reproducible chromatographic separation and high mass accuracy measurements along with data normalization methods29 to follow the changes in retention times and relative integrated peak intensities that reflect the changes in concentration of analytes in a perturbed biological system. Moreover, informatics algorithms are employed to facilitate the data analysis due to the large dataset that can be obtained from LC-MS experiments. Overall, this is highly effective in comparative proteomics due to its comprehensiveness and throughput. To date, label-free quantitative proteomics approach has been successfully applied to the quantitative proteome analysis of human serum,30,31 yeast,27,32 and Shewanella oneidensis33.
This study presents the use of label-free proteomics approach and stable isotope labeling to quantify differentially expressed proteins in cells during the course of viral infection. Specifically, we apply a non-linear approach to align and quantify proteins (without isotope labeling) expressed when measles virus infects cells. Measles, one of the typical viral diseases of childhood, is a ssRNA negative strand enveloped virus. Measles virus was used to test a new approach to protein quantitation that does not require chemical labeling of the proteins. Digests of whole cell lysates were examined directly with a high mass accuracy ESI-TOF mass spectrometer equipped with a separate reference sprayer. This dual spray configuration allows the introduction of reference ions through a separate ESI probe without signal suppression of the main ESI ions and allows for the mass correction that yields accurate mass within 5 ppm relative error. The ESI-TOF data was analyzed using a newly developed software package XCMS34 for non-linear alignment of the chromatograms and integration of the peak intensities. This allowed us to monitor the changes in levels of the measles structural protein, phosphoprotein P, during the course of cellular infection.
We also applied quantitative profiling approaches based on direct analysis of intensity using 18O and cICAT labeling in combination with nanoLC-MS/MS, FT-MS, DIOS-MS, and capillary LC-ESI TOF MS to profile the changes in protein and metabolite expression levels in Flock House Virus (FHV)-infected Drosophila cells. FHV is a small non-enveloped icosahedral insect positive strand nodavirus that replicates robustly in plants, insects, yeast, and mammalian cells.35–37 Its genome consists of two single stranded messenger-sense RNAs, RNA1 and RNA2. RNA1 encodes the viral replicase, protein A (112 kDa), and RNA2 encodes the coat protein alpha (44 kDa). Following assembly of the virus particle, most of the alpha protein subunits undergo an autocatalytic cleavage, which yields mature coat proteins beta (39 kDa) and gamma (4.4 kDa).38 Cell cultures infected with FHV also produce a subgenomic RNA, RNA3, which is derived from the 3′-end of RNA1 and is not packaged into the virions. It encodes the two non-structural proteins, B1 and B2 (10 kDa). Protein B2 functions to inhibit RNA silencing.39 We monitored expression of both structural and non-structural FHV proteins in Drosophila cells at various times after infection of sf9 Drosophila cells with virus. The rate at which viral proteins were produced during the course of infection was determined by monitoring the peak intensity ratio of at least one peptide and its isotopic analog as internal standard. This approach was validated by determining the absolute concentration of one of the viral proteins during the course of infection, using an internal, isotopically labeled standard. We also identified and quantified the differentially expressed host proteins following viral infection. A total of 1500 host proteins were identified and quantified, of which 150 were up-regulated while 66 were down-regulated in response to viral infection.
In addition to monitoring protein regulation, we developed a mass-based metabolite profiling approach to monitor changes in metabolite levels during the course of viral infection. The examination of the metabolite profile from whole cells was motivated by the improved capabilities of mass spectrometry technology, making quantitative endogenous metabolite information possible. These novel viral and cell protein analyses combined with metabolite profiling offer a comprehensive window into the process of viral infection (see Table 1).
Table 1.
Experimental approaches used in this study
| Separation | Mass Spectrometry | Label | Virus/Target | Purpose | Time points (hours) | Figure |
|---|---|---|---|---|---|---|
| 1D-nano LC/MS | ESI-ion trap | None | Measles virus proteins | Viral protein expression levels | 0, 18, 72 | 1 |
| 1D-capillary LC/MS | ESI-TOF | None | Measles virus proteins | Relative protein quantitation | 0, 18, 72 | 1 |
| None | DIOS | 16O/18O | FHV proteins α,B2,A expression kinetic | Viral protein expression kinetics | 0,4,8, 12, 16, 20, 22, 24 | 2 |
| 1D-nanoLC | ESI-ion trap | 16O/18O | FHV proteins α,B2,A expression kinetics | Viral protein expression kinetics | 0,4,8,12, 16, 20, 22, 24 | 3 |
| 2D-nanoLC | FT-MS | cICAT | FHV/Untargeted cellular proteins | Cellular protein expression changes | 0,8 | 4,5 |
| 1D-microLC/MS | ESI-TOF | None | FHV/Untargeted small molecules (100–1000 m/z) | Small molecule changes in response to viral infection | 0,12 | 6 |
Experimental Section
Materials and Reagents
All reagents were obtained in high purity from Sigma-Aldrich except when noted otherwise. Ammonium bicarbonate (NH4HCO3), ammonium formate (HCO2NH4), guanidine hydrochloride (Gdn·HCl), dibasic sodium phosphate (Na2HPO4), trizma hydrochloride, trizma base, ethylenediaminetetraacetic acid (EDTA), phosphate buffered saline solution (PBS), monobasic sodium phosphate (NaH2PO4), sodium chloride (NaCl), ammonium hydroxide (NH4OH), Tris, sodium fluoride (NaF), sodium orthovanadate (Na3VO4), iodoacetamide (IAA), Triton X-100, penicillin, streptomycin, and phenylmethylsulfonyl fluoride (PMSF) were purchased from Sigma (St. Louis, MO). Sodium sulfate (Na2SO4) was from ICN Biochemicals Inc. (Aurora, OH). Trifluoroacetic acid (TFA) and formic acid were from Fluka (Milwaukee, WI). Hydrochloric (HCl) acid and hydrofluoric (HF) acid were from Fisher Scientific (Fairlawn, NJ). HPLC grade acetonitrile (CH3CN) and methanol (CH3OH) were obtained from EM Science (Darmstadt, Germany). UltraLink™ immobilized monomeric avidin, Tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl), ImmunoPure D-biotin, and bicinchonic acid (BCA) protein assay reagent kit were purchased from Pierce (Rockford, IL). Water was purified by a Barnstead Nanopure system (Dubuque, IA). Cleavable ICAT reagents were from Applied Biosystems, Inc. (Foster City, CA). RapiGest, (heptadecafluoro-1,1,2,2-tetrahydrododecyl)dimethylchlorosilane, and trypsin, were obtained from Waters Corporation, Gelest, Inc., and Promega, respectively.
Cell Culture and Viruses
Drosophila melanogaster cells (Schneider’s line 1) were suspended at a density of 4 × 107 cells/mL in a Schneider’s insect medium supplemented with 15% fetal bovine serum, 100 units of penicillin/mL, and 100 μg of streptomycin/mL. The suspension was inoculated with FHV at a multiplicity of 20 plaque forming units/cell and allowed to attach for 1 hour at room temperature with gentle agitation. A volume of 2.5 mL (1 × 108 cells) was aliquoted into 100 mm tissue culture dishes containing 12.5 mL growth medium. The dishes were incubated without further agitation at 27°C. Cells were harvested and processed for protein analysis at 0, 4, 8, 12, 16, 20, 22, and 24 hours post-infection. A sample of uninfected cells served as a control. For measles virus, HeLa cells were used as previously reported, and cell proteins analyzed 0, 18, and 72 hours post-infection.40 Measles virus was propagated, purified, and quantitated as previously published.40,41
Measles virus
HeLa cells were infected with measles virus, and harvested at 18 or 72 hours post-infection, or mock-infected with buffer. Cells were lysed by freeze/thaw, clarified, and the total protein concentration was determined by Bradford assay. Proteins were digested with trypsin following denaturation in 6 M urea, reduction with DTT, and reaction with iodoacetamide. Proteins were identified using a chip-based nanoelectrospray LC-MS/MS with an ion trap (Agilent MSD Trap, HPLC Chip Cube). A total of 500 ng protein was injected, and the peptides were eluted with a gradient from 8–37% acetonitrile, at a flow rate of 500 nL/min over 20 minutes. MS/MS data was search against SwissProt database using Mascot. The search results show that individual ion scores above 32 indicate that the peptide based identification at 95% confidence level. Ion score of 48 was obtained for the phosphoprotein P peptides. A total of one microgram protein was also injected on a capillary flow reverse-phase HPLC column (Agilent 1100 system), using a reverse-phase column (Zorbax 300 SB-C18 with 0.5mm inner diameter and 150 mm length) at a flow rate of 7 μL/min. The resolving portion of the gradient was from 5 to 37% acetonitrile/0.1 % formic acid over 35 minutes. The mass spectrometer was an ESI-TOF (Agilent MSD) with a reference spray containing ions at m/z 121.0509 and 922.0098. The scan range was from 100–2000 m/z with a capillary voltage of 3500 V, and a fragmentator voltage of 120 V. The program XCMS was used to align the datasets,34 and Analyst was used to calculate the peak heights and areas for particular ions.
Protein Extraction
At various times after infection, medium was removed from the dishes and the cell monolayers were rinsed with PBS. The cells were washed off the tissue culture dish with 10 mL of PBS buffer, transferred to a 15 mL conical tube and pelleted by centrifugation at 670 × g for 5 minutes. The supernatant was removed and the cells were immediately frozen in a dry ice-ethanol bath and stored at −80°C pending analysis. Frozen cells were thawed at room temperature and then resuspended in 150 μL TNE buffer (10 mM Tris Buffer, 100 mM NaCl, 1 mM EDTA, pH 8) and lysed with 150 μL of 2% (wt/v) RapiGest in TNE buffer. Samples were mixed with a vortexer and incubated at room temperature for 60 minutes to extract proteins. The samples were then centrifuged at 20,000 × g for 10 minutes and supernatants were used for protein digestion.
Tryptic Protein Digestion for 18O labeling
Each time point sample was diluted 10 fold in 10 mM tris buffer to a final concentration of 0.1% RapiGest. The total protein concentration of each sample was measured by Bradford assay and 300 μL of each sample (~100 μg proteins) was used for protein digestion. Samples were boiled for 5 min. to denature the proteins followed by 5 min sonication, reduction and alkylation with 2 mM TCEP at 37°C for 30 min and 10 mM IAA at 37°C in the dark for 30 min, respectively. Proteins were then digested at 37°C with trypsin at a protein:enzyme ratio of 50:1 (w/w) overnight, followed by a second trypsin digestion under the same conditions. Each sample was brought to a final concentration of 50 mM (pH < 2) HCl to break down RapiGest. The samples were subsequently incubated at 37°C for 1 hour and centrifuged at 25,000 × g for 10 min.
Preparation of the 16O/18O Labeled FHV Digest
Labeling experiments were performed by digesting the proteins in H2O18 and by using Prolytica 18O labeling kit obtained from Strategene, Inc. 18O labeling efficiency of ~ 95% was obtained indicating efficient 18O incorporation to peptides. 16O/18O labeling with Prolytica kit was done using the protocol recommended by the manufacturer. Briefly, two 50 μL portions of the tryptic digest from each time point were dried in a SpeedVac. The dried peptides were reconstituted with 10 mM of Tris buffer followed by the addition of 10 μL of resuspended immobilized trypsin. The peptide/immobilized trypsin mixture was dried without heat in a SpeedVac for 1 hour. The dried mixture was resuspended in 40 μL H2O18 and 10 μL acetonitrile while the other dried portion in 40 μL water and 10 μL of acetonitrile. The mixtures were continuously agitated in a vortex mixer for 3–5 hours at room temperature. The reaction was stopped by adding 2.5 μL of concentrated formic acid. The samples were centrifuged at 13200 × g for 5 minutes and the supernatants were used for the analysis.
Internal Standards for 18O Labeling
Sucrose gradient purified FHV at a concentration of 0.93 mg/mL and a protein coat concentration of about 0.275 μg/mL as measured by Bradford assay and the 16-hour time point were used as internal standards in this study. One hundred microliters of each virus solution and 0.2% RapiGest in TNE buffer were mixed to dissolve the virus particles. These solutions were subsequently digested using the same digestion protocol described above.
Cleavable ICAT (cICAT) labeling
Equal amounts of Drosophila melanogaster proteins (1 mg each) were labeled either with the light (control, cICAT-13C0) or the heavy (8 hours post FHV infection, cICAT-13C9) isotopic versions of the cICAT reagent using a modified method from that recommended by the manufacturer. Briefly, 1 mg of each cell protein extract was dissolved in 80 μL of 6 M Gdn·HCl in 50 mM NH4CO3, pH 8.3. Each sample was chemically reduced by adding 1 μL of 100 mM TCEP·HCl followed by boiling in a water bath for 10 min. The reduced samples were transferred to vials containing either cICAT-13C0 or cICAT-13C9 dissolved in 40 μL of CH3CN and incubated at 37°C for 2 h. The two samples were combined, buffer exchanged into 50 mM NH4CO3, pH 8.3, using a D-Salt Excellulose desalting column (Pierce, Rockford, IL) and digested with trypsin overnight at 37°C using an enzyme to protein ratio of 1:50 (w/w). The digestion was quenched by boiling the samples in a water bath for 10 min and adding PMSF to a final concentration of 1 mM.
Affinity purification and cleaving of cICAT-labeled peptides
A 1.5 mL bed volume UltraLink immobilized monomeric avidin column was slurry packed in a glass Pasteur pipet and equilibrated with 2× PBS (0.2 M sodium phosphate, 0.3 M NaCl, pH 7.2). The column was blocked with 2 mM D-biotin in 2× PBS, pH 7.2, and the biotin was stripped from the reversible binding sites of the column as per manufacturer’s instructions and the column was re-equilibrated with 2× PBS, pH 7.2. The cICAT-labeled peptides were boiled for 5 min, cooled to room temperature, and loaded onto the avidin column and allowed to incubate for 15 min at ambient temperature. After washing the column with 10 bed volumes each of 2× PBS, pH 7.2, 1× PBS, pH 7.2, and 50 mM NH4CO3, pH 8.3/20% CH3CN, the cICAT-labeled peptides were eluted using 30% CH3CN/0.4% formic acid and lyophilized to dryness. The biotin moiety was cleaved from the cICAT-labeled peptides by treatment with the cleaving reagents provided by the manufacturer for 2 h at 37 °C, and lyophilized to dryness.
Strong cation exchange fractionation of cICAT labeled peptides
The lyophilized Drosophila melanogaster/FRV cICAT-labeled peptides were dissolved in 250 μL of 0.1% formic acid and injected onto a strong cation exchange liquid chromatography (SCXLC) column (1 mm × 150 mm, Polysulfoethyl A, PolyLC Inc., Columbia, MD). The following HCO2NH4/CH3CN multistep gradient was used to elute the cICAT-labeled peptides from the column at a flow rate of 50 μL/min: 3% mobile phase B for 5 min, followed a linear increase to 10% B in 35 min, a linear increase to 60% B in 46 min then a linear increase to 100% B in 1 min and maintained at 100% B for 9 min. Mobile phase A was 25% CH3CN, and mobile phase B was 25% CH3CN, 0.5 M HCO2NH4, pH 3.0. Fractions were collected every minute for 96 min. The SCXLC fractions were reconstituted in 20 μL of 0.1 % TFA and 5 μL of each combined into 16 total fractions (from 10 to 90 min) prior to mass spectral analysis.
Mass Spectrometry
DIOS-MS experiments were conducted on an Applied Biosystems Voyager STR mass spectrometer operated in the positive ion mode. This instrument was equipped with automated and multisampling capabilities for rapid sample analysis. DIOS chips were attached to a modified MALDI target plate with a conductive tape and samples were irradiated with a 337 nm N2 laser operated at 5 Hz. Five replicates of each time point sample were deposited on the DIGS chip with each spot analyzed twice. Mass spectra were generated by averaging 500 individual laser shots into a single spectrum. Each spectrum was accumulated from 10 shots at 50 different locations within the DIOS spot. The whole data collection process took less than 3 hours to complete. The ratios of the peak intensities of 16O and 18O labeled peptides, m/z 1073.6 and 1077.6 corresponding to LSQPGLAFLK and m/z 1935.1 and 1939.1 corresponding to the VVVTTTQTAPVPQQNVPR were used to calculate the FHV protein ratios between the time point samples and internal standard.
NanoLC-MS/MS experiments were performed on an Agilent MSD/trap ESI-ion trap mass spectrometer which was directly coupled to an Agilent 1100 nano-pump and micro-auto sampler for tandem LC-MS analysis. Mobile phases utilized for the experiment consisted of buffer A: 99.9% H2O (Burdick and Jackson, high purity solvent) + 0.1% formic acid (J.T. Baker) and buffer B: 99.9 % acetonitrile (Fisher, optima grade) + 0.1% formic acid which were pumped at a flow rate of 250 nL/min. Eight microliters of each time point sample mixed with internal standard at ratio of 2.5:1 were injected into a reversed phase fused silica column (100 μM ID, 15 cm long) pulled to a diameter of <5 μm and then packed with C18 stationary phase (Zorbax SB-C18, Agilent). The experiment was performed using a 120 minute reversed phase gradient. A blank run was performed between every two samples to ensure that there was no sample carry-over. The integrated intensities of the extracted ion chromatograms (EIC) of m/z 968.4 (+2 charge ion of VVVTTTQTAPVPQQNVPR), and 970.4 (+2 charge ion of the 18O-labeled VVVTTTQTAPVPQQNVPR), 944.5 (+2 charge ion of VFVDPLATTTTIQDPLR), 946.5 (+2 charge ion of the 18O-labeled VFVDPLATTTTIQDPLR), 584.4(+2 charge ion of LALIQELPDR), and 586.5 4 (+2 charge ion of the 18O-labeled LALIQELPDR) were used to calculate the coat protein, protein A, and protein B2 ratios between the time point samples and internal standard, respectively. MS/MS data were searched against SwissProt database using Mascot (MatrixScience, Ltd) to confirm that m/z 970.4, 946.5, 586.3 represented the peptides, VVVTTTQTAPVPQQNVPR, VFVDPLATTTTIQDPLR, and LALIQELPDR from FHV with 18O modification.
Nanoflow RPLC-FT-MS/MS of cICAT labeled peptides
Ten cm long nanoRPLC-ESI columns were coupled online with a hybrid linear ion-trap (LIT) Fourier transform ion cyclotron resonance mass spectrometer (LTQ-FT, ThermoElectron, San Jose, CA) to analyze the cICAT labeled peptides from the uninfected and FHV infected Drosophila melanogaster cells. To construct the nanoRPLC-ESI columns, 75 μm i.d. fused-silica microcapillaries (Polymicro Technologies, Phoenix, AZ) were flame-pulled to construct a 10 cm fine i.d. (i.e., 5–7 μm) tip against which 3 μm, 300 Å pore size C-18 silica-bonded stationary RP particles (Vydac) were slurry packed using a slurry packing pump (Model 1666, Alltech Associates, Deerfield, IL). The columns were connected via a stainless steel union to an Agilent 1100 nanoflow LC system (Agilent Technologies, Palo Alto, CA), which was used to deliver mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile). After loading 6 μL of sample, the cICAT-labeled peptides were eluted at a flow rate of ~200 nL/min using a linear step gradient of 2–40% B for 110 min and 40–98% B for 30 min. The hybrid LIT-FT-MS was operated in a data-dependent MS/MS mode in which the three most intense peptide molecular ions in a FT-MS scan were sequentially and dynamically selected for subsequent collision-induced dissociation (CID) in the LIT-MS using a normalized collision energy of 35%. The temperature for the capillary of the ion source was set at 160 °C.
Metabolite Extraction and μLC-ESI-TOF MS Analysis
Cells stored in the −80°C freezer were taken out to thaw for 5 minutes before 600 μL of ice cold acetone was added. Samples were sonicated for 20 minutes and subsequently transferred to 2 mL eppendorf vials. The original tubes were washed with 200 μL acetone and the washing solution were collected and transferred to the corresponding eppendorf vial. The samples were placed in the in −20°C freezer, and kept there for an additional 20 minutes after which they were centrifuged in a 5417C Eppendorf centrifuge at 16400 rpm. The supernatant was decanted and was placed in a new vial. The remaining pellet was subjected to a second extraction with 350 μL MeOH and 50 μL H2O, 1% formic acid. The procedure was repeated and the second supernatant was pooled with the first before drying and storage in −20°C freezer until analysis. Prior to analysis each extract was dissolved in 12 μL 95:5 ACN:H2O (0.1% formic acid) out of which 4 μL was injected in duplicate for each extraction. The LC separation was performed with an Agilent 1100 series fitted with capillary pump heads on a 150 × 0.5 mm (5 μm particle size) Agilent Zorbax column at a flow rate of 7 μL/minute. Gradient separation included a 4 minute hold at 5% ACN then a 26 minute linear gradient to 95% ACN which was held for 2 minutes before re-equilibration. MS Data was collected using an Agilent LC/MSD TOF, scanning m/z range 100 to 1000. Two internal calibrants at m/z 121.0509 and 922.0098, sprayed with a separate nebulizer were employed to improve mass accuracy. CID fragmentation patterns of the 496 and 524 m/z ions were obtained using a Q-TOF mass spectrometer (Waters/Micromass).
Peptide identification and quantitation for cICAT experiment
The raw MS/MS data acquired on the hybrid LIT-FT-MS were searched using SEQUEST against a combined Drosophila melanogaster/FHV proteome database (19477 protein entries) downloaded from the European Bioinformatics Institute (EBI) (http://www.ebi.ac.uk/proteome/index.html). Dynamic modifications for cysteinyl (Cys) residues were set by mass additions of the cleaved cICAT labels (227.13 Da for the light label, 236.16 Da for the heavy label) in a single search. SEQUEST criteria were set as Xcorr ≥ 1.9 for [M+H]1+ ions, ≥ 2.2 for [M+H]2+ ions and ≥ 2.9 for [M+H]3+ ions, and DeltaCn ≥ 0.08 for the identification of fully tryptic peptides within the cICAT-labeled samples. The identified peptides were quantified using XPRESS (ThermoElectron, San Jose, CA), which calculates the relative abundances (13C9/13C0, in this data set) of peptides based on the area of their extracted ion chromatograms (XIC). Significance thresholds for up and down regulation were determined by evaluating the normal equations:
| Equation 1.1 |
| Equation 1.2 |
A significant up- or down- regulation was considered to be 2σ-above or below the mean.
DIOS Chip Preparation
The details of DIGS chip preparation have been described elsewhere.42 Briefly, DIOS chips were prepared by etching low resistivity (0.005–0.02 Ω-cm) n-type Si(100) wafers (Silicon Quest) in 25% v/v HF/ethanol under white light illumination at a current density of 5 mA/cm2 for 2 minutes. Typically, photopatterning was performed to create 36 sample spots on each chip. Immediately after etching, the DIOS chip was rinsed with ethanol and dried in a stream of N2 to give an H-terminated surface, which was oxidized by exposure to ozone (flow rate of 0.5 g/h from an ozone generator directed at the surface for 30 seconds) and subsequently modified with (heptadecafluoro-1,1,2,2-tetrahydrododecyl)dimethylchlorosilane as silylating reagent. The silylation reaction was performed by adding 100 μL of the appropriate silylating reagent on the oxidized DIOS chip, which was placed in a glass Petri dish and incubated in an oven at 90°C for 30 minutes. The modified DIOS chip was then rinsed thoroughly with ethanol and was dried in a stream of NI. This simple silylating procedure generates a modified DIOS chip terminated with a perfluoroalkyl group.
Safety Considerations
Extreme care should be taken in handling hydrofluoric acid solutions because of their toxicity and corrosiveness. All inhalation, ingestion, or skin and eye contact should be strictly avoided. Etching of silicon wafers should be conducted in a ventilated fume hood using proper double-layered nitrile gloves, lab coat and goggles. Hydrofluoric acid solution spills and burns can be neutralized and treated with 2.5% calcium gluconate gel.
Results and Discussion
Monitoring unlabeled viral proteins using non-linear alignment of LC/MS data: Measles virus infection of HeLa cells
Antibodies to selected regions of the measles virus (MV) hemagglutinin (HA) react with the viral HA expressed on the plasma membrane of intact infected HeLa cells and transduce a membrane signal that selectively down-regulate the viral phosphoprotein inside the cell, thereby down-modulating viral transcription.40,43 In contrast, antibodies to non-MV or to HeLa cell surface antigens have no effect on the MV phosphoprotein inside the cell and on the modulation of viral transcription. We employed mass spectrometry to determine the down-regulation of MV phosphoprotein in HeLa cells in the presence of antibodies to MV and its normal expression in HeLa cells treated with the antibody to HeLa cell surface antigen during the course of MV infection. Figure 1 indicates the ability to identify the anticipated levels and to quantitate MV phosphoprotein under these various experimental conditions.
Figure 1.

Individual ion intensities for measles virus phosphoprotein P obtained from nanoESI-TOF experiment. Two ions at m/z 609.3 and 720.7 corresponding to the phosphoprotein P tryptic peptides, ASDVETAEGGEIHELLR and AGSSGLSKPCLSAIGSTEGGAPR, respectively, were measured for each measles virus infected cell sample, and the relative peak areas (arbitrary units) were compared. Inset: extracted ion chromatogram for ion m/z 720.7, comparing 18 and 72 hour time points from infected cells, using the XCMS program.
In this experiment, HeLa cells were infected with the Edmonston strain of measles virus, and harvested at two different time points, 18 and 72 hours. Uninfected cells at the same time points were analyzed as a control. Another experiment was designed to examine antibody-induced antigenic modulation in measles virus. HeLa cells were incubated with either a control antibody (antibody to HeLa cells) or an antibody against measles virus before infection.40 The relative quantitation of measles virus proteins was then examined. In the first experiment, measles virus proteins present in the cells were identified using nanoLC-MS/MS. A second experiment was performed for relative quantitation of the viral proteins. From the ion trap experiment, two ions at m/z 609.3 and 720.7 corresponding to the tryptic peptides, ASDVETAEGGEHELLR and AGSSGLSKPCLSAIGSTEGGAPR, respectively, derived from the measles phosphoprotein P were identified from the infected cells. The peptide, ASDVETAEGGEIHELLR, is a tryptic fragment of the 19 amino acid naturally processed and presented peptide, MV-P1, of the measles phosphoprotein.44 The phosphoprotein forms part of ribonucleoprotein (RNP) complex, and is involved in both replication and transcription, and also has a structural role in the assembled virus, surrounding the RNA. In the second experiment, the samples were analyzed using an ESI-TOF equipped with capillary LC. This instrument has the advantage of a wider dynamic range and very accurate mass measurement, and therefore has the potential for better quantitative measurements than the ion trap used for protein identification. Each sample was run in triplicate. The data were first analyzed using XCMS,34 after which the two ions at m/z 609.3 and 720.7, corresponding to the measles virus phosphoprotein were selected, and integrated using Analyst (Figure 1). For the uninfected cells at 18 and 72 hours, the integrated intensities of both ions are zero. At 18 and 72 hours, the ion currents allow comparison of the intensities at different time points, which correspond to the multiplication of the virus, and expression of the phosphoprotein. The ratio of ion intensities between 18 and 72 hours was 1.6 for the ion m/z 609.3 and 1.9 for the ion at m/z 720.7. This indicates that the relative level of measles virus phosphoprotein increased by a factor of between 1.6 – 1.9 between 18 and 72 hours after infection. When a control antibody was incubated with HeLa cells, the expression of phosphoprotein P progressed with the same kinetics as infected cells without antibody (Figure 1). However, when antibody against measles virus was added, expression of phosphoprotein P was almost completely suppressed. This experiment demonstrates that the process of antibody-induced antigenic modulation in measles virus can be monitored using mass spectrometry, and the result is in agreement with previous experiments, using methionine label and immunoprecipitation.40
Monitoring viral proteins via isotopic labeling: FHV-infected Drosophila cells
18O labeling of peptides during tryptic digestion is a simple, robust, and effective way to generate isotopic tags for protein quantitation. The approach incorporates two 18O atoms from water at the C terminus of every lysine or arginine residue, by exchange during a trypsin digestion, resulting in a mass difference of 4 Da between two differentially labeled peptides. To assess the viability of this approach in the quantitative analysis of protein expression in FHV-infected Drosophila cells, we determined its labeling efficiency and specificity. In the initial set of experiments, two identical digests of purified FHV particles, one performed in the presence of H2O18 and the other in the presence of H2O16, were mixed at a 1:1 ratio and analyzed by DIOS-MS and LC-MS/MS. Representative DIOS-MS data of a 1:1 mixture of 16O and 18O labeled FHV digests revealed peptide peaks derived from the tryptic cleavage of the coat proteins within 10 ppm mass accuracy (Figure 2A). The inset (right) shows that the isotope pairs were separated by 4 Da indicating the incorporation of two 18O atoms. Comparison of the relative peak intensities of the 18O and 16O labeled peptides demonstrated that 18O was efficiently incorporated into the peptides during proteolysis. The peptide mass fingerprint of this data set was searched against SwissProt protein database using Mascot (Matrix Science Ltd.). Search results indicated that protein scores greater than 52 are significant. A significant score (85 for the coat protein) was obtained for the digest, indicating greater than 95% confidence level in the match. Additional validation of the protein identification was obtained from LC-MS/MS analysis of the same 16O:18O mixture. An MS/MS spectrum (Figure 2B) of the doubly charged ion of the peptide VVVTTTQTAPVPQQNVPR with m/z of 968.2 Da showed characteristic b- and y- fragment ions that allowed straightforward interpretation of the MS/MS data for protein identification.
Figure 2.

A. DIOS-MS spectrum of a 1:1 mixture of 16O:18O FHV digest. Inset, center: Linearity of the relative quantitation using isotopically labeled FHV digests. Inset, right: the expanded view of the DIOS-MS spectrum of the 16O and 18O labeled peptide, VVVTTTQTAPVPQQNVPR, combined at a 1:1 ratio. B. MS/MS spectrum of the doubly charged ion of 16O and 18O-labeled FHV tryptic peptide, VVVTTTQTAPVPQQNVPR from LC-MS/MS analysis.
In order to determine the accuracy and sensitivity of the 18O labeling method to small variations in protein expression levels, the linearity of the method was examined. Differentially labeled 16O and 18O FHV digests were mixed at 16O:18O ratios ranging from 0.1 to 10 and analyzed by DIOS-MS and nanoLC-MS/MS. Using the peptide VVVTTTQTAPVPQQNVPR and its corresponding 18O analog as internal standard, the response of the peak intensity ratio as a function of the concentration ratio was monitored. The data shown in Figure 2A (inset, center) revealed a linear response over the working concentration range with a slope close to 1 and a R2 value of 0.9974. The precision of the measurement was within 15%, which was calculated using the percent relative standard deviation (RSD) of the concentration ratio. This implied that the approach was sensitive to small variations in protein concentration and that accurate quantitation was possible.
Monitoring viral protein expression kinetics: FHV-infected Drosophila cells
The applicability of the 18O labeling approach in quantitative protein profiling was examined in a time course experiment of viral protein synthesis in FHV-infected Drosophila cells. Previous pulse chase experiments showed that FHV proteins are expressed in different amounts at different times during the course of infection.38,45 The expression of the viral proteins as a function of time was determined using nanoLC-MS/MS, using the 18O-labeled peptides generated from infected cells at 16 hours post-infection as internal standard. Aliquots of cell lysates collected at eight time points after infection (see Experimental Section for details) and an uninfected control lysate were digested separately and then mixed in a 1:1 (v/v) ratio with the 18O-labeled peptides from the 16 hour time point sample. Each time point sample was analyzed in triplicate. MS/MS data generated from these samples were searched against SwissProt database using Mascot. The search results show that individual ion scores above 31 indicate that the peptide based identification at 95% confidence level. Peptide ion scores in this search were from 37 to 77 indicating unambiguous identification of the FHV coat protein, protein A, and protein B2. However, our search was not able to uniquely identify B1 because protein B1 is expressed in smaller amounts and more importantly, it is identical to the C-terminal portion of protein A. Thus, peptides identified from the C-terminal end of protein A may have contain a partial contribution from protein B1.
Based on these search results, we used three peptides (VVVTTTQTAPVPQQNVPR (m/z 968.4, +2 charge), VFVDPLATTTTIQDPLR (m/z 946.1, +2 charge) and LALIQELPDR (m/z 534.5, +2 charge)) and their 18O analogs to determine relative steady state levels of the coat protein, protein A, and protein B2 over the course of infection. As shown in Figure 3A and B, protein A was produced early, followed by protein B2, then the coat protein. Protein A could be detected within the first 3 hours after infection, and its relative concentration increased steadily thereafter and reached a maximum at 10 hours after which it plateaued. Similarly, protein B2 was produced early after infection but its level continued to increase until 20 hours before reaching a plateau. In contrast, synthesis of the coat protein was significantly delayed (Figure 3B). Its levels were low for the first 10 hours but increased rapidly thereafter until reaching maximum at 20 hours followed by a plateau, following sigmoidal kinetics. The viral protein expression kinetics that we observed is consistent with the respective functions of each protein. Protein A is the replicase, thus requiring early expression, and the coat protein is expressed at an initially slower rate, as its function is viral assembly and encapsidation. Protein B2 was found to exhibit the earliest expression kinetics of the three viral proteins observed in this experiment. This is consistent with the recently discovered function of B2,39 which is to inhibit RNA interference from the cell. It is reasonable to assume that earliest possible expression of B2 as a rapid response to cellular RNA silencing is advantageous for viral synthesis and function. These results are in excellent agreement with previous pulse-chase experiments demonstrating temporal control of FHV protein synthesis in Drosophila cells.46
Figure 3.

A. Relative rates of viral protein expression (FHV viral proteins A, B2) induced in FHV-infected Drosophila cells analyzed by nanoLC-MS/MS. B. FHV coat protein growth curve generated from DIOS and nanoLC-MS/MS analysis of proteolyzed cell lysate. A polynomial equation was fit to each set of data.
These results prompted us to determine the absolute concentration of the viral coat protein at various times after infection. Specifically, we monitored the change in concentration of the coat protein using known amounts of 18O-labeled pure FHV digest as internal standard. Aliquots from each of the eight time point samples and control were digested, mixed with the internal standard at a 16O:18O ratio of 2.5:1 (v/v), and then analyzed by LC-MS/MS and DIOS-MS. Each time point sample was run in triplicate for LC-MS/MS and in 10 replicates for DIOS-MS. The average ratio of 16O- and 18O-labeled peptides and the corresponding percent relative standard deviation (RSD) for each time point was obtained using the peptide VVVTTTQTAPVPQQNVPR. The measured percent RSD values were less than 10%, reflecting the excellent reproducibility of both types of analysis. Based on the known concentration of the internal standard, the absolute concentration of the coat protein was determined at each time point. LC-MS/MS and DIOS-MS data demonstrated that the expression kinetics of the coat protein is slow in the first 10 hours, rapidly increase between 10 and 20 hours, and then plateau (Figure 3B). From the known concentration of the coat protein, which was measured by the Bradford assay, and the known number of cells from which it had been obtained, we could determine the yield of coat protein per cell and, based on the known composition of FHV particles, the number of virions present at a given time point. This revealed that the number of virus particles per cell at 16 hours post-infection was greater than 6.0 × 104, consistent with the large paracrystalline arrays of virions observed in cells by electron microscopy late in infection.47,48 The growth curves of coat protein synthesis obtained by DIOS-MS and nanoLC-MS/MS were in good agreement. The DIOS-MS analysis was much more rapid, requiring only 3 hours, while nanoLC-MS/MS required 3 days of continuous automated analysis. This demonstrates the potential for DIOS-MS in the rapid absolute quantitation of viral proteins during the infection process.
Monitoring Host Protein Expression: FHV-infected Drosophila cells
The response of Drosophila to bacterial and fungal infection involves two signaling pathways, Toll and Imd, to activate transcription factor NF-kB family of proteins and the induction of pathways associated with the innate immune response.49 The response to infection with Drosophila virus C is distinct from the response to bacteria and fungi, and suggests the possibility of a conserved innate immunity pathway specific to viral infection, involving the Jak-STAT signaling pathway.50 In contrast, the response to infection with Drosophila X virus activates Toll, thus producing a response similar to that of bacteria.41 This indicates the importance of examining the response of Drosphila to additional viruses.
FHV-infected Drosophila cells collected at various times after infection represented ideal samples to identify and quantify differentially expressed host proteins and potentially gain insights into virus-host interactions. To reduce sample complexity, we chose the cICAT labeling strategy which involves affinity tagging of proteins at cysteine residues and isolation of proteolytically generated peptides by affinity chromatography. Peptides without cysteine residues are thus excluded, reducing complexity and permitting quantification of low abundance host proteins. The host protein expression profile was monitored by FT-MS at 0 and 8 hours post-infection. At 8 hours post-infection, FHV-infected Drosophila cells remain metabolically active and have had ample time to respond to the replication of the virus. A total of 1500 proteins were identified by searching the MS/MS data against a combined Drosophila melanogaster/FRV proteome database (19477 protein entries) downloaded from the European Bioinformatics Institute (EBI) (http://www.ebi.ac.uk/proteome/index.html). The host protein expression levels were quantified based on one or more distinct peptides unique to the differentially expressed Drosophila proteins (see Experimental Section for details). Significant expression level changes were determined by fitting a normal distribution curve to the cICAT ratios, and taking proteins at 2σ-above, a ratio of 1.69, or 2σ-below, a ratio of 0.63, the mean (Figure 4). Of the 1500 proteins that were identified and quantified in the cICAT experiment, 150 were up-regulated and 66 were down-regulated by these criteria (Table 2).
Figure 4.

Probability distribution of cICAT ratios obtained from the cICAT FT-MS experiment. The frequency distribution of cICAT ratios was calculated and distributed into bins. A normal distribution curve was fit to this data, and the mean (μ) and standard deviation (σ) were calculated. cICAT ratios corresponding to frequencies that were 2 σ above or below the mean were considered to be significantly up (1.69) or down (0.63) regulated.
Table 2.
Selected proteins and the response to viral infection between 0 and 8 hours. The complete list can be found as Supplementary Table 2.
| Accession Number | Protein Name | Average Ratio I8hours/I0hours |
|---|---|---|
| Up regulated Proteins in FHV-infected Drosophila Cells | ||
| Q9VM33 | Probable elongation factor G, mitochondrial precursor (mEF-G) | 157.2 |
| P30189 | DNA topoisomerase I (EC 5.99.1.2) | 54.6 |
| P02516 | Heat shock protein 23 | 33.8 |
| Q9V3I5 | CG6297-PA, isoform A (Cg6297-pb, isoform b) (Protein kinase JIL-1) | 15.7 |
| Q9VYD7 | Mitochondrial import inner membrane translocase subunit Tim9A | 6.0 |
| Q9VQ96 | CG4267-PA(RH13166p) | 4.5 |
| Q24572 | Chromatin assembly factor 1 P55 subunit | 3.7 |
| Q24208 | Eukaryotic translation initiation factor 2 gamma subunit | 3.7 |
| P52304 | Serine/threonine-protein kinase polo (EC 2.7.1.37) | 3.5 |
| Q8I8U7 | Transcription-associated protein 1 (dTRA1) | 2.6 |
| P40793 | Cdc42 homolog | 2.6 |
| P23573 | Cell division control protein 2 cognate (EC 2.7.1.37) | 2.3 |
| Q9Y1A3 | Mitochondrial import inner membrane translocase subunit Tim8 | 2.2 |
| P02518 | Heat shock protein 27 | 2.2 |
| Q9VHA0 | Polycomb protein Scm (Sex comb on midleg protein) | 2.1 |
| O61231 | 60S ribosomal protein L10 (QM protein homolog) (dQM) | 2.1 |
| P36958 | DNA-directed RNA polymerase II 15.1 kDa polypeptide (EC 2.7.7.6) | 2.0 |
| Q27367 | Croquemort protein (d-CD36) | 2.0 |
| Q9V9S8 | Ferrochelatase, mitochondrial precursor (EC 4.99.1.1) | 2.0 |
| Down regulated Proteins in FHV-infected Drosophila Cells | ||
| Q94901 | CG8597-PA, isoform A (Cg8597-pb, isoform b) (RNA-binding protein lark) | 0.6 |
| Q94535 | Splicing factor U2af 38 kDa subunit | 0.6 |
| O96840 | CG5788-PA (Ubiquitin-conjugating enzyme) (EC 6.3.2.19) | 0.6 |
| O46037 | Vinculin | 0.6 |
| Q9VTU3 | CG6811-PA(LD02491p) | 0.6 |
| Q7KNS3 | Lissencephaly-1 homolog (Lissencephalyl) (Dlis1) (DLis-1) | 0.5 |
| P29845 | Heat shock 70 kDa protein cognate 5 | 0.5 |
| Q9VNH6 | Probable exocyst complex component Sec8 | 0.5 |
| Q9VL00 | Ubiquitin thiolesterase otubain-like protein | 0.5 |
| Q9TVQ5 | Chromatin protein SPT4 (CG12372-PA) | 0.5 |
| P46824 | Kinesin light chain (KLC) | 0.5 |
| Q24306 | Apoptosis 1 inhibitor (Inhibitor of apoptosis 1) | 0.3 |
| Q9V498 | Calsyntenin-1 precursor | 0.3 |
| O77263 | Probable tRNA (guanine-N(7)-)-methyltransferase | 0.2 |
| Q9GP60 | Pol protein | 0.01 |
Proteins with expression levels that changed significantly were broadly categorized on the basis of their biochemical function. As can be seen in Figure 5a and b, differentially expressed host proteins fell into similar classes regardless of whether they were up- or down-regulated. Of particular interest with regard to viral infection was the significant up-regulation of heat shock proteins, particularly hsp23, whose levels were 34-fold increased, which are involved in folding of viral structural proteins and assistance in viral assembly.51 A more specific response to viral infection might be reflected in the up-regulation of proteins involved in cell death, e.g. croquemort protein and a corresponding down-regulation of proteins that inhibit cell death, e. g. inhibitor of apoptosis 1. By inducing cell death, the production and spread of virus in the host could be limited and thereby contribute to survival of the organism.
Figure 5.
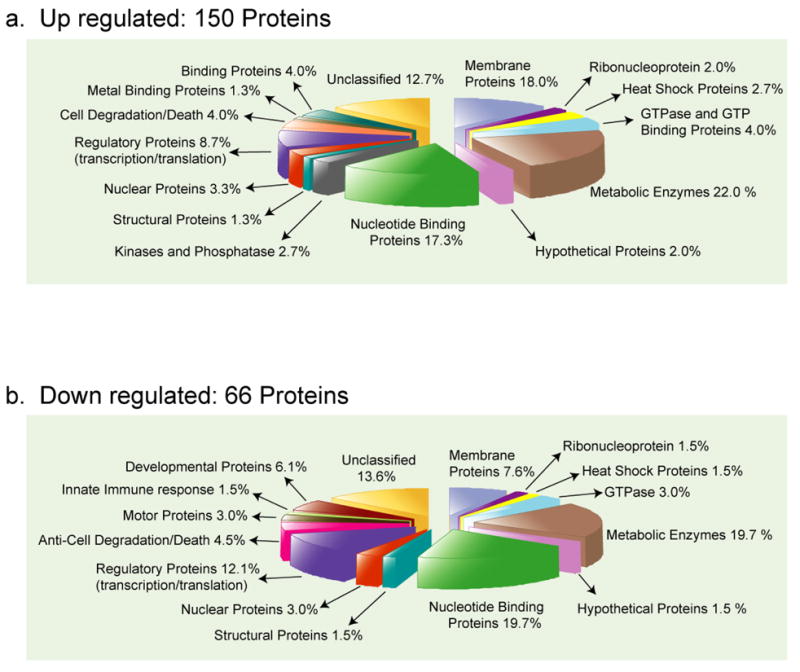
Functional categories of the differentially expressed host proteins obtained from cICAT FT-MS/MS experiments. Over 1500 proteins were identified and quantified of which (a) 150 proteins increased in expression and (b) 66 proteins decreased in expression. The 216 proteins were broadly classified based on their biochemical function by GO category. Numbers indicate the percentage fraction of the proteins in each category.
Metabolite Analysis: FHV-infected Drosophila cells
The metabolite profile in the FHV-infected Drosophila cells was also monitored during the course of infection. Since metabolite levels are regulated by gene expression and enzyme activity, monitoring these changes may lead to the identification of cellular components or pathways that are important to virus replication and the molecular signatures associated with viral diseases. To this end, using micro-scale RPLC separation, in conjunction with high mass accuracy ESI-TOF, the changes in metabolite concentrations within the cell during the course of viral infection were examined. The integrated areas of the molecular ion signals, retention time, and mass precision tolerance in parts per million (ppm) of endogenous metabolites were measured. Comparison of the total ion chromatograms (TIC) of the metabolite fractions from 0 and 12 hours post-infection shows significant differences in the intensities (Figure 6A). Typical metabolite fraction yields over 1000 unique ions after μLC-ESI TOF MS analysis. These molecular ions were characterized by its monoisotopic m/z, peak height/area, and retention time.
Figure 6.

A. μLC ESI-TOP TIC of the metabolite extract from 0 and 12 hours post-infection undigested samples. B. Average integrated areas of the molecular ions with m/z 496 (red) and 524 (black) from two separate injections. C. Fragmentation patterns and the corresponding structures of the lysophosphatidylcholines. The MS/MS fragmentation pattern shows product ions with m/z 104 and 184. Both metabolite ions undergo a significant change in their relative intensity during the course of infection.
An essential component required for comparative analysis of metabolite levels is to analyze the retention time and mass precision between replicate injections. This comparison allows the determination of the degree of reproducibility and specificity of the measurement. Our data reveals that the metabolite separation was reproducible within 0.16 minutes and the mass precision tolerance was below 10 ppm. This was obtained from a pairwise comparison of replicate injections of 14 selected molecular ions from two time point samples. These results imply that a good mass precision and a reproducible chromatography can be obtained which are useful in clustering the metabolites on the basis of their retention times and masses.
Analysis of the integrated area associated with each molecular ion revealed that over 30 metabolite ions were either up- or down-regulated. These metabolite ions undergo significant change in relative intensity during the course of infection and were targeted for identification by subsequent MS/MS analysis. Of particular interest were two molecular ions observed at m/z 496 and 524 which decrease markedly during the infection process (Figure 6B). MS/MS data of these metabolites (Figure 6C) showed a fragmentation pattern typical for the family of lysophosphatidylcholines (LPC). These ions were identified by tandem MS/MS in an ion trap mass spectrometer. Product ions were observed at m/z 184 and 104, a fragmentation pattern which is typical for all phosphocholine containing phospholipids.52 The product ions at m/z 184 and 104 correspond to the phosphocholine and the choline ion, respectively.
LPCs are derived from either plasma membrane phospholipids or lipoproteins as part of normal physiological activity or disease process. Under physiological conditions, LPCs exist as micelles, a component of oxidized low density lipoprotein, bound to serum proteins, and as ligands to G-protein receptors.53,54 Moreover, their biological role is diverse which includes modulating plasma membrane integrity, cell proliferation, transmembrane signal transduction, inhibiting viral membrane fusion elicited by a number of virus, enhancing resistance to phytopathogens, and immunoregulation.54–58 LPC has also been identified in apoptotic cell clearance by functioning as a soluble “find me” signal and as a membrane-bound “eat me” signal.59 Thus, monitoring the changes in LPC concentration in response to viral infection can provide information that is complementary to proteomics data. The observed down regulation of the LPCs (m/z 496 and 524) correlated with the up-regulation of proteins involved in apoptosis, e.g croquemort and the Cdc42 homolog, and the down-regulation of the proteins vinculin and CG6811-PA, which are associated with plasma membrane integrity and membrane phospholipid exchange, respectively. Thus, the down regulation of LPC further reflects the initial stages in FHV viral replication and cell death.
Conclusions
Mass spectrometry was used to monitor the regulation of protein and endogenous metabolite in response to two dissimilar viral infections. Isotope labeling strategies with mass spectrometry allowed for quantitative analysis of viral proteins, including the temporal control of viral protein synthesis in FHV-infected Drosophila cells and changes in the host cell proteins. Viral protein expression levels were monitored by one-dimensional tandem MS and DIOS-MS experiments with 16O/18O labeling. A more comprehensive profile of the cellular proteins was obtained by using two-dimensional tandem MS experiments with cICAT labeling. The cICAT was essential for the quantitative analysis and identification of the 1500 cellular proteins, of which more than 200 were either up- or down- regulated. Using an approach without protein labeling, relative quantitation of the measles virus phosphoprotein P during infection was monitored, showing the changes in its expression level and antibody-induced antigenic modulation in the MV infected cells. Endogenous metabolite levels at two different times during FHV infection of Drosophila cells were measured, showing temporal changes in over 30 metabolites. The down regulation of two LPCs correlated with changes in lipid-related proteins, providing complementary information between the metabolomic and proteomic data. Changes in metabolite and protein expression levels were associated with apoptosis, plasma membrane integrity, and membrane phospholipid exchange. The vast majority of proteins and metabolites may be structurally and functionally uncharacterized, suggesting the enormous potential of this approach for gaining fundamental biochemical and clinical understanding of biological systems.
Supplementary Material
A complete list of the differentially expressed host proteins and the response to viral infection between 0 and 8 hours (Supplementary Table 2).
Acknowledgments
This work was supported by NIH grant GM55775 and Strategene, Inc. to GS, and AI036222 to MBAO. A.N. is supported by a postdoctoral fellowship from The Swedish Research Council (VR).
References
- 1.Flint SJ. Principles of virology molecular biology, pathogenesis, and control of animal viruses. 2. ASM Press; Washington, D.C: 2004. [Google Scholar]
- 2.Mendez J, Audicana A, Isern A, Llaneza J, Moreno B, Tarancon ML, Jofre J, Lucena F. J Virol Methods. 2004;117(1):19–25. doi: 10.1016/j.jviromet.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Barry R, Soloviev M. Proteomics. 2004;4(12):3717–3726. doi: 10.1002/pmic.200300877. [DOI] [PubMed] [Google Scholar]
- 4.Shieh B, Li C. Retrovirology. 2004;1(1):11. doi: 10.1186/1742-4690-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fruh K, Simmen K, Luukkonen BG, Bell YC, Ghazal P. Drug Discov Today. 2001;6(12):621–627. doi: 10.1016/s1359-6446(01)01808-6. [DOI] [PubMed] [Google Scholar]
- 6.Piersanti S, Martina Y, Cherubini G, Avitabile D, Saggio I. Am J Pharmacogenomics. 2004;4(6):345–356. doi: 10.2165/00129785-200404060-00002. [DOI] [PubMed] [Google Scholar]
- 7.Snijders AM, Meijer GA, Brakenhoff RH, van den Brule AJ, van Diest PJ. Mol Pathol. 2000;53(6):289–294. doi: 10.1136/mp.53.6.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aebersold R. J Infect Dis. 2003;187:S315–S320. doi: 10.1086/374756. [DOI] [PubMed] [Google Scholar]
- 9.Flory MR, Gingras AC, Keller A, Lee H, Li XJ, Nesvizhskii A, Ranish J, Ye ML, Zhang H, Aebersold R. Eur J Cell Biol. 2004;83:88. [Google Scholar]
- 10.Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, Doyle MJ, Yi EC, Dai H, Thorsson V, Eng J, Goodlett D, Berger JP, Gunter B, Linseley PS, Stoughton RB, Aebersold R, Collins SJ, Hanlon WA, Hood LE. Mol Cell Proteomics. 2004;3(10):960–969. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Sechi S, Oda Y. Drug Discov Today. 2004;9(2):S41–S46. [PubMed] [Google Scholar]
- 12.Sechi S. Proteomics Nephrol. 2004;141:59–78. doi: 10.1159/000074590. [DOI] [PubMed] [Google Scholar]
- 13.Ferguson PL, Smith RD. Annu Rev Biophys Biomol, Struct. 2003;32:399–424. doi: 10.1146/annurev.biophys.32.110601.141854. [DOI] [PubMed] [Google Scholar]
- 14.Lill J. Mass Spectrom Rev. 2003;22(3):182–194. doi: 10.1002/mas.10048. [DOI] [PubMed] [Google Scholar]
- 15.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Nat Biotechnol. 1999;17(10):994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 16.Mann M. Nat Biotechnol. 1999;17(10):954–955. doi: 10.1038/13646. [DOI] [PubMed] [Google Scholar]
- 17.Brown KJ, Fenselau C. J Proteome Res. 2004;3(3):455–462. doi: 10.1021/pr0340835. [DOI] [PubMed] [Google Scholar]
- 18.Fenselau C, Reynolds K. Abstracts of Papers of the American Chemical Society. 2003;226:U336. [Google Scholar]
- 19.Heller M, Mattou H, Menzel C, Yao XD. J Am Soc Mass Spectrom. 2003;14(7):704–718. doi: 10.1016/S1044-0305(03)00207-1. [DOI] [PubMed] [Google Scholar]
- 20.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Anal Chem. 2001;73(13):2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 21.Yao XD, Afonso C, Fenselau C. J Proteome Res. 2003;2(2):147–152. doi: 10.1021/pr025572s. [DOI] [PubMed] [Google Scholar]
- 22.Hsu JL, Huang SY, Chow NH, Chen SH. Anal Chem. 2003;75(24):6843–6852. doi: 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- 23.Peters EC, Horn DM, Tully DC, Brock A. Rapid Commun Mass Spectrom. 2001;15(24):2387–2392. doi: 10.1002/rcm.517. [DOI] [PubMed] [Google Scholar]
- 24.Chakraborty A, Regnier FE. J Chromatogr A. 2002;949(1–2):173–184. doi: 10.1016/s0021-9673(02)00047-x. [DOI] [PubMed] [Google Scholar]
- 25.Goodlett DR, Keller A, Watts JD, Newitt R, Yi EC, Purvine S, Eng JK, von Haller P, Aebersold R, Kolker E. Rapid Commun Mass Spectrom. 2001;15(14):1214–1221. doi: 10.1002/rcm.362. [DOI] [PubMed] [Google Scholar]
- 26.Smith RD, Anderson GA, Lipton MS, Masselon C, Pasa-Tolic L, Udseth H, Belov M, Shen Y, Veenstra TD. Adv Protein Chem. 2003;65:85–131. doi: 10.1016/s0065-3233(03)01017-9. [DOI] [PubMed] [Google Scholar]
- 27.Smith RD, Shen Y, Tang K. Acc Chem Res. 2004;37(4):269–278. doi: 10.1021/ar0301330. [DOI] [PubMed] [Google Scholar]
- 28.Zimmer JS, Monroe ME, Qian WJ, Smith RD. Mass Spectrom Rev. 2006;25(3):450–482. doi: 10.1002/mas.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Callister SJ, Barry RC, Adkins JN, Johnson ET, Qian WJ, Webb-Robertson BJ, Smith RD, Lipton MS. J Proteome Res. 2006;5(2):277–286. doi: 10.1021/pr050300l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radulovic D, Jelveh S, Ryu S, Hamilton TG, Foss E, Mao Y, Emili A. Mol Cell Proteomics. 2004;3(10):984–997. doi: 10.1074/mcp.M400061-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Wang W, Zhou H, Lin H, Roy S, Shaler TA, Hill LR, Norton S, Kumar P, Anderle M, Becker CH. Anal Chem. 2003;75(18):4818–4826. doi: 10.1021/ac026468x. [DOI] [PubMed] [Google Scholar]
- 32.Silva JC, Denny R, Dorschel C, Gorenstein MV, Li GZ, Richardson K, Wall D, Geromanos SJ. Mol Cell Proteomics. 2006;5(4):589–607. doi: 10.1074/mcp.M500321-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Fang R, Elias DA, Monroe ME, Shen Y, McIntosh M, Wang P, Goddard CD, Callister SJ, Moore RJ, Gorby YA, Adkins JN, Fredrickson JK, Lipton MS, Smith RD. Mol Cell Proteomics. 2006;5(4):714–725. doi: 10.1074/mcp.M500301-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. Anal Chem. 2006;78(3):779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 35.Dasgupta R, Cheng LL, Bartholomay LC, Christensen BM. J Gen Virol. 2003;84:1789–1797. doi: 10.1099/vir.0.18938-0. [DOI] [PubMed] [Google Scholar]
- 36.Price BD, Rueckert RR, Ahlquist P. Proc Natl Acad Sci U S A. 1996;93(18):9465–9470. doi: 10.1073/pnas.93.18.9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price BD, Roeder M, Ahlquist P. J Virol. 2000;74(24):11724–11733. doi: 10.1128/jvi.74.24.11724-11733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneemann A, Reddy V, Johnson JE. Adv Virus Res, Vol 50. 1998;50:381–446. doi: 10.1016/s0065-3527(08)60812-x. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Li WX, Ding SW. Science. 2002;296(5571):1319–1321. doi: 10.1126/science.1070948. [DOI] [PubMed] [Google Scholar]
- 40.Fujinami RS, Oldstone MB. Nature. 1979;279(5713):529–530. doi: 10.1038/279529a0. [DOI] [PubMed] [Google Scholar]
- 41.Zambon RA, Nandakumar M, Vakharia VN, Wu LP. Proc Natl Acad Sci U S A. 2005;102(20):7257–7262. doi: 10.1073/pnas.0409181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen ZX, Thomas JJ, Averbuj C, Broo KM, Engelhard M, Crowell JE, Finn MG, Siuzdak G. Anal Chem. 2001;73(3):612–619. doi: 10.1021/ac000746f. [DOI] [PubMed] [Google Scholar]
- 43.Fujinami RS, Oldstone MB. J Immunol. 1980;125(1):78–85. [PubMed] [Google Scholar]
- 44.Johnson KL, Ovsyannikova IG, Poland GA, Muddiman DC. J Proteome Res. 2005;4(6):2243–2249. doi: 10.1021/pr0501416. [DOI] [PubMed] [Google Scholar]
- 45.Friesen P, Scotti P, Longworth J, Rueckert R. J Virol. 1980;35(3):741–747. doi: 10.1128/jvi.35.3.741-747.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friesen P, Rueckert R. J Virol. 1981;37(2):876–886. doi: 10.1128/jvi.37.3.876-886.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fisher AJ, McKinney BR, Wery JP, Johnson JE. Acta Crystallogr B. 1992;48(Pt 4):515–520. doi: 10.1107/s0108768192000053. [DOI] [PubMed] [Google Scholar]
- 48.Fisher AJ, McKinney BR, Schneemann A, Rueckert RR, Johnson JE. J Virol. 1993;67(5):2950–2953. doi: 10.1128/jvi.67.5.2950-2953.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Royet J, Reichhart JM, Hoffmann JA. Curr Opin Immunol. 2005;17(1):11–17. doi: 10.1016/j.coi.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 50.Dostert C, Jouanguy E, Irving P, Troxler L, Galiana-Arnoux D, Hetru C, Hoffmann JA, Imler JL. Nat Immunol. 2005;6(9):946–953. doi: 10.1038/ni1237. [DOI] [PubMed] [Google Scholar]
- 51.Chromy LR, Pipas JM, Garcea RL. Proc Natl Acad Sci U S A. 2003;100(18):10477–10482. doi: 10.1073/pnas.1832245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brugger B, Erben G, Sandhoff R, Wieland FT, Lehmann WD. Proc Natl Acad Sci U S A. 1997;94(6):2339–2344. doi: 10.1073/pnas.94.6.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabarowski JH, Zhu K, Le LQ, Witte ON, Xu Y. Science. 2001;293(5530):702–705. doi: 10.1126/science.1061781. [DOI] [PubMed] [Google Scholar]
- 54.Kabarowski JH, Xu Y, Witte ON. Biochem Pharmacol. 2002;64(2):161–167. doi: 10.1016/s0006-2952(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 55.Gunther-Ausborn S, Praetor A, Stegmann T. J Biol Chem. 1995;270(49):29279–29285. doi: 10.1074/jbc.270.49.29279. [DOI] [PubMed] [Google Scholar]
- 56.Gunther-Ausborn S, Stegmann T. Virology. 1997;235(2):201–208. doi: 10.1006/viro.1997.8699. [DOI] [PubMed] [Google Scholar]
- 57.Spivak SG, Kisel MA, Yakovleva GA. Russ J Plant Physl+ 2003;50(3):293–296. [Google Scholar]
- 58.Korotaeva AA, Cheglakov IB, Morozkin AD, Suslova IV, Prokazova NV. Membr Cell Biol. 1997;10(5):521–534. [PubMed] [Google Scholar]
- 59.Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Mol Cell. 2004;14(3):277–287. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A complete list of the differentially expressed host proteins and the response to viral infection between 0 and 8 hours (Supplementary Table 2).