

Abstract
Xenopus oocyte maturation requires the phosphorylation and activation of p42 mitogen-activated protein kinase (MAPK). Likewise, the dephosphorylation and inactivation of p42 MAPK are critical for the progression of fertilized eggs out of meiosis and through the first mitotic cell cycle. Whereas the kinase responsible for p42 MAPK activation is well characterized, little is known concerning the phosphatases that inactivate p42 MAPK. We designed a microinjection-based assay to examine the mechanism of p42 MAPK dephosphorylation in intact oocytes. We found that p42 MAPK inactivation is mediated by at least two distinct phosphatases, an unidentified tyrosine phosphatase and a protein phosphatase 2A–like threonine phosphatase. The rates of tyrosine and threonine dephosphorylation were high and remained constant throughout meiosis, indicating that the dramatic changes in p42 MAPK activity seen during meiosis are primarily attributable to changes in MAPK kinase activity. The overall control of p42 MAPK dephosphorylation was shared among four partially rate-determining dephosphorylation reactions, with the initial tyrosine dephosphorylation of p42 MAPK being the most critical of the four. Our findings provide biochemical and kinetic insight into the physiological mechanism of p42 MAPK inactivation.
INTRODUCTION
The mitogen-activated protein kinases (MAPKs) constitute a family of evolutionarily conserved protein kinases critical for cellular responses such as proliferation, differentiation, and stress adaptation (Ferrell, 1996a; Moriguchi et al., 1996; Robinson and Cobb, 1997; Lewis et al., 1998; Widmann et al., 1999). MAPKs also help monitor the internal status of the cell and regulate cell cycle progression (Minshull et al., 1994; Takenaka et al., 1997, 1998; Wang et al., 1997; Cross and Smythe, 1998; Guadagno and Ferrell, 1998).
p42 MAPK plays a central role in the progression of Xenopus oocytes through meiosis. Immature oocytes are arrested in a G2-like state with inactive p42 MAPK (the relevant MAPK) and inactive Cdc2/cyclin B (Figure 1). In response to progesterone, the oocytes activate their Cdc2, reenter meiosis 1, progress into meiosis 2, and then arrest spontaneously in metaphase of meiosis 2 (Figure 1). These mature metaphase-arrested oocytes can then be ovulated and fertilized. The Mos/MAPK kinase (MEK)/p42 MAPK cascade becomes activated concomitantly with Cdc2 during maturation (Figure 1) (Sagata et al., 1988; Ferrell et al., 1991; Kobayashi et al., 1991a,b; Matsuda et al., 1992), and this activation is essential for Cdc2/cyclin B activation and oocyte maturation. Interfering with p42 MAPK activation by microinjection of MEK antibodies (Kosako et al., 1994, 1996), antisense Mos oligonucleotides (Sagata et al., 1988), or the MAPK phosphatase CL100 (Gotoh et al., 1995) delays or prevents Cdc2 activation. Artificially activating p42 MAPK by microinjection of Mos (Sagata et al., 1989a; Yew et al., 1992), constitutively active MEK (Gotoh et al., 1995; Huang et al., 1995), or thiophosphorylated, active p42 MAPK (Haccard et al., 1995) can cause Cdc2 activation and oocyte maturation in the absence of progesterone. Thus p42 MAPK activation is of critical importance to the G2–M transition during Xenopus oocyte maturation.
Figure 1.

Activation and inactivation of p42 MAPK at the onset and completion of Xenopus oocyte maturation. p42 MAPK and Cdc2 become activated concomitantly before germinal vesicle breakdown (GVBD). p42 MAPK remains maximally active until ∼30–40 min after fertilization. Cdc2 decreases in activity during interkinesis, returns to maximal activity in the M2 phase, and then decreases in activity ∼5 min after fertilization.
p42 MAPK remains active throughout meiosis 1 and meiosis 2, and artificially inactivating it during this period delays the reactivation of Cdc2 and permits rereplication of DNA (Furuno et al., 1994). Thus, p42 MAPK activity is important for the transition from meiosis 1 to meiosis 2. In addition, immunodepleting Mos from cytostatic factor (CSF)-arrested egg extracts depletes the extracts of their CSF activity (Sagata et al., 1989b) as assayed by the cleaving blastomere assay (Masui and Markert, 1971). Likewise, addition of MAPK phosphatase-1 (MKP-1) to CSF-arrested extracts causes them to inactivate Cdc2, exit meiosis, and progress into interphase (Minshull et al., 1994). Maintaining p42 MAPK activity therefore appears to be essential for maintaining metaphase arrest in mature oocytes and eggs.
Xenopus oocytes and eggs are an attractive system for studying the biochemistry of p42 MAPK regulation. p42 MAPK activation and inactivation are particularly dramatic and well synchronized during maturation and after fertilization. Essentially all of the oocyte’s p42 MAPK becomes phosphorylated and activated during maturation, and it remains quantitatively activated until ∼30 min after fertilization. These marked changes in p42 MAPK phosphorylation must be brought about by large changes in the balance between p42 MAPK phosphorylation and dephosphorylation. Because of their size and the availability of concentrated cytoplasmic oocyte and egg extracts, the enzymes responsible for these changes can be studied with powerful, direct biochemical methods.
MAPKs are activated by the phosphorylation of a tyrosine and a threonine residue within a Thr-X-Tyr sequence motif (Thr-Glu-Tyr in the classical p42/p44 MAPKs) (Anderson et al., 1990; Payne et al., 1991; Posada and Cooper, 1992). Both phosphorylations are performed by dual-specificity MAP kinase kinases (termed MEKs, MAPKKs, or MKKs) (Crews et al., 1992; Kosako et al., 1992; Wu et al., 1992). Much less is known about the enzymes that perform the dephosphorylation and inactivation of p42 MAPK. Several protein phosphatases have been identified that can dephosphorylate and thereby inactivate p42 MAPK in vitro. The specificity of these phosphatases varies; some act only on phosphotyrosine, others act only on phosphothreonine, and still others act on both residues (reviewed by Clarke, 1994; Nebreda, 1994; Keyse, 1998). Examples of the first two families include CD45, a transmembrane tyrosine phosphatase isolated from hematopoietic cells, and protein phosphatase 2A (PP2A), a ubiquitously expressed serine/threonine phosphatase (Sturgill et al., 1988; Anderson et al., 1990). Members of the third family of MAPK-directed phosphatases, the dual-specificity phosphatases, are particularly intriguing. These proteins act via a catalytic mechanism analogous to that of tyrosine phosphatases (Ishibashi et al., 1992; Zhou et al., 1994; Denu et al., 1996a,b) and demonstrate a strong preference for MAPK family members as substrates (reviewed by Keyse, 1998).
In recent years, an ever-increasing number of phosphatases that can inactivate p42 MAPK in vitro have been identified and cloned. Despite this fact, the exact identities of the phosphatases that do inactivate p42 MAPK in vivo remain elusive. For example, MKP-1 (a dual-specificity MAPK phosphatase) rapidly inactivates p42 MAPK in vitro and inhibits MAPK activation in vivo in transfected cell lines (Charles et al., 1993; Sun et al., 1993); however, mkp-1 knock-out mice activate their p42 MAPK with normal kinetics and display no obvious phenotypic abnormalities (Dorfman et al., 1996). Moreover, biochemical studies of the MAPK phosphatase activities present in lysates from mammalian cell lines argue that traditional tyrosine-specific and serine/threonine-specific phosphatases are primarily responsible for the dephosphorylation of p42 and p44 MAPK (Alessi et al., 1995). However, it remains formally possible that these phosphatases do not have access to MAPK in situ. Genetic approaches offer a strong starting point for further biochemical studies and may ultimately identify the most relevant phosphatases (Doi et al., 1994; Wurgler-Murphy et al., 1997; Martin-Blanco et al., 1998; Sugiura et al., 1998), but these approaches are limited to genetically tractable organisms and may not succeed if the relevant MAPK phosphatases regulate multiple targets in addition to MAPKs. Consequently, a new biochemical strategy is needed to allow a mechanistically detailed analysis of p42 MAPK dephosphorylation in a physiologically relevant setting.
To this end we designed a biochemical assay to examine the dephosphorylation of microinjected 32P-labeled p42 MAPK in intact Xenopus oocytes, taking advantage of the large size of the oocyte and the ease of determining an oocyte’s cell cycle status. We have assessed whether the rate of dephosphorylation of either residue changes during oocyte maturation or after release of mature oocytes into the mitotic cell cycle, whether the dephosphorylation of the two residues is catalyzed by a single dual-specificity phosphatase or rather by separate phosphatases, whether the dephosphorylation is ordered or random and processive or distributive, and which reactions exert the most control over the overall rate of p42 MAPK dephosphorylation. We have also investigated the role of MEK in establishing the level of p42 MAPK activity throughout meiosis. Finally, we have used CSF-arrested egg extracts to characterize in greater detail the nature of the phosphatases involved and the mechanism of p42 MAPK dephosphorylation.
MATERIALS AND METHODS
Recombinant Proteins
A plasmid harboring the cDNA for a constitutively active, (His)6-tagged version of human MEK-1 (with Ser 218 replaced by Glu, Ser 222 replaced by Asp, and a deletion of amino acids 32–51, hereafter denoted MEK R4F) was provided by Natalie Ahn (University of Colorado, Boulder, CO) (Mansour et al., 1994, 1996). Plasmids containing the cDNAs for (His)6-tagged Xenopus p42 MAPK proteins (K57R, K57R/T188V, and K57R/Y190F) were derived from plasmids obtained from Jim Posada and Jonathan Cooper (Fred Hutchinson Cancer Research Center, Seattle, WA) (Posada and Cooper, 1992). All recombinant proteins were expressed in Escherichia coli and purified to homogeneity by nickel-chelate chromatography by Ramesh Bhatt (Stanford University, Stanford, CA).
Preparation of 32P-labeled MAPK
Radiolabeled, bisphosphorylated p42 MAPK protein (p42 MAPK*) was prepared by incubating purified recombinant (His)6-MAPK (either the inactive K57R mutant or the K57R/T188V and K57R/Y190F phosphorylation site mutants; 0.8–1 μM) with (His)6-MEK R4F (0.5–1 μM) in kinase buffer (50 mM Tris, pH 7.0, 100 mM NaCl, 0.1 mg/ml BSA, 10 mM MgCl2) plus ATP (67 μM; 0.33 μCi/μl carrier-free [γ-32P]ATP). The reaction was incubated for 2 h at room temperature, and excess salt and ATP were removed by centrifuging through 100 μl of Sephadex G-25 resin (Sigma, St. Louis, MO) preequilibrated with kinase buffer.
Oocyte Isolation and Microinjection
Xenopus ovarian tissue was surgically removed, and oocytes were defolliculated for 1–1.5 h at room temperature with 2.5 mg/ml collagenase and 0.5 mg/ml polyvinylpyrrolidone in Ca2+-free modified Barth’s solution (88 mM NaCl, 1 mM KCl, 0.82 mM MgSO4, 2.4 mM NaHCO3, 10 mM HEPES, pH 7.5). The oocytes were then washed four times with modified Barth’s solution. Stage VI oocytes were sorted manually and incubated at 16°C for at least 10 h in OR2 solution (82.5 mM NaCl, 2.5 mM KCl, 1 mM CaCl2, 1 mM Na2HPO4, 5 mM HEPES, pH 7.5) supplemented with 1 mg/ml BSA and 50 μg/ml gentamicin (Sigma).
Three types of oocytes were used for microinjection: immature G2-arrested oocytes (G2 oocytes), oocytes treated with 5 μg/ml progesterone and collected shortly after GVBD (M1 oocytes), and oocytes treated with progesterone overnight (at least double the time required for GVBD) and collected after full maturation (M2 oocytes).
Oocytes were microinjected, 6–10 per time point, with 50 nl of p42 MAPK* reaction mixture (∼1.6 ng of p42 MAPK*, yielding a nominal concentration of ∼40 nM) and transferred to fresh OR2 for the duration of the time course. Because we encountered variability in the activation of M2 oocytes after microinjection, we instead activated these oocytes by transfer to OR2 containing 5 μM ionophore A23187 (Boehringer Mannheim, Indianapolis, IN) for 10 min immediately after microinjection. The oocytes were then transferred into fresh OR2 for the remainder of the time course. Six oocytes per time point were collected, frozen on dry ice, and stored at −80°C.
Oocyte Lysis
Oocytes were thawed rapidly and lysed by pipetting up and down in 60 μl of ice-cold extraction buffer (EB: 0.25 M sucrose, 0.1 M NaCl, 2.5 mM MgCl2, 20 mM HEPES, pH 7.2) containing 10 mM EDTA, protease inhibitors (10 μg/ml leupeptin, 10 μg/ml pepstatin, 10 μg/ml aprotinin, 1 mM PMSF), and phosphatase inhibitors (50 mM β-glycerophosphate, 1 mM sodium orthovanadate, 2 μM microcystin). Samples were clarified by centrifugation for 3–5 min in a Beckman E microcentrifuge (Fullerton, CA) with a right-angle rotor. Crude cytoplasm was removed and immediately added to 0.2 vol of 6× Laemmli sample buffer for subsequent SDS-PAGE or diluted 1:2 in EB and 0.2% Triton X-100 plus inhibitors for immunoprecipitation.
CSF Extracts
CSF-arrested egg extracts were prepared as described (Murray, 1991). To verify the integrity of the extracts, 25-μl samples were supplemented with sperm chromatin in the presence or absence of CaCl2 (0.4 mM) for 1 h at room temperature, and nuclear morphology was assessed by phase-contrast and epifluorescence microscopy as described (Walter et al., 1997). Extracts were considered acceptable if they maintained mitotic nuclear morphology in the absence of CaCl2 and progressed into interphase in the presence of CaCl2. Extracts were pretreated for 10 min on ice with various phosphatase inhibitors, p42 MAPK* was added (1.5 μl per 25-μl extract, yielding a final p42 MAPK* concentration of ∼50 nM), and aliquots were taken at various times for immunoprecipitation and/or autoradiography and immunoblotting.
SDS-PAGE and Transfer to Blotting Membranes
All samples were processed on 10% SDS polyacrylamide gels (acrylamide:bisacrylamide, 100:1) and transferred to Immobilon P (Millipore, Bedford, MA) blotting membranes. 32P-labeled MAPK was detected by autoradiography and quantified with a Molecular Dynamics PhosphorImager (Sunnyvale, CA).
Immunoblotting
Samples were separated on 10% SDS polyacrylamide gels (acrylamide: bisacrylamide, 100:1), and the proteins were transferred to an Immobilon P (Millipore) blotting membrane. The membrane was blocked with 3% nonfat milk in Tris-buffered saline (150 mM NaCl, 20 mM Tris, pH 7.6) and incubated with primary antibody (MAPK antibody DC3 [Hsiao et al., 1994], 1:500 dilution; phospho-specific MAPK [New England Biolabs, Beverly, MA], 1:1000 dilution; MEK antibody 662 [Hsiao et al., 1994], 1:500 dilution) for 1 h (MAPK and MEK) or overnight (phospho-specific MAPK). Blots were washed five times with Tris-buffered saline and 0.5% Tween 20 and probed with an alkaline phosphatase–conjugated secondary antibody for detection by enhanced chemiluminescence (ECL) (Amersham, Arlington Heights, IL). For reprobing, blots were stripped by incubation with 100 mM Tris-HCl, pH 7.4, 100 mM 2-mercaptoethanol, and 2% SDS at 70°C for 40 min.
Phosphoamino Acid Analysis
p42 MAPK* bands were excised from blotting membranes and subjected to one-dimensional phosphoamino acid analysis at pH 3.5 (Boyle et al., 1991; Kamps, 1991). Radiolabeled p42 MAPK* proteins and their constituent phosphoamino acids were visualized by autoradiography and quantified with a Molecular Dynamics PhosphorImager.
Tryptic Analysis
p42 MAPK* bands were excised from blotting membranes, digested with l-1-tosylamide-2-phenylethylchloromethyl–treated trypsin (Worthington Biochemical, Freehold, NJ) in situ, and subjected to one-dimensional thin-layer electrophoresis at pH 8.9 as described (Boyle et al., 1991; Luo et al., 1991).
Immunoprecipitation with (His)6 Antibody
Lysates were prepared from oocytes or aliquots taken from extracts and diluted 1:30 (extracts) or 1:2 (oocyte lysates) in EB and 0.2% Triton X-100 plus inhibitors. The diluted lysates were added to 10 μl of washed protein-A agarose prebound to (His)6 antibody (Clontech, Palo Alto, CA). Samples were rotated for 3 h at 4°C to immunoprecipitate (His)6-p42 MAPK* proteins, washed three times with EB and 0.2% Triton X-100 plus inhibitors, and resuspended in Laemmli sample buffer. Control samples containing a comparable amount of either p42 MAPK* starting material (∼13 ng) or untreated extract (6 μl) were processed and subjected to immunoprecipitation in parallel.
MEK Immunoprecipitation and Activity Assay
Immunoprecipitations and linked MEK and MAPK assays were performed in duplicate to measure the activity of MEK. Oocytes were collected and lysed as described above. Crude cytoplasm (two oocyte equivalents) was removed, diluted 1:8 in EB containing 0.1% Triton X-100, and added to 10 μl of washed protein-A agarose prebound to MEK antibody 662 (Hsiao et al., 1994). After rotation for 3 h at 4°C, immunoprecipitates were washed twice with EB containing 0.1% Triton X-100 and once with MEK kinase buffer (20 mM HEPES, pH 7.5, 20 mM MgCl2, 10 mM 2-mercaptoethanol). MEK kinase buffer supplemented with 60 μM ATP and 2 μg/ml wild-type Xenopus (His)6-MAPK was then added, and the samples were incubated at 30°C. Ten minutes later, 10 μg of myelin basic protein (MBP) and 0.5 μCi of [γ-32P]ATP were added, and the reaction mixture was incubated at 30°C for an additional 5 min. The reaction was stopped by the addition of Laemmli sample buffer, and the proteins were separated on 12.5% SDS polyacrylamide gels (acrylamide:bisacrylamide, 100:1) and transferred to Immobilon P (Millipore) blotting membranes. Incorporation of 32P into MBP was visualized by autoradiography and quantified with a Molecular Dynamics PhosphorImager. To verify equal loading of MEK proteins, samples were immunoblotted with MEK antibody 662.
DNA Replication Assays
For DNA replication assays, 25 μl of extract was supplemented with either PP1 buffer or protein phosphase inhibitor-2 (PPI-2) (40 U/μl; New England Biolabs) and further incubated on ice for 10 min. Sperm chromatin (500 sperm/μl), [α-32P]dCTP (0.2 μCi/μl), and CaCl2 (0.4 mM) were added at room temperature, and 4.5-μl aliquots were removed periodically into 0.5 vol of 2× replication stop buffer. Samples were analyzed as described previously (Dasso and Newport, 1990) and quantified with a Molecular Dynamics PhosphorImager.
Curve Fitting
To generate the curves shown below (see Figures 2, 4, 6, 7, 9, and 11) and to estimate half-times and apparent rate constants for the dephosphorylation reactions, we fit the experimental dephosphorylation data to the following equation:
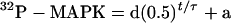 |
using KaleidaGraph (Abelbeck Software, Reading, PA). τ represents the half-time for the dephosphorylation, ln 2/τ is the apparent zero-order rate constant for the dephosphorylation, and a and d are empirically determined parameters.
Figure 2.

Dephosphorylation of p42 MAPK* in intact oocytes. (A) High-stoichiometry phosphorylation of K57R p42 MAPK* in vitro. The autoradiogram shows a one-dimensional tryptic analysis of 32P-labeled, in vitro–phosphorylated p42 MAPK*. Inactive (K57R) p42 MAPK was incubated with purified constitutively active MEK R4F and [γ-32P]ATP. The phosphorylated p42 MAPK* was subjected to SDS-PAGE, transferred to a polyvinylidene fluoride membrane, and digested in situ with l-1-tosylamide-2-phenylethylchloromethyl–treated trypsin. The tryptic digests were separated by thin-layer electrophoresis at pH 8.9 and subjected to autoradiography and PhosphorImager quantitation. Phosphopeptide spots were identified by comparison with phosphorylation site mutant p42 MAPKs (K57R/T188V, lane 1, and K57R/Y190F, lane 2) and with previous analyses (Ferrell and Bhatt, 1997). Lanes 3 and 4 show the tryptic peptides from two independent in vitro phosphorylation reactions with K57R p42 MAPK. The major spot present in the p42 MAPK* digest corresponded to the bisphosphorylated tryptic peptide (bis-phos. MAPK), implying a phosphorylation stoichiometry of ∼1.85 mol/mol. Minor spots corresponded to mono-Tyr-phosphorylated p42 MAPK [mono-phos. MAPK (pTyr)] and mono-Thr-phosphorylated p42 MAPK [mono-phos. MAPK (pThr)]. DNP, dinitrophenyl. (B) Autoradiogram showing a typical time course of p42 MAPK* dephosphorylation in G2 oocytes. p42 MAPK* was microinjected into oocytes, which were frozen at different times and subsequently lysed and subjected to SDS-PAGE and autoradiography. (C) Time course of p42 MAPK* dephosphorylation in G2, M1, and activated M2 oocytes. The data shown represent averages ± SEM for two (M1) or three (G2 and activated M2) independent experiments.
Figure 4.

Changes in MEK activity can account for the changes in the steady-state phosphorylation of p42 MAPK. Two uninjected oocytes (G2, M1, and activated M2) per time point were collected and lysed, and the endogenous MEK proteins were immunoprecipitated with antiserum 662. (A) A linked MEK and MAPK assay was performed, samples were analyzed by SDS-PAGE and autoradiography, and total incorporation of 32P into MBP was quantified. Data are from one experiment with assays performed in duplicate and are shown as averages ± ranges. Activities are normalized to the MEK activity in unactivated M2 oocytes (100%). (B) A p42 MAPK immunoblot of the same samples is shown.
Figure 6.

Calcium-independent dephosphorylation of p42 MAPK* in CSF extracts. (A) p42 MAPK* was added to CSF extracts simultaneously with either water or CaCl2 (0.4 mM), and aliquots were taken at the times indicated. Samples were analyzed by SDS-PAGE, autoradiography, and quantitation. Data are shown as means ± SEM for seven experiments. The calculated half-times for dephosphorylation were 5.7 ± 0.7 min (water) and 5.0 ± 0.4 min (0.4 mM CaCl2). (B) Dephosphorylation of p42 MAPK* on tyrosine and threonine in calcium-treated CSF extracts is shown. Samples from A were subjected to phosphoamino acid analysis and quantified. Data are shown as means ± SEM for four experiments. The calculated half-times for dephosphorylation were 4.0 ± 0.8 min (tyrosine) and 4.5 ± 0.7 min (threonine).
Figure 7.

The rate of dephosphorylation of p42 MAPK* does not change in Ca2+-treated CSF extracts. p42 MAPK* was added to a Ca2+-treated CSF extract at various times, and the dephosphorylation was monitored by SDS-PAGE, autoradiography, and quantitation. (A) Autoradiogram showing p42 MAPK* dephosphorylation for extracts in which p42 MAPK* was added either concomitantly with 0.4 mM CaCl2 (left) or 40 min after the CaCl2 (right). (B) Cumulative quantitative data from two (p42 MAPK* added at 10 or 20 min), four (p42 MAPK* added at 40 min), or seven (p42 MAPK* added at 0 min) experiments, expressed as means ± SEM.
Figure 9.

Rapid dephosphorylation of Y190F p42 MAPK*. Three MAPK proteins were added to CSF extracts: p42 MAPK* phosphorylated on tyrosine and threonine (K57R), p42 MAPK* phosphorylated only on tyrosine (T188V), or p42 MAPK* phosphorylated only on threonine (Y190F). CaCl2 (0.4 mM) was added simultaneously with the p42 MAPK* reaction mixture, and aliquots were taken at various times. Samples were analyzed by SDS-PAGE, autoradiography, and PhosphorImager quantitation. (A) Representative autoradiogram showing the time course of dephosphorylation of the K57R/T188V and K57R/Y190F proteins. (B) Graphical representation of p42 MAPK* dephosphorylation. Data are shown as mean values ± SEM for three experiments performed in duplicate. The calculated half-times for dephosphorylation were 4.8 ± 0.2 min (K57R), 7.1 ± 0.3 min (K57R/T188V), and 1.6 ± 0.1 min (K57R/Y190F).
Figure 11.

Effects of various inhibitors on p42 MAPK* dephosphorylation in CaCl2-treated CSF extracts. CSF extracts were preincubated with okadaic acid (1 μM; A–C), I-2 (40 U/μl; D), or EGTA (10 mM; E) for 5–10 min on ice. CaCl2 (0.4 mM) and p42 MAPK* (50 nM) were added simultaneously (CaCl2 was replaced with water for EGTA-treated extracts), and aliquots were taken at various times. Samples were analyzed by SDS-PAGE and autoradiography. Data are shown as mean values ± SEM for n experiments as indicated below. (A) Okadaic acid–induced inhibition of p42 MAPK* dephosphorylation (n = 4) is shown. (B and C) Okadaic acid inhibits p42 MAPK* threonine dephosphorylation (C) but does not affect tyrosine dephosphorylation (B). Samples were subjected to phosphoamino acid analysis and quantified (n = 4). (D) PP1 activity is not required for p42 MAPK* dephosphorylation (n = 3). I-2 (40 U/μl) was used to inhibit PP1. The same concentration of I-2 inhibited DNA replication (our unpublished results). (E) EGTA does not inhibit p42 MAPK* dephosphorylation (n = 3).
To calculate several curves (see Figure 10), we numerically solved the rate equations for the following dephosphorylation reactions (see Figure 10I, schematic):
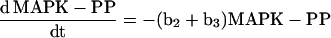 |
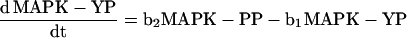 |
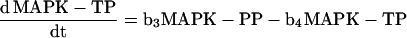 |
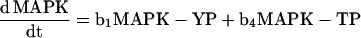 |
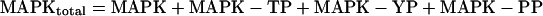 |
using Mathematica 2.2.2 (Wolfram Research, Champaign, IL). PP denotes biphosphorylated MAPK; YP denotes mono-Tyr-phosphorylated MAPK; and TP denotes mono-Thr-phosphorylated MAPK.
Figure 10.

Levels of bis-, mono-, and nonphosphorylated p42 MAPK*. p42 MAPK* was microinjected into G2 oocytes, and its dephosphorylation was monitored by tryptic peptide analysis. (A) Overall levels of p42 MAPK* phosphorylation. p42 MAPK* phosphorylation was monitored by SDS-PAGE, autoradiography (A), and quantitation (our unpublished results). (B) One-dimensional tryptic mapping of phosphorylated p42 MAPK*. Samples of monophosphorylated p42 MAPK* phosphorylation site mutants (lanes 1 and 2), bisphosphorylated p42 MAPK* starting material (lane 3), and p42 MAPK* after microinjection into oocytes (lanes 4–8) were subjected to exhaustive tryptic digestion followed by thin-layer electrophoresis at pH 8.9. (C–H) The measured levels of bisphosphorylated p42 MAPK* (C), mono-Tyr-phosphorylated p42 MAPK* (D), mono-Thr-phosphorylated p42 MAPK* (E), nonphosphorylated p42 MAPK* (F), and total tyrosine (G)- and threonine (H)-phosphorylated p42 MAPK* are expressed as mean values ± SEM for two experiments (one performed in duplicate and one single determination). The curves shown in C–H were calculated from the rate equations for p42 MAPK dephosphorylation (see MATERIALS AND METHODS) assuming the values of the rate constants shown in I. (I) A simple kinetic scheme for the in vivo dephosphorylation of p42 MAPK.
RESULTS
A Direct In Vivo Assay for MAPK-directed Phosphatase Activity
We capitalized on the ease of microinjecting Xenopus oocytes to assess p42 MAPK–directed phosphatase activities in intact cells. Our strategy was to microinject pools of oocytes with 32P-labeled p42 MAPK, collect the oocytes at different times after microinjection, and determine how much radiolabel remained in the p42 MAPK by SDS-PAGE and PhosphorImager quantitation. By using pools of oocytes from different cell cycle stages, we were able to obtain a panoramic view of p42 MAPK–directed phosphatase activity throughout meiosis.
Recombinant, catalytically inactive (K57R) Xenopus p42 MAPK protein was phosphorylated to high stoichiometry on the activating tyrosine and threonine residues in vitro using recombinant, constitutively active MEK-1 R4F and [γ-32P]ATP. One-dimensional tryptic analysis of the p42 MAPK* indicated that >85% of the protein was bisphosphorylated (Figure 2A). The specific radioactivity of the p42 MAPK* (counts per minute/mole) was sufficiently high to allow us to microinject relatively small amounts of p42 MAPK* (yielding concentrations of ∼40 nM or ∼13% that of the endogenous p42 MAPK [Ferrell, 1996b]) and still to use small numbers of oocytes (six oocytes per time point). The use of the catalytically inactive K57R MAPK protein ensured that the oocyte’s level of MAPK activity would not be perturbed by the microinjection.
Figure 2B shows the results of a typical experiment with the radiolabeled p42 MAPK* microinjected into G2 oocytes. The MAPK* was readily detectable after PAGE and autoradiography. Over the course of a 30-min incubation there was a marked decrease in the radiolabeled MAPK, with a half-time of ∼5 min (Figure 2, B and C).
In principle, the decrease in radiolabel in p42 MAPK* could be caused by either dephosphorylation or degradation. We therefore examined whether the concentration of microinjected p42 MAPK* remained constant or decreased after microinjection into G2, M1, and activated M2 oocytes. Oocytes were collected at various times, lysed, and subjected to immunoprecipitation with an anti-(His)6 antibody, which recognized the microinjected His-tagged p42 MAPK* but not the endogenous p42 MAPK. Immunoprecipitates were then immunoblotted with p42 MAPK antiserum. As shown in Figure 3 (bottom), there was no detectable loss of the injected p42 MAPK* protein. Therefore, the observed decrease in p42 MAPK* labeling (Figures 3, top, and 2, B and C) was caused by dephosphorylation.
Figure 3.

The phosphorylation state of microinjected p42 MAPK* rapidly adopts that of endogenous p42 MAPK. Oocytes were microinjected, collected, frozen at various times, and lysed, and the p42 MAPK* proteins were immunoprecipitated with (His)6 antibodies. Uninjected oocytes (UN) were processed in parallel as controls to ensure that endogenous p42 MAPK did not coprecipitate with the injected p42 MAPK*. Top, the time course of disappearance of p42 MAPK* microinjected into G2, M1, and activated M2 oocytes is shown. The p42 MAPK* was detected by autoradiography after (His)6 immunoprecipitation (IP). MEK R4F, being (His)6-tagged, was also immunoprecipitated by the (His)6 antibody. Middle, the blot shown above was probed with phospho-specific p42 MAPK antibodies followed by ECL detection. This shows the activation state of the injected p42 MAPK* throughout maturation and after activation. Bottom, the same blot was stripped and reprobed with the MAPK antiserum DC3 followed by ECL detection to show the level and activation state of the injected p42 MAPK*.
The overall rate of p42 MAPK* dephosphorylation was similar in oocytes at all stages of the meiotic cell cycle (Figure 2C). Thus, changes in p42 MAPK–directed phosphatase activity apparently contribute to neither the abrupt activation of p42 MAPK before GVBD nor the abrupt inactivation of p42 MAPK after parthenogenetic activation of mature oocytes.
The Phosphorylation State of the Injected p42 MAPK* Reflects That of Endogenous p42 MAPK
If the microinjected p42 MAPK* were behaving similarly to the endogenous p42 MAPK, then we would expect the following: 1) in G2 oocytes, where essentially none of the endogenous p42 MAPK is phosphorylated, the loss of 32P from p42 MAPK* should be accompanied by a shift of p42 MAPK* from its electrophoretically retarded phosphorylated form to the faster-migrating nonphosphorylated form and a loss in recognition by phospho-MAPK antibodies; 2) in M1 oocytes, where essentially all of the endogenous p42 MAPK is phosphorylated, the 32P in the p42 MAPK* should be rapidly replaced by nonradioactive phosphate, and the p42 MAPK* should remain shifted and phospho-MAPK reactive; and 3) in activated M2 oocytes, where the endogenous p42 MAPK remains phosphorylated for ∼30 min and then becomes dephosphorylated, the 32P in the p42 MAPK* initially should be replaced by nonradioactive phosphate for ∼30 min, and then all phosphate should be lost. On the other hand, if the injected p42 MAPK* were behaving differently from endogenous p42 MAPK, we might expect p42 MAPK* phosphorylation to be too high in G2 oocytes or too low in M1 oocytes or to differ from endogenous p42 MAPK in the timing of its shift to the dephosphorylated form after activation of M2 oocytes.
We microinjected p42 MAPK* into G2, M1, and M2 oocytes, immunoprecipitated the recombinant protein at each time point, and analyzed the results by SDS-PAGE followed by transfer to a blotting membrane and PhosphorImager quantitation. We subjected the same blot to immunoblot analysis with a monoclonal antibody specific for the bisphosphorylated, active form of p42 MAPK and then stripped the blot and reprobed it with the anti-p42 MAPK antiserum DC3. This allowed us to determine the phosphorylation state of the recombinant protein at each time point. Consistent with expectation, the recombinant p42 MAPK* protein maintained or rapidly adopted the phosphorylation state of the endogenous protein in all cell cycle stages (Figure 3, middle and bottom). In G2 oocytes, the p42 MAPK* shifted to the nonphosphorylated form when it lost its radiolabel; in M1 oocytes, the radiolabel was rapidly replaced with nonradioactive phosphate, and the p42 MAPK* remained phosphorylated; and in activated M2 oocytes, the p42 MAPK* remained phosphorylated until 30 min after activation (Figure 3, middle and bottom). These results suggest that the oocyte does not distinguish between the endogenous and recombinant forms of p42 MAPK with respect to their phosphorylation and dephosphorylation.
MEK Activity Parallels p42 MAPK Activity during Oocyte Maturation and Early Embryogenesis
Because of the apparently constitutive nature of p42 MAPK–directed phosphatase activity in G2, M1, and activated M2 oocytes, we asked whether regulated activity of the MAPK activator MEK sufficed to explain the tight regulation of p42 MAPK activity seen during the meiotic cell cycle. To test this, nonmicroinjected G2, M1, and activated M2 oocytes were collected and lysed, the endogenous MEK protein was immunoprecipitated, and a linked kinase assay was performed to measure the level of MEK activity in each sample. In addition, p42 MAPK phosphorylation was assessed by immunoblotting.
Comparable levels of MEK were immunoprecipitated in each sample (our unpublished results). The activity of the immunoprecipitated MEK protein, however, varied dramatically throughout the cell cycle (Figure 4A), climbing at least 16-fold from baseline levels in G2 oocytes to maximum levels in M2 oocytes and falling back to baseline levels in M2 oocytes ∼40 min after oocyte activation. The pattern of MEK activation and inactivation (Figure 4A) closely resembled that seen for p42 MAPK (Figure 4B), in good agreement with previous studies (Matsuda et al., 1992). Because of the rapidity with which p42 MAPK* is dephosphorylated in vivo, the changes in MEK activity measured here appear sufficient to account for the overall pattern of p42 MAPK regulation observed during maturation and after fertilization.
The Tyrosine and Threonine Residues Are Dephosphorylated at Similar Rates
We next examined the individual rates of dephosphorylation of the tyrosine and threonine residues on p42 MAPK*. In principle, dephosphorylation of p42 MAPK in vivo could be accomplished either by a single dual-specificity phosphatase, by one or more tyrosine- and threonine-specific phosphatases, or by a combination of both. As a first step toward distinguishing between these possibilities, we determined whether the dephosphorylation of either residue was affected by vanadate, which inhibits tyrosine-specific and dual-specificity phosphatases, or by okadaic acid, which inhibits several types of serine/threonine-specific phosphatases. We also examined whether the rates of dephosphorylation of the individual residues varied in G2, M1, and activated M2 oocytes.
We microinjected p42 MAPK* plus vanadate (nominal oocyte concentration of ∼1 mM), okadaic acid (nominal oocyte concentration of ∼1 μM), or no inhibitor, collected oocytes at various times, and separated the p42 MAPK* bands by SDS-PAGE followed by transfer to a blotting membrane. We excised the p42 MAPK* bands and performed partial acid hydrolysis and one-dimensional phosphoamino acid analysis. Figure 5A shows representative phosphoamino acid analysis data from the experiment depicted in Figure 2B; quantitative data from two (M1) or three (G2 and activated M2) experiments are depicted graphically in Figure 5, B and C.
Figure 5.

Dephosphorylation of p42 MAPK* on tyrosine and threonine during oocyte maturation. p42 MAPK* was subjected to partial acid hydrolysis and analyzed by one-dimensional thin-layer electrophoresis at pH 3.5. (A) Autoradiogram showing the dephosphorylation of tyrosine and threonine residues on p42 MAPK* in G2 oocytes in either the absence or presence of 1 mM sodium orthovanadate. (B) Tyrosine dephosphorylation of p42 MAPK* in G2 oocytes (left), M1 oocytes (middle), and activated M2 oocytes (right). Data are shown as mean values ± SEM from two (M1) or three (G2 and activated M2) independent experiments. Note that vanadate (VO4; □) slowed tyrosine dephosphorylation, and okadaic acid (OA; ○) had no effect. (C) Threonine dephosphorylation of p42 MAPK* in G2 oocytes (left), M1 oocytes (middle), and activated M2 oocytes (right). Data are shown as mean values ± SEM from two (M1) or three (G2 and activated M2) independent experiments. Note that both vanadate (□) and okadaic acid (○) slowed threonine dephosphorylation.
In the absence of phosphatase inhibitors, the rates of tyrosine and threonine dephosphorylation closely approximated one another for all three sets of oocytes. The average half-times for removal of the tyrosine and threonine phosphates, estimated by nonlinear least squares curve fitting, were 5.2 ± 1.0 and 3.8 ± 0.5 min, respectively.
Okadaic Acid Slows the Threonine Dephosphorylation of p42 MAPK*
Coinjection of okadaic acid significantly slowed the rate of threonine dephosphorylation in both G2 and M1 oocytes (Figure 5C) while leaving the rate of tyrosine dephosphorylation unaffected (Figure 5B). These results implicate a PP1- or PP2A-like phosphatase in the dephosphorylation of the threonine residue and suggest that tyrosine dephosphorylation is not contingent on threonine dephosphorylation. These results also indicate that at least two phosphatases participate in the dephosphorylation of p42 MAPK* in oocytes: an okadaic acid–sensitive threonine phosphatase and an okadaic acid–insensitive tyrosine phosphatase. This eliminates the possibility that a single dual-specificity phosphatase is responsible for the bulk of the p42 MAPK* dephosphorylation.
Okadaic acid had a paradoxical effect on p42 MAPK* dephosphorylation in activated M2 oocytes, accelerating the overall rate of dephosphorylation. It also frequently caused the oocytes to exhibit a morbid “gray puffball” appearance. We did not analyze the effects of okadaic acid on the individual rates of tyrosine and threonine dephosphorylation in activated M2 oocytes but will return to the question using egg extracts later in this article.
Vanadate Slows Both the Tyrosine and Threonine Dephosphorylation of p42 MAPK*
Coinjection of vanadate with p42 MAPK* inhibited tyrosine dephosphorylation in G2, M1, and activated M2 oocytes relative to that seen in control oocytes (Figure 5B), as expected. In addition, vanadate inhibited p42 MAPK* threonine dephosphorylation (Figure 5C). This finding indicates either that threonine dephosphorylation is accomplished by a vanadate-sensitive dual-specificity phosphatase (in addition, perhaps, to the okadaic acid–sensitive serine/threonine phosphatase inferred above) or that the vanadate-inhibited dephosphorylation of tyrosine on p42 MAPK* is a prerequisite for efficient threonine dephosphorylation.
Constitutive Dephosphorylation of p42 MAPK* in Egg Extracts
To dissect the mechanism of p42 MAPK dephosphorylation further, we turned to a Xenopus egg extract system, cytostatic factor (CSF) extracts prepared from unfertilized eggs. This cell-free system offers greater experimental manipulability than the in vivo oocyte system while faithfully recapitulating key aspects of the cell cycle. CSF extracts remain arrested in metaphase of meiosis 2 until exposure to calcium triggers their exit from meiosis and progression into the first mitotic cell cycle.
We first tested the ability of CSF extracts to dephosphorylate p42 MAPK*. p42 MAPK* was dephosphorylated in CSF extracts at a rate similar to that seen in intact oocytes (Figure 6A, ○; cf. Figures 2C and 5, B and C). Next, we added calcium to the extracts to release them from CSF arrest and to determine whether fertilization-induced changes in intracellular calcium might be expected to stimulate p42 MAPK–directed phosphatase activity. As Figure 6A shows, the rate of dephosphorylation was similar in the presence or absence of added calcium. The rates of tyrosine and threonine dephosphorylation were indistinguishable from one another (half-times of 4.0 ± 0.8 and 4.5 ± 0.7 min, respectively; Figure 6B) and from those measured in intact oocytes (5.2 ± 1.0 and 3.8 ± 0.5 min, respectively).
Because p42 MAPK undergoes an abrupt change in its phosphorylation state ∼30–40 min after fertilization of eggs or addition of calcium to CSF extracts, we were particularly interested in examining whether there was an increase in MAPK-directed phosphatase activity at that time. To this end, we added radiolabeled p42 MAPK* to CSF extracts at various times after calcium treatment. As shown in Figure 7, the rate of dephosphorylation was similar irrespective of when the p42 MAPK* was added to the extract. Thus, the abrupt dephosphorylation of p42 MAPK* is achieved without any increase in the activity of the MAPK-directed phosphatases.
To ensure that the behavior of recombinant p42 MAPK* protein in these experiments mimicked that of the endogenous protein, we repeated our control experiments from Figure 3 using CSF extracts. Figure 8 (top) illustrates the calcium-independent dephosphorylation of p42 MAPK* in these extracts. Again, as assessed by phospho-MAPK (Figure 8, middle) and DC3 (Figure 8, bottom) immunoblotting, the phosphorylation state of the recombinant protein conformed to that of the endogenous protein throughout the time course. The p42 MAPK* remained phosphorylated and active in the absence of calcium while ultimately reverting to its dephosphorylated, inactive form in the presence of calcium. Thus, the phosphorylation state of recombinant p42 MAPK* protein paralleled that of the endogenous protein in CSF extracts.
Figure 8.

The phosphorylation state of p42 MAPK* mimics that of endogenous p42 MAPK in CSF extracts. CSF extracts were supplemented with p42 MAPK* in the presence or absence of CaCl2 as described in Figure 6, aliquots were taken at 0, 5, 15, and 30 min, and the p42 MAPK* protein was immunoprecipitated with (His)6 antibodies. Samples with no added p42 MAPK* (left lane) were processed in parallel as controls to ensure that endogenous p42 MAPK did not coprecipitate with the injected p42 MAPK*. Top, the time course of disappearance of p42 MAPK* from the extracts is shown. The p42 MAPK* was detected by autoradiography after (His)6 immunoprecipitation. MEK R4F, being (His)6-tagged, was also immunoprecipitated by the (His)6 antibody. Middle, the blot shown above was probed with phospho-specific p42 MAPK antibodies followed by ECL detection. This shows the activation state of the added p42 MAPK* during CSF arrest and after treatment with CaCl2. Bottom, the same blot was stripped and reprobed with the MAPK antiserum DC3 followed by ECL detection to show the level and activation state of the added p42 MAPK*.
Tyrosine Dephosphorylation of p42 MAPK* Increases the Rate of Threonine Dephosphorylation
Two possibilities are suggested by the observation that vanadate inhibits threonine dephosphorylation in oocytes (Figure 5C) and CSF extracts (our unpublished results). One is that the threonine dephosphorylation of p42 MAPK* is contingent on previous dephosphorylation of tyrosine. An alternative possibility is that a vanadate-sensitive dual-specificity phosphatase contributes to the dephosphorylation of both residues, whereas an okadaic acid–sensitive phosphatase contributes only to the dephosphorylation of the threonine.
We examined these possibilities using recombinant p42 MAPK proteins in which the activating tyrosine or threonine residue had been mutated to a nonphosphorylatable phenylalanine (Y190F) or valine (T188V). We phosphorylated these activation site mutants in vitro with constitutively active MEK-1 and [γ-32P]ATP, removed the excess ATP, and added them to CSF extracts in the presence of calcium. As depicted in Figure 9B, tyrosine dephosphorylation of T188V p42 MAPK protein was similar in rate to that seen for the K57R protein, indicating that the tyrosine residue is dephosphorylated with similar kinetics regardless of the phosphorylation state of the adjacent threonine residue. In contrast, threonine dephosphorylation of the Y190F protein was much faster than that seen for the K57R protein (Figure 9, A and B), suggesting that dephosphorylation of threonine in the K57R protein is hindered by concomitant phosphorylation of tyrosine. Moreover, dephosphorylation of the Y190F protein was insensitive to vanadate but highly sensitive to okadaic acid (our unpublished results), arguing against the involvement of a dual-specificity phosphatase. Similar results were obtained from microinjection of singly phosphorylated p42 MAPK proteins into oocytes (our unpublished results). If we assume that the behavior of the recombinant phosphorylation site mutants mirrors that of their endogenous monophosphorylated counterparts, these data support the hypothesis that tyrosine dephosphorylation increases the rate of threonine dephosphorylation; that is, dephosphorylation of threonine occurs much more rapidly in the absence of an adjacent phosphotyrosine.
Tryptic Analysis of p42 MAPK* Dephosphorylation
To corroborate and extend these observations, we analyzed the dephosphorylation of p42 MAPK* in oocytes by a different method, tryptic peptide analysis. These experiments served two purposes. First, they provided a way of quantifying the rates of tyrosine and threonine dephosphorylation that did not rely on partial acid hydrolysis (and the assumption that hydrolysis yields were invariant in any single experiment). Second, they allowed us to measure directly how much monophosphorylated p42 MAPK* accumulated during the time course of p42 MAPK* dephosphorylation. From this information and some simple kinetic modeling, we were able to estimate apparent rate constants for each of the four dephosphorylation reactions (Figure 10I), thereby determining whether there was a single rate-determining step for p42 MAPK* dephosphorylation or whether the control of p42 MAPK* dephosphorylation was distributed among multiple steps.
Bisphosphorylated p42 MAPK* was microinjected into G2 oocytes (to allow dephosphorylation to proceed unopposed by phosphorylation). The p42 MAPK* bands (Figure 10A) were quantified, excised, and subjected to exhaustive digestion with trypsin. The tryptic peptides were separated by one-dimensional thin-layer electrophoresis, thereby allowing the levels of bisphosphorylated p42 MAPK*, mono-Tyr-phosphorylated p42 MAPK*, and mono-Thr-phosphorylated p42 MAPK* to be quantified individually (Figure 10, B–E). We were also able to infer the levels of nonphosphorylated p42 MAPK* (Figure 10F) and the total levels of tyrosine- and threonine-phosphorylated p42 MAPK* (Figure 10, G and H).
As shown in Figure 10C, the level of bisphosphorylated MAPK dropped with a half-time of ∼4 min. This was accompanied by the accumulation of modest amounts of mono-Tyr-phosphorylated p42 MAPK* (Figure 10D) but essentially no mono-Thr-phosphorylated p42 MAPK* (Figure 10E). The lack of mono-Thr-phosphorylated p42 MAPK* is consistent with the observation that the dephosphorylation of the Y190F p42 MAPK* mutant is rapid (Figure 9); evidently any mono-Thr-phosphorylated p42 MAPK* that forms is rapidly dephosphorylated, so that little accumulates. This finding implies that the apparent first-order rate constant for the removal of a tyrosine phosphate from bisphosphorylated p42 MAPK* (b3 in Figure 10I) must be small compared with the rate constant for dephosphorylation of mono-Thr-phosphorylated p42 MAPK* (b4 in Figure 10I). Likewise, the accumulation of a nonnegligible amount of mono-Tyr-phosphorylated p42 MAPK* implies that b1 and b2 must be more similar in magnitude, ensuring that the rates of mono-Tyr-phosphorylated MAPK* production and removal are more nearly balanced.
We then determined how well a simple first-order kinetic scheme (Figure 10I) could account for the populations of bis-, mono-, and nonphosphorylated p42 MAPK* inferred from the tryptic analysis. We set b1 = 0.10 min−1 (taking the apparent rate constant b1 to be ln 2/t1/2, where t1/2 = 7 min), on the basis of the data from the T188V mutant (Figure 9), and b4 = 0.45 min−1 (t1/2 = 1.5 min), on the basis of the data from the Y190F mutant (Figure 9), and varied the assumed values of b2 and b3. We then calculated the expected levels of all of the experimentally determined forms of p42 MAPK* and determined what values of b2 and b3 yielded good fits to the experimental data. As shown in Figure 10, C–H, when b2 and b3 were both taken to be equal to ∼0.08 min−1 (t1/2 = 8.7 min), a good fit was obtained to the tryptic data. In particular, the experimentally observed paucity of mono-Thr-phosphorylated p42 MAPK* (Figure 10E), the higher levels of mono-Tyr-phosphorylated p42 MAPK* (Figure 10D), and the overall rates of tyrosine and threonine dephosphorylation (Figure 10, G and H) were all accounted for. Thus, it seems that both “legs” of the basic dephosphorylation scheme (the tyrosine-then-threonine leg represented by b2 and b1 and the threonine-then-tyrosine leg represented by b3 and b4) contribute to the overall rate of p42 MAPK* dephosphorylation in vivo.
PP2A, not PP1, Is the Major Threonine-directed MAPK Phosphatase
Finally, we sought to identify the okadaic acid–sensitive phosphatase responsible for the dephosphorylation of the threonine residue on p42 MAPK*. To determine whether such an activity existed in CSF extracts, we followed the kinetics of dephosphorylation of our p42 MAPK* protein in extracts pretreated with okadaic acid. In agreement with our in vivo results, dephosphorylation of p42 MAPK* was slowed in the presence of 1 μM okadaic acid (Figure 11A). Increasing the okadaic acid concentration to 5 μM had no apparent additional inhibitory effect (our unpublished results). Moreover, as observed in G2 and M1 oocytes, okadaic acid treatment inhibited only threonine dephosphorylation (Figure 11C) while leaving tyrosine dephosphorylation unaffected (Figure 11B).
Two major classes of okadaic acid–sensitive serine/threonine phosphatases, PP2A and PP1, function in eukaryotic cells. Although PP2A activity is inhibited at 10- to 100-fold lower concentrations of okadaic acid than is PP1 activity, both activities may be affected at the concentration of okadaic acid used in our experiments (1 μM) (Cohen, 1989; Shenolikar, 1994; Tournebize et al., 1997). Therefore, to distinguish between these two activities, we used the PP1 inhibitor I-2, a 22.8-kDa protein that specifically binds to and inhibits the PP1 catalytic subunit (Cohen, 1989; Shenolikar, 1994). To ensure that the I-2 protein had inhibited PP1, we assessed DNA replication, which is PP1-dependent (Walker et al., 1992). Pretreatment of extracts with 40 U/μl I-2 (1.5 μM) had no effect on the rate of p42 MAPK* dephosphorylation (Figure 11D) but blocked DNA replication (our unpublished results). Thus, p42 MAPK* threonine dephosphorylation does not depend on PP1-like activities.
We next tested whether any of the threonine dephosphorylation of p42 MAPK* could be attributed to the okadaic acid–insensitive, Ca2+-dependent phosphatase or calcineurin (PP2B). Extracts were pretreated with EGTA (10 mM) to chelate calcium and prevent calcineurin activation. EGTA failed to slow, and in fact slightly accelerated, the rate of p42 MAPK* dephosphorylation (Figure 11E). Thus there is no evidence of a role for calcineurin in p42 MAPK* dephosphorylation in this system.
Taken together, these data suggest that PP2A or a PP2A-like enzyme is the major threonine-specific p42 MAPK phosphatase functioning during meiosis.
DISCUSSION
Activation of p42 MAPK can cause mitogenesis, cell transformation, and cell fate changes in various eukaryotic systems, emphasizing the importance of maintaining strict control over the activity of this kinase. Much is known about the kinases responsible for activating MAPKs; much less is known about the physiologically relevant MAPK-directed phosphatases. We have taken a direct biochemical approach to characterizing both the nature and mechanism of p42 MAPK dephosphorylation in intact cells and have exploited the Xenopus oocyte system for this purpose. The results presented here elucidate the mechanism of p42 MAPK inactivation and thereby contribute to a more complete understanding of p42 MAPK regulation in vivo.
The p42 MAPK–directed Phosphatases Are Constitutively Active
One important result to emerge from our studies concerns the constitutive nature of p42 MAPK–directed phosphatase activity during meiotic maturation and early embryogenesis. A priori, it seems reasonable that p42 MAPK–directed phosphatases would be highly active in G2-phase immature oocytes and in fertilized eggs, to help ensure that p42 MAPK remains dephosphorylated and inactive during these times (Figure 1), and that the phosphatase activity would be turned off during the several hours between germinal vesicle breakdown and fertilization, when p42 MAPK is high in activity (Figure 1). Decreased activity of the p42 MAPK–directed phosphatases during this period would allow the oocyte to maintain a high p42 MAPK activity at less of an energy cost. However, we found no evidence that either the tyrosine or threonine dephosphorylation of p42 MAPK slowed down during this period; dephosphorylation of the tyrosine and threonine phosphates continued to proceed with half-times of ∼5 min. These findings indicate that p42 MAPK activation is continuously dependent on MEK activity throughout maturation.
We found no sign that the oocyte adapts to the activation of p42 MAPK by upregulating a p42 MAPK–directed phosphatase activity, unlike the situation in some somatic cells (Grumont et al., 1996; Brondello et al., 1997). In the oocyte, p42 MAPK activity remains both responsive to MEK and dependent on MEK throughout maturation.
Our findings shed light on several previous studies of Mos, MEK, and p42 MAPK function during meiotic maturation and early embryogenesis. Immunodepletion of the MAPKKK Mos from CSF-arrested extracts depletes these extracts of their cytostatic activity as assayed by the cleaving blastomere assay (Sagata et al., 1989b). Because thiophosphorylated p42 MAPK alone can score as CSF in this assay, this result implies that both MEK and p42 MAPK become rapidly inactivated when Mos is inactivated. Likewise, blocking Mos synthesis after GVBD results in rapid inactivation of p42 MAPK (Furuno et al., 1994; Roy et al., 1996), again consistent with constitutively high levels of p42 MAPK- and MEK-inactivating enzymes.
Is MEK Sufficiently Active to Maintain High p42 MAPK Activity in the Face of Such High Dephosphorylation Rates?
Given how rapidly the phosphates in p42 MAPK* are removed, even when the kinase is fully active, the question arises as to whether the specific activity of MEK is high enough to maintain p42 MAPK activity. The concentration of MEK in oocytes has been estimated to be ∼1.3 μM (Ferrell and Bhatt, 1997), and the pseudofirst-order rate constant for phosphorylation of p42 MAPK by MEK in vitro has been estimated to be 1–10 μM−1 min−1 (Alessi et al., 1994; Dent et al., 1994; Mansour et al., 1996), implying a rate of 1.3–13 phosphates transferred per minute per molecule of p42 MAPK. The dephosphorylation rate we observe is ∼0.1 phosphate removed per minute per molecule of p42 MAPK (ln 2/∼5 min ≈ 0.1 min−1). Thus, active MEK is sufficiently high in activity to overwhelm the MAPK-directed phosphatase activity present in an oocyte; in this situation there is no need to invoke auxiliary factors or scaffolds (Schaeffer et al., 1998) to explain the in vivo activity of MEK.
A PP2A-like Activity Is Responsible for Much of the Threonine Dephosphorylation of p42 MAPK*
Our studies provide direct evidence that a PP2A-like activity dephosphorylates, or regulates the dephosphorylation of, the threonine residue of p42 MAPK* in egg extracts and intact oocytes. Several previous reports have identified PP2A as a candidate p42 MAPK phosphatase. PP2A (but not PP1) can dephosphorylate the threonine residue on p42 MAPK and thereby inactivate the kinase in vitro (Sturgill et al., 1988; Anderson et al., 1990;). In somatic cells, interaction with the oncogenic SV40 small t antigen stimulates cell proliferation, at least in part, by compromising the ability of PP2A to inactivate both MEK-1 and the MAPK ERK-1 (Sontag et al., 1993). Similarly, Alessi et al. (1995) have identified PP2A as the primary threonine phosphatase acting on MAPK in pheochromocytoma 12 cell extracts. In Xenopus, microinjection of mRNA encoding the homeobox protein HOX11, a PP2A- and PP1-binding oncoprotein, into G2-arrested oocytes results in a modest inhibition of phosphorylase a–directed phosphatase activity and stimulation of GVBD (Kawabe et al., 1997). Likewise, expression of the Bβ′ regulatory subunit of PP2A in Xenopus oocytes increases the endogenous PP2A activity and inhibits progesterone-induced maturation (Iwashita et al., 1997).
PP2A is active during mitosis and plays an important role in maintaining the short steady-state length of microtubules in mitotic Xenopus egg extracts (Tournebize et al., 1997). It is curious, then, that recent work has shown that p42 MAPK, implicated here as a PP2A substrate, is also active during mitosis and is also important for maintaining short mitotic microtubules (Gotoh et al., 1991; Guadagno and Ferrell, 1998). It would be of interest to determine whether the particular PP2A-like enzyme responsible for p42 MAPK inactivation is active during mitosis, whether some of the cell’s p42 MAPK is protected from PP2A during mitosis, and whether PP2A isoforms exert both positive and negative effects on mitotic microtubule dynamics.
Recently a number of novel serine/threonine phosphatases have been identified that are distinct from PP2A but have okadaic acid sensitivities similar to that of PP2A (Honkanen et al., 1991; Brewis et al., 1993; Chen et al., 1994; Armstrong et al., 1995). We cannot eliminate the possibility that one of these could contribute to the PP2A-like activity seen in Xenopus oocytes and eggs.
A Distinct Tyrosine-directed p42 MAPK Phosphatase Is Responsible for the Tyrosine Dephosphorylation of p42 MAPK
The exact identity of the tyrosine-directed p42 MAPK phosphatase remains a mystery, but several candidate proteins exist. Sarcevic et al. (1993), for example, purified a tyrosine-specific p42 MAPK phosphatase of ∼47 kDa from Xenopus interphase egg extracts; unfortunately, the identity of this phosphatase has not been reported. Similarly, a Xenopus homologue of the dual-specificity phosphatases CL100/MKP-1 has been cloned, and the protein has been shown to be present throughout oocyte maturation (Lewis et al., 1995). In view of our observation that Xenopus p42 MAPK* dephosphorylation occurs via separate tyrosine- and threonine-directed phosphatase activities, it is intriguing to hypothesize that XCL100 may act strictly as a tyrosine phosphatase in vivo. In support of this hypothesis, the human dual-specificity phosphatase VHR, when assayed using a bisphosphorylated MAPK-derived peptide, dephosphorylated phosphotyrosine much more rapidly than phosphothreonine (with second-order rate constants of 32,000 and 14 M−1 s−1, respectively) (Denu et al., 1995). This dephosphorylation reaction was ordered, with rapid tyrosine dephosphorylation being followed by slower threonine dephosphorylation. Likewise, VHR dephosphorylates only the tyrosine residue of full-length activated ERK2 with a second-order rate constant of 40,000 M−1 s−1 and catalyzes most, if not all, of the rapid ERK inactivation seen in anisomycin-treated COS-1 cells (Todd et al., 1999). It is therefore possible that either XCL100 or a Xenopus VHR homologue is responsible for dephosphorylating only the tyrosine residue on p42 MAPK in oocytes and eggs, with the responsibility of threonine dephosphorylation then falling to a dedicated threonine phosphatase such as PP2A.
In addition, Pulido et al. (1998) have recently characterized two mammalian tyrosine phosphatases as being strong candidates for MAPK-inactivating proteins in vivo. These two phosphatases, PTP-SL and STEP, exist in both transmembrane and cytosolic forms. Interestingly, their cytosolic forms are similar in size (∼45 kDa) to the Xenopus tyrosine phosphatase cloned by Sarcevic et al. (1993), raising the intriguing possibility that either PTP-SL or STEP and the as yet unidentified ∼47-kDa tyrosine phosphatase from Xenopus are one and the same. The receptor tyrosine phosphatase CD45 can also dephosphorylate the tyrosine residue on active p42 MAPK in vitro, thereby inactivating the kinase (Sturgill et al., 1988; Anderson et al., 1990), although the physiological significance of this result is uncertain. Finally, it could be the case that different tyrosine phosphatases function, in either an overlapping or nonoverlapping manner, at different times during meiosis to downregulate p42 MAPK activity. For instance, the yeast pheromone-responsive MAPKs Fus3 and Kss1 are regulated by both tyrosine-specific (PTP2 and PTP3) and dual-specificity (MSG5) phosphatases in vivo (Zhan et al., 1997; Keyse, 1998).
In Oocytes, p42 MAPK Threonine Dephosphorylation Is Partially Contingent on Tyrosine Dephosphorylation
We have shown that the efficiency of threonine dephosphorylation is increased by previous tyrosine dephosphorylation, although the oocyte does possess a more slowly acting mechanism for the dephosphorylation of threonine in the presence of phosphotyrosine. This observation explains why threonine dephosphorylation is partially inhibited by vanadate; vanadate blocks the b3 + b4 route (Figure 10I) to threonine dephosphorylation by blocking the b3 step, and the b3 + b4 route is a major contributor to the overall rate of threonine dephosphorylation.
The behavior seen here is the reverse of that seen by Alessi et al. (1995) in extracts from somatic cells. On the basis of their studies, they concluded that MAPK threonine dephosphorylation was rate-limiting, with tyrosine dephosphorylation being delayed by the presence of phosphothreonine. Thus there may be different mechanisms for MAPK dephosphorylation in different cell types.
Control and Sensitivity in MAPK Dephosphorylation
Our results show that there is no single rate-determining step in the dephosphorylation of p42 MAPK in oocytes and extracts. At least two distinct enzymes, two dephosphorylation “routes” (MAPK-PP → MAPK-TP → MAPK and MAPK-PP → MAPK-YP → MAPK), and each of the four individual dephosphorylation reactions (Figure 10I) share in the control of MAPK dephosphorylation. However, they do not share equally. The tyrosine dephosphorylation of bisphosphorylated p42 MAPK seems to be particularly important and to exert the greatest degree of control over the dephosphorylation of MAPK. This step is relatively slow (t1/2 ≈ 8.7 min) and sets up the MAPK for a rapid (t1/2 ≈ 1.5 min) second dephosphorylation. Consequently, changes in the rate of the initial tyrosine dephosphorylation step would be expected to have a higher impact on the overall rate of MAPK dephosphorylation than would changes in the other three steps. This makes the identification of the enzyme responsible for this step a particularly important goal.
ACKNOWLEDGMENTS
We thank Natalie Ahn, Jonathan Cooper, and Jim Posada for providing plasmids used in this work, Ramesh Bhatt for providing purified recombinant proteins, and Sarah Walter and Joanne Westendorf for critical reading of this manuscript. This work was supported by National Institutes of Health grant GM-46383 (to J.E.F.) and a Howard Hughes Medical Institute predoctoral fellowship (to M.L.S.).
REFERENCES
- Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol. 1995;5:283–295. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U, Ashworth A, Marshall CJ, Cowley S. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13:1610–1619. doi: 10.1002/j.1460-2075.1994.tb06424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson NG, Maller JL, Tonks NK, Sturgill TW. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature. 1990;343:651–653. doi: 10.1038/343651a0. [DOI] [PubMed] [Google Scholar]
- Armstrong CG, Mann DJ, Berndt N, Cohen PT. Drosophila PPY, a novel male specific protein serine/threonine phosphatase localized in somatic cells of the testis. J Cell Sci. 1995;108:3367–3375. doi: 10.1242/jcs.108.11.3367. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- Brewis ND, Street AJ, Prescott AR, Cohen PT. PPX, a novel protein serine/threonine phosphatase localized to centrosomes. EMBO J. 1993;12:987–996. doi: 10.1002/j.1460-2075.1993.tb05739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brondello JM, Brunet A, Pouyssegur J, McKenzie FR. The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade. J Biol Chem. 1997;272:1368–1376. doi: 10.1074/jbc.272.2.1368. [DOI] [PubMed] [Google Scholar]
- Charles CH, Sun H, Lau LF, Tonks NK. The growth factor-inducible immediate-early gene 3CH134 encodes a protein-tyrosine-phosphatase. Proc Natl Acad Sci USA. 1993;90:5292–5296. doi: 10.1073/pnas.90.11.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MX, McPartlin AE, Brown L, Chen YH, Barker HM, Cohen PT. A novel human protein serine/threonine phosphatase, which possesses four tetratricopeptide repeat motifs and localizes to the nucleus. EMBO J. 1994;13:4278–4290. doi: 10.1002/j.1460-2075.1994.tb06748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PR. Signal transduction. Switching off MAP kinases. Curr Biol. 1994;4:647–650. doi: 10.1016/s0960-9822(00)00144-5. [DOI] [PubMed] [Google Scholar]
- Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. 1989;58:453–508. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–480. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- Cross DA, Smythe C. PD 98059 prevents establishment of the spindle assembly checkpoint and inhibits the G2-M transition in meiotic but not mitotic cell cycles in Xenopus. Exp Cell Res. 1998;241:12–22. doi: 10.1006/excr.1998.4023. [DOI] [PubMed] [Google Scholar]
- Dasso M, Newport JW. Completion of DNA replication is monitored by a feedback system that controls the initiation of mitosis in vitro: studies in Xenopus. Cell. 1990;61:811–823. doi: 10.1016/0092-8674(90)90191-g. [DOI] [PubMed] [Google Scholar]
- Dent P, Chow YH, Wu J, Morrison DK, Jove R, Sturgill TW. Expression, purification and characterization of recombinant mitogen-activated protein kinase kinases. Biochem J. 1994;303:105–112. doi: 10.1042/bj3030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu JM, Lohse DL, Vijayalakshmi J, Saper MA, Dixon JE. Visualization of intermediate and transition-state structures in protein-tyrosine phosphatase catalysis. Proc Natl Acad Sci USA. 1996a;93:2493–2498. doi: 10.1073/pnas.93.6.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu JM, Stuckey JA, Saper MA, Dixon JE. Form and function in protein dephosphorylation. Cell. 1996b;87:361–364. doi: 10.1016/s0092-8674(00)81356-2. [DOI] [PubMed] [Google Scholar]
- Denu JM, Zhou G, Wu L, Zhao R, Yuvaniyama J, Saper MA, Dixon JE. The purification and characterization of a human dual-specific protein tyrosine phosphatase. J Biol Chem. 1995;270:3796–3803. doi: 10.1074/jbc.270.8.3796. [erratum (1995) 270, 10358]. [DOI] [PubMed] [Google Scholar]
- Doi K, Gartner A, Ammerer G, Errede B, Shinkawa H, Sugimoto K, Matsumoto K. MSG5, a novel protein phosphatase promotes adaptation to pheromone response in S. cerevisiae. EMBO J. 1994;13:61–70. doi: 10.1002/j.1460-2075.1994.tb06235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman K, Carrasco D, Gruda M, Ryan C, Lira SA, Bravo R. Disruption of the erp/mkp-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene. 1996;13:925–931. [PubMed] [Google Scholar]
- Ferrell JE., Jr MAP kinases in mitogenesis and development. Curr Top Dev Biol. 1996a;33:1–60. doi: 10.1016/s0070-2153(08)60336-1. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996b;21:460–466. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, Jr, Bhatt RR. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J Biol Chem. 1997;272:19008–19016. doi: 10.1074/jbc.272.30.19008. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, Jr, Wu M, Gerhart JC, Martin GS. Cell cycle tyrosine phosphorylation of p34cdc2 and a microtubule-associated protein kinase homolog in Xenopus oocytes and eggs. Mol Cell Biol. 1991;11:1965–1971. doi: 10.1128/mcb.11.4.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuno N, Nishizawa M, Okazaki K, Tanaka H, Iwashita J, Nakajo N, Ogawa Y, Sagata N. Suppression of DNA replication via Mos function during meiotic divisions in Xenopus oocytes. EMBO J. 1994;13:2399–2410. doi: 10.1002/j.1460-2075.1994.tb06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh Y, Masuyama N, Dell K, Shirakabe K, Nishida E. Initiation of Xenopus oocyte maturation by activation of the mitogen-activated protein kinase cascade. J Biol Chem. 1995;270:25898–25904. doi: 10.1074/jbc.270.43.25898. [DOI] [PubMed] [Google Scholar]
- Gotoh Y, Nishida E, Matsuda S, Shiina N, Kosako H, Shiokawa K, Aikyama T, Ohta K, Sakai H. In vitro effects on microtubule dynamics of purified Xenopus M phase activated MAP kinase. Nature. 1991;349:251–254. doi: 10.1038/349251a0. [DOI] [PubMed] [Google Scholar]
- Grumont RJ, Rasko JE, Strasser A, Gerondakis S. Activation of the mitogen-activated protein kinase pathway induces transcription of the PAC-1 phosphatase gene. Mol Cell Biol. 1996;16:2913–2921. doi: 10.1128/mcb.16.6.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadagno TM, Ferrell JE., Jr Requirement for MAPK activation for normal mitotic progression in Xenopus egg extracts. Science. 1998;282:1312–1315. doi: 10.1126/science.282.5392.1312. [DOI] [PubMed] [Google Scholar]
- Haccard O, Lewellyn A, Hartley RS, Erikson E, Maller JL. Induction of Xenopus oocyte meiotic maturation by MAP kinase. Dev Biol. 1995;168:677–682. doi: 10.1006/dbio.1995.1112. [DOI] [PubMed] [Google Scholar]
- Honkanen RE, Zwiller J, Daily SL, Khatra BS, Dukelow M, Boynton AL. Identification, purification, and characterization of a novel serine/threonine protein phosphatase from bovine brain. J Biol Chem. 1991;266:6614–6619. [PubMed] [Google Scholar]
- Hsiao K-M, Chou S-y, Shih S-J, Ferrell JE., Jr Evidence that inactive p42 mitogen-activated protein kinase and inactive Rsk exist as a heterodimer in vivo. Proc Natl Acad Sci USA. 1994;91:5480–5484. doi: 10.1073/pnas.91.12.5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Kessler DS, Erikson RL. Biochemical and biological analysis of Mek1 phosphorylation site mutants. Mol Biol Cell. 1995;6:237–245. doi: 10.1091/mbc.6.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Bottaro DP, Chan A, Miki T, Aaronson SA. Expression cloning of a human dual-specificity phosphatase. Proc Natl Acad Sci USA. 1992;89:12170–12174. doi: 10.1073/pnas.89.24.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashita J, Shima H, Nagao M, Sagata N. cDNA cloning of a novel B subunit of Xenopus protein phosphatase 2A and its biological activity in oocytes. Biochem Biophys Res Commun. 1997;232:218–222. doi: 10.1006/bbrc.1997.6259. [DOI] [PubMed] [Google Scholar]
- Kamps MP. Determination of phosphoamino acid composition by acid hydrolysis of protein blotted to Immobilon. Methods Enzymol. 1991;201:21–27. doi: 10.1016/0076-6879(91)01005-m. [DOI] [PubMed] [Google Scholar]
- Kawabe T, Muslin AJ, Korsmeyer SJ. HOX11 interacts with protein phosphatases PP2A and PP1 and disrupts a G2/M cell-cycle checkpoint. Nature. 1997;385:454–458. doi: 10.1038/385454a0. [DOI] [PubMed] [Google Scholar]
- Keyse SM. Protein phosphatases and the regulation of MAP kinase activity. Semin Cell Dev Biol. 1998;9:143–152. doi: 10.1006/scdb.1997.0219. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Golsteyn R, Poon R, Stewart E, Gannon J, Minshull J, Smith R, Hunt T. Cyclins and their partners during Xenopus oocyte maturation. Cold Spring Harb Symp Quant Biol. 1991a;56:437–447. doi: 10.1101/sqb.1991.056.01.051. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Minshull J, Ford C, Golsteyn R, Poon R, Hunt T. On the synthesis and destruction of A- and B-type cyclins during oogenesis and meiotic maturation in Xenopus laevis. J Cell Biol. 1991b;114:755–765. doi: 10.1083/jcb.114.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosako H, Akamatsu Y, Tsurushita N, Lee KK, Gotoh Y, Nishida E. Isolation and characterization of neutralizing single-chain antibodies against Xenopus mitogen-activated protein kinase kinase from phage display libraries. Biochemistry. 1996;35:13212–13221. doi: 10.1021/bi960956f. [DOI] [PubMed] [Google Scholar]
- Kosako H, Gotoh Y, Matsuda S, Ishikawa M, Nishida E. Xenopus MAP kinase activator is a serine/threonine/tyrosine kinase activated by threonine phosphorylation. EMBO J. 1992;11:2903–2908. doi: 10.1002/j.1460-2075.1992.tb05359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosako H, Gotoh Y, Nishida E. Mitogen-activated protein kinase kinase is required for the mos-induced metaphase arrest. J Biol Chem. 1994;269:28354–28358. [PubMed] [Google Scholar]
- Lewis T, Groom LA, Sneddon AA, Smythe C, Keyse SM. XCL100, an inducible nuclear MAP kinase phosphatase from Xenopus laevis: its role in MAP kinase inactivation in differentiated cells and its expression during early development. J Cell Sci. 1995;108:2885–2896. doi: 10.1242/jcs.108.8.2885. [DOI] [PubMed] [Google Scholar]
- Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- Luo K, Hurley TR, Sefton BM. Cyanogen bromide cleavage and proteolytic peptide mapping of proteins immobilized to membranes. Methods Enzymol. 1991;201:149–152. doi: 10.1016/0076-6879(91)01014-s. [DOI] [PubMed] [Google Scholar]
- Mansour SJ, Candia JM, Matsuura JE, Manning MC, Ahn NG. Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry. 1996;35:15529–15536. doi: 10.1021/bi961854s. [DOI] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 1998;12:557–570. doi: 10.1101/gad.12.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool. 1971;177:129–145. doi: 10.1002/jez.1401770202. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Kosako H, Takenaka K, Moriyama K, Sakai H, Akiyama T, Gotoh Y, Nishida E. Xenopus MAP kinase activator: identification and function as a key intermediate in the phosphorylation cascade. EMBO J. 1992;11:973–982. doi: 10.1002/j.1460-2075.1992.tb05136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull J, Sun H, Tonks NK, Murray AW. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell. 1994;79:475–486. doi: 10.1016/0092-8674(94)90256-9. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Gotoh Y, Nishida E. Roles of the MAP kinase cascade in vertebrates. Adv Pharmacol. 1996;36:121–137. doi: 10.1016/s1054-3589(08)60579-7. [DOI] [PubMed] [Google Scholar]
- Murray AW. Cell cycle extracts. Methods Cell Biol. 1991;36:581–605. [PubMed] [Google Scholar]
- Nebreda AR. Inactivation of MAP kinases. Trends Biochem Sci. 1994;19:1–2. doi: 10.1016/0968-0004(94)90163-5. [DOI] [PubMed] [Google Scholar]
- Payne DM, Rossomando AJ, Martino P, Erickson AK, Her J-H, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase) EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada J, Cooper JA. Requirements for phosphorylation of MAP kinase during meiosis in Xenopus oocytes. Science. 1992;255:212–215. doi: 10.1126/science.1313186. [DOI] [PubMed] [Google Scholar]
- Pulido R, Zuniga A, Ullrich A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998;17:7337–7350. doi: 10.1093/emboj/17.24.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- Roy LM, Haccard O, Izumi T, Lattes BG, Lewellyn AL, Maller JL. Mos proto-oncogene function during oocyte maturation in Xenopus. Oncogene. 1996;12:2203–2211. [PubMed] [Google Scholar]
- Sagata N, Daar I, Oskarsson M, Showalter SD, Vande Woude GF. The product of the mos proto-oncogene as a candidate “initiator” for oocyte maturation. Science. 1989a;245:643–646. doi: 10.1126/science.2474853. [DOI] [PubMed] [Google Scholar]
- Sagata N, Oskarsson M, Copeland T, Brumbaugh J, Vande Woude GF. Function of c-mos proto-oncogene product in meiotic maturation in Xenopus oocytes. Nature. 1988;335:519–525. doi: 10.1038/335519a0. [DOI] [PubMed] [Google Scholar]
- Sagata N, Watanabe N, Vande Woude GF, Ikawa Y. The c-mos proto-oncogene product is a cytostatic factor responsible for meiotic arrest in vertebrate eggs. Nature. 1989b;342:512–518. doi: 10.1038/342512a0. [DOI] [PubMed] [Google Scholar]
- Sarcevic B, Erikson E, Maller JL. Purification and characterization of a mitogen-activated protein kinase tyrosine phosphatase from Xenopus eggs. J Biol Chem. 1993;268:25075–25083. [PubMed] [Google Scholar]
- Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade [see comments] Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- Shenolikar S. Protein serine/threonine phosphatases—new avenues for cell regulation. Annu Rev Cell Biol. 1994;10:55–86. doi: 10.1146/annurev.cb.10.110194.000415. [DOI] [PubMed] [Google Scholar]
- Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the MAP kinase pathway and induces cell proliferation. Cell. 1993;75:887–897. doi: 10.1016/0092-8674(93)90533-v. [DOI] [PubMed] [Google Scholar]
- Sturgill TW, Ray LB, Erikson E, Maller JL. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature. 1988;334:715–718. doi: 10.1038/334715a0. [DOI] [PubMed] [Google Scholar]
- Sugiura R, Toda T, Shuntoh H, Yanagida M, Kuno T. pmp1+, a suppressor of calcineurin deficiency, encodes a novel MAP kinase phosphatase in fission yeast. EMBO J. 1998;17:140–148. doi: 10.1093/emboj/17.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- Takenaka K, Gotoh Y, Nishida E. MAP kinase is required for the spindle assembly checkpoint but is dispensable for the normal M phase entry and exit in Xenopus egg cell cycle extracts. J Cell Biol. 1997;136:1091–1097. doi: 10.1083/jcb.136.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka K, Moriguchi T, Nishida E. Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science. 1998;280:599–602. doi: 10.1126/science.280.5363.599. [DOI] [PubMed] [Google Scholar]
- Todd JL, Tanner KG, Denu JM. Extracellular regulated kinases (ERK)1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway. J Biol Chem. 1999;274:13271–13280. doi: 10.1074/jbc.274.19.13271. [DOI] [PubMed] [Google Scholar]
- Tournebize R, Andersen SS, Verde F, Doree M, Karsenti E, Hyman AA. Distinct roles of PP1 and PP2A-like phosphatases in control of microtubule dynamics during mitosis. EMBO J. 1997;16:5537–5549. doi: 10.1093/emboj/16.18.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DH, DePaoli-Roach AA, Maller JL. Multiple roles for protein phosphatase 1 in regulating the Xenopus early embryonic cell cycle. Mol Biol Cell. 1992;3:687–698. doi: 10.1091/mbc.3.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter SA, Guadagno TM, Ferrell JE., Jr Induction of a G2-phase arrest in Xenopus egg extracts by activation of p42 MAP kinase. Mol Biol Cell. 1997;8:2157–2169. doi: 10.1091/mbc.8.11.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XM, Zhai Y, Ferrell JE., Jr A role for mitogen-activated protein kinase in the spindle assembly checkpoint in XTC cells. J Cell Biol. 1997;137:433–443. doi: 10.1083/jcb.137.2.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Wu J, Michel H, Rossomando A, Haystead T, Shabanowitz J, Hunt DF, Sturgill TW. Renaturation and partial peptide sequencing of mitogen-activated protein kinase (MAP kinase) activator from rabbit skeletal muscle. Biochem J. 1992;285:701–705. doi: 10.1042/bj2850701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurgler-Murphy SM, Maeda T, Witten EA, Saito H. Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol Cell Biol. 1997;17:1289–1297. doi: 10.1128/mcb.17.3.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew N, Mellini ML, Vande Woude GF. Meiotic initiation by the mos protein in Xenopus. Nature. 1992;355:649–652. doi: 10.1038/355649a0. [DOI] [PubMed] [Google Scholar]
- Zhan XL, Deschenes RJ, Guan KL. Differential regulation of FUS3 MAP kinase by tyrosine-specific phosphatases PTP2/PTP3 and dual-specificity phosphatase MSG5 in Saccharomyces cerevisiae. Genes Dev. 1997;11:1690–1702. doi: 10.1101/gad.11.13.1690. [DOI] [PubMed] [Google Scholar]
- Zhou G, Denu JM, Wu L, Dixon JE. The catalytic role of Cys124 in the dual specificity phosphatase VHR. J Biol Chem. 1994;269:28084–28090. [PubMed] [Google Scholar]