

Abstract
N5-Carboxyaminoimidazole ribonucleotide mutase (N5-CAIR mutase or PurE) from Escherichia coli catalyzes the reversible interconversion of N5-CAIR to carboxyaminoimidazole ribonucleotide (CAIR) with direct CO2 transfer. Site-directed mutagenesis, a pH rate profile, DFT calculations, and X-ray crystallography together provide new insight into the mechanism of this unusual transformation. These studies suggest that a conserved, protonated histidine (His45) plays an essential role in catalysis. The importance of proton transfers is supported by DFT calculations on CAIR and N5-CAIR analogs in which the ribose-5’-phosphate is replaced with a methyl group. The calculations suggest that the non-aromatic tautomer of CAIR (isoCAIR) is only 3.1 kcal/mol higher in energy relative to its aromatic counterpart, implicating this species as a potential intermediate in the PurE catalyzed reaction. A structure of wild-type PurE co-crystallized with 4-nitroaminoimidazole ribonucleotide (NO2-AIR, a CAIR analog) and structures of H45N and H45Q PurEs soaked with CAIR have been solved and provide the first insight into the binding of an intact PurE substrate. A comparison of 19 available structures of PurE and PurE mutants in apo and nucleotide bound forms reveal a common, buried carboxylate/CO2 binding site for CAIR and N5-CAIR in a hydrophobic pocket in which the carboxylate/CO2 interacts with backbone amides. This work has led to a mechanistic proposal in which the carboxylate orients the substrate for proton transfer from His45 to N5-CAIR to form an enzyme-bound aminoimidazole ribonucleotide (AIR) and CO2 intermediate. Subsequent movement of the aminoimidazole moiety of AIR, re-orients it for addition of CO2 at C4 to generate isoCAIR. His45 is now in a position to remove a C4 proton to produce CAIR.
INTRODUCTION
Biochemical studies and genomic analyses of the Escherichia coli and Gallus gallus carboxyaminoimidazole ribonucleotide synthases (PurEs) indicate that these purine biosynthetic enzymes provide an unusual example of evolutionary divergence in a highly conserved, primary metabolic pathway (1-4). Class I PurEs, typified by the E. coli protein and found in most prokaryotes and fungi, catalyze the reversible transfer of a CO2 group from the carbamate of N5-carboxyaminoimidazole ribonucleotide (N5-CAIR) to C4, yielding 4-carboxyaminoimidazole ribonucleotide (CAIR) (Figure 1). On the other hand, class II PurEs, typified by the G. gallus enzyme and found in higher eukaryotes, form CAIR by reversible transfer of CO2 to aminoimidazole ribonucleotide (AIR) (4). Our laboratories are interested in the structures of class I and II PurEs, the mechanisms of these transformations, and the evolutionary relationship between the two classes of PurEs.
Figure 1.

A possible mechanism for PurE-catalyzed conversion of N5-CAIR to CAIR. Note the proposed intermediates AIR/CO2 and isoCAIR. The role of the protein is based on examination of 19 structures of PurEs from different sources and with different nucleotides bound. The aminoimidazole numbering scheme is shown on N5-CAIR. R = ribose-5’-phosphate.
Studies of the E. coli PurE (EcPurE) have been challenging because of the instability of N5-CAIR (kdecomp= 0.75 min-1 at pH 7.8 and 30°C) (1) and CAIR (kdecomp of 0.016 min-1 at pH 7, 50°C) (5). Our early mechanistic studies using [4,7-13C]- N5-CAIR showed that the CO2 from the carbamate is transferred directly to C4, presumably through an AIR and CO2 intermediate, without the latter dissociating into solution (6). Support for an AIR intermediate was provided by studies of Alenin et al. which demonstrated that both N5-CAIR and CAIR can be generated non-enzymatically from AIR and CO2 (7). N5-CAIR is generated rapidly (second to minute timescale), while CAIR was detected on a much slower time scale (days), presumably an example of kinetic and thermodynamic control, respectively. The kcat values for EcPurE for conversion of N5-CAIR to CAIR and for the reverse reaction are both ~103 min-1 (1, 8). Thus, the enzyme has evolved to control the fate of CO2, rather than dramatically altering the decarboxylation rates.
The co-crystal structure of EcPurE with CAIR (EcPurE-AIRx) revealed a disordered nucleotide(s) in the active site (AIRx) with only the positions of the ribose-5’- phosphate and aminoimidazole being unambiguous (3). This information, in conjunction with sequence alignments of many PurEs, provided a model for how the Class I mutase and the Class II PurE might differ in the catalysis of their respective transformations. From this sequence analysis, two common conserved regions were identified between the classes: one includes the P-loop (G15SxxD) providing the binding site for the phosphate of the nucleotide and the other includes the 40s loop (S43AH45R46/K). All class I enzymes contain an arginine in this loop, while class II enzymes contain a lysine. Our initial studies suggested that this lysine was not functioning as a CO2 carrier (6). A third region, the 70s loop, is conserved within each class and is distinct between classes. This loop was thus postulated to play a major role in governing the differences in substrate specificity between the class I and class II PurEs (3).
While the nucleotide electron density in the EcPurE-AIRx structure was partially disordered, the structure eliminated a mechanistic possibility in which AIR and N5-CAIR could bind simultaneously in a single active site for intermolecular CO2 transfer (3). The structure also suggested that binding of the N5-CAIR carbamate in the “hydrophobic” pocket (composed of Ala44 from the 40s loop and Ala70 and Leu76 from the 70s loop) along with the intrinsic instability of N5-CAIR might be sufficient to catalyze the 103 rate acceleration observed for decarboxylation. Previous model studies on the effects of solvent polarity and desolvation on decarboxylation supported this model (9-11)
Recent studies from the Kappock laboratory on the class I PurE from Acetobacter aceti (AaPurE) have provided additional insight into potential mechanisms for this mutase reaction (12). The AaPurE was shown to have a pH optimum of 7, despite the fact that the intracellular environment of this organism can drop as low as pH 4 (13). Two observations obtained from their studies have important mechanistic implications for all class I PurEs. First, mutagenesis studies of two conserved histidines in the vicinity of the active site indicated that only His59 (His45 in EcPurE) played an important role in catalysis (12). Second, crystals of an inactive H59N AaPurE at pH 5.4 soaked with CAIR yielded a structure with electron density interpreted as isoCAIR, a proposed intermediate in the mutase reaction (Figure 1) (12).
Enzymes have evolved a number of strategies to catalyze decarboxylation and carboxylation reactions on structurally diverse substrates (14). While many enzymes use organic (thiamin, pyridoxal, biotin) or inorganic cofactors (Mg2+, Mn2+), others, such as the PurEs, are cofactor independent. In the conversion of CAIR to N5-CAIR, recent structures of the thiamin-dependent decarboxylase, pyruvate oxidase (POX), are particularly relevant to the mechanism of PurE. With a POX active site mutant (F479W), the authors were able to observe an equivalent of isoCAIR, that is, 2-lactyl-thiamin diphosphate (LThDP) (15). The carboxylate group of LThDP was oriented perpendicular to the plane of the thiazolium ring of ThDP in an appropriate position to maximize p orbital overlap with the π system of ThDP subsequent to C-C bond cleavage. The carboxylate was found in a hydrophobic pocket between the side chains of Trp479 and Ile480, hydrogen-bonded with Glu483 and the amide backbone of Gly35. In POX, decarboxylation of LThDP is rate-limiting as LThDP has been shown to accumulate on the enzyme in solution (16).
In the N5-CAIR to CAIR direction, the best protein models for PurE involve the decarboxylation of N-carboxy-biotin (17). Unfortunately there are at present no particularly informative structures of these enzymes with biotin bound and the mechanisms of decarboxylation are still being debated (18-20). In addition, the N5-CAIR carbamate is significantly more labile than carboxybiotin (t1/2 > 100 min) (17).
In the studies with EcPurE reported herein, we have used structure, mutagenesis, a pH rate profile, and DFT-calculations to provide additional insight into the E. coli mutase reaction. Site-directed mutagenesis of His45 indicates that this residue is essential for catalysis, while titrations with CAIR monitoring fluorescence changes suggest that binding is not altered by mutations at this position. Computational methods have been used to define potential roles for proton transfer in the decarboxylation of N5-CAIR and CAIR. Finally, several new crystal structures of PurE have been obtained: a co-crystal structure with 4-nitroaminoimidazole ribonucleotide (NO2-AIR), a sub-micromolar inhibitor E. coli PurE (4, 21), and structures with H45N and H45Q mutant PurEs soaked with CAIR. These are the first PurE structures containing an intact substrate. These studies together with re-analysis of earlier structural studies (3) have allowed us to propose a mechanism for this unusual mutase reaction (Figure 1).
EXPERIMENTAL
Materials and Methods
Ampicillin, kanamycin, and all reagents were obtained from Sigma. CAIR, AIR and NO2-AIR were prepared by published procedures and quantified using published extinction coefficients (1, 8, 21). WT E. coli PurE (53 U/mg), E. coli N5-carboxyaminoimidazole ribonucleotide synthetase (EcPurK, 28 U/mg), and E. coli succinoaminoimidazole carboxamide ribonucleotide synthetase (EcPurC, 56 U/mg) were purified as previously described (8). All enzyme assays were carried out on a Cary 3 UV-Vis specotrophotometer with temperature regulation using a Lauda water bath. One unit of enzyme activity corresponds to 1 μmol of product formed min-1.
Preparation of PurE mutants
Primers for preparation of H45N, H45Q, and H45W mutant EcPurEs were made by the MIT Biopolymers Facility or Gibco BRL Lifetech (Supplemental Materials, Table 1). Mutagenesis was carried out using the Kunkel method (22) on pNC2, which encodes both EcPurE and EcPurK under an IPTG-inducible promoter (6). The desired mutation in each plasmid was confirmed by DNA sequencing at the MIT Biopolymers Facility.
Expression and Purification of PurE mutants
PurE mutants were expressed in PCO135 E. coli (purE-, CGSC#5403, Yale E. coli Genetic Stock Center). Competent PCO135 E. coli were prepared using the CaCl2 method (23) and co-transformed with pNC2-H45N, -H45Q, or –H45W and pGP1-2, which encodes the T7 RNA polymerase under control of the temperature-sensitive λ repressor (24). Colonies were selected for growth on LB/agar plates at 37°C containing 50 μg/mL ampicillin and 35 μg/mL kanamycin present in all growth media.
A single colony was used to inoculate a culture (100 mL of LB) that was grown overnight at 37°C with shaking at 200 rpm. A portion of this culture (10 mL) was then collected by centrifugation at 3000 rpm for 10 min at 4°C, washed with ice-cold PBS, and used to inoculate 1 L of LB in a 2 L flask containing a stir bar. This culture was grown at 37°C at 200 rpm until an OD600 of 1.0 was reached, at which point an equal volume of LB at 49°C was added while vigorously stirring the media. The cells were then grown for an additional 4 h at 37°C with shaking at 200 rpm. Cells (~ 10 g/L media) were collected by centrifugation and frozen at -80°C.
Purification of PurE Mutants
All purification steps were carried out at 4°C. Cells (~9 g) were resuspended in 5 mL/g Buffer A (100 mM Tris pH 7.8) to which 200 U of DNase I (2000 U/mg, Roche), 200 U of RNase (30 U/mg, Roche), 0.1% (w/v) phenylmethylsulfonyl fluoride, and 6 mM β-mercaptoethanol were added. The cells were lysed by 2 passes through a French press at 14,000 psi. Cell debris was removed by centrifugation at 19,000 rpm for 20 min in a Beckman JA25.5 rotor. Streptomycin sulfate (6% w/v, 0.2 volumes) was then added to the supernatent over 20 min and the solution was stirred for an additional 20 min before centrifugation as above. The mutant proteins were then precipitated with (NH4)2SO4 and purified by DEAE Sepharose and hydroxyapatite chromatography as previously described for wt PurE (8). Protein concentration was determined using the calculated extinction coefficient (ε280=13,940 M-1 cm-1 for wt PurE, ProtParam, www.expasy.ch), and typical yields of the mutant PurEs were 10 mg/g cells. New hydroxyapatite and Sephadex G-25 resin were used in purifying each mutant PurE, and DEAE Sepharose FF resin was cleaned thoroughly between uses to avoid contaminating PurEs from previous runs. The identity of each mutant was confirmed by ESI-MS (MIT Biopolymers Facility).
Enzyme Assay for N5-CAIR to CAIR
A modified version of the coupled assay of Davisson was used (25). The assay buffer contained in a final volume of 600 μL: 50 mM HEPES (pH 7.5), 10 mM MgCl2, 2 mM PEP, 0.5 mM ATP, 10 mM L-aspartate, 1.2 U pyruvate kinase (451 U/mg, Sigma P-7768), 2.5 U EcPurK, 2 U EcPurC, and variable amounts of PurE (60—600 μM for mutant PurEs, 1-10 nM for wt PurE) at 23°C. After pre-incubation for 2 min, the reaction was initiated by addition of 37 μM AIR, and succinoaminoimidazole carboxamide ribonucleotide (SAICAR) formation was monitored directly (ε282 = 8,607 M-1 cm-1).
Enzyme Assay for CAIR to N5-CAIR
The assay buffer contained in a final volume of 600 μL: 100 mM Tris pH 8.0, 2-1,000 μM CAIR, and variable amounts of PurE (60—600 μM for mutant PurEs, 1-10 nM for wt PurE) at 37°C. The reaction was monitored directly by the disappearance of CAIR (Δε250 = 7,710 M-1 cm-1 at pH 8.0) as previously described (8). The kinetic parameters were calculated using Equation 1 and non-linear least squares fitting of the data using Kaleidagraph software (Synergy). Cuvettes with different path lengths were required at high concentrations of CAIR and PurE.
| (1) |
pH Rate Profile for Conversion of CAIR to N5-CAIR by wt PurE
A three component, constant ionic strength, buffer system was used at 37°C (26). In the pH range 5.1 to 8.8 the components were 100mM Tris/50 mM MES/50 mM acetic acid, and the pH was adjusted by addition of 12 M HCl or 10 M NaOH. At pH 9.2 and 9.5, the buffer components were 100 mM ACES/52 mM Tris/52 mM ethanolamine, and the pH was adjusted by addition of 10 M NaOH. Before and after each experiment, the pH of the assay solution was verified using a glass semi-microelectrode (9103BN, Thermo Orion).
In order to calculate PurE activity at each pH, Δε250 values were obtained at each pH derived from endpoint assays. CAIR was quantified using ε250 = 10, 980 M-1 cm-1 at pH 8.0 in 100 mM Tris (8). A known concentration of CAIR (100 μM) was then added to 0.6 mL of the three component buffer system at each pH, and the absorbance spectrum was immediately recorded. Sufficient wt PurE (5 U/mL) was then added to the cuvette to rapidly consume all of the CAIR. The final A250 was subtracted from the initial A250, and the result was divided by the initial CAIR concentration to yield the Δε250 at each pH (Supplemental Materials, Table 2).
At each pH, initial velocities were determined using the conditions described above with CAIR being varied from 7—350 μM. Background rates of non-enzymatic CAIR decarboxylation at each pH were determined and substracted from the initial velocities (Table 2, Supplemental Materials). At each pH, two independent experiments were carried out and kcat and kcat/Km were determined by independent fits to Equation 1. These values obtained were then averaged and used in Equation 2 with non-linear least squares fitting with Kaleidagraph (Synergy) to obtain pK1 and pK2 where kcat or kcat/Km = kobs and kmax is the pH-independent parameter for kcat or kcat/Km.
Table 2.
Refinement Statistics.
| EcPurE-
NO2-AIR |
EcPurE-
H45N-CAIR |
EcPurE-
H45Q-CAIR |
EcPurE-
H45Q- NO2-AIR |
|
|---|---|---|---|---|
| resolution (Å) | 1.8 | 2.0 | 2.3 | 1.8 |
| total # of nonhydrogen atoms | 1373 | 1341 | 1281 | 1347 |
| # of protein atoms | 1197 | 1197 | 1197 | 1202 |
| # of ligand atoms | 22 | 22 | 22 | 22 |
| # of water atoms | 154 | 122 | 62 | 123 |
| # of reflections in refinement | 12357 | 10410 | 6763 | 12061 |
| # of reflections in test set | 667 | 1082 | 697 | 1347 |
| R factor (%)a | 15.2 | 18.9 | 19.8 | 18.8 |
| Rfree (%)b | 20.2 | 23.3 | 24.5 | 22.1 |
| RMS deviation from ideal geometry bonds (Å) | 0.001 | 0.007 | 0.016 | 0.015 |
| angles (°) | 1.8 | 1.4 | 1.5 | 1.5 |
| Average B-factor (Å2) | 14.9 | 31.7 | 39.6 | 38.3 |
| Ramachandran plot most favored region (%) | 92.2 | 92.0 | 89.9 | 92.1 |
| additional allowed region (%) | 7.8 | 8.0 | 10.1 | 7.9 |
R factor = Σhkl | | Fobs| - k | Fcal | | /Σhkl | Fobs | , where Fobs and Fcal are observed and calculated structure factors, respectively.
Rfree the sum is extended over a subset of reflections that were excluded from all stages of refinement.
| (2) |
Fluorescence Titrations to Monitor NO2-AIR and CAIR Binding to wt and mutant PurEs
Fluorescence measurements were made using a QuantaMaster 4 SE fluorimeter (Photon Technology International) at 23°C. Samples were excited at 295 nm and the emission was monitored 310-500 nm at a scan rate of 0.1 s/nm with 5 nm slits. Titrations were carried out by addition of 1 μL aliquots of 0.4 mM NO2-AIR to wt PurE (750 nM monomer) in 10 mM KPi (pH 7.8). Titrations of the mutant PurEs (3 μM monomer) in 10 mM KPi (pH 7.8) were carried out by addition of 1 μL aliquots of 1 mM CAIR. Spectra were corrected for both CAIR or NO2-AIR fluorescence background and enzyme dilution.
Fluorescence intensities at the emission λmax (335 nm) were plotted as (Fo-F)/Fo, where Fo is the starting fluorescence intensity before addition of ligand and F is the observed fluorescence at each ligand concentration. The data were then fit to Equation 3 using Kaleidagraph software (Synergy) where L is the concentration of CAIR or NO2-AIR, E is the concentration of PurE monomer, and Kd is the dissociation constant.
| (3) |
Characterization of the Mutant PurEs Quaternary Structure by Sedimentation Velocity Analytical Ultracentrifugation (SV-AUC)
SV-AUC experiments were carried out on an Optima XL-1 analytical ultracentrifuge (Beckman Coulter, Fullerton, CA) at the Biophysical Instrumentation Facility at MIT). Before each experiment, protein samples (wt, H45N, H45Q, or H45W PurE, 0.5 mM) were dialyzed against PBS for 24 h at 4°C in a Slide-A-Lyzer cassette with a 10 kDa MWCO membrane (Pierce). Samples were diluted to 18 μM with the dialysis buffer and placed in double-sector Epon centerpieces with quartz windows in an An60Ti 4-hole rotor. Sedimentation at 30,000 rpm was monitored for 24 h at 25°C by continuous scanning at 280 nm along the length of the cell.
SEDNTERP software was used to calculate buffer density (1.00399 g/mL), viscosity (0.010183 P), and protein partial specific volume from the amino acid content (0.7421 mL/g for wt PurE) (27). Data (~200 traces for each experiment) were fit using SEDFIT88 from 1-20 S using a continuous distribution of sedimentation coefficients (C(s)) derived from solutions to the Lamm equations (28). The C(s) results were converted to a molecular weight distribution (C(m)) by SEDFIT88, and data were integrated using Kaleidagraph software (Synergy).
Complementation of PCO135 purE- E. coli by Mutant PurEs
PCO135 cells were transformed with pNC2, pNC2-H45N, pNC2-H45Q, or pNC2-H45W and pGP1-2 as described above. A single colony from a fresh transformation was then used to inoculate 5 mL of LB with 50 μg/mL ampicillin and 35 μg/mL kanamycin for 8 h at 37°C. The cells were collected by centrifugation at 3,000 rpm at 4°C for 10 min and washed with 2 × 5 mL ice-cold M9 minimal medium. The cells were then resuspended in 1 mL of M9 medium and 10 μL was spread onto M9 agar plates with or without 1 mM adenine and 1 mM guanine. The plates were incubated at either 30 or 37°C, and cell growth was monitored after 16, 24, 48, and 72 h.
DFT Calculations of Tautomerization Energies of AIR, CAIR, and N5-CAIR Analog
The ribose-5’- phosphate groups of AIR, CAIR, and N5-CAIR were replaced by methyl groups in all computational studies. Ground state geometry optimizations were then carried out using Gaussian 03 (29) with the B3LYP hybrid functional (30, 31) on the N1-methyl derivatives and their tautomers. For C, H, O, and N the 6-311+G(d) basis set was used. Due to the use of diffuse functions of this basis set, a tight self-consistent field was employed to provide the most accurate energy values. All calculated structures were verified to be local minima (all positive Eigenvalues) for ground state structures by frequency calculations. The energies obtained by DFT for each N1-methyl derivative and its tautomer were then compared.
DFT Calculations on C-CO2 and N-CO2 Bond Lengths in Protonated CAIR and N5-CAIR Analogs
The effect of protonation at N3, C4, or N5 on bond length of C4 or N5 to CO2 of each N1-methyl derivative and its tautomer was studied using Gaussian 03 and the ground state optimized structures determined above. A proton was added to each position in silico and the resulting structures were then re-optimized using the same procedures as described above.
Crystallization of wt PurE with NO2-AIR
PurE was exchanged into 100 mM Tris pH 8.0 buffer using a Micro Bio-Spin chromatography column from Biorad. The protein was diluted to a concentration of 20 mg/mL (1.1 mM) and mixed with 10 mM NO2-AIR. The PurE: NO2-AIR complex was then crystallized using 24 % PEG400, 0.2 M Mg(NO3)2, and 100 mM Tris buffer, pH 8.0 as the reservoir solution as previously described (3). Crystals were flash-frozen in liquid N2 before data collection using 20% ethylene glycol in the mother liquor as a cryoprotectant.
Crystallization of H45N and H45Q PurE followed by soaking with NO2-AIR and CAIR
H45N and H45Q PurEs were exchanged into 100 mM Tris pH 8.0 buffer using a Micro Bio-Spin chromatography column. Diffraction quality H45N and H45Q PurE crystals were obtained using conditions identical to those previously described for the wt PurE (3). The mutant crystals were soaked in a mother liquor solution containing either 10 mM CAIR or 10 mM NO2-AIR for 1 h prior to freezing in liquid N2 with 20% ethylene glycol in the mother liquor as a cryoprotectant.
Data collection and processing
Data for the EcPurE-NO2-AIR complex were collected at the beamline X12C of the National Synchrotron Light Source with a BRANDEIS CCD detector, and a 1° oscillation step, 60 s exposure time and 170 mm crystal to detector distance. Data for the EcPurE-H45N-CAIR complex were collected with a rotating anode X-ray generator using a R-AXIS IV detector with 0.5° oscillation step, 15 min exposure time and 300 mm crystal to detector distance. Data for both EcPurE-H45Q-CAIR and EcPurE-H45Q-NO2-AIR complexes were collected at the beamline 24ID of the Advanced Photon Source. For both data sets, a Quantum 315 detector was used to record diffraction images with a 1° oscillation step and a 1 s exposure time.
The HKL2000 program suite was used to integrate and scale all data (32). All four data sets were processed in the tetragonal body-centered space group, I422, with one molecule per asymmetric unit. The cell dimensions were a = 112.5 Å, c = 49.8 Å for the EcPurE-NO2-AIR complex with solvent content of 44%; a = 111.3 Å, c = 49.2 Å for the EcPurE-H45N-CAIR data, a = 111.3 Å and c = 49.4 Å for the EcPurE-H45Q-CAIR data set, and a = 111.7 Å, c = 49.4 Å for the EcPurE-H45Q-NO2-AIR data with 43% solvent content for all the mutants. The data processing statistics are shown in Table 1.
Table 1.
Summary of data collection and processing statistics.
| EcPurE-
NO2-AIR |
EcPurE-
H45N-CAIR |
EcPurE-
H45Q-CAIR |
EcPurE-
H45Q- NO2-AIR |
|
|---|---|---|---|---|
| PDB ID | 2ATE | 2NSL | 2NSJ | 2NSH |
| resolution (Å) | 30-1.8 | 50-2.0 | 50-2.3 | 50-1.8 |
| wavelength (Å) | 0.981 | 1.542 | 0.979 | 0.979 |
| space group | I422 | I422 | I422 | I422 |
| # of reflections | 34010 | 81423 | 53069 | 96435 |
| # of unique refl | 13604 | 10623 | 8526 | 14561 |
| Redundancy | 2.5 | 7.7(7.5) | 6.2(6.3) | 6.6(5.4) |
| completeness(%) | 89.9(69.1) | 98.7(96.4) | 95.3(99.7) | 99.2(100) |
| Rsym (%) | 7.2(24.9) | 10(55.5) | 13(26.6) | 9.0(44.9) |
| I/σ | 16.0 | 21.8(4.5) | 12.5(4.5) | 17.9(3.7) |
Values for the highest resolution shell are given in parentheses.
Rsym=ΣΣi|Ii - <I> | /Σ<I>, where <I> is the mean intensity of the N reflections with intensities Ii and common indices h,k,l.
Structure solution and refinement
All structures were determined by molecular replacement using the published EcPurE structure as a search model (PDB ID 1QCZ) (3). Rigid body refinement from the CNS program suite was used as the starting point (33). Subsequent refinement included several rounds of simulated annealing, B factor refinement, minimization in CNS, and Refmac5 refinement in CCP4i, followed by manual model refinement in COOT (34, 35). The ligands were modeled in based on clear Fo-Fc density. Water molecules were included in the later rounds of refinement. Quality of the final models was assessed using PROCHECK (36). The refinement statistics are summarized in Table 2.
Preparation of Figures
Figures 6-7 and Supplemental Figures 6-10 were prepared using Pymol (37).
Figure 6.

(A) Stereoview of the wt EcPurE active site bound to NO2-AIR with Fo-Fc density (2σ) shown. (B) Stereoview of H45Q EcPurE active site bound to CAIR with Fo-Fc density (2σ) shown.
Figure 7.
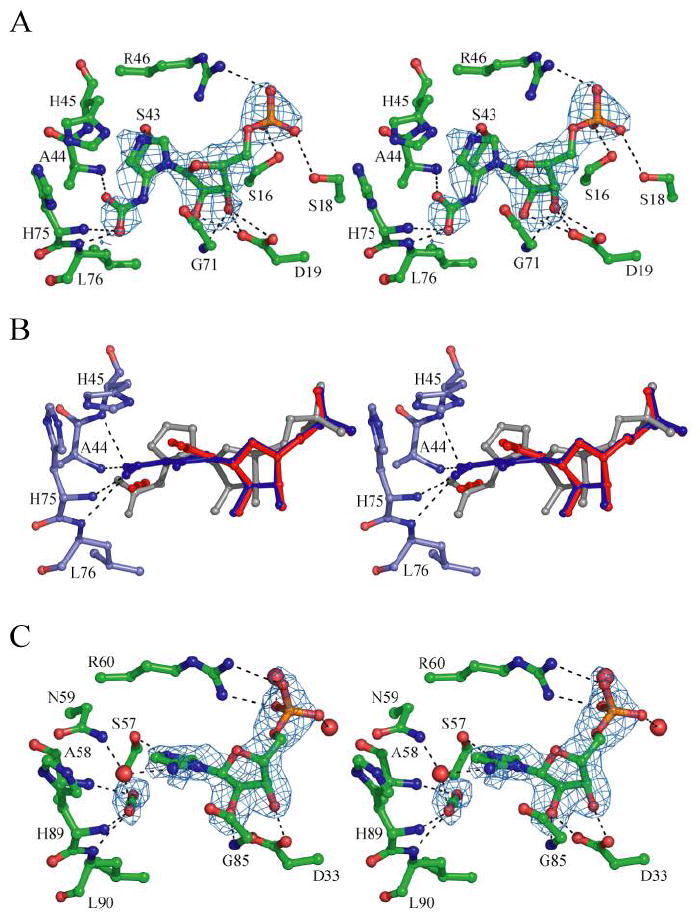
(A) Modeling of N5-CAIR into the EcPurE-AIRx structure (1D7A) (3) after use of four-fold symmetry averaging to improve the signal. The initial Fo-Fc density (3.5σ) is shown. (B) Superposition of wt PurE with NO2-AIR (blue), AIR/CO2 from the re-interpreted AaPurE-isoCAIR structure described in panel C (red), and N5-CAIR modeled from the PurE-AIRx structure described in panel A (grey). The positions of the carboxylate groups stay the same in all structures and are located in an amide backbone pocket. (C) Re-interpretation of the AaPurE-isoCAIR complex structure (2FWP) (12). Initial Fo-Fc density (3σ) after exclusion of isoCAIR is shown with the final refined model of AIR and CO2 in place of isoCAIR.
RESULTS
His45 is an Essential Residue in PurE
Crystal structures of the octameric E. coli and A. aceti PurEs soaked or co-crystallized with CAIR suggest that His45 (or its equivalent) is the only likely residue in the active site to function as a general acid/base catalyst (3, 12). This residue is absolutely conserved in both class I and II PurEs. We have made mutants of His45 in E. coli PurE. To avoid contamination of these mutants with endogenous wt PurE present in μM levels inside the cell (38), our mutants were expressed from a plasmid in the PCO135 E. coli strain, a purine auxotroph with a purE genotype (39). Recent sequence analysis of the purE gene in this strain revealed that the genotype contains a stop codon at Trp151, potentially resulting in formation of a truncated and inactive PurE (12). By expressing PurE mutants in this E. coli strain, background levels of PurE activity were reduced such that His45 mutants with activity 1/104 that of the wild type PurE could be studied. PurE mutants in this strain were readily expressed and purified to homogeneity by modifications of our previously published procedure (15% SDS-PAGE, Supplemental Materials, Figure 1) and their identity confirmed by ESI-MS (Supplemental Materials, Table 3).
The assays for PurE in the forward and reverse directions and the complications, in large part associated with the instability of N5-CAIR and CAIR, have been described previously in detail (8, 25). The results of the assays in each direction for wt and mutant PurEs are summarized in Table 3. They suggest that His45 plays an essential role in catalysis with the H45N mutants possessing activity < ~104 wt-PurE activity in both the forward and reverse directions. H45Q-PurE, on the other hand, had very low, but detectable, activity. These results are comparable to the recent studies reported with AaPurE. In those studies, activity was measured in the CAIR to N5-CAIR direction for the H45 equivalent (H59N) and was < 0.00008 s-1, while the activity for the H45Q mutant was 1/104 the wt activity (12).
Table 3.
Kinetic Parameters for WT and Mutant PurEs.
| kcat(s-1)
N5-CAIR→CAIR |
kcat(s-1)
CAIR→N5-CAIR |
Km CAIR
(μM) |
Kd CAIR
(μM) |
Kd NO2-
AIR (nM) |
|
|---|---|---|---|---|---|
| WT | 15.5 ± 0.9 | 15.6 ± 0.2 | 22.2 ± 4.6 | ND | 86 ± 31 |
| H45N | < 0.0003 | < 0.00008 | NDa | 20.9 ± 1.9 | ND |
| H45Q | 0.0021 ± 0.0003 | 0.0012 ± 0.0004 | ND | 16.3 ± 2.6 | ND |
| H45W | < 0.0003 | 0.0004 ± 0.0002 | ND | ND | ND |
ND = not determined
In vivo Complementation
The PCO135 E. coli strain was used to investigate whether these His45 mutant PurEs could complement purine auxotrophy. Cells co-transformed with a pNC2-based plasmid containing a mutant purEgene as well as purK (which encodes the amidoligase required to generate N5-CAIR) and pGP1-2 were selected for growth on plates containing minimal media with and without the addition of adenine and guanine. To our surprise, all of the cells containing PurE mutants were able to grow in the absence of added purines at rates similar to cells with wt purE added in the same vector. A control in which PCO135 cells were transformed with a plasmid containing purK but not purE, failed to grow (data not shown), indicating that a purEgene is essential for g rowth. These results are similar to those recently reported with the AaPurE. (12). Interestingly, in the case of AaPurE, complementation was only observed in the presence of AaPurK, indicating that the AaPurE cannot accept substrate from the endogenous EcPurK, implying an interaction between the proteins in vivo (12).
A possible explanation for mutant activity in vivo is that mixed PurE octamers are formed from the truncated purE gene product and the overexpressed PurE mutants. We propose that this heterooligomer possesses sufficient CAIR activity to support bacterial growth.
Analysis of CAIR Binding to Inactive PurE Mutants by Fluorescence Titration
EcPurE contains two tryptophans, and the complexed structures with NO2-AIR and CAIR described below suggest that Trp20, adjacent to Asp19 which serves as a ligand for both the 2’ and 3’ hydroxyls of the nucleotide, might serve as a probe for substrate binding. Fluorescence studies of the inactive PurE mutants showed that they possessed an intrinsic fluorescence emission at 335 nm. We examined the possibility that titration of the PurE mutants with CAIR might alter the fluorescence emission and allow measurement of the Kd of CAIR. No fluorescence change was observed when CAIR was added to H45W PurE. However with both H45N and H45Q PurEs, large decreases in fluorescence were observed upon CAIR addition and an additional blue-shift in fluorescence from 345→335 nm was observed with H45N (Supplemental Materials, Figure 2). Data were fit to Equation 3 to give a Kd of 16.3 ± 2.6 μM and 20.9 ± 1.9 μM, for H45Q and H45N, respectively (Figure 2 and Table 3). Similar experiments with AaPurE have been carried out (12) and several distinctions between the Ec and AaPurEs are apparent. The EcPurE data were best fit using a model for full occupancy of the PurE active sites, in agreement with the full occupancy observed in the H45N-CAIR and H45Q-CAIR structures described below. The AaPurE data gave results that were best fit to a model with partial occupancy of the active sites, consistent with the partial occupancy observed in several AaPurE crystal structures (12). The Kds for CAIR binding to the H59N and H59Q AaPurEs are twenty fold lower those measured with EcPurE and small fluorescence increases rather than large fluorescence decreases with EcPurE were observed upon CAIR titration (12). The basis for the differences in binding and subunit occupancy between EcPurE and AaPurE are not clear.
Figure 2.
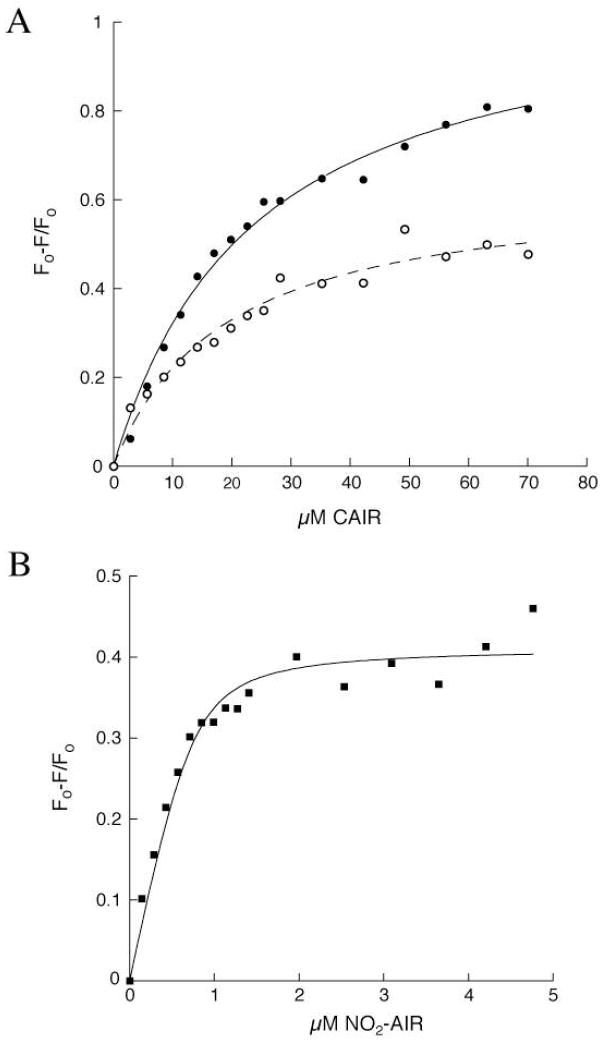
Fluorescence titrations of PurE with NO2-AIR and CAIR (A) Observed changes in fluorescence at 335 nm for H45N (●) and for H45Q (○) PurE upon titration with CAIR (Kd = 20.9 ± 1.9 μM, R2 = 0.99 and Kd = 16.3 ± 2.6 μM, R2 = 0.98 for H45N and H45Q, respectively). (B) Observed changes in fluorescence at 335 nm upon titration of wt PurE with NO2-AIR (Kd =86 ± 31 nM, R2 = 0.96).
Previous studies by Davisson have shown that NO2-AIR is a competitive inhibitor of EcPurE with a Kis of 0.5 μM (4, 21). NO2-AIR is proposed to be a CAIR analog, and we have used this analog with wt-PurE in crystallization studies described below. In addition, Davisson’s laboratory has shown that this analog binds 103 more tightly to a class II G. gallus PurE than the class I E. coli PurE (4, 21) suggesting that its mode of binding might be mechanistically informative. Titration of wt-PurE with NO2-AIR also shows a decrease in intrinsic tryptophan fluorescence (λmax = 335 nm) giving rise to a Kd of 86 ± 31 nM (Figure 2B and Supplemental Materials, Figure 2). No fluorescence changes were observed upon titration of wt PurE with either AIR or ribose-5’-phosphate, indicating that that the structure of the base is important for binding events and conformational changes that give rise to a fluorescent signal (data not shown).
SV-AUC Experiments to Define the Quaternary Structure of the His45 Mutant PurEs
The active site of PurE lies at the interface of three monomers (3). It was therefore important to determine that the His45 mutations do not affect the protein’s quaternary structure. SV-AUC was used to determine the molecular weight distribution of oligomers. As shown in Supplemental Materials Figure 3 and Table 4, the predominant species in solution for all the PurEs is the octamer, indicating that loss of activity with these mutants is not due to disruption of the quaternary structure.
pH Rate Profile
In an effort to obtain further insight into the role of His45 in catalysis, a pH-rate profile for CAIR decarboxylation catalyzed by wt EcPurE was determined. As noted above, the instabilities of N5-CAIR and CAIR as well as the changes in their extinction coefficients as a function of pH, have made this analysis challenging, especially at low pH. Assays at pH < 5.1 are not possible due to the rate of non-enzymatic CAIR decarboxylation relative to the observed enzyme activity. Both the kcat and kcat/Km profiles appear to be bell-shaped and are shown in Figure 3. The data for kcat vs. pH profile were fit to Equation 2 and gave pK1 = 5.9 ± 0.4 and pK2 8.6 ± 0.4. These values are very similar to those recently obtained for the AaPurE, pK1 = 5.1 ± 0.2 and pK2 = 8.4 ± 0.1 (12). The data for the kcat/Km versus pH-rate profile were also fit to Equation 2 and gave pK1 = 6.7 ± 1.6 and pK2 = 7.5 ± 1.5. Unlike the AaPurE enzyme, the best fit for EcPurE is associated with a single ionization event on the acidic side of the profile. The values are, however, similar to those observed in AaPurE, in which data were fit to a model containing two indistinguishable ionization events for pK1 (pK1 = 6.0 ± 0.1, pK2 = 7.2 ± 0.1) (12).
Figure 3.
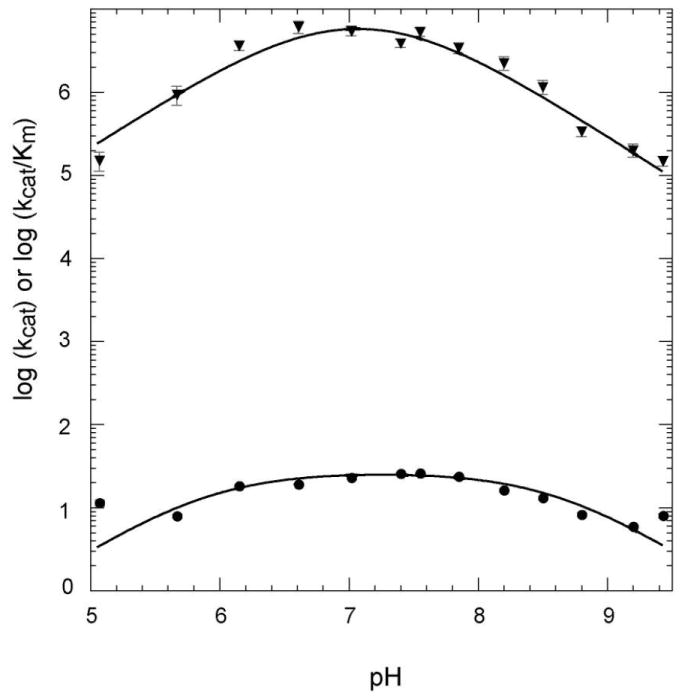
pH-rate profiles for wt EcPurE monitoring the decarboxylation of CAIR. The solid line represents the best fit to Equation 2. The error bars for each point are shown. The kcat vs. pH profile (●) was fit to pK1 = 5.9 ± 0.4 and pK2 = 8.6 ± 0.4 (R2 = 0.94). The kcat/Km vs. pH profile (▼) was fit to pK1 = 6.7 ± 1.6 and pK2 = 7.5 ± 1.5 (R2 = 0.95).
The pKa on the acidic side of the kcat/Km profile could be associated with protonation of N3 of CAIR as a pKa of 6.3 has been previously reported (5). The kcat profile on the acidic limb is difficult to rationalize. The instability of the nucleotides and inability to measure extinction coefficients for N5-CAIR at pH 6 and lower, suggest that any analysis on this limb should be viewed with caution. The basic side of the pH profiles is proposed to be associated with protonated His45, which we postulate is directly or indirectly (through a water) required to form an isoCAIR intermediate. Analysis of the pH rate profile of CAIR with AaPurE has come to a similar conclusion, that is the basic limb of the profile was proposed to be associated with His59 (12). With the AaPurE studies, additional support for this assignment was made based on the pH profile of an active PurE mutant, H59D PurE (12).
Efforts to Understand how Protonation Affects Decarboxylation using DFT calculations on N1-methyl-aminoimidazole Derivatives
To provide insight into the role protonation of CAIR and N5-CAIR could play in decarboxylation, DFT calculations have been carried out in which the ribose-5’- phosphate in each molecule has been replaced by a methyl group. Reports of a possible isoCAIR intermediate in the H59N AaPurE structure (12) suggested that C4 protonation can be catalyzed by the enzyme. Initially, tautomeric forms of the N1-methyl derivatives were examined. In the case of Me-CAIR (1), the tautomer (t-1) is only 3.1 kcal/mol higher in energy than the aromatic starting state. Tautomerization of Me-AIR (2) has a similar energetic barrier (Figure 4). In contrast, the tautomer of Me-N5-CAIR (3), t-3, is unfavorable by 10.0 kcal/mol. Given that t-1 is the surrogate of isoCAIR, the small energy difference between the tautomers of CAIR suggest that protonation at C4 is energetically reasonable prior to loss of CO2. NMR studies on AIR riboside (AIRs) support the ease of tautomerization. Solvent exchange of the C4 proton of AIRs for deuterium occurs with a kexchange of 0.9 min-1, presumably through the imino tautomer (40). Although efforts to detect this tautomer using 13C NMR (expected chemical shift of 110 (for aromatic carbon) vs 40-50 ppm (for tetrahedral carbon)) were unsuccessful (40), observation of moderately rapid exchange supports the ease of protonation at C4 of aminoimidazoles.
Figure 4.
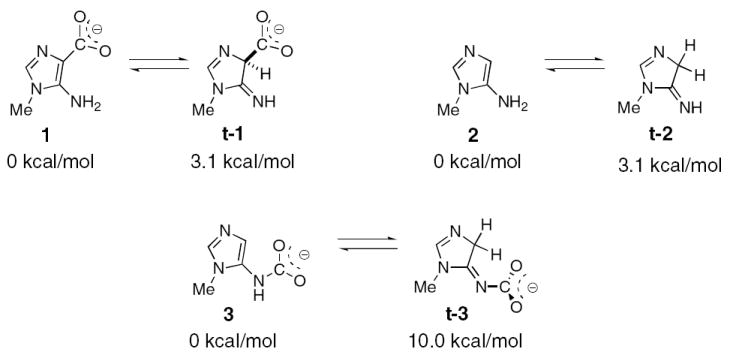
Structures of the AIR, N5-CAIR, and CAIR derivatives used in the computational studies. The calculated relative energies of the derivatives and the non-aromatic tautomers are shown.
Our mutagenesis studies on His45 and pH rate profile of the wt PurE suggest the importance of a protonated histidine in the decarboxylation of CAIR. The effect of proton transfer to either N3, C4, or N5 on loss of CO2 was studied using ground state optimized structures for 1, t-1, 3, and t-3 and by re-optimization of the structures after proton addition to these positions. For the CAIR analog 1, protonation at either N3 or N5 does not change the length of the C4-C (carboxy) bond (1.52 Å) (Figure 5A). However, in the latter case a new hydrogen bond is introduced between the carboxylate and the protonated amine. Loss of CO2 occurred only after protonation of C4. For the isoCAIR analog t-1, loss of CO2 only occurred after protonation at N5. N3 protonation resulted in only a slight lengthening of the C4-C (carboxy bond) from 1.61 to 1.66 Å (Supplemental Materials, Figure 4), demonstrating that imidazole N3 protonation does not contribute significantly to catalysis and thus is a strategy unlikely to be used by PurE .
Figure 5.
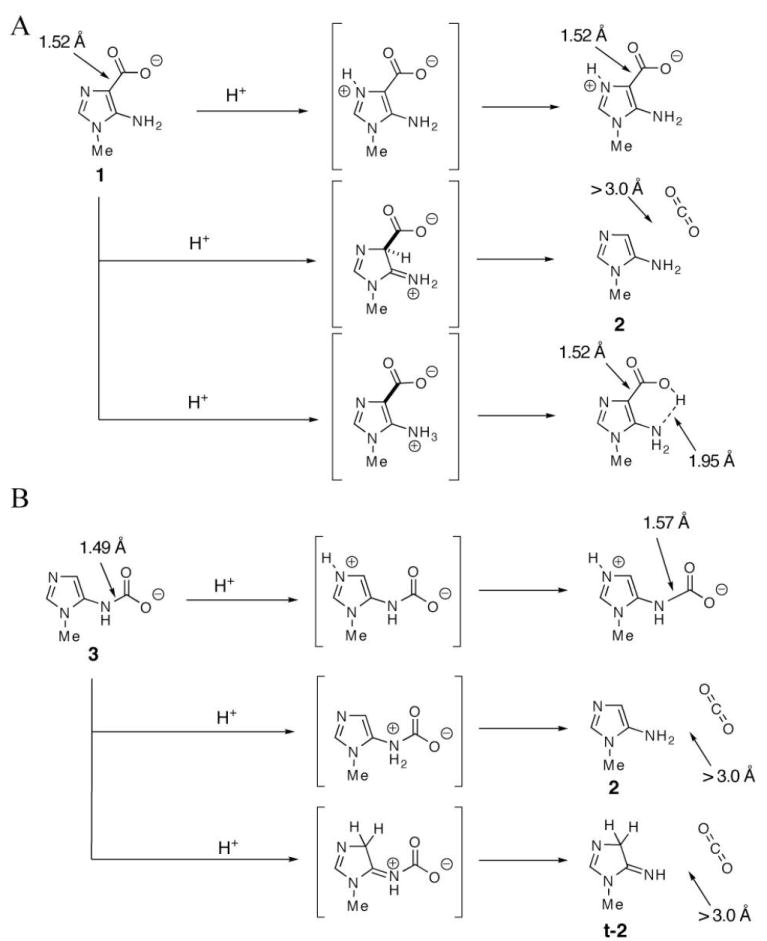
Computational studies on the effects of protonation of compounds 1 (A) and 3 (B) on loss of CO2 and bond lengths. Protonation of 1 at C4 or 3 at N5 results in bond cleavage and CO2 dissociation.
For the N5-CAIR analog, 3, protonation at either C4 or N5 leads to bond cleavage and loss of CO2. Protonation of N3 leads only to a slight lengthening of the N5-C (carbamate) bond from 1.49 to 1.57 Å (Figure 5A) . Similar results were obtained by protonation of either N3 or N5 in species t-3 (Supplemental Materials, Figure 4).
While these computational studies have been carried out on a much simpler system than the PurE- nucleotide complex, they provide a starting point for mechanistic thinking. These calculations suggest that protonation at either N5 of 3 or C4 of 1 leads to loss of CO2. Extending this observation to PurE indicates that the enzyme may be able to catalyze decarboxylation of either CAIR or N5-CAIR by use of general acid catalysis. In the former case, protonation at C4 of CAIR leads to an isoCAIR+ intermediate or transition state that could decompose directly to AIR and CO2. In the latter case, protonation of the N5 of N5-CAIR, perhaps concomitant with loss of CO2, would lead to AIR. Additional computational studies are needed on these nucleotides in the presence of the complete, solvated PurE active site.
Crystallization and Structure Determination
In order obtain insight into the location of the C4 position of CAIR relative to His45 and the location of the carboxylate relative to the hydrophobic pocket (Ala44/Ala70/Leu76), wt EcPurE was crystallized with a stable CAIR analog, NO2-AIR. In addition, H45Q mutant crystals were soaked with CAIR or NO2-AIR, and the H45N mutant crystals were soaked with CAIR. These experiments resulted in structures of four different complexes with resolutions ranging between 1.8-2.3 Å (Table 1). All of the structures were determined by molecular replacement using the previously published EcPurE structure (1QCZ) as the search model. Of the 169 residues contained in an EcPurE monomer, all of the structures are missing the first seven residues due to lack of clear electron density. The rest of the residues are present and located in the most favored and additionally allowed regions of the Ramachandran plot. The quality of the final models was verified using PROCHECK. The final results including the R factor values are summarized in Table 2.
NO2-AIR Binding
NO2-AIR was co-crystallized with PurE and found to occupy all eight active sites of the octamer. These results contrast with our previous co-crystallization results with CAIR and wt EcPurE and the recent soaking experiments of AaPurE crystals with CAIR (12). The electron density for NO2-AIR is shown in Figure 6A. It binds close to the surface of PurE, and a large portion of the nucleotide remains solvent accessible (Supplemental Materials, Figure 5). The ribose-5’- phosphate moiety of the nucleotide has extensive interactions with PurE which are similar to those previously reported with the partially disordered EcPurE-AIRx co-crystal structure. The phosphate hydrogen bonds with Ser16 and Ser18 of the P loop, Arg46 of the 40s loop, and several water molecules. The 2’ and 3’ ribose hydroxyl groups are hydrogen bonded to the carboxylate of Asp19 and the backbone amide hydrogen of Gly71.
The N3 of the NO2-imidazole of the nucleotide is hydrogen bonded to Ser43, indicating that it is not protonated. This is expected given its pKa of 0.3 (41). The NO2 group hydrogen bonds to the amides of Ala44 (2.7 Å), His45 (3.3 Å), His75 (3.1 Å), and Leu76 (3.3 Å) with reasonable geometry. An interaction is also observed between the exocyclic amine and the backbone amide carbonyl of Gly71 (3.1 Å). Ser43 and Ala44 are strictly conserved among all class I and class II PurE’s, while Gly71 and His75 are present in only class I enzymes. The hydrophobic pocket, largely consisting of conserved residues Ala 44, Ala70, and Leu76, is unoccupied.
His45 is located parallel to the nitro-aminoimidazole ring, and appears to be stacked above C4 and the NO2 group. Unfortunately the lack of observable hydrogen bonds to His45 indicates that its protonation state cannot be determined from this structure. Nevertheless, the close proximity of the Nδ atom of His 45 to the C4 position of NAIR (3.6 Å) supports a model in which His45 could function as the general acid catalyst for CAIR decarboxylation. A minimal rotation of the imidazole ring around its χ1 torsion angle in its protonated state is required to effectively deliver the proton to C4.
Comparison of the apo-wt PurE structure with the NO2-AIR wt-PurE structure reveals that NO2-AIR binding causes no conformational changes in the enzyme (Supplemental Materials, Figure 6). An overlay of both backbone and side chain atoms in the structures reveals an RMSD of 0.21 Å (as determined by Swiss PDB-Viewer). Most of the protein motions that are observed are limited to the P-loop and Arg46, which interact directly with the phosphate group of NO2-AIR. A slight rotation (~19°) of the His45 imidazole (or possibly imidazolium) ring towards a more parallel orientation with NO2-AIR is also observed. A structure obtained of H45Q PurE soaked with NO2-AIR is similar to that obtained for the wt enzyme (Supplemental Materials, Figure 7).
CAIR Binding
Previous attempts to co-crystallize CAIR with wt PurE resulted in partial occupancy of the active sites and ambiguous electron density that could be interpreted as either AIR, CAIR, N5-CAIR, or a mixture of species (hence AIRx) (3). Similarly, efforts to crystallize the acidophilic AaPurE with CAIR resulted in structures that contained either AIR or electron-density interpreted as isoCAIR (Figure 1) (12). H59Q(N)-AaPurE mutants are inactive, but efforts to co-crystallize and observe CAIR with each of these mutants were unsuccessful (12). Success has finally been achieved when H45N and H45Q EcPurE crystals at pH 8.0 were soaked with CAIR (10 mM) at the same pH. At this pH, the rate of CAIR decomposition is minimal.
The structures of H45N and H45Q PurEs are nearly identical and revealed intact CAIR bound for the first time (Figure 6B and Supplemental Figure 8). As in the case of NO2-AIR, Ser43 is H bonded to the N3 of CAIR indicating it is not protonated and the backbone carbonyl of Gly71 interacts with its exocyclic amine. Hydrogen bonding interactions are also observed between the carboxylate of CAIR and the backbone amide of Ala44 (2.7 Å), Gln/Asn45 (3.0 Å), His75 (2.6 Å), and Leu76 (3.0 Å). The two NO2-AIR complexes and the two CAIR complexes superimpose within experimental error with only small structural differences. The hydrogen bonding pattern is the same in all four complexes with each oxygen atom of the nitro/carboxylate group forming two hydrogen bonds with amide groups. Thus, despite the charge difference between the zwitterionic NO2-group of NO2-AIR and the anionic carboxylate of CAIR, interactions between the C4 substituent of the nucleotide and the protein appear to be similar.
In both cases, the Asn or Gln side chain at position 45 is located above the C4 position of CAIR. In the H45Q structure, Gln45 appears in a similar conformation as His45 in the NO2-AIR structure described above. No hydrogen bonding interactions are observed between Gln45 and either the protein or substrate. The tilt in the amide side chain relative to the amino imidazole ring is more pronounced in the H45N structure, in which the angle between Cγ, Nδ of Asn45 and C4 of CAIR is ~124°. As with the NO2-AIR structure, very few protein motions are observed upon nucleotide binding in either structure and these are mostly limited to P-loop residues. The rmsd values for all atoms in the H45N-CAIR and H45Q-CAIR structures compared to the wt apo enzyme are 0.23 Å and 0.28 Å, respectively.
Comparison of the NO2-AIR/CAIR Complexes to Apo and Nucleotide bound PurEs: Evidence for a Common Carboxylate (CO2) Binding Site and Aminoimidazole Movement
EcPurE-AIRx
In our published structure of wt EcPurE cocrystallized with CAIR, the carboxylate density at C4 was missing and unexplained electron density was observed near N5 (3). An AIR-like species (AIRx), most likely N5-CAIR, was observed. In addition, only four of the eight possible binding sites of the PurE octamer were occupied. At the time, we attributed this observation to PurE mediated conversion of CAIR to N5-CAIR. However, due to experimental uncertainty, only AIR was included in the final model. To gain insight into the mechanism of PurE, we have re-examined the EcPurE-AIRx structure.
We recalculated the electron density for EcPurE-AIRx using four-fold symmetry averaging to improve the signal. We then modeled N5-CAIR into the active site using the electron density and the geometric constraints based on the AIR molecule from the original EcPurE structure (1D7A). A PurE-N5-CAIR structure was generated (Figure 7A). Several interesting observations can be made from a comparison of this model with the CAIR(NO2-AIR)-PurE structures. First, the carboxylate of N5-CAIR superimposes well with the carboxylate (nitro) substituents of CAIR (NO2-AIR). Second, the remaining atoms of aminoimidazole of N5-CAIR are significantly shifted relative to the other nucleotide bound PurEs (Figure 7B and Supplemental Materials, Figure 9A). Both of these observations have important mechanistic implications.
The repositioning of the aminoimidazole of AIRx relative to CAIR takes place through a change in the sugar pucker, a rotation of the glycosidic torsion angle, and a shift in the ribose position. AIRx has a symmetric O4’-endo-C1’-exo twist sugar pucker favoring C1’-exo, a maximum torsion angle (νmax) of 37° and an average glycosidic torsion angle (C2-N1-C1’-O4’) of 1°. CAIR and NO2-AIR also have a O4’-endo-C1’-exo sugar pucker and a more pronounced νmax of 55° (averaged over the four CAIR/NO2-AIR complex structures). The glycosidic torsion angle for the CAIR/NO2-AIR molecules ranges between 51°-68°. In the CAIR/NO2-AIR complexes the ribose makes a more conventional pair of H-bonds with Asp19, Oδ1-O2’ and Oδ2-O3’, whereas in the AIRx complex, the O2’ hydroxyl group donates a bifurcated hydrogen bond to the carboxylate oxygen atoms of Asp19 (2.9 Å to Oδ1 and 2.7 Å to Oδ2) and the O3’-hydroxyl group donates an additional hydrogen bond (2.9 Å) to the Oδ2 of the carboxylate group.
AaPurE-AIR
A comparison of the EcPurE-CAIR (NO2-AIR) structures with the published AaPurE-AIR complexes (12) has also been made. A superposition of these structures reveals that the aminoimidazole ring and the ribose puckering mode of AIR is similar to that of CAIR (NO2-AIR) (12) (Supplemental Materials, Figure 10). The average νmax of 48° and the average glycosidic torsion angle of 64° are similar to those of CAIR/NO2-AIR.
AaPurE-isoCAIR
A comparison between the EcPurE-CAIR (NO2-AIR) complexes and the AaPurE-isoCAIR structure (12) also reveals that the aminoimidazole moiety and the sugar pucker conformations are nearly identical with a comparable νmax of 51° for the isoCAIR (Supplemental Materials, Figure 9B). The hydroxyl groups of the ribose moiety in the AaPurE-AIR and the AaPurE-isoCAIR structures make a pair of hydrogen bonds with Asp33 (19 in EcPurE).
The C4-carboxylate groups of CAIR and isoCAIR and the NO2 group of NO2-AIR are found in approximately the same position in all of the structures and in all cases make hydrogen bonds only with the backbone amides (Figure 7B and Supplemental Materials, Figures 9 and 10). The carboxylate binding site thus appears to be conserved. Finally, these different PurE structures show virtually no differences in the conformations of the active site residues. The major difference in the structures is the position of the base of N5-CAIR relative to that of CAIR, NO2-AIR, AIR, and isoCAIR.
ApoPurEs
Further evidence in support of a common carboxylate binding site has been obtained by examining apo-PurE structures from four different organisms (3, 42-44). Twenty-five monomers with no ligands bound were selected and examined for conserved waters (Supplemental Materials, Table 5). The two most highly conserved water molecules occupy positions corresponding to the carboxylate oxygen atoms of isoCAIR in the AaPurE-isoCAIR complex. However, based on EcPurE-NO2-AIR and the mutant EcPurE-CAIR structures, there is no obvious space for a water molecule once nucleotide is bound and this space is occupied by the CO2 (NO2) group. These observations strongly suggest that these conserved water molecules occupy the CO2 binding site.
Reanalysis of AaPurE-isoCAIR
In the original report of isoCAIR complexed with AaPurE, the observed electron density was not well fit by the isoCAIR model (12). Thus we reanalyzed this the X-ray structure to determine if it might be a mixture of species: isoCAIR, AIR, and CO2 intermediates proposed in the reaction mechanism (Figure 1). During our re-analysis at higher contour levels (3σ) in difference Fourier maps, the connection between the C4 of the aminoimidazole and the carboxylate disappears and the carboxylate density appears to be linear (Figure 7C). Based on this re-analysis we believe that the electron density is indicative of one molecule of AIR and one molecule of CO2, although a small amount of isoCAIR may be present in equilibrium with these molecules. Refinement of this model generated a structure with reasonable geometry and good refinement statistics. As seen with the isoCAIR model (12), a slightly irregular shape for the imidazole density is still present when modeled with AIR/CO2. As revealed in Figure 7B, the linear CO2 superimposes on the carboxylate binding site and the aminoimidazole moiety superimposes on this moiety observed in all of the other nucleotide bound PurE structures with the exception of EcPurE-AIRx.
DISCUSSION
The mutase reaction catalyzed by the class I PurE is deceptively simple, but unprecedented enzymatically (Figure 1). The instability of N5-CAIR suggests that this nucleotide functions as a CO2 carrier similar to carboxy-biotin, and that the major function of PurE is to sequester the CO2 generated from N5-CAIR decarboxylation for CAIR formation. Bacteria and fungi may have evolved this strategy to avoid the difficult task of directly binding CO2 at the expense of utilizing an equivalent of ATP and an amidoligase (PurK) to form a labile carbamate from AIR and HCO3-. The instabilities of N5-CAIR and CAIR have made this reaction difficult to investigate mechanistically. The structures of PurEs have played an important role in our working model (Figure 1).
Nineteen crystal structures are available including unliganded structures of PurE’s from E. coli (3), A. aceti (42), Thermotoga maritima (43), and Bacillus anthracis (44) and nucleotide bound structures (CAIR, NO2-AIR, AIR, AIRx, and isoCAIR) of EcPurE, AaPurE, and mutants (3, 12). These structures together suggest several features that might contribute to catalysis including the location of a conserved histidine to act as a general acid/base, conformational changes leading to repositioning of the nucleotide during catalysis, and a buried carboxylate/CO2 binding site formed primarily by amide groups and shielded from the solvent by the nucleotide itself. This sequestered site suggests how the CO2 from N5-CAIR is transferred, without dissociation from the active site, to the C4 position in CAIR (6). The structures also suggest that isoCAIR, AIR, and CO2 can be accommodated as intermediates in the PurE catalyzed reaction.
Our working model, based on the structures reported herein and reanalysis of previously published data, is shown in Figure 1. Formation of the enzyme/N5-CAIR complex results primarily from hydrogen bonding interactions between Asp19 and the ribose hydroxyl groups, Arg46 and the 5’-phosphate, and the amide groups of Ala44, His75, and Leu76 and the carboxylate group. The Leu76 amide group occurs at the N-terminus of an α-helix which provides additional interaction with the carboxylate via the helix dipole. His45 is then positioned to protonate N5, leading to the formation of AIR and CO2. The conversion to CAIR is facilitated by a shift in the nucleotide position, a change in the maximum torsion angle (νmax), and rotation about the glycosidic bond. During this shift, the hydrogen bonds to Asp19 and Arg46 are retained; however, the imidazole is repositioned so that its C4 atom is nearest the CO2 carbon atom. The CO2 molecule may also rotate in the amide backbone pocket towards the His75 and His45 amides, facilitating C-C bond formation with AIR. The resulting tetrahedral intermediate (isoCAIR) is now positioned so that His45 can serve as the general base to form CAIR. In the product complex, hydrogen bonds to Asp19 and Arg46 are retained and the carboxylate group of CAIR now hydrogen bonds to the amide groups of Ala44, His45, His75, and Leu76.
The role of His45 in EcPurE as a general acid/base catalyst is supported by the site-directed mutagenesis studies, the pH rate profile analysis, and the DFT calculations. Similar studies carried out on AaPurE have come to similar conclusions (12).
The structures and biochemical studies suggest that the movement of the aminoimidazole ring is key to catalysis (Figure 7B) and may be linked to the carboxylate moiety. The importance of the carboxylate is underscored by our fluorescence titration data with EcPurE and CAIR, NO2-AIR, AIR, and ribose-5’-phosphate. Only CAIR and NO2-AIR binding cause fluorescence changes, the carboxylate (nitro group) presumably triggering conformational change(s). To assess the nature of this (these) conformational change(s) during catalysis the active site residues of all available PurE structures were compared (Supplemental Materials, Table 5). As noted above, they show minimal differences (Supplemental Materials, Figure 6) with the exception of His45 and Arg46. Arg46, which hydrogen bonds to the 5’-phosphate of the nucleotide, moves away from the active site when nucleotide is absent and towards the active site when nucleotide is bound. Arg46 is adjacent to the essential His45 that shows predominantly a single conformation. However, in two structures a rotation of the χ1 torsion angle of His45 is detected. The χ1 angle of His45 in the wtEcPurE-NO2-AIR complex is -62°, while it is -45° in one monomer of H89F AaPurE (2FWB) and +22° in the empty monomer of wt AaPurE-AIR (2FWJ). These rotations are in a direction that brings the His45 sidechain significantly closer to the imidazole of the nucleotide, where it could protonate either N5 or C4 of N5-CAIR or CAIR, respectively.
Finally, the structures have also provided insight into the proposed intermediacy of AIR/CO2 and isoCAIR. When H59N-AcPurE crystals were soaked with CAIR at pH 5.4, density associated with isoCAIR was reported (12). Under these conditions CAIR is protonated at N3 (pKa of 6.3), and it is possible that this form of the substrate binds to the enzyme. Thus despite the fact that the native proton source, His59, has been removed in the H59N mutant, a proton could be introduced into the active site via N3, which could then be used to protonate C4 to generate isoCAIR. Our DFT calculations suggest that despite the non-aromatic structure of isoCAIR, a single hydrogen bond (3 kcal/mol) might be sufficient to stabilize the structure. The rapid, non-enzymatic exchange of a proton from C4 in AIR also suggests that this intermediate is moderately stable (40).
Our re-analysis of the H59N-AcPurE-isoCAIR structure (Figure 7C) suggests that the observed electron density might be associated predominantly with AIR/CO2 in equilibrium with a smaller amount of isoCAIR. Recently a structure of a mutant POX was solved with 2-lactyl thiamin diphosphate (LThDP) in its active site (15). This enzyme utilizes ThDP to catalyze decarboxylation of pyruvate, a reaction very similar to the decarboxylation of CAIR. In the structure, clear electron density was observed between lactyl group appended to thiamin and the carboxylate leaving group. The environment of the carboxylate is very similar to that observed in PurE. The carboxylate group is located in a hydrophobic pocket and forms hydrogen bonds with a glutamate and the amide of a glycine. Using a difference Fourier map (Fo-Fc), negative electron density was observed near the carboxylate group, which was interpreted to indicate that a fraction of the carboxylate had undergone C-C bond cleavage and generated linear CO2. Given the similarities in chemistry and the mechanism of catalysis between this system and PurE, the postulated crystallographic model for a mixture of AaPurE-AIR/CO2 and isoCAIR is reasonable. It should be noted, however, that while the CO2 in PurE is protected from solvent by the nucleotide, in POX, the CO2 is solvent accessible allowing it to escape and making the reaction irreversible.
The data obtained thus far can be accommodated within the mechanism proposed in Figure 1. Given the difficulties of carrying out more conventional mechanistic studies on the PurE reaction, further mechanistic insight will likely be obtained only by creative crystallography and computational methods.
Supplementary Material
A table of primers used in EcPurE mutagenesis, a Coomassie-stained gel of PurE proteins expressed in PCO135 E. coli, fluorescence titration data, a table of Δ250 values used in calculating the pH-rate profile, ESI-MS results for the EcPurE mutants, distribution of species obtained after SEDFIT analysis of SV-AUC data, a table summarizing SV-AUC results, a figure showing a surface representation of EcPurE-NO2-AIR, a figure showing conformational changes observed upon NO2-AIR binding, figures showing the EcPurE-H45Q-NO2-AIR and H45N-CAIR complexes, a figure comparing the EcPurE-NO2-AIR and AaPurE-AIR structures, and a table listing all available PurE structures. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems at MIT (NSF-0070319 and NIH GM68762) is gratefully acknowledged. We thank Tadhg Begley for helpful discussions and the staffs of MacCHESS supported by NIH grant RR-01646 and NE-CAT supported by NIH grant RR-15301 for assistance with data collection.
This work was supported by the NIH grants RR15301 and GM073220 (SEE) and NIH Grant GM32191 (AAH and JS). SEE is indebted to the W. M. Keck Foundation and the Lucille P. Markey Charitable Trust. AAH was supported by a NSF predoctoral fellowship. TEB was supported, in part, by an ACS Organic Division Award (sponsored by Novartis Pharmaceuticals).
ABBREVIATIONS
- PurE
N5-carboxyaminoimidazole ribonucleotide mutase or carboxyaminoimidazole ribonucleotide synthase
- AaPurE
Acetobacter aceti PurE
- EcPurE
Escherichia coli PurE
- PurK
N5-carboxyaminoimidazole ribonucleotide synthetase
- EcPurC
Escherichia coli succinoaminoimidazole carboxamide ribonucleotide synthetase
- POX
pyruvate oxidase
- PK
pyruvate kinase
- AIR
aminoimidazole ribonucleotide
- N5-CAIR
N5-carboxyaminoimidazole ribonucleotide
- CAIR
4-carboxyaminoimidazole ribonucleotide
- NO2-AIR
4-nitroaminoimidazole ribonucleotide
- SAICAR
succinoaminoimidazole carboxamide ribonucleotide
- ATP
adenosine 5’-triphosphate
- PEP
phosphoenol pyruvate
- ThDP
thiamin diphosphate
- LThDP
2-lactyl-ThDP
- Tris
tris(hydroxymethyl)aminomethane
- KPi
potassium phosphate
- MES
4-morpholineethanesulfonic acid
- ACES
N-(2-acetamido)-2-aminoethanesulfonic acid
- U
unit of enzyme activity in μmol product/min
- DFT
density functional theory
- asu
asymmetric unit
- RMSD
root mean square deviation
- wt
wild type
- EcPurE-AIRx
EcPurE crystal structure co-crystallized with CAIR showing partially disordered nucleotide
Footnotes
The coordinates of the EcPurE structures have been deposited in the Protein Data Bank under accession numbers 2ATE for PurE-NO2-AIR, 2NSH for H45Q PurE-NO2-AIR, 2NSJ for H45Q PurE-CAIR, and 2NSL for H45N PurE-CAIR.
References
- 1.Mueller EJ, Meyer E, Rudolph J, Davisson VJ, Stubbe J. N5-carboxyaminoimidazole ribonucleotide: evidence for a new intermediate and two new enzymatic activities in the de novo purine biosynthetic pathway of Escherichia coli. Biochemistry. 1994;33:2269–78. doi: 10.1021/bi00174a038. [DOI] [PubMed] [Google Scholar]
- 2.Firestine SM, Davisson VJ. Carboxylases in de novo purine biosynthesis. Characterization of the Gallus gallus bifunctional enzyme. Biochemistry. 1994;33:11917–26. doi: 10.1021/bi00205a030. [DOI] [PubMed] [Google Scholar]
- 3.Mathews II, Kappock TJ, Stubbe J, Ealick SE. Crystal structure of Escherichia coli PurE, an unusual mutase in the purine biosynthetic pathway. Structure Fold Des. 1999;7:1395–406. doi: 10.1016/s0969-2126(00)80029-5. [DOI] [PubMed] [Google Scholar]
- 4.Firestine SM, Poon SW, Mueller EJ, Stubbe J, Davisson VJ. Reactions catalyzed by 5-aminoimidazole ribonucleotide carboxylases from Escherichia coli and Gallus gallus: a case for divergent catalytic mechanisms. Biochemistry. 1994;33:11927–34. doi: 10.1021/bi00205a031. [DOI] [PubMed] [Google Scholar]
- 5.Litchfield GJ, Shaw G. Purines, Pyrimidines, and Imidazoles. Part XXXVIII. A Kinetic Study of the Decarboxylation of 5-Amino-1-β-D-ribofuranosylimidazole-4-carboxylic Acid 5’-Phosphate and Related Compounds. J Chem Soc (B) 1971:1474–1484. [Google Scholar]
- 6.Meyer E, Kappock TJ, Osuji C, Stubbe J. Evidence for the direct transfer of the carboxylate of N5-carboxyaminoimidazole ribonucleotide (N5-CAIR) to generate 4-carboxy-5-aminoimidazole ribonucleotide catalyzed by Escherichia coli PurE, an N5-CAIR mutase. Biochemistry. 1999;38:3012–8. doi: 10.1021/bi9827159. [DOI] [PubMed] [Google Scholar]
- 7.Alenin VV, Kostikova TR, Domkin VD. Chemical Synthesis of N1-Substituted 5-Aminoimidazoles and the Formation of N-Carboxylation Products in Aqueous-Solutions of Potassium Bicarbonate. Zh Obstic Khimii. 1987;57:692–701. [Google Scholar]
- 8.Meyer E, Leonard NJ, Bhat B, Stubbe J, Smith JM. Purification and characterization of the purE, purK, and purC gene products: identification of a previously unrecognized energy requirement in the purine biosynthetic pathway. Biochemistry. 1992;31:5022–32. doi: 10.1021/bi00136a016. [DOI] [PubMed] [Google Scholar]
- 9.Kemp DS, Cox DD, Paul KG. Physical organic chemistry of benzisoxazoles. IV. Origins and catalytic nature of the solvent rate acceleration for the decarboxylation of 3-carboxybenzisoxazoles. J Amer Chem Soc. 1975;97:7312–7318. [Google Scholar]
- 10.Crosby J, Stone R, Lienhard GE. Mechanisms of thiamine-catalyzed reactions. Decarboxylation of 2-(1-carboxy-l-hydroxyethyl)-3,4-dimethylthiazolium chloride. J Amer Chem Soc. 1970;92:2891–2900. doi: 10.1021/ja00712a048. [DOI] [PubMed] [Google Scholar]
- 11.Acevedo O, Jorgensen WL. Medium Effects on the Decarboxylation of a Biotin Model in Pure and Mixed Solvents from QM/MM Simulations. J Org Chem. 2006;71:4896–4902. doi: 10.1021/jo060533b. [DOI] [PubMed] [Google Scholar]
- 12.Constantine CZ, Starks CM, Mill CP, Ransome AE, Karpowicz SJ, Francois JA, Goodman RA, Kappock TJ. Biochemical and structural studies of N5-carboxyaminoimidazole ribonucleotide mutase from the acidophilic bacterium Acetobacter aceti. Biochemistry. 2006;45:8193–208. doi: 10.1021/bi060465n. [DOI] [PubMed] [Google Scholar]
- 13.Menzel U, Gottschalk G. The internal pH of Acetobacterium wieringae and Acetobacter aceti during growth and production of acetic acid. Arch Microbiol. 1985;143:47–51. [Google Scholar]
- 14.Liu A, Zhang H. Transition Metal-Catalyzed Nonoxidative Decarboxylation Reactions. Biochemistry. 2006;45:10407–11. doi: 10.1021/bi061031v. [DOI] [PubMed] [Google Scholar]
- 15.Wille G, Meyer D, Steinmetz A, Hinze E, Golbik R, Tittmann K. The catalytic cycle of a thiamin diphosphate enzyme examined by cryocrystallography. Nat Chem Biol. 2006;2:324–8. doi: 10.1038/nchembio788. [DOI] [PubMed] [Google Scholar]
- 16.Tittmann K, Wille G, Golbik R, Weidner A, Ghisla S, Hubner G. Radical phosphate transfer mechanism for the thiamin diphosphate- and FAD-dependent pyruvate oxidase from Lactobacillus plantarum. Kinetic coupling of intercofactor electron transfer with phosphate transfer to acetyl-thiamin diphosphate via a transient FAD semiquinone/hydroxyethyl-ThDP radical pair. Biochemistry. 2005;44:13291–303. doi: 10.1021/bi051058z. [DOI] [PubMed] [Google Scholar]
- 17.Knowles JR. The mechanism of biotin-dependent enzymes. Annu Rev Biochem. 1989;58:195–221. doi: 10.1146/annurev.bi.58.070189.001211. [DOI] [PubMed] [Google Scholar]
- 18.Wendt KS, Schall I, Huber R, Buckel W, Jacob U. Crystal structure of the carboxyltransferase subunit of the bacterial sodium ion pump glutaconyl-coenzyme A decarboxylase. EMBO J. 2003;22:3493–502. doi: 10.1093/emboj/cdg358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Yang Z, Shen Y, Tong L. Crystal structure of the carboxyltransferase domain of acetyl-coenzyme A carboxylase. Science. 2003;299:2064–7. doi: 10.1126/science.1081366. [DOI] [PubMed] [Google Scholar]
- 20.Attwood PV, Wallace JC. Chemical and catalytic mechanisms of carboxyl transfer reactions in biotin-dependent enzymes. Acc Chem Res. 2002;35:113–20. doi: 10.1021/ar000049+. [DOI] [PubMed] [Google Scholar]
- 21.Firestine SM, Davisson VJ. A Tight Binding Inhibitor of 5-Aminoimidazole Ribonucleotide Carboxylase. J Med Chem. 1993;36:3484–3486. doi: 10.1021/jm00074a033. [DOI] [PubMed] [Google Scholar]
- 22.Kunkel TA. Rapid and Efficient Site-Specific Mutagenesis without Phenotypic Selection. Proc Natl Acad Sci U S A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning A Laboratory Manual. Second. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. [Google Scholar]
- 24.Tabor S, Richardson CC. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci U S A. 1985;82:1074–8. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Firestine SM, Misialek S, Toffaletti DL, Klem TJ, Perfect JR, Davisson VJ. Biochemical role of the Cryptococcus neoformans ADE2 protein in fungal de novo purine biosynthesis. Arch Biochem Biophys. 1998;351:123–34. doi: 10.1006/abbi.1997.0512. [DOI] [PubMed] [Google Scholar]
- 26.Ellis KJ, Morrison JF. Methods in Enzymology. Academic Press, Inc; 1982. Buffers of Constant Ionic Strength for Studying pH-Dependent Processes; pp. 405–426. [DOI] [PubMed] [Google Scholar]
- 27.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding SE, Rowe AJ, Horton JC, editors. Royal Society of Chemistry; Cambridge, UK: 1992. pp. 90–125. [Google Scholar]
- 28.Schuck P. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophysical J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frisch MJT, G W, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 30.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 31.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37 doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project-Number 4. The CCP-4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. COOT: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 37.DeLano WL. The PyMOL Molecular Graphics Systems 2002 [Google Scholar]
- 38.Rudolph J. PhD Thesis. Department of Chemistry, Massachusetts Institute of Technology; Cambridge, Massachusetts: 1993. [Google Scholar]
- 39.Stouthamer PC, de Haan PG, Nijkamp HJ. Mapping of purine markers in Escherichia coli K 12. Genet Res. 1965;6:442–453. doi: 10.1017/s0016672300004328. [DOI] [PubMed] [Google Scholar]
- 40.Schendel FJ. PhD Thesis. Department of Biochemistry, University of Wisconsin-Madison; Madison, WI: 1986. [Google Scholar]
- 41.Blazevic N, Kajfez F, Sunjic V. σ values of some nitroimidazoles. J Heterocycl Chem. 1970;7:227–229. [Google Scholar]
- 42.Settembre EC, Chittuluru JR, Mill CP, Kappock TJ, Ealick SE. Acidophilic adaptations in the structure of Acetobacter aceti N5-carboxyaminoimidazole ribonucleotide mutase (PurE) Acta Crystallogr D Biol Crystallogr. 2004;60:1753–60. doi: 10.1107/S090744490401858X. [DOI] [PubMed] [Google Scholar]
- 43.Schwarzenbacher R, Jaroszewski L, vo Delft F, Abdubek P, Ambing E, Biorac T, Brinen LS, Canaves JM, Cambell J, Chiu HJ, Dai X, Deacon AM, DiDonato M, Elsiliger MA, Eshagi S, Floyd R, Godzik A, Grittini C, Grzechnik SK, Hampton E, Karlak C, Klock HE, Koesema E, Kovarik JS, Kreusch A, Kuhn P, Lesley SA, Levin I, McMullun D, McPhillips TM, Miller MD, Morse A, Moy K, Ouyang J, Page R, Quijano K, Robb A, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, Vincent J, Wang X, West B, Wolf G, Xu Q, Hodgson KO, Wooley J, Wilson IA. Crystal structure of a phosphoribosylaminoimidazole mutase PurE (TM0446) from Thermotoga maritima at 1.77-Å resolution. Proteins. 2004;55:474–8. doi: 10.1002/prot.20023. [DOI] [PubMed] [Google Scholar]
- 44.Boyle MP, Kalliomaa AK, Levdikov V, Blagova E, Fogg MJ, Brannigan JA, Wilson KS, Wilkinson AJ. Crystal structure of PurE (BA0288) from Bacillus anthracis at 1.8 Å resolution. Proteins. 2005;61:674–6. doi: 10.1002/prot.20599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A table of primers used in EcPurE mutagenesis, a Coomassie-stained gel of PurE proteins expressed in PCO135 E. coli, fluorescence titration data, a table of Δ250 values used in calculating the pH-rate profile, ESI-MS results for the EcPurE mutants, distribution of species obtained after SEDFIT analysis of SV-AUC data, a table summarizing SV-AUC results, a figure showing a surface representation of EcPurE-NO2-AIR, a figure showing conformational changes observed upon NO2-AIR binding, figures showing the EcPurE-H45Q-NO2-AIR and H45N-CAIR complexes, a figure comparing the EcPurE-NO2-AIR and AaPurE-AIR structures, and a table listing all available PurE structures. This material is available free of charge via the Internet at http://pubs.acs.org.