

Abstract
Progressive multifocal leukoencephalopathy (PML) is a fatal demyelinating disorder of the CNS caused by infection of glial cells with the polyomavirus, JCV. Here we report that genomic stability and DNA repair are significantly dysregulated by JCV infection of human astrocytes. Metaphase spreads exhibited increased ploidy correlating with duration of infection. Increased micronuclei formation and phospho-Histone2AX expression also indicated DNA damage. Western blot analysis revealed perturbation in expression of some DNA repair proteins including a large elevation of Rad51. Immunohistochemistry on clinical samples of PML showed robust labeling for Rad51 in nuclei of bizarre astrocytes and inclusion body-bearing oligodendrocytes that are characteristic of JCV infection. Finally, in vitro end-joining DNA repair was altered in extracts prepared from JCV-infected human astrocytes. Alterations in DNA repair pathways may be important for the life cycle of JCV and the pathogenesis of PML.
Keywords: JCV, DNA repair, DNA damage, PML, polyomavirus
INTRODUCTION
The fatal demyelinating disease, progressive multifocal leukoencephalopathy (PML), is caused by the cytolytic destruction of oligodendrocytes in the brain as a result of the replication of the human gliotropic polyomavirus, JC virus (JCV). JCV is very common in the human population. However since the virus is readily controlled by the immune system, infection is usually subclinical and JCV enters a state of latency which is poorly understood but is generally defined as the presence of viral genome in the absence of viral gene expression. Only severe impairment in the functioning of the immune system will allow active replication of the virus and development of PML and the disease is diagnosed mostly in AIDS patients (Berger and Houff, 2006). The prominent histopathological findings in PML are multiple foci of myelin loss in the CNS, oligodendrocytes with enlarged eosinophilic nuclei containing viral inclusion bodies and enlarged bizarre astrocytes with lobulated hyperchromatic nuclei (reviewed in Del Valle and Piña-Oviedo, 2006; Khalili et al., 2006).
JCV is a member of the polyomavirus family of small DNA viruses with circular genomes that transform cells in culture and induce tumors in experimental animals. One feature of polyomaviruses is their ability to induce genomic instability. JCV is mutagenic for established cultured cell lines and human peripheral blood lymphocytes (Theile and Grabowski, 1990). Antibody titres to JCV have been correlated to chromosomal aberrations occurring in lymphocytes (Lazutka et al., 1996; Neel et al., 1996) and JCV infection of human colonic cells induces chromosomal instability and changes in ploidy (Ricciardiello et al., 2003). In the case of PML, stoichiometric analysis of cellular DNA content using the Feulgen technique indicated the occurrence of hyperploidy in inclusion-bearing oligodendrocytes and bizarre astrocytes (Ariza et al, 1996). There is evidence that the closely related polyomaviruses BKV and SV40 also induce genomic instability (reviewed by White et al., 2005). Previously, we have examined the effects of ectopic expression of two JCV regulatory proteins on cellular processes including DNA repair. These studies examined the viral early protein, large T-antigen, and agnoprotein, a small protein encoded in the late region. Large T-antigen, when expressed alone in cells, was found to inhibit the high fidelity pathway of double-strand break (DSB) repair, homologous recombination directed DNA repair (HRR), and caused the accumulation of mutations in the affected cells (Trojanek et al., 2006). Cells expressing T-antigen were found to be much more sensitive in their ability to recover from γ-irradiation or cisplatin treatment than T-antigen-negative controls and were impaired in an assay in which HRR mediated DNA repair leads to the reconstruction of wild type green fluorescent protein (GFP) from two non-functional heteroallelic fragments of GFP cDNA delivered into cells by transfection (Trojanek et al., 2006). The mechanism for this impairment was found to involve the cellular DNA repair protein, Rad51.
Ectopic expression of agnoprotein alone in cells was also found to affect the response of cells to DNA damage. Cells expressing agnoprotein were more sensitive to the cytotoxic effects of cisplatin and exhibited increased chromosome fragmentation, micronuclei formation and an accumulation of aneuploid cells (Darbinyan et al., 2004). However, in the case of agnoprotein, the mechanism of action was found to be due to an inhibition of the low fidelity pathway of DSB repair, non-homologous end-joining (NHEJ) through a mechanism involving the cellular DNA repair protein, Ku70 (Darbinyan et al., 2004).
The studies discussed above involved the introduction of a single viral protein, large T-antigen or agnoprotein, into cells by transfection. In the present studies, we have examined the occurrence of DNA damage, chromosome instability and changes in DNA repair during the course of JCV infection of astrocytes where both early and late proteins are present. We also have performed immunohistochemistry of clinical samples of PML and control brains to compare changes in our infected cell cultures to those that occur in infected astrocytes and oligodendrocytes in vivo.
RESULTS
To examine molecular and cellular events that occur during JCV infection, we have developed a cell culture system where JCV replicates efficiently (Radhakrishnan et al., 2003, 2004). This consists of purified primary human astrocytes and the Mad-1 strain of JCV, which was derived from a patient with PML. In some experiments, we have used the Mad-1/SVEdelta (Vacante et al, 1989), which is a chimeric virus containing the Mad-1 genome and part of the SV40 origin. This virus allows more efficient and timely performance of experiments since Mad-1/SVEdelta shows enhanced replication in culture (Liu et al, 1998).
In the first series of experiments to investigate the effect of JCV infection on cellular DNA, we analyzed colcemid-induced metaphase spreads from Mad-1 JCV-infected astrocytes and uninfected controls. As expected, nearly all of the uninfected cells contained a diploid (2n) content of chromosomes as shown in Fig. 1A (see panels 1 and 6). However the JCV-infected cultures contained a significant number of cells that were aneuploid/hyperploid. Some of these were tetraploid (4n) as shown in Fig. 1A, panel 4, while others contained more than a tetraploid (>4n) content of chromosomes, e.g., Fig. 1A, panel 3. In each of cultures, 70 metaphase spreads were scored as either being diploid or aneuploid/hyperploid and this data is shown in Fig. 1b. In the uninfected cultures, ≧ 95% of the cells contained 2n chromosomes while in the JCV-infected cultures, ~20% of the cells were aneuploid/hyperploid in the early stage of infection increasing to 38% in the late stage of infection. To further analyze changes in cellular DNA upon Mad-1 JCV infection, micronuclei (MIN) formation assays were performed. This assay allows the detection of micronuclei that arise from chromosomal fragments that are not incorporated into daughter nuclei at mitosis and thus reflect genomic disruption (Heddle 1973; Kalweit et al., 1999; Kersten et al., 2002). The proportion of binucleated cells with micronuclei was determined at two different concentrations of cytochalasin B (0.8 and 1.0 μg/ml) on day 14 post-infection. As shown in Fig. 1c, MIN formation was ≤2% in uninfected cultures. However in the case of the JCV-infected astrocytes, MIN formation was ~ 17% at both concentrations of cytochalasin B. This indicates that JCV is inducing genomic disruption in these astrocytes.
Figure 1. Genetic instability in astrocytes infected with the Mad-1 strain of JCV.
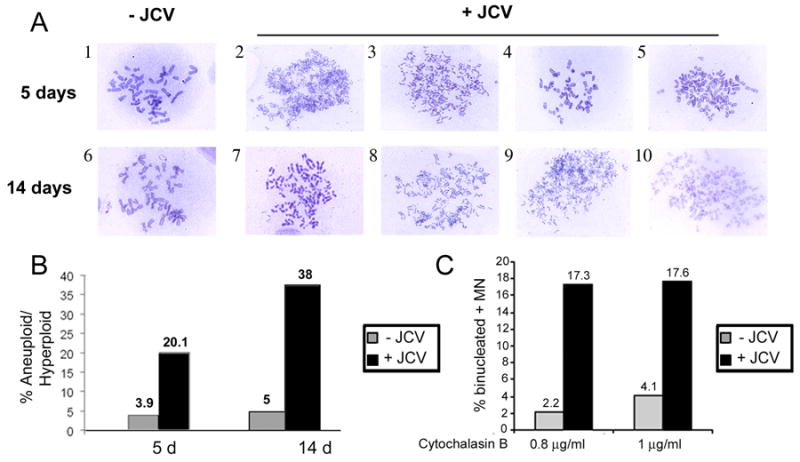
A. Metaphase spreads were prepared from JCV-infected astrocytes and uninfected controls at 5 days and 14 days post-infection. Representative photomicrographs are shown. B. For each condition, 70 metaphases were scored as being either diploid or aneuploid/hyperploid (≧ 4n), and the percentage of aneuploid/hyperploid cells plotted for each time point. C. Micronuclei formation was scored as described in Materials and Methods and the results plotted for two concentrations of cytochalasin B.
When mammalian cells incur double-strand break (DSB) DNA damage, the histone variant H2AX becomes phosphorylated on serine 139 (Rogakou et al., 1998). This phosphorylated histone, designated γH2AX, is found to cover megabase chromatin domains surrounding DSBs (Rogakou et al., 1999), increases in amount linearly with the extent of DSBs (Rogakou et al., 1999) and is thought to function in the recruitment of repair factors to nuclear foci after DNA damage (Paull et al., 2000). To examine H2AX phosphorylation during JCV infection, primary astrocytes were infected with Mad-1/SVEdelta JCV and monitored the course of viral infection during this experiment by Western blot for T-antigen, which is an early product, VP1 and agnoprotein, which are late products (Fig. 2A). Tubulin was used as a loading control. We permeabilized these Mad-1/SVEdelta JCV-infected cells and uninfected controls (days 4, 8 and 12) and stained with FITC-conjugated antibody to γH2AX and also with propidium iodide so that the cell cycle could be examined. γH2AX was increased after JCV infection, peaking at 8 days (Fig. 2B). Of note for the cell cycle, the number of cells in the G2/M phases of the cell cycle was increased at the 12-day time point for the infected cells compared to the uninfected controls. Also in the infected cells at day 12, there was an increase in the number of cells undergoing apoptosis (but not in the control cultures).
Figure 2. Flow cytometric analysis of cell cycle and γH2AX expression.
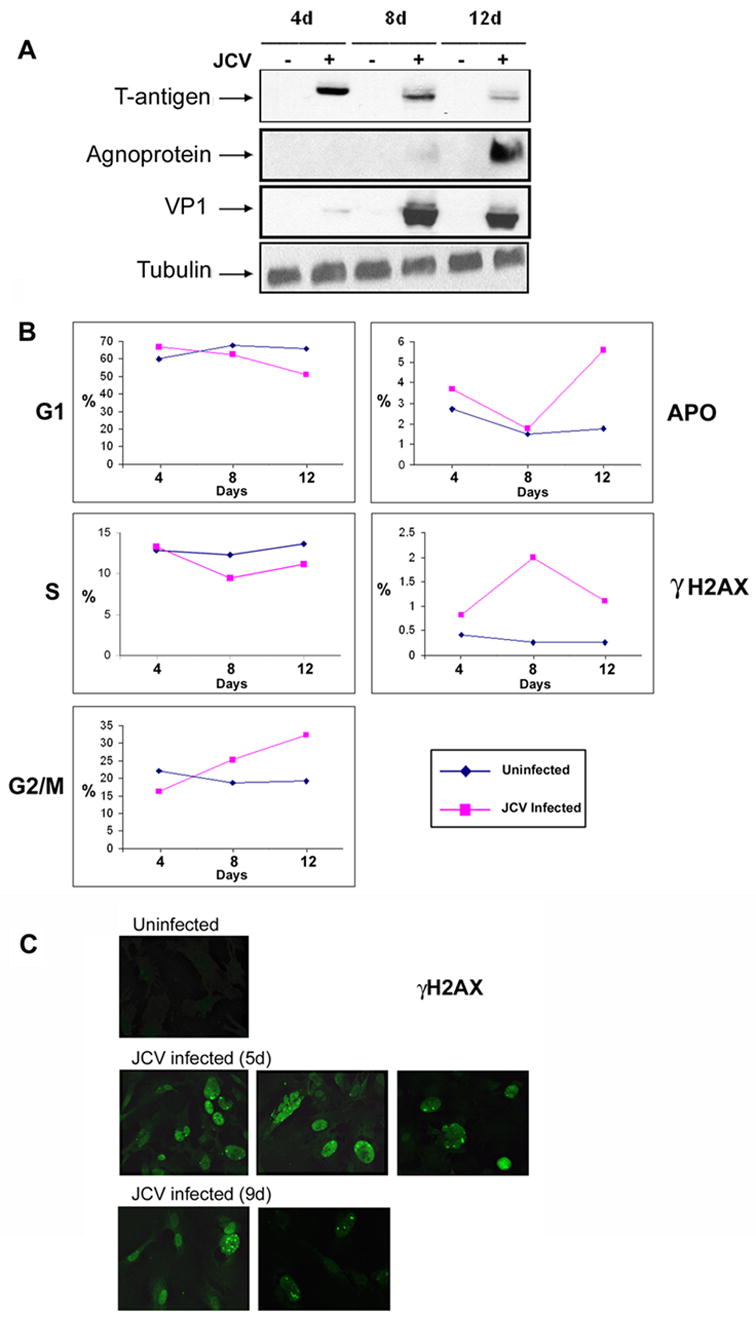
A. Cells were harvested from Mad-1/SVEdelta JCV-infected astrocytes and uninfected controls at 4, 8 and 12 days post-infection and stained with FITC-conjugated antibody to phospho-Histone H2AX (Ser139) and with propidium iodide and subject to flow cytometry analysis as described in Materials and Methods. The peaks corresponding to each of the different stages of the cell cycle are plotted against time with the subdiploid peak defined as the apoptotic peak (APO) and FITC fluorescence used to define the phosphorylation of histone H2AX (γH2A). B. Western blot of the cultures shown in Panel A to verify the expression of viral proteins during the course of infection. These experiments were conducted three times. C. Cells cultures from the 4 day and 8 day time points were replated on glass coverslips and analyzed by immunocytochemistry using an antibody specific for phospho-H2AX (anti-γH2AX) at days 5 and 9 after infection respectively.
The induction of H2AX phosphorylation was also investigated by immunofluorescence. Cell cultures from the 4 day and 8 day time points were replated on glass coverslips and analyzed by immunocytochemistry using an antibody specific for phospho-H2AX (anti-γH2AX) at days 5 and 9 after infection respectively. As shown in Fig. 2C, immunocytochemistry confirmed the FACS data that H2AX phosphorylation occurs upon Mad-1/SVEdelta JCV infection. Phosphorylation of H2AX was also demonstrated by cell fractionation/western blotting where cells were lysed in Triton X-100 to extract soluble proteins followed by DNaseI/high salt extraction to isolate a chromatin-associated fraction. In this experiment, γH2AX appeared in the chromatin-associated fraction of astrocytes during Mad-1/SVEdelta JCV infection (Fig. 3). H1 and Grb2 were used as markers for the chromatin-associated and soluble fractions respectively. Interestingly, H1 was increased in the chromatin-associated fraction of JCV-infected cells. H1 has been found to modulate the DNA ligase IV-dependent ligation of DSBs and is phosphorylated by DNA-dependent protein kinase (Kysela et al., 2005). Infection of cells was verified by Western blot with antibody against the JCV capsid protein VP1.
Figure 3. Immunocytochemistry and Western blot analysis of γH2AX expression.
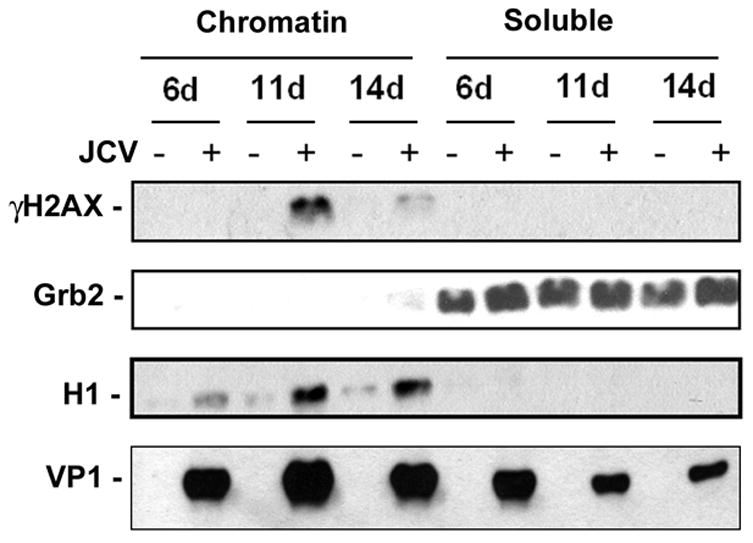
Mad-1/SVEdelta JCV-infected astrocytes and uninfected controls were harvested at 6, 11 and 14 days post-infection and fractionated into chromatin-associated and soluble components as described in Materials and Methods. γH2AX was measured by Western blot. H1 and Grb2 were used as fractionation and loading controls. Infection was verified by Western blot to VP1.
Next, we analyzed the level of expression of DNA repair proteins in the cytoplasmic and nuclear fractions of Mad-1/SVEdelta JCV-infected astrocytes isolated by a two step lysis procedure designed to separate these two subcellular compartments (Fig. 4A). The most dramatic difference was observed for the HR DNA repair protein Rad51. Rad51 was barely detectable in the cytoplasm (lane 7) and nuclei (lane 8) of uninfected astrocytes but was massively induced at all time points following JCV infection and was found at high levels in both the nuclei and cytoplasm. Densitometric analysis allowed a quantitation of the differences in Rad51 levels in Fig. 4A and revealed that the induction of Rad51 at days 10 and 15 was 30 to 40-fold when normalized to the expression of Rad51 in the nuclei of uninfected cells (Fig. 4B). α-Tubulin and lamin A/C were used as markers for the integrity of the cytoplasmic and nuclear fractions respectively. Interestingly a band corresponding to cleaved lamin (28 kDa), which is associated with nuclear membrane disintegration, appeared at the 15-day time point of Mad-1/SVEdelta JCV infection (Fig. 4A, lane 5, labeled with an asterisk). This is consistent with the data from Fig. 2A indicating an increase in apoptosis late in JCV infection. T-antigen was used to monitor the course of infection. No dramatic changes were observed for Ku70, Ku80 and NF2. As expected JCV infection induced the level of p53, which was localized in the nucleus and peaked at 10 days post-infection. Interestingly, Nibrin was also induced in nuclei of infected cells.
Figure 4. Expression of DNA repair proteins in the cytoplasmic and nuclear subcellular compartments.
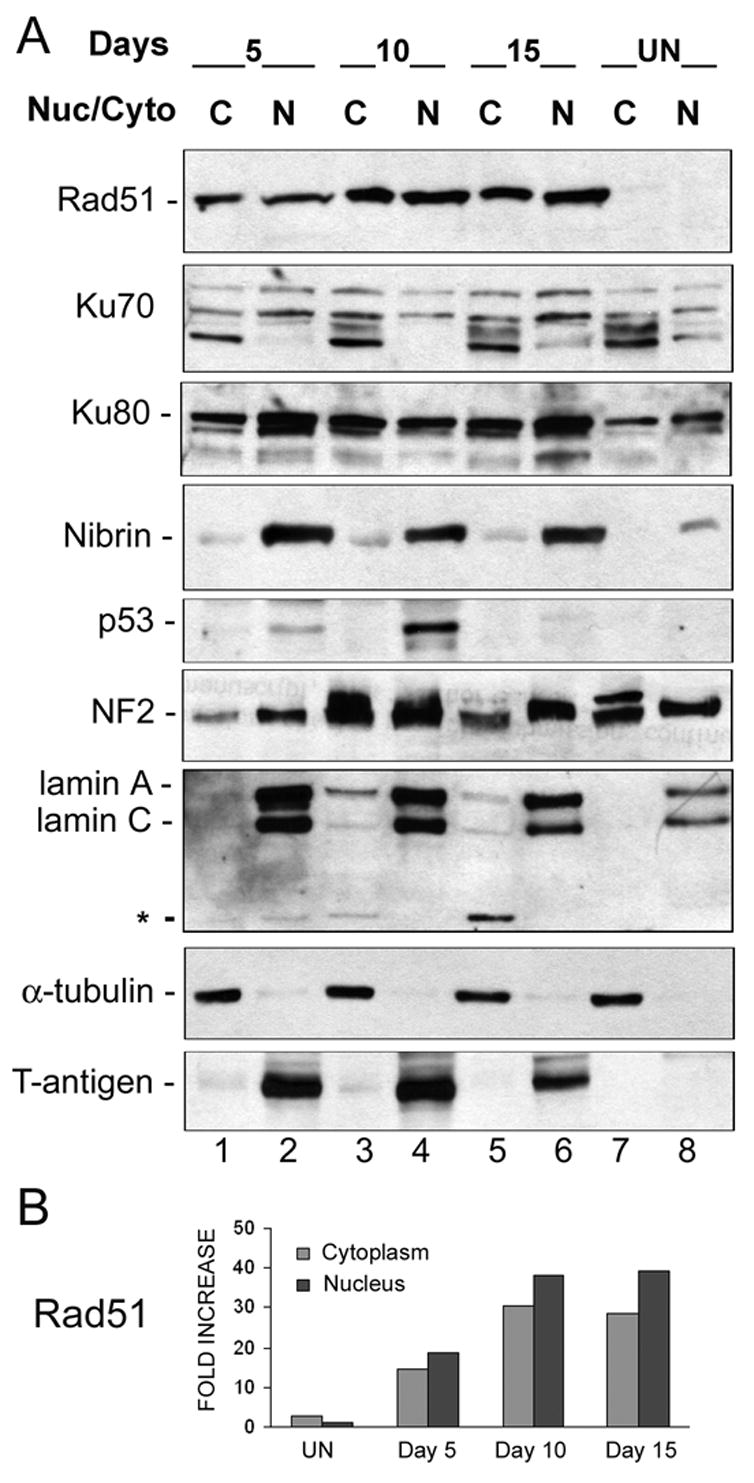
A. Astrocytes infected with Mad-1/SVEdelta JCV for 5, 10 and 15 days and uninfected controls were subject to subcellular fractionation and cytoplasmic (C) and nuclear (N) proteins were harvested. These were subject to Western blot analysis for DNA repair proteins. To assess the purity of the fractions, α-tubulin was used as a marker for cytoplasm and lamin A/C as a marker for the nucleus. The asterisk indicates a band corresponding to the cleaved form of lamin (28 kDa). T-antigen was used as a marker for infection. Ku70 runs as multiple bands, which may be due to phosphorylation. B. Quantitation of the differences in Rad51 levels in Panel A was performed by densitometric analysis of the X-ray film scanned on a Molecular Imager FX with the Quantity One Program (Bio-Rad, Hercules CA). The results were normalized relative to expression of Rad51 in the nuclei of uninfected cells and are presented as a histogram.
Next, we investigated the expression of DNA repair protein but this time using an extraction/separation protocol that allows the isolation of soluble cellular proteins, which are released by mild detergent, from chromatin-associated proteins, which are extracted by high salt and DNase treatment (Fig. 5). Again, we observed a large induction of Rad51 in Mad-1/SVEdelta JCV-infected cells that was observed at all time points following JCV infection. Interestingly there was a moderate increase in the amount of Ku70 in the chromatin-associated fraction at day 11 following JCV infection (compare lane 3 and 4). Changes were also observed for the DSB repair protein Artemis (Moshous et al., 2001). A novel higher molecular weight band appeared in the chromatin-associated fraction at 11 and 14 days following JCV infection (lanes 4 and 6, labeled with an asterisk). The nature of this band, which runs with an apparent molecular weight of about 100 kDa compared to 97 kDa for full-length Artemis, is not known but other reports indicate that Artemis can become heavily phosphorylated in response to DNA damage and that phosphorylated Artemis migrates more slowly (Soubeyrand et al, 2006; Zhang et al, 2004).
Figure 5. Expression of DNA repair proteins in chromosome-associated and soluble fractions.
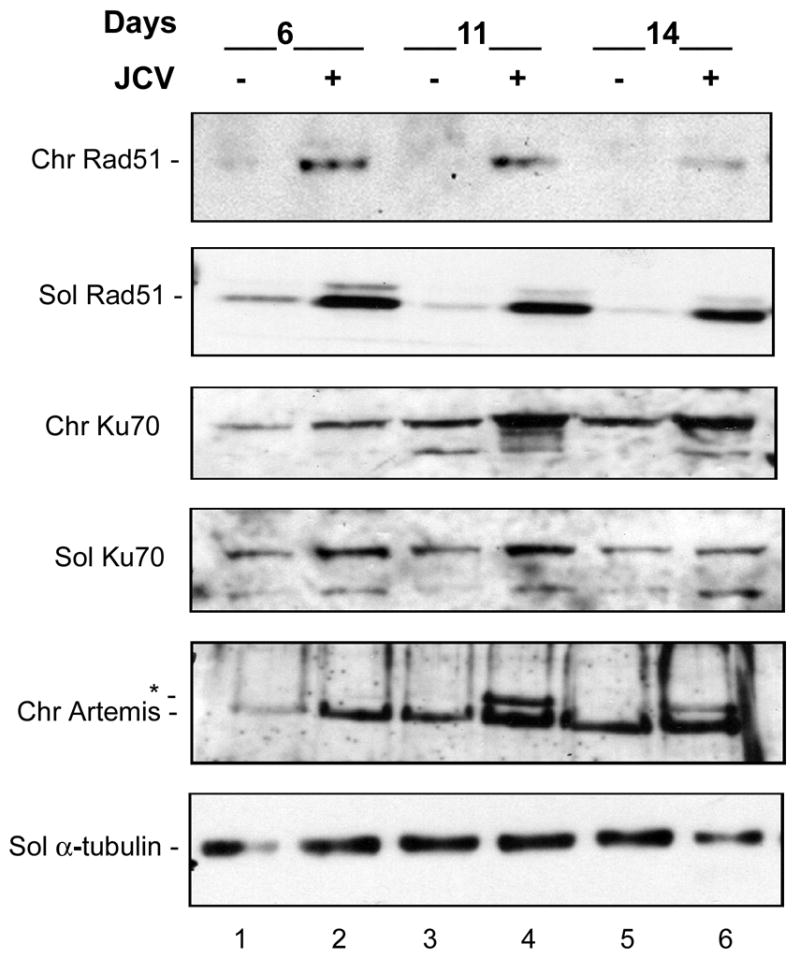
Astrocytes infected with Mad-1/SVEdelta JCV for 6, 11 and 14 days and uninfected controls were subject to a fractionation procedure to separate soluble proteins (released by mild detergent) from proteins associated with chromatin (released by high salt and DNase treatment). These are labeled as “Sol” and “Chr” respectively. These were subject to Western blot analysis for DNA repair proteins. The asterisk denotes a band in the Artemis Western blot which runs at about 100 kDa compared to 97 kDa for full-length Artemis. Ku70 runs as multiple bands, which may be due to phosphorylation.
The induction of Rad51 by Mad-1/SVEdelta JCV and subcellular distribution of Rad51 was confirmed by immunocytochemistry (Fig. 6). JCV-infected astrocytes expressed agnoprotein predominantly in the perinuclear region and T-antigen and VP1 were mainly located in the nucleus. Rad51 was much higher than in uninfected controls and was found in both the nuclear and the cytoplasmic compartments of these cells (Fig. 6, center panels).
Figure 6. Immunocytochemical detection of Rad51 expression.
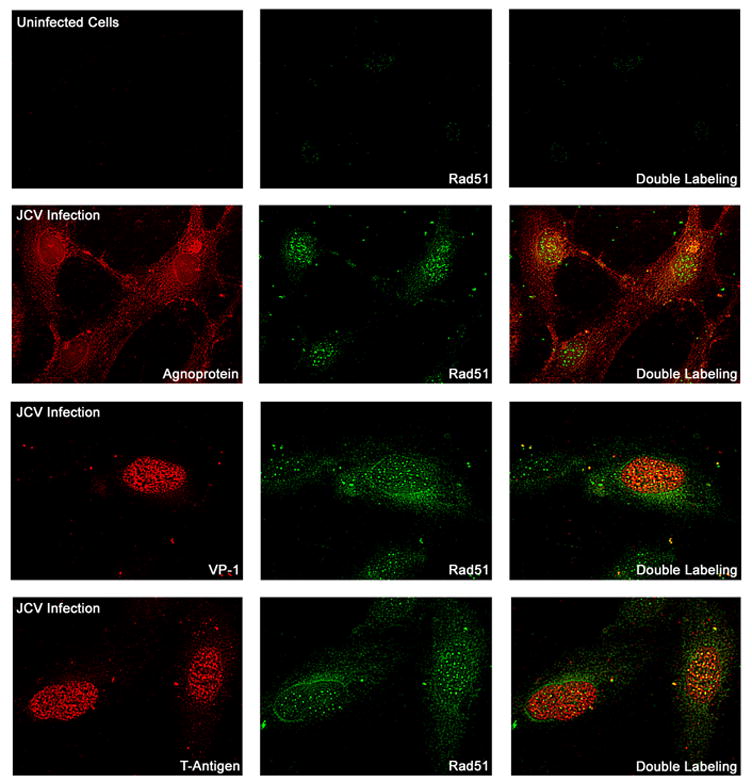
Primary human astrocytes were prepared, plated on glass cover slips and infected with Mad-1/SVEdelta JCV or used as uninfected controls. Immunocytochemistry was performed with labeling for viral proteins as indicated (left hand panels, red), Rad51 (center panels, green) and double labeling (right hand panels).
Rad51 is a component of the HR DNA repair machinery. To measure HR DNA requires the introduction of a stably transfected reporter cassette which is not possible in primary astrocytes because of their proliferative capacity. For this reason, we decided to prepare nuclear extracts to repair DNA substrates in vitro and then sequence the repaired products, i.e., this is essentially a NHEJ assay followed by product isolation and sequencing. We utilized nuclear extracts isolated from Mad-1/SVEdelta JCV-infected astrocytes and uninfected controls to assay NHEJ activity. As shown in Fig. 7, uninfected astrocytes exhibited a measurable rate of NHEJ activity which was increased at 5 days post-infection but then declined at 11 and 14 days post-infection. To address the question of the fidelity of these DNA repair processes, we isolated the multimeric products of NHEJ that were executed by JCV-infected astrocyte nuclear extracts and those derived from uninfected controls. After PCR amplification using T7 and T3 primers, the end-joined products were cloned and 10 clones for each set were sequenced. As shown in Table 1, two kinds of end-joined product were found. Both of these products contained the DNA sequence shown in Table 1, Panel A joined at its right hand side (marked with an asterisk) to the DNA sequence shown in Table 1, Panel B joined at its left-hand side (marked with an asterisk). This corresponds to the joining of the 5′ four nucleotide overhang from digestion at the BamHI site of pBlueScript(KS+) substrate (shown in Panel A) to the EcoRV blunt end at the other end of the pBlueScript(KS+) substrate (shown in Panel B). In the first type of product, all genetic information is intact, i.e., the BamHI 5′ four nucleotide overhang (Panel A) is completely filled in and blunt end ligated to the EcoRV blunt end (Panel B). In the second type of product, genetic information is lost and three nucleotides of the BamHI site 5′ four nucleotide overhang are missing from the product (nucleotides shown in white on black font in Panel A). Presumably this triplet is removed by single-strand exonuclease digestion of the last three nucleotides of the 5′ overhang and addition of one nucleotide prior to ligation to the EcoRV blunt end, although since the trinucleotide “ATC” is repeated at the site of joining, the possibility of the occurrence of removal of three nucleotides by double-strand exonuclease digestion of the EcoRV end prior to ligation cannot be excluded. The two products were designated “triplet intact” and “triplet deleted” and their sequence is shown in Table 1, Panel C. All of the products repaired by the nuclear extracts from JCV-infected astrocytes were “triplet deleted” whereas most products for the uninfected cells were “triplet intact” (Table 1, Panel C) indicating an important change in DNA repair caused by JCV infection. It is important to note that HRR cannot occur in these assays since no recombinational template for the plasmid substrate is present.
Figure 7. Non-homologous end-joining DNA repair assay.
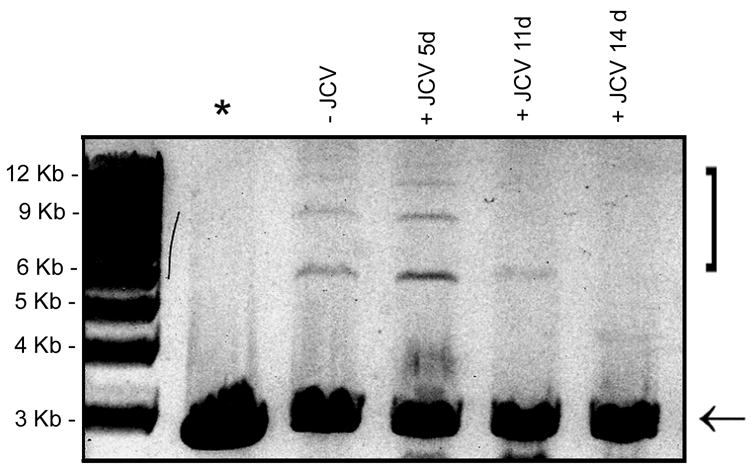
Nuclear extracts were prepared from uninfected astrocytes and from astrocytes infected with Mad-1/SVEdelta JCV for 5, 11 and 14 days. NHEJ assays were performed with a linearized plasmid substrate as described in Materials and Methods and the reactions run on an ethidium bromide/agarose gel. The position of the products are indicated by the bracket. The substrate is indicated with an arrow. The lane labeled “*” indicates the negative control (no nuclear extract).
TABLE 1.
Next, we investigated if the changes that were observed in JCV infection of primary astrocytes in culture were reflected in PML patients infected with JCV in vivo. Brain samples were obtained from three HIV-positive PML patients and controls from three HIV-positive patients without PML and three HIV-negative patients without PML. A series of immunohistochemical studies were performed on these archival PML samples. Expression of Rad51 was absent in normal astrocytes (identified by labeling with GFAP) in a representative non-PML control brain sample (Figure 8, Panels A–C). In contrast a GFAP positive bizarre astrocyte within a demyelination plaque in a case of PML (Panel D, Rhodamine), shows robust and abundant nuclear foci of Rad51 (Panels E, fluorescein and F, double-labeling). On the other hand, enlarged oligodendrocytes harboring intra-nuclear inclusion bodies, labeled with VP1 (Panel G, rhodamine), show a discrete number of Rad51 foci (Panels H, fluorescein and G, double-labeling). An elevation of Rad51 in HIV+ PML+ samples relative to HIV+ PML− and HIV− PML− samples was confirmed by Western blot analysis of proteins extracted from homogenates of the frozen brain patient samples (data not shown). Detection of Ku70 was found in the cytoplasm of bizarre astrocytes of PML brain (Figure 9, Panel A), and JCV-infected oligodendrocytes (Panel B). Although bizarre astrocytes are characteristic, their glial nature was corroborated by double-labeling with GFAP and Ku70 (Panel C). On the other hand, Ku80 was detected in the nuclei of both phenotypes, bizarre astrocytes (Panel D), and oligodendrocytes harboring inclusion bodies (Panel E), as corroborated with double labeling for GFAP and Ku80 (Panel F). Expression of Ku70 and Ku80 was undetectable in regions of the same brain samples outside of the PML plaque lesion and in immunohistochemistry of non-PML brain controls (data not shown). Finally, other DNA repair proteins were detected in the glial cells affected by JCV in cases of PML. Artemis was found in the cytoplasm of bizarre astrocytes (Figure 10, Panel A), Mre11 was present also in the cytoplasm of bizarre astrocytes (Panel B), Nibrin was detected in the cytoplasm of both phenotypes, bizarre astrocytes and enlarged oligodendrocytes (Panel C), and Chk1 was found expressed in the cytoplasm of bizarre astrocytes and in the nuclei of oligodendrocytes harboring inclusion bodies (Panel D). Expression of Mre11, Nibrin and Chk1 was undetectable in regions of the same brain samples outside of the PML plaque lesion and in immunohistochemistry of non-PML brain controls (data not shown).
Figure 8. Detection of the DNA repair protein Rad51 in glial cells of PML.
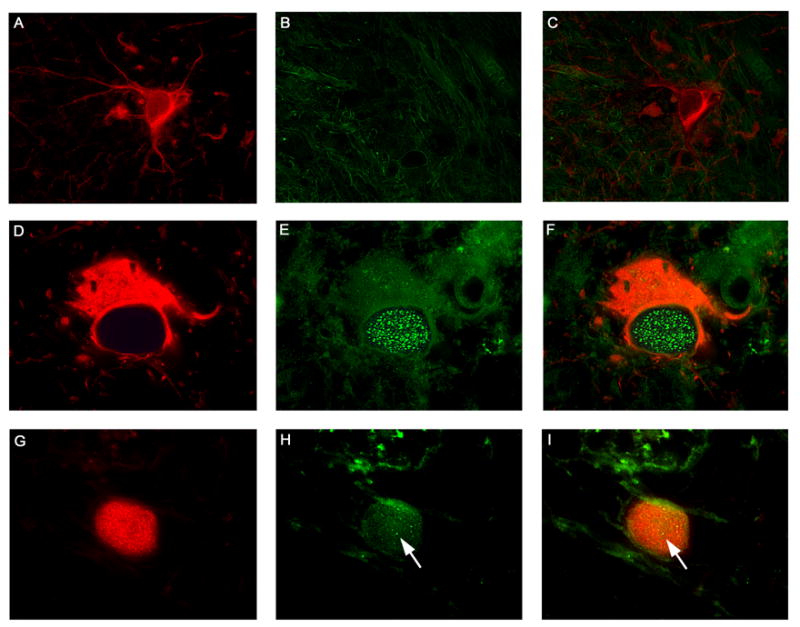
Samples were obtained from HIV-negative patients without PML (HIV −, PML −), HIV-positive patients without PML (HIV +, PML −) and HIV-positive patients with PML (HIV +, PML +) and analyzed by immunohistochemistry as described in Materials and Methods. Normal astrocytes in the cortex of a control brain were labeled with Glial Fibrillary Acidic Protein (Panel A, rhodamine), and showed no expression of Rad51 (Panels B, fluorescein and C, double labeling). Bizarre astrocytes within demyelination plaques in cases of Progressive Multifocal Leukoencephalopathy, also labeled for GFAP (Panel D, rhodamine), demonstrated numerous foci of Rad51 in the nuclei (Panels E, fluorescein and F, double labeling). JCV infected oligodendrocytes were labeled with VP1 (Panel G, rhodamine), highlighting the intranuclear viral inclusion body, and showed minimal foci of Rad51 in the same nuclear location (indicated by arrows in Panels H, fluorescein, and I, double labeling). All panels original magnification ×1000.
Figure 9. Detection and cellular localization of DNA repair enzymes Ku70 and Ku80 in cases of PML.
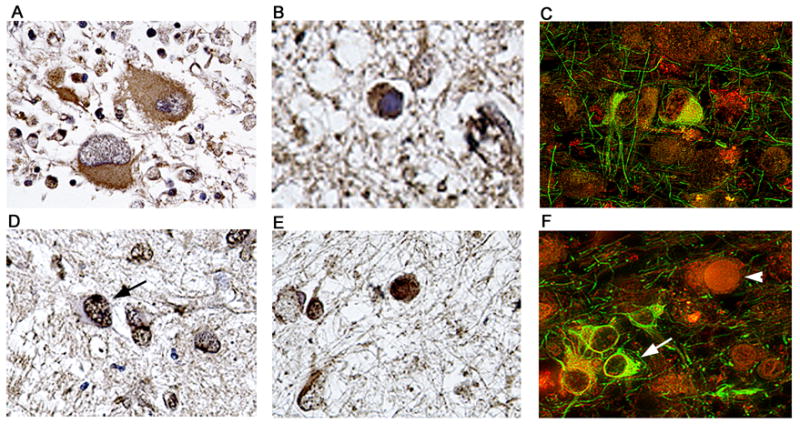
Immunohistochemistry against the DNA repair enzyme Ku70 demonstrated its presence in the cytoplasm of bizarre atypical astrocytes of PML (Panel A), and weakly in the cytoplasm of JCV infected oligodendrocytes harboring intranuclear inclusion bodies (Panel B). Double labeling with GFAP (fluorescein) demonstrates the astrocytic nature of the cells expressing Ku70 (Panel C, rhodamine). Immunohistochemistry for Ku80 showed that it was robustly expressed in the nuclei of bizarre astrocytes (indicated by an arrow in Panel D) and in the nuclei and cytoplasm of enlarged oligodendrocytes (Panel E). Double labeling (Panel F) with GFAP (fluorescein) shows the location of Ku80 (rhodamine) in astrocytes (arrow) and the presence in the nucleus of a non-labeled JCV infected oligodendrocyte (arrowhead). Panels A and D, original magnification ×400; all other panels original magnification ×1000. Labeling for Ku70 or Ku80 was not observed in areas outside of the PML lesions or in brain samples from the control patients without PML (data not shown).
Figure 10. Immunohistochemical detection of other DNA repair proteins in cases of PML.
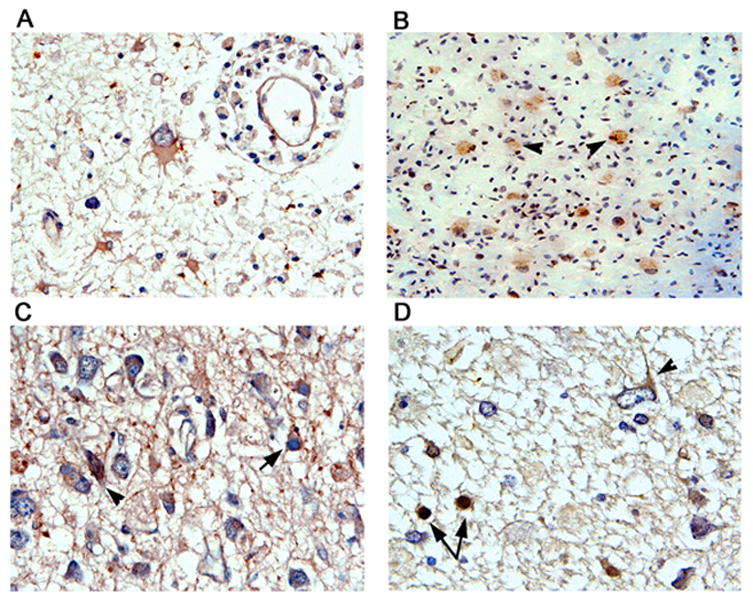
Bizarre astrocytes within demyelinated plaques of PML show cytoplasmic immunoreactivity for Artemis (Panel A). MRE11 was weakly detected in the cytoplasm of bizarre astrocytes from PML (Panel B). Immunohistochemistry for Nibrin (Panel C) showed that it was found in the cytoplasm of both bizarre astrocytes (arrowhead) and enlarged oligodendrocytes harboring inclusion bodies (arrow). Immunohistochemistry for Chk1 (Panel D) shows cytoplasmic reactivity in bizarre astrocytes (arrowhead) and in the nuclei of oligodendrocytes (arrows). All panels original magnification ×400 except Panel B ×200. Immunolabeling for MRE11, Nibrin or Chk1 was not observed in areas outside of the PML lesions or in brain samples from the control patients without PML (data not shown).
DISCUSSION
We have found that JCV infection of primary human fetal astrocytes induces chromosomal instability and DNA damage as evidenced by the occurrence of aneuploidy/hyperploidy in metaphase spreads which increases with time of infection, induction of micronuclei which are an indicator of chromosomal fragmentation (Heddle 1973; Kalweit et al., 1999; Kersten et al., 2002) and the phosphorylation of the histone H2AX on serine 139 (γH2AX) which is a marker of double strand DNA breaks (Paull et al., 2000; Rogakou et al., 1998; 1999). γH2AX induction by JCV infection was detectable by FACS analysis, immunocytochemistry and Western blot. As described in the introduction, it has previously been reported that JCV transformation of non-permissive cells, such as white blood cells and colon cells, is associated with the induction of genomic instability (Lazutka et al., 1996; Neel et al., 1996; Ricciardiello et al., 2003; Thiele and Grabowski 1990). Now we have documented the occurrence of these changes during the course of productive infection of permissive primary cells that support the active replication of JCV, a virus of narrow tropism.
In previous reports, we have examined the effects of JCV large T-antigen and agnoprotein on cellular DNA repair. Both viral proteins impaired the ability of cells to respond to DNA damage: large T-antigen was found to inhibit HR DNA repair while agnoprotein was found to inhibit NHEJ. However, there are two important differences between the experimental contexts of these studies and the present studies. Firstly, the previous studies utilized cell lines that expressed the particular viral protein by itself. In this study, viral infection is used and all viral proteins are expressed and in differing amounts in the early and late stages of infection. Secondly, the previous studies utilized established cell lines that constitutively express the viral protein. In the case of T-antigen, JCV T-antigen positive and negative mouse medulloblastoma cell lines were compared (Trojanek et al., 2006) while for agnoprotein, agnoprotein positive and negative mouse 3t3 fibroblasts were used (Darbinyan et al., 2004). In both cases, selection has been made in the cell cultures for cells that are able to survive and grow progressively. In this situation, chronic changes in cellular physiology might be expected to occur that permit the cell to tolerate the viral protein. While this is very important in understanding the mechanisms of JCV-mediated tumorigenesis, there may be important differences to the context of viral infection. In this study we are interested in understanding the acute events occurring during viral replication where the end point is cell death and virus release. In other words, the differences observed in the perturbations in DNA repair between the experiments reported here and our earlier reports may be due one or more important differences in the experimental settings including cell type (human astrocytes versus rodent fibroblast) and the nature of expression (acute expression of multiple viral proteins versus chronic expression of a single viral protein).
The chromosome abnormalities resulting from JCV infection occur early in infection with 20% cells being hyperploid/aneuploid after 5 days suggesting that a viral early protein is involved. In the case of the closely related polyomavirus SV40, it has long been known to induce chromosomal abnormalities (Marshak et al., 1973) and this function is associated with large T-antigen (Ray et al., 1990; 1992; Stewart and Bacchetti 1991). Attempts to map the function to subdomains within the SV40 T-antigen protein using mutants have yielded conflicting results implicating the N-terminal pRb-binding domain (Woods et al., 1994) or the p53-binding domain (Ray et al., 1998). JCV T-antigen is also able to bind to pRb and p53 (Krynska et al., 1997; White et al., 2006). Yet other reports have implicated the interaction of SV40 T antigen with other proteins for the induction of genetic instability. These include Nbs1 in the disruption of DNA replication control (Wu et al., 2004), Mre11 in nuclear DNA-repair foci (Digweed et al., 2003) and Bub1, the spindle assembly checkpoint protein that is part of the machinery that ensures the correct segregation of chromosomes at mitosis (Cotskiki et al., 2004).
Infection of astrocytes with JCV also affects their cell cycle profiles (Fig. 2). The percentage of cells in G2/M is increased especially late in infection. This could be due to induction of the G2/M checkpoint in these cells. This checkpoint serves to delay the entry of cells into mitosis in response to DNA damage so as to allow time for DNA repair. Thus the DNA damage induced by JCV may be responsible for the increase in the number of cells in the G2/M phase. The cell cycle profiles also reveal that the number of cells undergoing apoptosis is increased late in infection. Interestingly, this is correlated with the appearance of cleaved lamin, a marker for apoptosis (Fig. 4). Remarkably, cultures in the late stage of infection exhibit a profound cytopathic effect and have severe DNA damage but less than 6% of them are apoptotic (Fig. 2). In this regard, we have recently found that JCV infection induces expression of the inhibitor of apoptosis protein, Survivin (L. Del Valle, submitted for publication).
Western blot analysis of various subcellular fractions from JCV-infected and uninfected astrocytes show the perturbation in the levels of expression of some important DNA repair proteins. The most notable of these is a very large induction in the expression of the HR-DNA repair protein, Rad51. Induction was seen in nuclear and cytoplasmic fractions (Fig. 4), in chromatin-associated and soluble fractions (Fig. 5) and by immunocytochemistry (Fig. 6). In clinical brain samples from PML patients relative to non-PML brain controls, Rad51 induction was detected by Western blot (Fig. 8) and immunohistochemistry where Rad51 was detected in the bizarre giant astrocytes that are characteristic of PML (Fig. 9). Also of interest, a higher molecular weight protein band was observed in Western blots of Artemis in JCV-infected astrocyte cultures but not controls. This runs at about 100 kDa compared to 97 kDa for full-length Artemis and may be due to phosphorylation of Artemis in response to DNA damage (Soubeyrand et al, 2006; Zhang et al, 2004).
The relationship of the occurrence of DNA damage to changes in DNA repair proteins during JCV infection poses a chicken and egg question. On the one hand, it is possible that DNA damage is caused by JCV infection and then the cell responds by inducing DNA repair proteins. On the other hand, it is possible that JCV infection induces DNA repair proteins inappropriately and that this subsequently leads to defective DNA repair and DNA damage. In this regard, we recently reported that transient transfection of human cells with a plasmid expressing JCV T-antigen strongly stimulates transcription from the Rad51 promoter (White et al., 2006). This induction was observed in the p53-null Saos2 cell line and hence is independent of p53, which is a repressor of the Rad51 promoter (Arias-Lopez et al., 2006). However T-antigen stimulation of the Rad51 promoter was not abrogated by cotransfection of a plasmid expressing p53 (White et al., 2006). Rad51 overexpression has been shown to promote alternative double-strand break repair pathways leading to genomic instability, chromosomal breaks and aneuploidy (Richardson et al., 2004). Paradoxically when Rad51 is overexpressed it reduces DSB-induced HR DNA repair perhaps because excess Rad51 blocks DNA strand exchange (Kim et al., 2001). Indeed, despite its role in maintaining genomic integrity, the aberrant increase in Rad51 expression that is found in tumor cells may contribute to genomic instability by promoting abnormal recombination between short repetitive elements and homologous sequences (Radershall et al., 2002; Richardson et al., 2004; Xia et al., 1997). Thus it is possible that the Rad51 promoter is induced by T-antigen early in the infection of astrocytes by JCV and that excess Rad51 promotes subsequent genetic instability. Products from DNA substrates joined in vitro by extracts from the nuclei of JCV-infected astrocytes differ significantly from those from uninfected cells (Table 1) although it is uncertain if excess Rad51 is involved in this phenomenon.
The alteration in DNA repair pathways that we have reported may be important for the life cycle of JCV and the pathogenesis of PML. Rad51 may have a direct role in viral replication or may indirectly promote the virus life cycle indirectly through effects on other cellular factors. Additionally when cells are non-permissive or semi-permissive for JCV replication, cells that are not killed by the virus may become transformed by T-antigen. T-antigen-induced Rad51 expression in these cells may cause genomic instability that may contribute to the early stages of progression of JCV-associated tumors.
MATERIALS AND METHODS
Antibodies and Western blotting
The following antibodies were used for Western blotting: Ku70 - AHP316 (Serotec, Raleigh, NC), Ku80 - AHP317 (Serotec), Grb2 -610111 (BD Biosciences, San Jose, CA), Rad51 - clone 3C10 (Upstate Biotechnology, Lake Placid, NY), p53 –Ab1 (Oncogene Science, clone 421), JCV T-antigen – Pab2 (Oncogene Research Products, clone PAb 416), α-tubulin (T6074; Sigma, St. Louis, MO), Histone H1 - AE-4 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), Anti-phospho-H2A.X (Ser139) - Upstate biotechnology (clone JBW301), Artemis – (Abcam, Cambridge, MA), Nibrin – (Santa Cruz), Lamin A/C, Chk1, phospho-Chk2 (Thr68) – (Cell Signaling, Danvers MA), NF2 – sc-332 (Santa Cruz). We have previously described rabbit polyclonal antibody against JCV agnoprotein and VP1 (Del Valle et al., 2002). Western blotting was performed as described earlier (Darbinyan et al., 2004).
JCV infection of astrocytes
Primary cultures of human astrocytes were prepared and infected with either JCV strain Mad-1 or with the chimeric Mad-1/SVEdelta virus as described previously (Radhakrishnan et al., 2003, 2004) at an moi = 1. Cells were freshly prepared and purified for each experiment. At 5–6, 10–11 and 14–15 days after infection, cells were harvested and analyzed as described below together with uninfected control cultures. Note that in infected cultures, virus production precedes cytopathic effect: we have found virus in the supernatant of infected cultures at a level of 106 and 107 viral copies/ml at day 5 and day 15 post-infection respectively (Radhakrishnan et al, 2004). The mechanism of viral release is thought to be via endosome formation similar to SV40 (Clayson et al., 1989).
Metaphase spreads
Metaphase spreads were prepared as we have previously described (Darbinyan et al., 2004). Briefly, cells were treated for 2 hours with 100 ng/ml colcemid. After mitotic arrest, cells were harvested, treated with 0.075 M KCl and fixed with methanol:acetic acid (3:1). The cell suspension was dried on a glass slide, stained in 0.4% Giemsa, soaked in xylene and mounted in SP15–100 Permount (Fisher, Pittsburgh, PA). Photomicrographs were taken under an Olympus light microscope with a magnification of 700X.
Micronuclei formation assay
Micronuclei formation was assayed as we have previously described (Darbinyan et al., 2004). Cells were treated for 24 hours with cytochalasin B (0.8 and 1.0 μg/ml) to block cytoplasmic but not nuclear division, washed, fixed in methanol, allowed to air dry and stained with 0.4% Giemsa. The proportion of binucleated cells with micronuclei was determined.
Flow cytometric analysis
Cells were harvested, washed with PBS and fixed in suspension in 1% methanol-free formaldehyde in PBS on ice for 20 min. Cells were then resuspended in 73% ethanol for at least 16–20 h at −20°C. Cells were washed with PBS and gently resuspended in 0.2% Triton X-100 in PBS/1%BSA for 30 min. Following low speed centrifugation, cells were incubated with anti-phospho-Histone H2A.X (Ser139)-FITC-conjugated antibody (mouse monoclonal, Upstate Biotechnologies, #16–202A) in 1% BSA in PBS overnight at 4°C. This antibody is also known as anti-γH2AX and only recognizes H2AX that is phosphorylated on the serine at position 139. Cells were then washed with PBS and stained with propidium iodide in PBS/RNase A. Flow cytometry was performed with a COULTER® EPICS ® FACScan flow cytometer to determine the cell cycle distribution and measure the green fluorescence.
Immunocytochemistry for γH2AX
Primary human fetal astrocytes were prepared and seeded on polylysine-coated glass chamber slides and left uninfected or infected with JCV (Mad-1/SVEdelta). After 5 days or 9 days after infection, cells were fixed with 4% paraformaldehyde. Fixed cells were blocked with 5% BSA in PBS for 2 h and incubated with anti-phospho-Histone H2A.X (Ser139)-FITC-conjugated antibody (mouse monoclonal, Upstate Biotechnologies, #16–202A) for 16 hr. Cells were then washed three times with PBS-0.01% Tween-20 at 10 min intervals. Slides were washed three times with PBS, mounted, and examined by fluorescence microscopy.
Preparation of nuclear and cytoplasmic fractions
Cell cultures were extracted to give nuclear and cytoplasmic fractions using the NE-PER nuclear and cytoplasmic reagents (Pierce Biotechnology, Rockford, IL) as we have previously described (White et al., 2006).
Preparation of chromatin-associated and soluble fractions
Chromatin-associated and soluble fractions were prepared based on the method of Mirzoeva and Petrini (2003). Briefly, astrocytes were harvested by trypsinization, resuspended in growth media, and counted. Cells were lysed in ice-cold CSK buffer [10 mM PIPES (pH 6.8), 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100 supplemented with 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mM ATP, 10 mM sodium-orthovanadate] for 10 min at 2.5 × 107 cells/ml. The supernatant from low-speed centrifugation (5 min, 1500 × g, 4°C) constituted the soluble fraction that was subsequently clarified by high-speed centrifugation (15 min at 14,000 × g, 4°C). The low-speed pellet (intact nuclei, verified microscopically), was washed once with ice-cold CSK buffer (5 min, 1500 × g, 4°C) and incubated in CSK buffer containing 50 units of RNase-free DNase I (Roche Biochemicals, Basel, Switzerland) at 1 × 108 nuclei/ml for 30 min at 37°C. DNase-treated nuclei were collected by centrifugation (15 min, 14,000 × g, 4°C) and the supernatant was retained. The DNase pellet was washed once in 1 ml of ice-cold CSK buffer (5 min, 16,000 × g, 4°C) and incubated in ice-cold CSK buffer containing 500 mM NaCl for 10 min at 4°C. This extract was clarified by centrifugation (15 min, 14,000 × g, 4°C) and pooled with the DNase-treated fraction, constituting the chromatin-associated, or insoluble, fraction. The protein concentration was determined by Bradford assay and 25 μg for soluble fraction and 5 μg for insoluble were resolved by 10% SDS-PAGE and Western blotted as described previously (Darbinyan et al., 2004).
Double-labeling immunocytochemistry for Rad51 and JC viral proteins
5 days post-infection with JCV (Mad-1/SVEdelta), primary cell cultures and uninfected controls were fixed with cold acetone for 3 minutes and rinsed with PBS. Cells were blocked with normal horse or goat serum for 1 hour at room temperature and incubated with primary antibodies overnight. Primary antibodies for viral proteins included a mouse monoclonal anti SV40 T-antigen, which cross-reacts with JCV T-antigen (Ab2, Clone PAb416, 1:100 dilution, Oncogene), a mouse monoclonal anti-VP1 (1:2000 dilution, generously provided by Dr. Walter Atwood, Brown University, Providence, Rhode Island), and a rabbit polyclonal anti-agnoprotein (1:1000 dilution) (previously described in Del Valle et al., 2002). After rinsing thoroughly with PBS, a rhodamine-tagged secondary anti-mouse or anti-rabbit antibody was incubated for 1 hour at room temperature in the dark. Then the cultures were treated with a basic buffer solution containing 0.02% Triton X-100 and re-blocked with normal horse serum for 1 hour, after which a Rad51 antibody (Ab-2, Clone 51RAD01, Calbiochem, 1:50 dilution) was incubated overnight. After rinsing with PBS an anti-mouse fluorescein-tagged secondary antibody was incubated for 1 hour in the dark. Finally the chambers were detached, the slides were mounted with an aqueous-based mounting media (Vector Laboratories) and visualized with a UV fluorescent microscope (Nikon), and images were processed using deconvolution software (Slidebook 4.0, Intelligent Imaging, Denver, Colorado).
Nonhomologous end joining assay for double strand break DNA repair
NHEJ assays were as we have previously described (Darbinyan et al., 2004). Nuclear extracts were prepared from control and Mad-1/SVEdelta JCV-infected astrocytes (5, 11 and 14 days post-infection) using hypotonic lysis buffer followed hypertonic nuclear lysis buffer and 3 rapid cycles of freezing-thawing. The nuclear lysates were cleared by centrifugation, dialyzed and NHEJ assays performed using a DNA substrate consisting of plasmid pBlueScript(KS+) linearized by restriction endonuclease digestion with BamHI and EcoRV. This DNA substrate is thus an approximately 3 Kb linear molecule with a 4 nucleotide 5′ overhang at one end (BamHI) and the other end blunt (EcoRV). The resulting 3 Kb linear DNA substrate fragment was added to end-joining reactions containing nuclear extracts and were incubated for 1 hour at 37°C. DNA products were deproteinized by treatment with proteinase K and analyzed by electrophoresis through a 0.7% agarose gel.
Sequence analysis of DNA products resulting from NHEJ reactions
NHEJ reactions were performed as described in the previous section and the multimeric products purified from the agarose gel for each of the NHEJ reactions. PCR was performed using standard T7 and T3 primers which flank the end-joined region of the DNA. The PCR products were cloned into the pCR2.1 plasmid (Invitrogen) and ten plasmids containing PCR inserts were selected for each set and analyzed by sequencing.
Clinical brain samples
A total of 12 archival cases of formalin fixed, paraffin embedded PML cases with their respective controls were collected from the NIH National Tissue Consortium at the Manhattan Brain Bank, Mount Sinai School of Medicine, New York, New York. Frozen portions of parieto-occipital lobe from three HIV negative, three HIV positive (no pathology) and three HIV-related PML cases were also obtained from the archives of the Manhattan Brain Bank. Patients were at age 41–52 years old males.
Immunohistochemistry for brain samples
Immunohistochemistry was performed as we have previously described (Enam et al., 2005) using the avidin-biotin-peroxidase system according to the manufacturer’s instructions (Vector Laboratories). Our modified protocol includes deparaffination in xylene, re-hydration in alcohol up to water, non-enzymatic antigen retrieval in citrate buffer, pH 6.0 at 95°C for 30 minutes. After a cooling period, endogenous peroxidase quenching was performed with 0.3% H2O2 in methanol for 20 minutes. Sections were rinsed with PBS and blocked with normal horse or goat serum (for mouse monoclonal or rabbit polyclonal antibodies respectively), and incubated with primary antibodies overnight at room temperature in a humidified chamber. Primary antibodies utilized included rabbit polyclonal antibodies against Artemis (AbCam, ab18319, 1:250 dilution), Mre11 (Santa Cruz Biotechnology, H300, 1:500 dilution), Chk1 (Santa Cruz Biotechnology, (FL-476, 1:00 dilution), and a goat polyclonal anti-Nibrin (Santa Cruz Biotechnology, C-19, 1:200 dilution). For the Mre11 antibody we utilized frozen sections. After rinsing with PBS a biotinylated secondary antibody was incubated for 1 hour, then avidin-biotin-peroxidase complexes (ABC Kit, Vector Laboratories), then sections were developed with diaminobenzidine (Sigma), and finally sections were counterstained with hematoxylin and mounted. For detection of Rad51, we modified our protocol to include a basic buffer containing Triton X-100 in each step. We utilized a mouse monoclonal antibody against human Rad51 (Ab-2, Clone 51RAD01, 1:50 dilution), and we labeled with a fluorescein-tagged secondary antibody as described for immunocytochemistry.
Acknowledgments
We thank past and present members of the Center for Neurovirology for their insightful discussion and sharing of ideas and reagents. We also wish to thank C. Schriver for editorial assistance. This work was supported by grants awarded by the NIH to KK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, Ashworth A, Silva A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006;7:219–224. doi: 10.1038/sj.embor.7400587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariza A, Mate JL, Serrano S, Isamat M, Keysers U, Aracil C, Navas-Palacios JJ. DNA amplification in glial cells of progressive multifocal leukoencephalopathy: an image analysis study. J Neuropathol Exp Neurol. 1996;55:729–733. doi: 10.1097/00005072-199606000-00007. [DOI] [PubMed] [Google Scholar]
- Berger JR, Houff S. Progressive multifocal leukoencephalopathy: lessons from AIDS and natalizumab. Neurol Res. 2006;28:299–305. doi: 10.1179/016164106X98198. [DOI] [PubMed] [Google Scholar]
- Clayson ET, Brando LV, Compans RW. Release of simian virus 40 virions from epithelial cells is polarized and occurs without cell lysis. J Virol. 1989;63:2278–2288. doi: 10.1128/jvi.63.5.2278-2288.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, Freire R, Entwistle A, Golemis EA, Roberts TM, Jat PS, Gjoerup OV. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci USA. 2004;101:947–952. doi: 10.1073/pnas.0308006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbinyan A, Siddiqui KM, Slonina D, Darbinian N, Amini S, White MK, Khalili K. Role of JC virus agnoprotein in DNA repair. J Virol. 2004;78:8593–600. doi: 10.1128/JVI.78.16.8593-8600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle L, Piña-Oviedo S. HIV disorders of the brain: pathology and pathogenesis. Front Biosci. 2006;11:718–732. doi: 10.2741/1830. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Gordon J, Enam S, Delbue S, Croul S, Abraham S, Radhakrishnan S, Assimakopoulou M, Katsetos CD, Khalili K. Expression of human neurotropic polyomavirus JCV late gene product Agnoprotein in human medulloblastoma. J Natl Cancer Inst. 2002;94:267–273. doi: 10.1093/jnci/94.4.267. [DOI] [PubMed] [Google Scholar]
- Digweed M, Demuth I, Rothe S, Scholz R, Jordan A, Grotzinger C, Schindler D, Grompe M, Sperling K. SV40 large T-antigen disturbs the formation of nuclear DNA-repair foci containing MRE11. Oncogene. 2002;21:4873–4878. doi: 10.1038/sj.onc.1205616. [DOI] [PubMed] [Google Scholar]
- Enam S, Sweet TM, Amini S, Khalili K, Del Valle L. Evidence for involvement of transforming growth factor beta1 signaling pathway in activation of JC virus in human immunodeficiency virus 1-associated progressive multifocal leukoencephalopathy. Arch Pathol Lab Med. 2005;128:282–291. doi: 10.5858/2004-128-282-EFIOTG. [DOI] [PubMed] [Google Scholar]
- Heddle JA. A rapid in vivo test for chromosomal damage. Mutat Res. 1973;18:187–190. doi: 10.1016/0027-5107(73)90035-3. [DOI] [PubMed] [Google Scholar]
- Kalweit S, Utesch D, von der Hude W, Madle S. Chemically induced micronucleus formation in V79 cells-comparison of three different test approaches. Mutat Res. 1999;439:183–190. doi: 10.1016/s1383-5718(98)00191-0. [DOI] [PubMed] [Google Scholar]
- Kersten B, Kasper P, Brendler-Schwaab SY, Müller L. Use of the photo-micronucleus assay in Chinese hamster V79 cells to study photochemical genotoxicity. Mutat Res. 2002;519:49–66. doi: 10.1016/s1383-5718(02)00113-4. [DOI] [PubMed] [Google Scholar]
- Khalili K, Gordon J, White MK. The polyomavirus, JCV and its involvement in human disease. Adv Exp Med Biol. 2006;577:274–287. doi: 10.1007/0-387-32957-9_20. [DOI] [PubMed] [Google Scholar]
- Kim PM, Allen C, Wagener BM, Shen Z, Nickoloff JA. Overexpression of human RAD51 and RAD52 reduces double-strand break-induced homologous recombination in mammalian cells. Nucl Acids Res. 2001;29:4352–4360. doi: 10.1093/nar/29.21.4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krynska B, Gordon J, Otte J, Franks R, Knobler R, DeLuca A, Giordano A, Khalili K. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J Cell Biochem. 1997;67:223–230. doi: 10.1002/(sici)1097-4644(19971101)67:2<223::aid-jcb7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Kysela B, Chovanec M, Jeggo PA. Phosphorylation of linker histones by DNA-dependent protein kinase is required for DNA ligase IV-dependent ligation in the presence of histone H1. Proc Natl Acad Sci USA. 2005;102:1877–1882. doi: 10.1073/pnas.0401179102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazutka JR, Neel JV, Major EO, Dedonyte V, Mierauskine J, Slapsyte G, Kesminiene A. High titers of antibodies to two human polyomaviruses, JCV and BKV, correlate with increased frequency of chromosomal damage in human lymphocytes. Cancer Lett. 1996;109:177–83. doi: 10.1016/s0304-3835(96)04448-5. [DOI] [PubMed] [Google Scholar]
- Liu CK, Hope AP, Atwood WJ. The human polyomavirus, JCV, does not share receptor specificity with SV40 on human glial cells. J Neurovirol. 1998;4:49–58. doi: 10.3109/13550289809113481. [DOI] [PubMed] [Google Scholar]
- Marshak MI, Varshaver NB, Shapiro NI. Induction of gene mutations and chromosomal aberrations by simian virus 40 in cultured mammalian cells. Mutat Res. 1975;30:383–396. [PubMed] [Google Scholar]
- Mirzoeva OK, Petrini JH. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res. 2003;1:207–218. [PubMed] [Google Scholar]
- Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- Neel JV, Major EO, Awa AA, Glover T, Burgess A, Traub R, Curfman B, Satoh C. Hypothesis: “Rogue cell”-type chromosomal damage in lymphocytes is associated with infection with the JC human polyoma virus and has implications for oncopenesis. Proc Natl Acad Sci USA. 1996;93:2690–2695. doi: 10.1073/pnas.93.7.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002;62:219–225. [PubMed] [Google Scholar]
- Radhakrishnan S, Otte J, Enam S, Del Valle L, Khalili K, Gordon J. JC virus-induced changes in cellular gene expression in primary human astrocytes. J Virol. 2003;77:10638–10644. doi: 10.1128/JVI.77.19.10638-10644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan S, Gordon J, Del Valle L, Cui J, Khalili K. Intracellular approach for blocking JCV gene expression using RNA interference during viral infection. J Virol. 2004;78:7264–7269. doi: 10.1128/JVI.78.13.7264-7269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray FA, Peabody DS, Cooper JL, Cram LS, Kraemer PM. SV40 T antigen alone drives karyotype instability that precedes neoplastic transformation of human diploid fibroblasts. J Cell Biochem. 1990;42:13–31. doi: 10.1002/jcb.240420103. [DOI] [PubMed] [Google Scholar]
- Ray FA, Meyne J, Kraemer PM. SV40 T antigen induced chromosomal changes reflect a process that is both clastogenic and aneuploidogenic and is ongoing throughout neoplastic progression of human fibroblasts. Mutat Res. 1992;284:265–273. doi: 10.1016/0027-5107(92)90011-p. [DOI] [PubMed] [Google Scholar]
- Ray FA, Waltman MJ, Lehman JM, Little JB, Nickoloff JA, Kraemer PM. Identification of SV40 T-antigen mutants that alter T-antigen-induced chromosome damage in human fibroblasts. Cytometry. 1998;31:242–250. [PubMed] [Google Scholar]
- Ricciardiello L, Baglioni M, Giovannini C, Pariali M, Cenacchi G, Ripalti A, Landini MP, Sawa H, Nagashima K, Frisque RJ, Goel A, Boland CR, Tognon M, Roda E, Bazzoli F. Induction of chromosomal instability in colonic cells by the human polyomavirus JC virus. Cancer Res. 2003;63:7256–7262. [PubMed] [Google Scholar]
- Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004;23:546–553. doi: 10.1038/sj.onc.1207098. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubeyrand S, Pope L, De Chasseval R, Gosselin D, Dong F, de Villartay JP, Hache RJ. Artemis phosphorylated by DNA-dependent protein kinase associates preferentially with discrete regions of chromatin. J Mol Biol. 2006;358:1200–1211. doi: 10.1016/j.jmb.2006.02.061. [DOI] [PubMed] [Google Scholar]
- Stewart N, Bacchetti S. Expression of SV40 large T antigen, but not small t antigen, is required for the induction of chromosomal aberrations in transformed human cells. Virology. 1991;180:49–57. doi: 10.1016/0042-6822(91)90008-y. [DOI] [PubMed] [Google Scholar]
- Theile M, Grabowski G. Mutagenic activity of BKV and JCV in human and other mammalian cells. Arch Virol. 1990;113:221–233. doi: 10.1007/BF01316675. [DOI] [PubMed] [Google Scholar]
- Trojanek J, Croul S, Ho T, Wang JY, Darbinyan A, Nowicki M, Del Valle L, Skorski T, Khalili K, Reiss K. T-antigen of the human polyomavirus JC attenuates faithful DNA repair by forcing nuclear interaction between IRS-1 and Rad51. J Cell Physiol. 2006;206:35–46. doi: 10.1002/jcp.20425. [DOI] [PubMed] [Google Scholar]
- Vacante DA, Traub R, Major EO. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology. 1989;170:353–361. doi: 10.1016/0042-6822(89)90425-x. [DOI] [PubMed] [Google Scholar]
- White MK, Gordon J, Reiss K, Del Valle L, Croul S, Giordano A, Darbinyan A, Khalili K. Human polyomaviruses and brain tumors. Brain Res Brain Res Rev. 2005;50:69–85. doi: 10.1016/j.brainresrev.2005.04.007. [DOI] [PubMed] [Google Scholar]
- White MK, Skowronska A, Gordon J, Del Valle L, Deshmane SL, Giordano A, Croul S, Khalili K. Analysis of a mutant p53 protein arising in a medulloblastoma from a mouse transgenic for the JC virus early region. Anticancer Res. 2006 In Press. [PubMed] [Google Scholar]
- Woods C, LeFeuvre C, Stewart N, Bacchetti S. Induction of genomic instability in SV40 transformed human cells: sufficiency of the N-terminal 147 amino acids of large T antigen and role of pRB and p53. Oncogene. 1994;9:2943–2950. [PubMed] [Google Scholar]
- Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, Heng H, Livingston D. SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev. 2004;18:1305–1316. doi: 10.1101/gad.1182804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia SJ, Shammas MA, Shmookler Reis RJ. Elevated recombination in immortal human cells is mediated by HsRAD51 recombinase. Mol Cell Biol. 1997;17:7151–7158. doi: 10.1128/mcb.17.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Succi J, Feng Z, Prithivirajsingh S, Story MD, Legerski RJ. Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response. Mol Cell Biol. 2004;24:9207–9220. doi: 10.1128/MCB.24.20.9207-9220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]