

Abstract
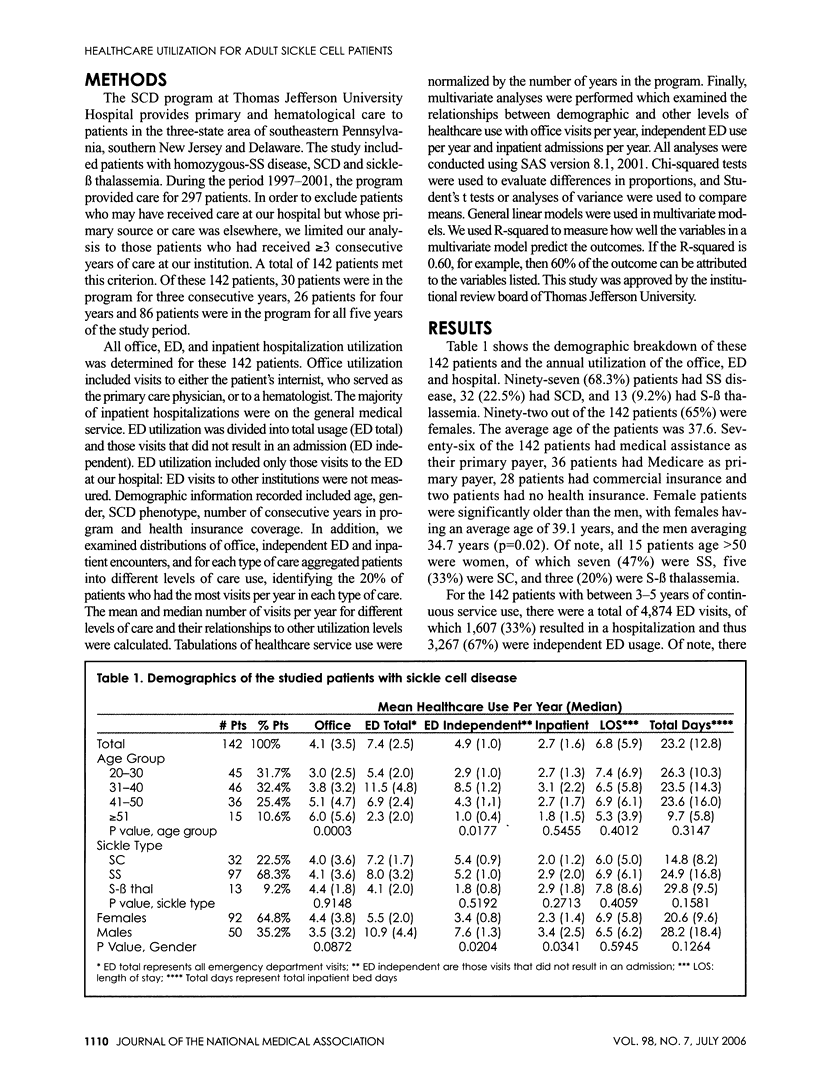
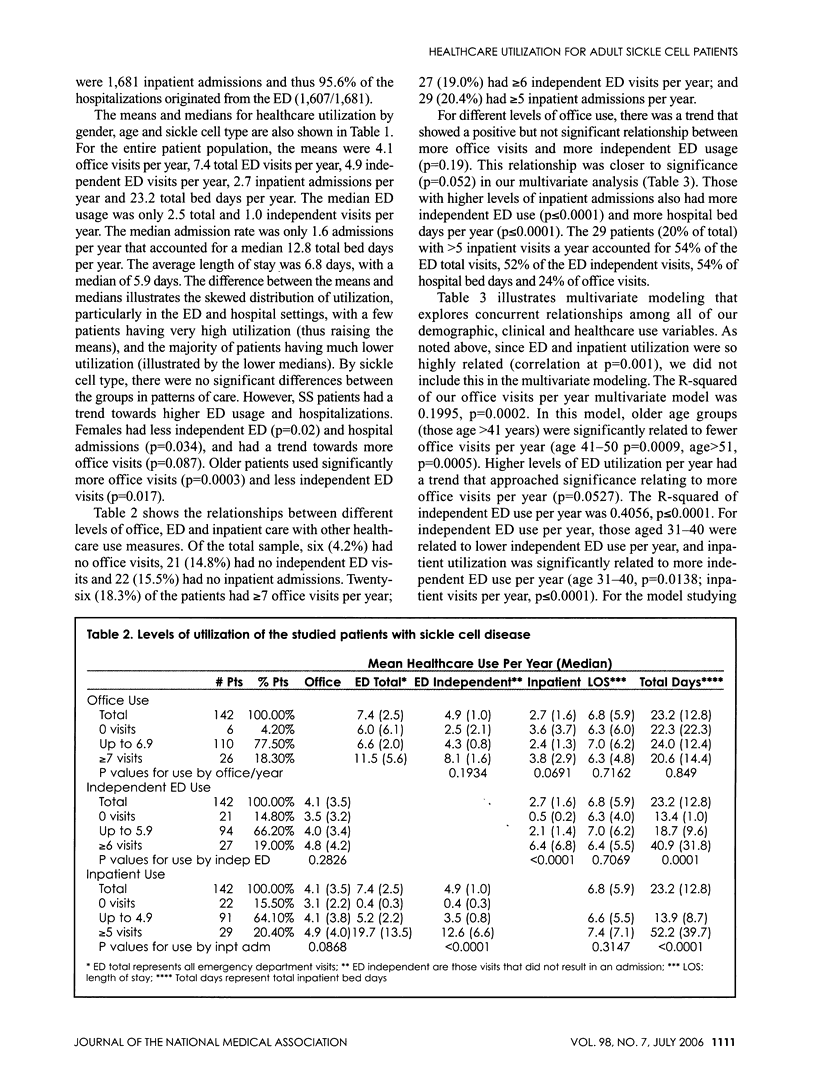
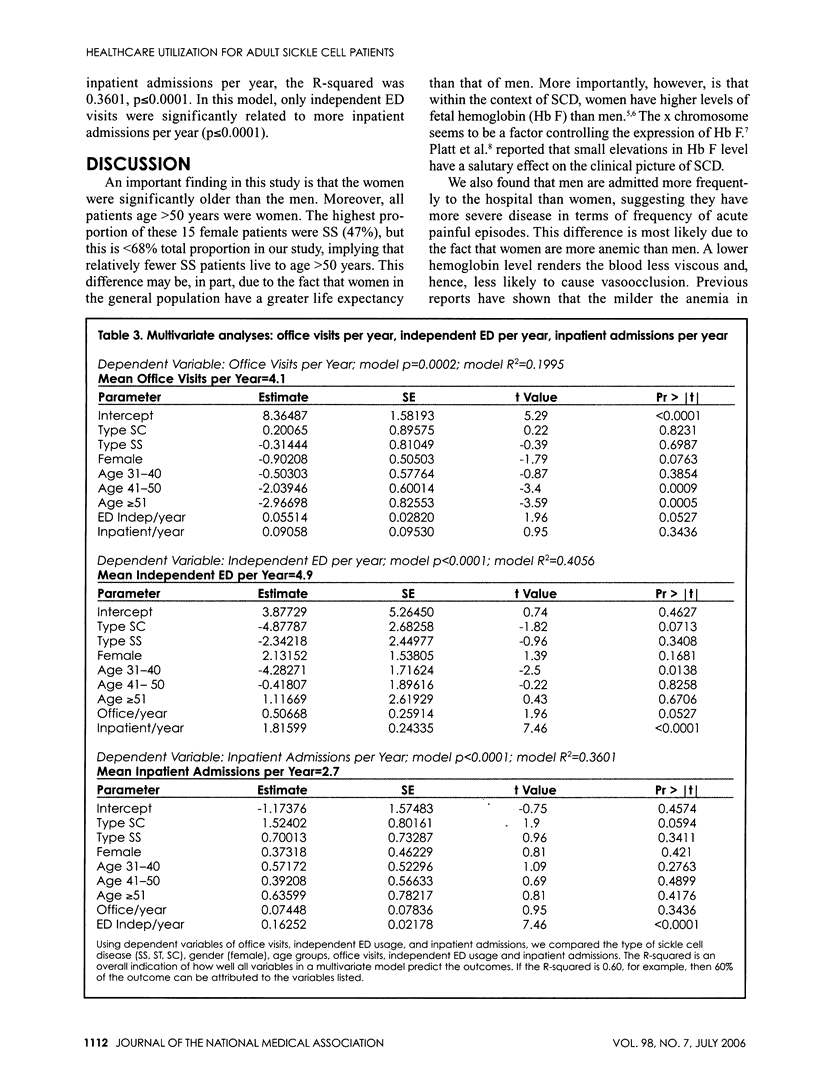
Sickle cell disease (SCD) is a hematological disorder that is manifested primarily by severe pain and chronic organ damage. Little normative data exists on what the usual healthcare utilization is of a population of SCD patients, especially adults. Our study analyzed the office, emergency department (ED) and hospital use data for 142 patients who received care for three consecutive years. Relationships between health service use, patient age, gender and sickle cell phenotype were described. Multivariate analyses studied relationships between demographic and clinical characteristics and levels of office, independent ED and inpatient encounters over a five-year period (1997-2001). We found female patients were older and had less ED and hospital admissions. The 20% highest inpatient utilizers accounted for 54% of the ED total visits, 52% of the ED independent visits, 54% of hospital bed days and 24% of office visits. The ED was a common place for utilization, with a mean of 7.4 visits per patient year, a third of which resulted in a hospital admission. The healthcare utilization of our adult sickle cell population is very complex, with a subset of our patients accounting for a majority of the resources used and female patients living longer but with less ED and hospital admissions.
Full text
PDF

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Baum K. F., Dunn D. T., Maude G. H., Serjeant G. R. The painful crisis of homozygous sickle cell disease. A study of the risk factors. Arch Intern Med. 1987 Jul;147(7):1231–1234. [PubMed] [Google Scholar]
- Benjamin L. J., Swinson G. I., Nagel R. L. Sickle cell anemia day hospital: an approach for the management of uncomplicated painful crises. Blood. 2000 Feb 15;95(4):1130–1136. [PubMed] [Google Scholar]
- Bilenker J. H., Weller W. E., Shaffer T. J., Dover G. J., Anderson G. F. The costs of children with sickle cell anemia: preparing for managed care. J Pediatr Hematol Oncol. 1998 Nov-Dec;20(6):528–533. doi: 10.1097/00043426-199811000-00003. [DOI] [PubMed] [Google Scholar]
- Davis H., Moore R. M., Jr, Gergen P. J. Cost of hospitalizations associated with sickle cell disease in the United States. Public Health Rep. 1997 Jan-Feb;112(1):40–43. [PMC free article] [PubMed] [Google Scholar]
- Dover G. J., Smith K. D., Chang Y. C., Purvis S., Mays A., Meyers D. A., Sheils C., Serjeant G. Fetal hemoglobin levels in sickle cell disease and normal individuals are partially controlled by an X-linked gene located at Xp22.2. Blood. 1992 Aug 1;80(3):816–824. [PubMed] [Google Scholar]
- Nagel R. L. Severity, pathobiology, epistatic effects, and genetic markers in sickle cell anemia. Semin Hematol. 1991 Jul;28(3):180–201. [PubMed] [Google Scholar]
- Okpala Iheanyi, Thomas Veronica, Westerdale Neil, Jegede Tina, Raj Kavita, Daley Sadie, Costello-Binger Hilda, Mullen Jean, Rochester-Peart Collis, Helps Sarah. The comprehensiveness care of sickle cell disease. Eur J Haematol. 2002 Mar;68(3):157–162. doi: 10.1034/j.1600-0609.2002.01523.x. [DOI] [PubMed] [Google Scholar]
- Platt O. S., Thorington B. D., Brambilla D. J., Milner P. F., Rosse W. F., Vichinsky E., Kinney T. R. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991 Jul 4;325(1):11–16. doi: 10.1056/NEJM199107043250103. [DOI] [PubMed] [Google Scholar]
- Steinberg M. H., Hsu H., Nagel R. L., Milner P. F., Adams J. G., Benjamin L., Fryd S., Gillette P., Gilman J., Josifovska O. Gender and haplotype effects upon hematological manifestations of adult sickle cell anemia. Am J Hematol. 1995 Mar;48(3):175–181. doi: 10.1002/ajh.2830480307. [DOI] [PubMed] [Google Scholar]
- Telfair Joseph, Haque Akhlaque, Etienne Marc, Tang Shenghui, Strasser Sheryl. Rural/urban differences in access to and utilization of services among people in Alabama with sickle cell disease. Public Health Rep. 2003 Jan-Feb;118(1):27–36. doi: 10.1093/phr/118.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]