

Abstract
The human immunodeficiency virus (HIV) fusion peptide (HFP) is the N-terminal apolar region of the HIV gp41 fusion protein and interacts with target cell membranes and promotes membrane fusion. The free peptide catalyzes vesicle fusion at least to the lipid mixing stage and serves as a useful model fusion system. For gp41 constructs which lack the HFP, high-resolution structures show trimeric protein and suggest that at least three HFPs interact with the membrane with their C-termini in close proximity. In addition, previous studies have demonstrated that HFPs which are cross-linked at their C-termini to form trimers (HFPtr) catalyze fusion at a rate which is 15−40 times greater than non-cross-linked HFP. In the present study, the structure of membrane-associated HFPtr was probed with solid-state nuclear magnetic resonance (NMR) methods. Chemical shift and intramolecular 13CO-15N distance measurements show that the conformation of the Leu-7 to Phe-11 region of HFPtr has predominant helical conformation in membranes without cholesterol and β strand conformation in membranes containing ∼30 mol% cholesterol. Interstrand 13CO-13CO and 13CO-15N distance measurements were not consistent with an in-register parallel strand arrangement but were consistent with either: (1) parallel arrangement with adjacent strands two-residues out-of-register; or (2) antiparallel arrangement with adjacent strand crossing between Phe-8 and Leu-9. Arrangement (1) could support the rapid fusion rate of HFPtr because of placement of the apolar N-terminal regions of all strands on the same side of the oligomer while arrangement (2) could support the assembly of multiple fusion protein trimers.
Keywords: HIV, fusion peptide, cholesterol, membranes, NMR, trimer
Enveloped viruses such as human immunodeficiency virus (HIV) are surrounded by a membrane and infect cells through fusion between the viral membrane and the target cell membrane (1, 2). This process is mediated by envelope proteins which traverse the viral membrane (3). For HIV, the gp41 envelope protein has a ∼170-residue ectodomain region which lies outside the viral membrane and contains a N-terminal ∼20-residue apolar “fusion peptide” (HFP). The HFP interacts with the target cell membrane and plays an essential role in membrane fusion (4). Peptides composed of the HFP sequence induce fusion between large unilamellar vesicles (LUVs) and between erythrocytes (5, 6). There are similar mutation/fusion activity relationships for HFP-induced fusion and HIV-induced fusion which suggests that HFP is a useful model system to understand some aspects of viral/target cell fusion (7-10). There is also evidence for participation of other regions of gp41 in membrane fusion (11-14). There are atomic-resolution structures of the “soluble ectodomain” of gp41 which does not contain the ∼30 N-terminal residues of gp41 (including the HFP) (2, 15-19). These structures are believed to correspond to the conformation after fusion has occurred and perhaps during some fusion steps (20). The gp41 molecules form a trimer in the soluble ectodomain structure with the three N-termini in close proximity at the end of an in-register helical coiled-coil. It has therefore been hypothesized that during viral/target cell fusion, at least three HFPs insert into the target cell membrane with their C-termini in close proximity. There is some antibody-based evidence for this hypothesis (21).
A variety of experimental methods have shown that membrane-associated HFP can assume either helical or nonhelical structure (5, 7, 22-32). Models for the helical structure have been developed based on nuclear magnetic resonance (NMR), electron spin resonance (ESR), infrared (IR), and circular dichroism (CD) data, as well as computer simulations (33-39). A β hairpin model for non-helical structure has been proposed based on IR and surface activity measurements in membranes (40).
Fluorescence, ESR, IR, and solid-state NMR data also suggest that the nonhelical HFPs in membranes form oligomeric structures (10, 41-44). These oligomeric structures may be important as evidenced by envelope protein trimerization and by experiments and modeling which indicate that the fusion site contains multiple trimers and a corresponding high HFP concentration (17, 18, 45). Additionally, the V2E mutation in gp41 dominantly interferes with HIV fusion and infectivity and suggests that oligomeric HFP is important in viral/target cell fusion (8, 10).
In an effort to mimic the oligomeric HFP topology suggested by the gp41 soluble ectodomain structures, HFP oligomers in dimeric (HFPdm) and trimeric form (HFPtr) have been synthesized with chemical cross-linking at their peptide C-termini. Cross-linking was accomplished with either cysteine or lysine residues. Comparative measurements were made between HFPdm and HFPtr and monomeric HFP (HFPmn) (28, 46). All three constructs contained additional C-terminal lysines to inhibit intermolecular self-association in aqueous solution and buffer conditions were found with minimal self-association. The rate of peptide-induced intervesicle lipid mixing was 15−40 times higher for HFPtr relative to HFPmn and suggests that oligomeric HFP topology plays a significant role in membrane fusion. The HFPdm rate was intermediate between the rates of HFPmn and HFPtr. Enhanced fusogenicity has also been observed for other oligomeric constructs which contain fusion peptides and support the hypothesis that oligomeric fusion peptide topology is one factor which contributes to fusion (47-49).
Solid-state NMR structural experiments have been carried out on membrane-associated HFPmn, HFPdm, and HFPtr. Backbone 13C carbonyl chemical shift and 2D exchange measurements suggest a predominant β strand conformation for HFPmn associated with membranes whose lipid headgroup and cholesterol composition is comparable to that found in host cells of the virus (25). In addition, the Phe-8 carbonyl chemical shift of HFPdm and HFPtr in the same host cell-like membrane composition is upfield of the random coil value and is more consistent with β strand conformation than with helical conformation (28, 46). For HFPtr associated with membranes which do not contain cholesterol, the Phe-8 chemical shift is downfield of the random coil value and is more consistent with helical conformation. In the present study, membrane-associated HFPtr was investigated using solid-state NMR methods including 13C chemical shifts and 13C-15N distance measurements to probe local peptide conformation and 13C-13C and 13C-15N distance measurements to probe interpeptide β strand arrangement. It had previously been proposed that the increased lipid mixing rate induced by β strand HFPdm and HFPtr was related to predominant parallel strand arrangement enforced by the C-terminal cross-linking (28). For this arrangement, the apolar N-terminal regions of the three strands would be on the same side of the oligomer and the resulting large apolar volume would cause significant membrane perturbation and rapid fusion rate. This structure would differ from the mixture of parallel and antiparallel arrangements observed for membrane-associated HFPmn (43). In this paper, an in-register parallel strand model for HFPtr was tested using a solid-state NMR 13CO-13CO distance measurement approach which had been previously applied to probe structure in β amyloid fibrils (50, 51).
The 13C-15N distances were determined with the rotational-echo double-resonance (REDOR) technique which is an approach to measure heteronuclear dipolar couplings under magic angle spinning (MAS) (52-60). MAS reduces solid-state NMR linewidths and increases signal-to-noise and the dipolar coupling dCN depends on rCN distance as (rCN)−3. The 13C-13C distances were determined with a constant-time double-quantum build-up with finite pulses (fpCTDQBU) technique which is an approach to measure homonuclear dipolar couplings under MAS (51, 61-65). The dipolar coupling dCC depends on rCC distance as (rCC)−3. This fpCTDQBU method is a finite pulse variant of the CTDQBU technique developed by Bennett et al. and incorporation of finite pulses increases the signal-to-noise, as will be discussed in the Results section (64). The utility of finite pulses was first demonstrated by Ishii in the related constant-time finite-pulse radiofrequency-driven recoupling (fpRFDR-CT) method and the two techniques are robust methods for determination of dCC with weak dependences on chemical shift differences, chemical shift anisotropy (CSA), resonance offset, spin relaxation, and rf inhomogeneity (51, 65).
Quantitative analysis of the REDOR and fpCTDQBU data requires explicit computer simulation of the experiments as a function of molecular geometry parameters. The SIMPSON computer program was used for this purpose and allows input of the geometric parameters as well as the NMR parameters used in the experiment (66). The HFPtr results in the present paper also rely on careful calibration of the REDOR and fpCTDQBU experiments with model compounds for which the 13C-15N and 13C-13C distances are known and comparable to distances in HFPtr structural models.
MATERIALS AND METHODS
Materials
Rink amide resin and Wang Resin were purchased from Advanced Chemtech (Louisville, KY). N-α-fluorenylmethoxycarbonyl (FMOC)-N'-ε-4-methyltrityl-l-lysine (FMOC-Lys(Mtt)) was purchased from Calbiochem-Novabiochem (La Jolla, CA), and FMOC amino acids and FMOC-β-Ala-Wang resin were obtained from Peptides International (Louisville, KY). For the labeled peptide syntheses, labeled amino acids were purchased from Icon Services Inc (Summit, NJ) and were FMOC-protected using literature methods (67, 68). In addition, FMOC-1-13C glycine was purchased from Sigma-Aldrich (St. Louis, MO). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-(1-glycerol) (POPG), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), phosphatidylinositol (PI), sphingomyelin, 1,2-di-O-tetradecyl-sn-glycero-3-phosphocholine (DTPC), 1,2-di-O-tetradecyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DTPG), and 1,2-di-O-phytanyl-sn-glycero-3-phosphoethanolamine (DPhPE) were purchased from Avanti Polar Lipids (Alabaster, AL). N-(2-hydroxyethyl)piperazine-N'-2-ethanesulfonic acid (HEPES) was obtained from Sigma-Aldrich (St. Louis, MO). The buffer solution used in the study contained 5 mM HEPES (pH 7.0) with 0.01% NaN3.
HFPtr synthesis
The “HFPtr-L7CF11N” fusion peptide trimer was synthesized with sequence (AVGIGALFLGFLGAAGSTMGARSWKKKKKK)3-Aβ and with a 13CO label at Leu-7 and a 15N label at Phe-11 on each strand. The related “HFPtr-F8CL9N” trimer was synthesized with sequence (AVGIGALFLGFLGAAGSTWMGARSKKKKKK)3-Aβ and with a 13CO label at Phe-8 and a 15N label at Leu-9 on each strand. Residues in the N-termini of HFPtr correspond to the 23 N-terminal residues (sequence AVGIGALFLGFLGAAGSTMGARS) of the LAV1a strain of the HIV-1 gp41 envelope fusion protein. HFPtr synthesis began with coupling of FMOC-Lys(Mtt) and activated β-Ala-Wang resin (46). A lysine trimer backbone was then formed with coupling of FMOC-Lys(Mtt) followed by coupling with N-α-FMOC-N'-ε-tert-butoxycarbonyl-l-lysine (FMOC-Lys(Boc)). Prior to the second and third couplings, the Mtt group on the lysine side chain was removed with 1% trifluoroacetic acid (TFA) in dichloromethane (DCM). The rest of the trimer was then synthesized using a peptide synthesizer (ABI 431A, Foster City, CA) equipped for standard FMOC chemistry. Cleavage of HFPtr from the resin was done in a 3 h reaction with a mixture of TFA-H2O-phenol-thioanisole-ethanedithiol in a 33:2:2:2:1 volume ratio and purification was done with reversed-phase HPLC and a preparative C18 column (Vydac, Hesperia, CA) and a water-acetonitrile gradient containing 0.1% TFA. Mass spectroscopy was used for peptide identification.
N-acetyl-L-leucine (NAL)
1-13C, N-13C doubly labeled NAL (D-NAL) was synthesized as a setup compound for fpCTDQBU experiments (69). Synthesis of D-NAL began with addition of 1-13C, 15N leucine to glacial acetic acid and heating to 100 °C. 1-13C acetic anhydride was then added, the solution was cooled to 80 °C, and water was added to react with any excess acetic anhydride. The D-NAL product was washed with cyclohexane, and the cyclohexane was subsequently removed under vacuum. Residual solvents were removed by water aspirator vacuum, drying in a vacuum dessicator and dissolution in water followed by lyophilization. Solution 1H and 13C NMR confirmed the identity, purity and labeling of D-NAL. The NAL sample for solid-state NMR was prepared by aqueous dissolution of a 1:9 mixture of D-NAL and unlabeled NAL (ICN, Aurora, OH) followed by slow evaporation of the water. The polycrystalline solid was ground with a mortar and pestle. The intramolecular 13C-13C distance of D-NAL in the crystal is 3.1 Å (70).
Glycyl-L-phenylalanyl-L-phenylalanine (GFF)
Doubly labeled GFF (D-GFF) was synthesized with13CO labels at Gly-1 and Phe-3 and was a model compound for the fpCTDQBU experiments. D-GFF synthesis began with dissolution of FMOC-1-13C-Phe in a N-methylpyrrolidone (NMP)/DCM mixture followed by 2 h coupling of the amino acid to unsubstituted Wang resin using a mixture of 1.0 M N,N'-dicyclohexylcarbodiimide in NMP and 0.1 M 4-dimethylaminopyridine in dimethylformamide. Unreacted amino groups on the resin were benzoylated with benzoic anhydride. Standard FMOC chemistry was used for the remaining synthesis with 4 h and 8 h coupling times for FMOC-Phe and FMOC-1-13C-Gly, respectively. D-GFF was cleaved from the resin in a 3 h reaction with a mixture of TFA-water-phenol in a 33:2:2 volume ratio and precipitated by addition of t-butyl methyl ether (TBME). The yellowish-white precipitate was washed with TBME, centrifuged, lyophilized, dissolved in water, and passed through a 4−8 μm filter. After slow evaporation of the water, colorless and needle-like D-GFF hemihydrate crystals were harvested. A small aliquot of the crystals was dissolved in dimethylsulfoxide-d6 and subsequent 1H and 13C NMR spectroscopy confirmed the purity and labeling of D-GFF. The solid-state NMR sample of GFF was prepared by aqueous dissolution of D-GFF and unlabeled GFF (Sigma-Aldrich) in a 1:49 ratio followed by slow evaporation of solvent. The polycrystalline solid was ground with a mortar and pestle. The intramolecular 13C-13C distance of D-GFF in the crystal is 5.40 Å (71).
I4 peptide
A 17-residue acetylated and amidated “I4” peptide with sequence Ac-AEAAAKEAAAKEAAAKA-NH2 was synthesized with a 13CO label at Ala-9 and a 15N label at Ala-13. The solid-state NMR I4 peptide sample was lyophilized from aqueous solution and is predominantly (83 ± 6%) α helical at Ala-9 (72). The two labeled nuclei should have a 13C-15N internuclear distance of ∼4.1 Å in the helical structure.
Lipid preparation
HFPtr samples were prepared using three lipid/cholesterol mixtures: (a) “PC-PG” had POPC and POPG in a 4:1 mol ratio and has a headgroup composition similar to that used in previous fusion peptide and fusion protein biophysical studies (73-76); (b) “LM3” had POPC, POPE, POPS, sphingomyelin, PI, and cholesterol in a 10:5:2:2:1:10 mol ratio and reflects the lipid headgroup and cholesterol composition of cells infected by HIV (77, 78); and (c) “LM3e” had DTPC, DTPG, DPhPE and cholesterol in a 9:3:3:10 mol ratio and was similar to LM3 in lipid headgroup and cholesterol composition but contained ether-linked rather than ester-linked lipids to eliminate natural abundance lipid 13CO signals. Lipid and cholesterol powders were dissolved in chloroform. The chloroform was removed under a stream of nitrogen followed by overnight vacuum pumping. Lipid dispersions were formed by addition of 5 mM pH 7.0 HEPES buffer followed by homogenization with 10 freeze-thaw cycles. Large unilamellar vesicles (LUVs) were prepared by extrusion through a filter with 100 nm diameter pores (79).
Solid-state NMR sample preparation
The samples were made in a manner similar to that used for doing functional fusion assays (46). HFPtr (0.1−0.2 μmol as determined by A280) was dissolved in ∼2 mL of 5 mM HEPES buffer and LUVs (∼30 μmol total lipid) were prepared in ∼2 mL of buffer. The peptide and LUV solutions were mixed and kept at room temperature overnight followed by ultracentrifugation at 35000 rpm for 5 h at 4 °C. The peptide/lipid pellet formed after ultracentrifugation was transferred by spatula to a 4 mm diameter MAS NMR rotor. Unbound HFPtr is predominantly monomeric under these conditions and does not pellet. For the HFPtr and other samples, the rotor volume of ∼40 μL was filled.
Solid-state NMR spectroscopy
Experiments were done on a 9.4 T spectrometer (Varian Infinity Plus, Palo Alto, CA) using a MAS probe in double resonance 13C/1H configuration for fpCTDQBU experiments and in triple resonance 13C/1H/15N configuration for REDOR experiments. Relative to the triple resonance configuration, the double resonance configuration has 1.5 times higher 13C sensitivity. The NMR detection channel was tuned to 13C at 100.8 MHz, the decoupling channel was tuned to 1H at 400.8 MHz, and for REDOR the third channel was tuned to 15N at 40.6 MHz. 13C shifts were externally referenced to the methylene resonance of adamantane at 40.5 ppm which allowed direct comparison with 13C shifts of proteins in aqueous solution (80, 81). 15N shifts were referenced to (15NH4)2SO4 at 20 ppm. Experiments used a MAS frequency of 8000 ± 2 Hz and spacers were used to restrict samples to the central 2/3 rotor volume (∼40 μL) in which rf field variation was less than 10%.
Experiments were performed at −50 °C rather than room temperature in order to achieve more efficient cross-polarization (CP) and greater signal per 13C nucleus. There are similar 13C backbone chemical shifts at both low temperature and room temperature, suggesting that cooling the sample does not cause significant peptide structural changes (29). The 1H, 13C and 15N pulse lengths and the CP matching condition were approximately obtained by running direct pulse and CP experiments on adamantane and (15NH4)2SO4. Further parameter optimization was made with model compounds. Recycle delays were between 1 and 2 s.
REDOR spectroscopy
The “all-but-one 15N π pulse” version was used to suppress natural abundance 13C signals and selectively detect the labeled Phe-8 13CO signal of HFPtr-F8CL9N (27, 52). An initial 2 ms CP was done with a ramped 40−50 kHz 13C rf field and a ∼60 kHz 1H rf field and was followed by a 1 ms dephasing period and then 13C detection. A single ∼50 kHz 13C refocusing π pulse was placed at the midpoint of the dephasing period, and two-pulse phase-modulation (TPPM) 1H decoupling of ∼90 kHz was applied during dephasing and detection (82). The 13C transmitter was set to ∼155 ppm and the 15N transmitter was set to ∼115 ppm. An “S0” and an “S1” acquisition were obtained. The dephasing period during the S1 acquisition contained a 40 kHz 15N π pulse at the midpoint and at the end of each rotor cycle except for the fourth and the eighth cycles. XY-8 phase cycling (0, 90, 0, 90, 90, 0, 90, 0) was used for the 15N pulses (83). The 15N pulses disrupted averaging of the 13C-15N dipolar coupling by MAS and led to selective attenuation of the Phe-8 13CO signal. The S0 acquisition did not contain 15N pulses and 13C-15N dipolar coupling was efficiently averaged by MAS and did not lead to 13C signal attenuation. The selective spectrum of Phe-8 13CO was obtained by subtracting the S1 signal from the S0 signal.
13CO-15N distances were measured with an “alternating 15N/13C π pulse” version of REDOR (53, 84). For the S1 acquisition, the dephasing period of length τi included a ∼40 kHz 15N π pulse at the midpoint of each rotor cycle and a ∼50 kHz 13C π pulse at the end of each rotor cycle except for the last cycle. The S0 acquisition did not include 15N π pulses during the dephasing period. TPPM 1H decoupling of ∼90 kHz was applied during dephasing and detection. 13C signals were attenuated by 13C-15N dipolar coupling in the S1 acquisition but not in the S0 acquisition. Spectra were acquired for different τi and the difference between the 13CO S0 and S1 signal intensities divided by the S0 signal intensity (ΔS/S0)i was the experimental parameter used to determine 13CO-15N dipolar couplings and distances. XY-8 phase cycling was used for the 15N π pulses and for all of the 13C π pulses except the final pulse. Individual S0 or S1 transients were added with phase cycling: 1H π/2, 0, 180, 0, 180; 1H CP, 90, 90, 90, 90; 13C CP 270, 270, 180, 180; final 13C π pulse, 270, 270, 180, 180; receiver, 180, 0, 90, 270.
The S0 signal intensity of the I4 peptide was used to optimize 1H π/2, 1H decoupling, 13C π, and CP rf fields, and the ΔS/S0 values of the I4 peptide were used to optimize the 15N π rf field.
fpCTDQBU spectroscopy
The NMR sequence has the form CP90+ζ – (fpRFDR)L – π/2ζ – π/20 – (fpRFDR)M – π/2180 – π/290 – (fpRFDR)N – acquisition where L, M, and N refer to the number of rotor cycles in each fpRFDR period and the other subscripts refer to the rf phases (64). Generation of transverse 13C magnetization during CP was followed by fpRFDR periods which contained finite 13C π pulses and during which the magnetization may be reduced by net 13C-13C dipolar coupling. The transverse magnetization was then detected during the acquisition period. For each value of ζ = 0, 90, 180, and 270, a distinct spectrum was obtained. The π/2180 - π/290 pulse pair served to refocus 13C-13C dipolar coupling so that there was no net coupling over the period which began N rotor cycles prior to the pulse pair and ended at the beginning of acquisition. For ζ = 90 or 270, there was similarly no net dipolar coupling over the period which began after CP and ended L rotor cycles after the π/2ζ - π/20 pulse pair. By contrast, for ζ = 0 or 180, there was net dipolar coupling during the dephasing period of duration τi = 2L × τR where τR is the rotor period. For each value of τi, S0 and S1 signals were acquired with S0 = Sζ=90 + Sζ=270 and S1 = Sζ=0 + Sζ=180. The parameter (ΔS/S0)i was calculated as in the REDOR experiment and served as the experimental constraint for determination of 13C-13C dipolar couplings and distances.
A single value of M and a single value of L + M + N were used for all τi values in a given experiment. The transverse magnetization therefore evolved for a single “constant-time” for all τi and the contribution to (ΔS/S0)i from effects other than the 13C-13C dipolar couplings was approximately independent of τi (85). Experiments were initially attempted with the non-constant-time and higher signal-to-noise sequence CP90+ζ – (fpRFDR)L – π/2ζ – π/20 – (fpRFDR)L – acquisition but the (ΔS/S0)i values obtained with this sequence for model compounds fitted poorly to simulations based on known 13CO-13CO distances. Constant-time was critical for quantitative interpretation of the data.
Other experimental details included: (1) the 13C transmitter was set to ∼177 ppm; (2) the CP period had 2 ms duration, ramped 40−50 kHz 13C rf field and a ∼54 kHz 1H rf field; (3) 13C π/2 pulses had ∼43 kHz rf field; (4) fpRFDR periods had one 13C π pulse every two rotor cycles and the pulse was centered in the two cycle increment; and (5) continuous-wave 1H decoupling at ∼95 kHz and ∼60 kHz were applied during the fpRFDR and acquisition periods, respectively. XY-8 phase cycling was used for fpRFDR π pulse train and the other pulses had phases: 1H π/2, 180, 0, 0, 180; 1H CP, 90, 90, 90, 90; 13C CP 270, 180, 270, 180. The corresponding values of ζ were 0, 90, 180, 270. Calibration of rf fields was done using the D-NAL sample. Both the D-NAL and D-GFF samples were used to study the effects of “finite” 13C π pulses, i.e. pulses with longer duration and concomitant smaller rf field.
Experimental data analysis
Integration of signal intensities in the isotropic carbonyl regions of the S0 and S1 spectra for a particular dephasing time τi yielded values denoted as S0i. and S1i. (ΔS/S0)iexp is a normalized dephasing parameter:
| (1) |
For each pair of S0i, S1i spectra, an experimental uncertainty σ was calculated as the root mean squared deviation of integrated intensities in 24 regions of the spectra without signal. The uncertainty in (ΔS/S0)iexp is denoted σiexp and was calculated (86):
| (2) |
Determination of distances between labeled nuclei relies on comparision of (ΔS/S0)iexp and (ΔS/S0)isim where sim ≡ simulated. The (ΔS/S0)isim only consider the labeled spins whereas (ΔS/S0)iexp has contributions from natural abundance nuclei and other effects. The (ΔS/S0)iexp values were therefore adjusted to remove these other contributions and the resulting corrected (ΔS/S0)i cor values were compared to (ΔS/S0)isim to obtain distances between labeled nuclei. We briefly discuss determination of (ΔS/S0)i cor for the REDOR measurements on the HFPtr-L7CF11N samples. The Supporting Information section provides a detailed description of the derivation of (ΔS/S0)icor for all of the samples and experiments.
Determination of (ΔS/S)icor relies on consideration of contributions to S0 and S1 from 13CO nuclei in different environments. The following parameters/approximations are used for REDOR of HFPtr-L7CF11N: (1) There is 99% labeling of the Leu-7 13CO and Phe-11 15N sites. S1 = S0 for a labeled Leu-7 13CO in a peptide strand with a Phe-11 14N. (2) S1 = 0 for a labeled Leu-713CO separated by one or two bonds from a natural abundance 15N at Phe-8 or Leu-7. The Leu-7 S1 is not affected by other natural abundance 15N. (3) S1 = 0 for natural abundance backbone 13COs at Gly-10 or Phe-11 which are separated by one or two bonds from the labeled Phe-11 15N. S1 = S0 for other natural abundance backbone 13CO sites. Criteria (1) and (2) are based on the close distance (≤ 2.5 Å) and consequent strong (≥ 200 Hz) dipolar coupling of 13CO and 15N nuclei separated by one or two bonds.
As described in the Supporting Information section, an expression for (ΔS/S0)icor can be derived:
| (3) |
where UC and UN are the fractional abundances of Leu-7 12CO and Phe-11 14N, respectively, AC and AN are the fractional 13C and 15N natural abundances, respectively, and n is the average number of unlabeled CO sites per peptide strand. For the HFPtr samples, values of UC, UN, AC, AN, and n are 0.01, 0.01, 0.011, 0.0037, and 29.33, respectively. Eq. 3 leads to (ΔS/S0)icor /(ΔS/S0)iexp ratios of ∼1.3. The value of σ cori was calculated by multiplying σ expi by the prefactor for (ΔS/S0)iexp in Eq. 3 (86).
Simulations
(ΔS/S0)isim were calculated as a function of internuclear distances between two or three labeled nuclei and as a function of τi. The simulations were done with the SIMPSON program and incorporated the MAS frequency and the 13C and 15N rf fields, pulse lengths, timing, and phases, as well as 13C resonance offsets and CSA principal values and axis directions (66). The REDOR simulations were based on a single 13CO/15N spin pair and did not consider the 15N chemical shift or CSA. The fpCTDQBU simulations were based on either two or three 13CO nuclei and did not consider 15N spins. In the experiments, the effects of 1H and 14N spins were largely removed by 1H decoupling, constant-time, and/or MAS, and these spins were not incorporated in any of the simulations.
Orientation-dependent simulation input parameters included 13CO CSA principal values, the Euler angles which relate the 13CO CSA principal axis system(s) to a fixed crystal axis system, and the Euler angles which relate the 13CO-15N or 13CO-13CO internuclear vectors to the crystal axis system. Although distance determination by REDOR and fpCTDQBU experiments is not strongly dependent on these parameters, an effort was made to use reasonable values for the parameters. The (δ11, δ22, δ33) CSA principal values for the I4 Ala-9, HFPtr-L7CF11N/PC-PG Leu-7, and HFPtr-L7CF11N/LM3e Leu-7 13COs were set to (246, 203, 86), (248, 202, 85) and (242, 195, 83) ppm, respectively, and were based on experimentally measured isotropic chemical shifts and literature CSA values (87). For GFF, the Gly-1 and Phe-3 CSA principal values were (255, 168, 91) and (243, 193, 106) ppm, respectively, and were determined by fitting experimental centerband and spinning sideband intensities with the Herzfeld-Berger method (88).
The Euler angles which relate a 13CO CSA principal axis system to the crystal axis system can be considered in terms of the relative orientation of the principal axis system and the 13CO chemical bonds and the structure and orientation of the molecule relative to the crystal axes. For I4 Ala-9, HFPtr Leu-7, and GFF Gly-1 13COs, the principal axis/chemical bond orientations were: (1) the δ33 axis perpendicular to the peptide plane; and (2) the δ22 axis tilted 130° from the CO-N bond (87). GFF Phe-3 has COO− rather than amide functionality and its δ33 axis was perpendicular to the OCO plane and its δ11 axis bisected the OCO angle (89, 90).
The crystal frame Euler angles for the 13CO principal axis systems and the internuclear vectors were calculated using a Mathematica program whose inputs were atomic coordinates from high-resolution structures and the previously detailed 13CO CSA principal axis directions. The atomic coordinates for GFF were obtained from its crystal structure while for parallel β strand and antiparallel β strand structures, coordinates were obtained respectively from the crystal structure of cutinase (PDB file name 1cex) and from the crystal structure of human gamma-D crystalline R58H mutant (PDB file name 1h4a) (71, 91, 92). The latter two structures have been refined to 1.0 and 1.15 Å resolution, respectively. For REDOR simulations of the I4 and HFPtr-L7CF11N/PC-PG samples, the Ala-56 CO and Glu-60 N coordinates of cutinase were used. These residues are in a helical region of cutinase and the labeled 13CO chemical shifts of the two solid-state NMR samples correlate with helical conformation. The REDOR simulation of the HFPtr-L7CF11N/LM3e sample used coordinates of Leu-114 CO and Gly-118 N in a β strand region of cutinase because the 13CO shift of this sample correlated with β strand conformation. The two-spin fpCTDQBU simulation of the HFPtr-L7CF11N/LM3e sample used cutinase Ile-37 CO and Ala-116 CO coordinates, and the three-spin fpCTDQBU simulation of the HFPtr-L7CF11N/LM3e sample used cutinase Ile-37 CO, Ala-116 CO and Thr-144 CO coordinates. In cutinase, these are in-register residues in a parallel β strand region and this was our initial structural model for β strand HFPtr.
Signal-to-noise
Experiments and simulations were carried out to assess the effect of the 13C π rf field on the GFF fpCTDQBU signal-to-noise ratio. Comparison was made between the experimentally-based signal-to-noise parameter:
| (4) |
and simulation:
| (5) |
where k is an adjustable parameter to provide overall comparison between (Signal/Noise)icor and (Signal/Noise)isim values for a set of τi.
Fitting of internuclear distances
The distance between labeled nuclei was determined by χ2 fitting:
| (6) |
where T is the number of τi and d is the dipolar coupling. In units of Å, the 13C-13N distance rCN = (3110 Hz/dCN)1/3 and the 13C-13C distance rCC = (7720 Hz/dCC)1/3 (93). The best-fit d is the one for which χ2 has global minimum value χ2min and the uncertainty σd is set by the values of d corresponding to χ2 = χ2min + 1 (86). According to statistics theory, the most likely value of χ2min is the number of degrees of freedom of the fit, ν, and ν = T−1 in the REDOR and fpCTDQBU analyses (94). The χ2min values in the fittings were close to ν and were consistent with reasonably accurate evaluation of the σ cori in Eq. 6.
RESULTS
13CO chemical shifts
Liquid-state and solid-state NMR studies on proteins have shown an empirical correlation between the 13CO chemical shift and local conformation with higher shifts correlating with helical structure and lower shifts correlating with β strand structure (81, 95, 96). In the present study, 13CO shifts at Leu-7 and Phe-8 were measured for membrane-associated HFPtr and it was observed that there is a strong dependence of shifts on membrane composition. The Phe-8 chemical shift in membrane-associated HFPtr-F8CL9N was specifically detected using REDOR filtering (Fig. 1a,b) and peak shifts of 178.4 and 172.5 ppm were observed in PC-PG and LM3 membranes, respectively. These values correlate with database distributions of Phe 13CO shifts for helical (177.1 ± 1.4 ppm) and β strand (174.2 ± 1.6 ppm) conformations (81). As displayed in Fig. 1c, the REDOR-filtered peak Leu-7 13CO shift in HFPtr-L7CF11N/PC-PG was 178.8 ppm. This shift agreed better with the database distribution of Leu 13CO in helical conformations (178.5 ± 1.3 ppm) than with the distribution in β strand conformations (175.7 ± 1.5 ppm).
Figure 1.
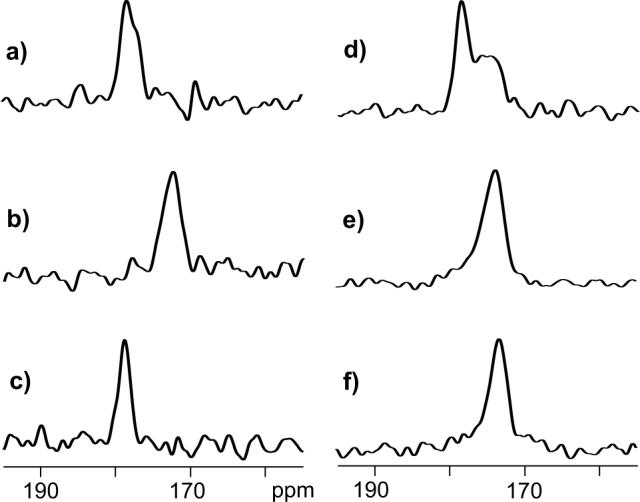
13C spectra of HFPtr-F8CL9N associated with (a) PC-PG and (b) LM3 membranes and of HFPtr-L7CF11N associated with (c, d) PC-PG, (e) LM3, and (f) LM3e membranes. The HFPtr:lipid mol ratio was ∼0.003 in the samples used to obtain spectra a and b and ∼0.007 in the samples used to obtain spectra c-f. Spectra a, b, and c are REDOR-filtered with τi = 1.0, 1.0, and 32.25 ms, respectively, and have 13CO peak chemical shifts of 178.4 (Phe-8), 172.5 (Phe-8), and 178.8 ppm (Leu-7), respectively. Spectra d, e, and f are REDOR S0 spectra with 32.25 ms dephasing period and have 13CO peak chemical shifts of 178.6 ppm, 173.8 ppm, and 173.4 ppm, respectively. Each spectrum was processed with 100 Hz Gaussian line broadening and baseline correction. The MAS frequency was 8000 Hz and the numbers of scans used to obtain spectra a, b, c, d, e, and f are 118784, 132864, 46048, 23024, 21760, and 98720, respectively.
In the HFPtr-L7CF11N/LM3 sample, there were relatively small values of ΔS/S00 and the Leu-7 13CO peak was not observed in the REDOR-filtered spectrum. REDOR S0 spectra were therefore used to obtain additional chemical shift information for the HFPtr-L7CF11N samples. The S0 spectrum of the HFPtr-L7CF11N/PC-PG sample (Fig. 1d) is a combination of a sharper signal peaked at 178.6 ppm and a broader signal centered near 176 ppm. Comparison with the REDOR-filtered spectrum (Fig. 1c) suggests that the downfield signal is primarily due to the labeled Leu-7 13CO while spin-counting and published spectra of 13CO labeled lipid samples suggest that the upfield signal is primarily due to natural abundance lipid 13CO (28). For the HFPtr-L7CF11N/LM3 sample (Fig. 1e), there is a single signal which is a superposition of intensity from the labeled Leu-7 and natural abundance lipid 13COs with a smaller contribution from natural abundance HFPtr 13COs. The HFPtr-L7CF11N/LM3e sample contains ether-linked rather than ester-linked lipids with consequent elimination of natural abundance 13CO lipid signals in the spectrum (Fig. 1f). The HFPtr-L7CF11N/LM3e spectrum is narrower than the HFPtr-L7CF11N/LM3 spectrum but the peak shift of the LM3e spectrum (173.4 ppm) is very similar to the peak shift of the LM3 spectrum (173.8 ppm) and suggests that the HFPtr conformation is insensitive to ether- vs. ester-linked lipids. The peak shifts are more consistent with Leu chemical shifts in β strand conformations (175.7 ± 1.5 ppm) than in helical conformations (178.5 ± 1.3 ppm).
Overall, the Leu-7 and Phe-8 peak HFPtr 13CO chemical shifts correlate with local helical conformation in PC-PG and with β strand conformation in LM3 and LM3e. Other studies of HIV and influenza fusion peptides suggest that the absence of cholesterol in PC-PG and its presence in LM3 and LM3e is an important determinant of peak chemical shifts and local conformations (27, 76).
13CO-15N distances
HFPtr-L7CF11N conformation was probed more quantitatively with REDOR determination of the distance between the labeled Leu-7 13CO and Phe-11 15N nuclei. If the Leu-7 to Phe-11 region has regular α helical conformation, the distance will be ∼4.1 Å while if the region has regular β strand conformation, the distance will be ∼11 Å. These distances correspond to dipolar couplings (dCN) of ∼45 Hz and ∼3 Hz, respectively.
The REDOR experiment was first validated using the helical I4 peptide with Ala-9 13CO and Ala-13 15N labels (Fig. 2a, b and Fig. 3). As τi was increased from 8.25 to 32.25 ms, there was a regular decrease in S1iexp/S0iexp values and concomitant increase in (ΔS/S0)iexp values. Using the approach and equations described in the Materials and Methods and Supplemental Material sections, the (ΔS/S0)icor were calculated from the (ΔS/S0)iexp and then fitted to the (ΔS/S0)isim calculated for an array of dCN values. The resulting dCN = 44.78 ± 0.22 Hz and rCN = 4.110 ± 0.007 Å are consistent with α helical structure between Ala-9 and Ala-13 and serve to validate the REDOR experiment.
Figure 2.
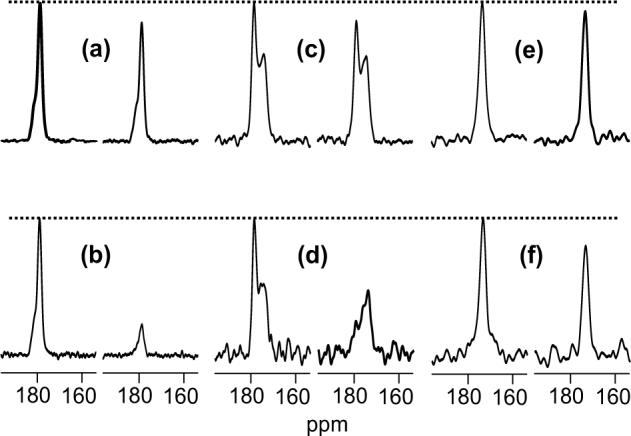
13C REDOR spectra of the (a, b) I4 peptide and HFPtr-L7CF11N associated with (c, d) PC-PG and (e, f) LM3e membranes. For each lettered pair of spectra, the S0 spectrum is on the left and the S1 spectrum is on the right. The MAS frequency = 8000 Hz, τR = 125 μs, and τi = 8.25 ms (spectra a, c, e) or τi = 32.25 ms (spectra b, d, f). Dotted lines are drawn at the peak S0 intensities. The I4 spectra were processed with 50 Hz Gaussian line broadening, the HFPtr spectra were processed with 100 Hz Gaussian line broadening, and baseline correction was applied to all spectra. The numbers of scans used to obtain each spectrum in panels a, b, c, d, e, and f are 32, 32, 37710, 23024, 40528, and 98720, respectively.
Figure 3.

(a) REDOR (ΔS/S0)icor (filled squares) and (ΔS/S0)isim (crosses) vs. dephasing time for the I4 peptide. Each (ΔS/S0)icor was based on a (ΔS/S0)iexp determined by integrations of 1 ppm regions in the S0 and S1 spectra. The integration region was centered at 178.8 ppm which is the peak shift in the S0 spectra. For each τi, there were 64 total (S0 + S1) scans. The values of σ cori are ∼0.005 and the heights of the black squares are approximately equal to the average value of 2 × σ cori. (b) A plot of χ2 vs. dCN yields dCN = 44.78 ± 0.22 Hz which corresponds to rCN = 4.110 ± 0.007 Å for the Ala-9 13CO/Ala-13 15N labeled pair. The uncertainty was determined using the approach described in the Materials and Methods section. The (ΔS/S0)isim values in plot a were calculated with the best-fit dCN.
REDOR experiments were then performed on the HFPtr-L7CF11N/PC-PG sample. For the Leu-7 13CO peak at 178.6 ppm, spectra with τi at 8.25 and 32.25 ms yielded S1iexp /S0iexp values which were in semi-quantitative agreement with the S1iexp/S0iexp values from the I4 sample (Fig. 2a-d). In the HFPtr spectra, the upfield natural abundance lipid 13CO signals near 175 ppm have (S1iexp/S0iexp ∼ 1 for all τi which is consistent with the expected large 13CO-15N distances and corresponding small dCN values for these nuclei. Fitting of the Leu-7 (ΔS/S0)icor to (ΔS/S0)isim yielded dCN = 44.8 ± 2.4 Hz and rCN = 4.11 ± 0.08 Å for the Leu-7 13CO/Phe-11 15N labeled pair (Fig. 4). These values are consistent with α helical structure between the labeled nuclei and correlate with the previously discussed downfield Leu-7 13CO chemical shift.
Figure 4.

(a) REDOR (ΔS/S0)icor (open squares with error bars) and (ΔS/S0)isim (crosses) vs. dephasing time for the HFPtr-L7CF11N/PC-PG sample. Each (ΔS/S0)icor was based on a (ΔS/S0)iexp determined from spectral integrations over a 1 ppm region centered at 178.6 ppm, the Leu-7 13CO peak chemical shift. The total (S0 + S1) numbers of scans used to obtain the (ΔS/S0)icor values for τi = 8.25, 16.25, 24.25, and 32.25 ms were 75420, 52832, 41568, and 46048, respectively. (b) A plot of χ2 vs. dCN yields dCN = 44.8 ± 2.4 Hz which corresponds to r = 4.11 ± 0.08 Å for the Leu-7 13CO/Phe-11 15N labeled pair. The (ΔS/S0)isim values in plot a were calculated with the best-fit dCN.
REDOR spectra of the HFPtr-L7CF11N/LM3e sample yielded S1iexp/S0iexp values which were significantly smaller than the S1iexp/S0iexp values for the I4 sample and suggest that the region between Leu-7 and Phe-11 has non-helical conformation (Fig. 2a, b, e, f). Fitting of (ΔS/S0)icor to (ΔS/S0)isim yielded dCN = 15.8 ± 1.8 Hz which corresponds to rCN = 5.8 ± 0.3 Å for the Leu-7 13CO/Phe-11 15N labeled pair (Fig. 5). This distance is longer than the ∼4.1 Å distance expected in a α helical conformation and shorter than the ∼11 Å distance expected in a β strand conformation. This result is discussed after presentation of the fpCTDQBU analyses; briefly, structural models suggest that the REDOR data reflect an interpeptide distance and provide information about the register of residues in adjacent β strands.
Figure 5.

(a) REDOR (ΔS/S0)icor (open squares with error bars) and (ΔS/S0)isim (crosses) vs. dephasing time for the HFPtr-L7CF11N/LM3e sample. Each (ΔS/S0)icor was based on a (ΔS/S0)iexp determined from spectral integrations over a 1 ppm region centered at 173.4 ppm, the peak shift in the S0 spectra. The total (S0 + S1) numbers of scans used to obtain the (ΔS/S0)0cor values for τi = 8.25, 16.25, 24.25, and 32.25 ms were 81056, 76288, 172512, and 197440, respectively. (b) A plot of χ2 vs. dCN yields dCN = 15.8 ± 1.8 Hz which corresponds to rCN = 5.8 ± 0.3 Å for the Leu-7 13CO/Phe-11 15N labeled pair. The (ΔS/S0)isim values in plot a were calculated with dCN = 15.8 Hz.
13CO-13CO distances
An in-register parallel β strand model for the HFPtr-L7CF11N/LM3e sample was tested with fpCTDQBU measurements of adjacent interstrand Leu-7 13CO/Leu-7 13CO dipolar couplings and distances. If the model is correct, the distance will be ∼4.8 Å with dCC ∼ 70 Hz.
The fpCTDQBU experiment was first validated using the GFF sample for which there is a ∼2% fraction of D-GFF molecules with 13CO labels at Gly-1 and Phe-3. D-GFF has an intramolecular 13CO/13CO distance of 5.40 Å and dCC ∼ 49 Hz. The GFF fpCTDQBU spectra have Phe-3, Phe-2 and Gly-1 13CO peaks at 180.3, 176.4, and 170.9 ppm, respectively (Fig. 6a-d). As τi was increased from 16 to 80 ms, there was a general decrease in S1iexp/S0Iexp values for the Phe-3 and Gly-1 peaks. This is consistent with the ∼65% contribution to these signals from D-GFF molecules for which there is significant Gly-1 13CO/Phe-3 13CO dipolar coupling. By contrast, the Phe-2 peak has S1iexp/S0iexp ∼ 1 for all τi which is consistent with the ∼98% contribution to this signal from unlabeled GFF molecules for which there are large 13CO/13C distances and corresponding small dCC values.
Figure 6.
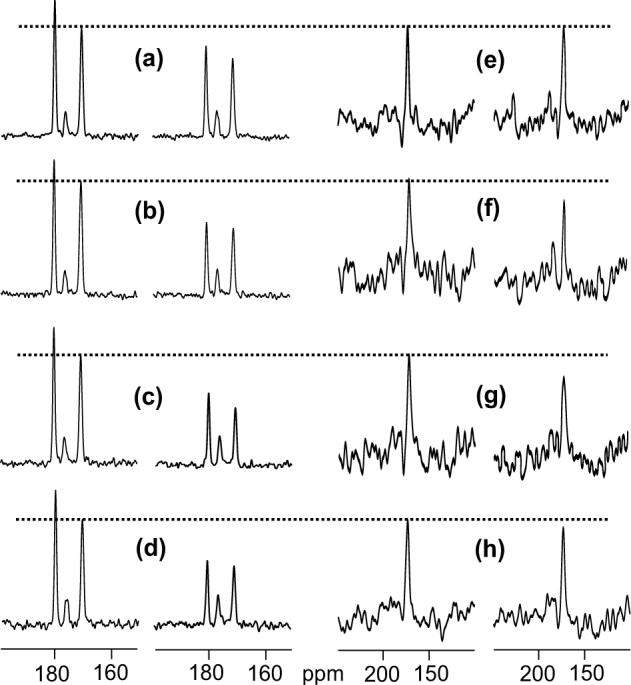
fpCTDQBU spectra of (a-d) the GFF sample and (e-h) the HFPtr-L7CF11N/LM3e sample. For each lettered pair of spectra, the S0 spectrum is on the left and the S1 spectrum is on the right. The MAS frequency = 8000 Hz, τR = 125 μs, the 13C π pulse rf field = 10 kHz, M = 336, and the total constant-time = (L + M + N) × τR = 84 ms. The values of L, N, and τi are: 128, 208, 32 ms (spectra a, e); 192, 144, 48 ms (spectra b, f); 256, 80, 64 ms (spectra c, g); 320, 16, 80 ms (spectra d, h). From left-to-right, each GFF spectrum has Phe-3, Phe-2 and Gly-1 13CO peaks at 180.3, 176.4, and 170.9 ppm, respectively. For each set of GFF and HFPtr spectra with the same τi, a dotted line is drawn at the peak S0 intensities of Gly-1 (GFF) and Leu-7 (HFPtr). The GFF spectra were processed with 50 Hz Gaussian line broadening, the HFPtr spectra were processed with 250 Hz Gaussian line broadening, and baseline correction was applied to all spectra. For some of the HFPtr spectra, there is a small glitch at ∼178 ppm which is due to DC offset in the data. The total numbers of scans used to obtain each spectrum in panels a, b, c, d, e, f, g, and h are 10240, 10240, 10240, 8192, 77056, 80736, 102432, and 152064, respectively.
As the magnitude of the constant-time parameter increases, transverse relaxation causes a decrease in 13CO signal intensity for all values of τi and consequent lower signal-to-noise ratio. It was therefore desirable to use the smallest constant-time which allowed observation of the full (ΔS/S0)i buildup for a ∼5 Å 13CO-13CO distance. Because RFDR is affected by 13C pulse duration, an investigation was made of the effect of the 13C π pulse rf field on the (ΔS/S0)i buildup rate and signal-to-noise ratio of the GFF sample (65). For the Gly-1 13CO signal, smaller 13C π pulse rf fields yielded faster buildup in both experiment and simulation (Fig. 7a) but also led to smaller S0 and S1 signal intensities, perhaps because of larger resonance offsets and poorer refocusing of chemical shift evolution. A 13C π pulse rf field of ∼10 kHz gave the best overall (ΔS/S0)i buildup rate and signal-to-noise ratio (Fig. 7b). Good agreement between (ΔS/S0)i cor and (ΔS/S0)isim at 10 and 43 kHz fields was obtained using dCC = 42 Hz. Good agreement between experimental and simulated signal-to-noise ratios at 10 kHz field was obtained using a single value of k (Eq. 5) while at 43 kHz field, this k value resulted in simulated signal-to-noise ratios about double those observed in experiment. Because the 13C π pulses at 43 kHz field are shorter than those at 10 kHz field, the 43 kHz experimental pulses are more poorly approximated by the ideal rectangular pulses of the simulations. This may result in poorer refocusing performance of the 43 kHz experimental 13C π pulses and lower signal-to-noise ratios relative to simulation.
Figure 7.

Dependence of fpCTDQBU of GFF Gly-1 13CO on 13C π pulse rf field: (a) (ΔS/S0)icor and (ΔS/S0)isim vs. dephasing time; and (b) signal-to-noise ratio of (ΔS/S0)icor and (ΔS/S0)isim vs. dephasing time. The MAS frequency = 8000 Hz, δ13C transmitter = 175.7 ppm, M = 336, and the constant-time = 84 ms. The symbol legend is: squares, cor, 10 kHz 13C π pulse rf field; crosses, sim, 10 kHz field; diamonds, cor, 43 kHz field; circles, sim, 43 kHz field. Uncertainties are displayed for the cor points in plot a and lines are drawn between like symbols in plot b. Each (ΔS/S0)icor was based on a (ΔS/S0)iexp determined from spectral integrations over a 0.5 ppm region centered at the Gly-1 13CO peak chemical shift. Each (ΔS/S0)icor value was determined using intensities from 4096 total (S0 + S1) scans. The S0isim and S1isim were calculated with dCC = 42 Hz which yielded the best overall agreement between (ΔS/S0)icor and (ΔS/S0)isim values. The value of k (Eq. 5) was set to give the best agreement at 10 kHz field between the sim and cor signal-to-noise ratios.
Subsequent fpCTDQBU experiments were done with a 10 kHz 13C π pulse rf field. Fig. 8 displays comparison and fitting of GFF Gly-1 13CO (ΔS/S0)i cor and (ΔS/S0)i sim over a wide range of τi. The analysis focused on Gly-1 13CO because it is has carbonyl rather than carboxyl functionality and is thus more relevant to the HFPtr sample. The fitting yielded dCC = 42.3 ± 0.5 Hz and corresponding rCC = 5.67 ± 0.02 Å for the Gly-1 13CO/Phe-3 13CO labeled pair. Variation of 13CO CSA axis orientations relative to the GFF atomic geometry did not greatly affect (ΔS/S0)isim and the fitted values of dCC. For example, different sets of (ΔS/S0)isim were calculated with each set having a randomly generated principal axis orientation. Each set of (ΔS/S0)isim was then fitted to (ΔS/S0)icor and the resulting dCC was always within a range of 37−47 Hz.
Figure 8.

(a) fpCTDQBU (ΔS/S0)icor (open squares with error bars) and (ΔS/S0)isim (crosses) vs. dephasing time for GFF Gly-1 13CO. The MAS frequency = 8000 Hz, the 13C π pulse rf field = 10 kHz, δ13C transmitter = 175.7 ppm, M = 336, and the constant-time = 84 ms. Each (ΔS/S0)icor was based on a (ΔS/S0)iexp determined from spectral integrations over a 0.5 ppm region centered at the Gly-1 13CO peak chemical shift. Each (ΔS/S0)icor value for τi = 16, 24, 32, 40, 48, 56, and 64 ms was determined using intensities from 20480 total (S0 + S1) scans and the (ΔS/S0)icor value for τi = 72 ms was determined using intensities from 16384 total scans. (b) A plot of χ2 vs. dCC yields dCC = 42.3 ± 0.5 Hz which corresponds to rCC = 5.67 ± 0.02 Å for the Gly-1/Phe-3 labeled pair. The (ΔS/S0)isim values in panel a were calculated with d = 42.3 Hz. (c) Plot of (ΔS/S0)icor(open squares with error bars) and (ΔS/S0)isim (crosses) calculated with dCC = 49 Hz which corresponds to rCC = 5.40 Å, the Gly-1 CO/Phe-3 CO distance in the X-ray structure. The (ΔS/S0)icor were fitted to (ΔS/S0)isim as a function of the D-GFF:unlabeled GFF ratio and the displayed (ΔS/S0)icor were calculated with the best-fit value of the ratio (1:71) for which χ2 = 7.
In Fig. 8a, the only systematic disagreement between (ΔS/S0)icor and (ΔS/S0)isim occurs at large values of τi for which (ΔS/S0)icor decreases more rapidly than (ΔS/S0)isim. The best-fit rCC is ∼5% larger than the 5.40 Å crystallographic distance (71). Uncertainty in the D-GFF:unlabeled GFF ratio in the sample is one possible reason for this discrepancy because this ratio affects the (ΔS/S0)iexp → (ΔS/S0)icor conversion (cf. Materials and Methods and Supporting Information). The potential effect of this uncertainty was tested by calculation of (ΔS/S0)isim with d = 49 Hz (rCC = 5.40 Å) and then fitting (ΔS/S0)icor to (ΔS/S0)isim as a function of the D-GFF:unlabeled GFF ratio (Fig. 8c). Good fits were obtained with ratios of ∼1:70 which is close to the ratio of ∼1:50 derived from weights of D-GFF and unlabeled GFF in the sample and by the ratio of the Gly-1:Phe-2 13CO intensities. Overall, the GFF studies demonstrate that 13C-13C distances in the ∼5 Å range can be reasonably accurately determined with the fpCTDQBU method.
The fpCTDQBU spectra of the HFPtrL7CF11N/LM3e sample are displayed in Fig. 6e-h. Relative to the Gly-1 and Phe-3 13CO peaks in the GFF spectra (Fig. 6a-d), the HFPtr spectra have larger S1i/S0i ratios which are consistent with a dCC smaller than in D-GFF and a Leu-7 13CO/Leu-7 13CO distance longer than that in D-GFF. The τi = 32, 48, and 64 ms points were fitted in the context of a “two-spin” or a “three-spin” model (Fig. 9). The 80 ms point was not included in the fitting because of the systematic disagreement between (ΔS/S0)icor and (ΔS/S0)isim at large values of τi in the GFF analysis (Fig. 8a). The two-spin (three-spin) model has two (three) adjacent in-register parallel β strands and the simulations are based on two (three) 13COs, each of which is at the same residue position in its respective strand. The interstrand distance and corresponding dCC value were input parameters for the simulations. In the three-spin model, the two outside strands were always equidistant from the central strand.
Figure 9.

fpCTDQBU (ΔS/S0)icor and (ΔS/S0)isim vs. dephasing time for the HFPtr-L7CF11N/LM3e sample. The MAS frequency = 8000 Hz, the 13C π pulse rf field = 10 kHz, δ13C transmitter = 178.4 ppm, M = 336, and the constant-time = 84 ms. The (ΔS/S0)icor values are open squares with error bars. Each (ΔS/S0)icor was based on a (ΔS/S0)iexp determined from spectral integrations over a 1 ppm region centered at 173.4 ppm, the peak shift in the S0 spectra. The total (S0 + S1) numbers of scans used to obtain the (ΔS/S0)icor values for τi = 32, 48, and 64 ms were 154112, 161472, and 204864, respectively. The (ΔS/S0)isim values were calculated with two-spin and three-spin models in plots a and b, respectively. In plot a, the up triangles, crosses, and down triangles correspond to dCC = 10, 15 and 20 Hz, respectively and in plot b, they correspond to dCC = 8, 13, and 18 Hz, respectively. The best-fit values of dCC are ∼15 Hz and ∼13 Hz for the two-spin and three-spin models, respectively, and correspond to interstrand Leu-7 13CO-13CO distances of 8.0 Å and 8.4 Å. Reasonable upper limits on dCC in the two-spin and three-spin models are ∼20 Hz and ∼18 Hz respectively, and correspond to distances of 7.3 Å and 7.5 Å.
The fitting is based on three points with relatively large uncertainties and visual comparison was used to assess best-fit d and its uncertainty. The best-fit values of dCC in the two-spin and three-spin models are ∼15 Hz and ∼13 Hz, respectively, and correspond to rCC of 8.0 Å and 8.4 Å. Reasonable upper limits on dCC in the two-spin and three-spin models are ∼20 Hz and ∼18 Hz, respectively, and correspond to rCC of 7.3 Å and 7.5 Å. The lower limits on dCC and upper limits on rCC are more difficult to assess because of the uncertainties in (ΔS/S0)>icor and weak dependence of (ΔS/S0)isim on dCC at small values of dCC. Overall, experimental values of dCC and rCC do not support the in-register parallel β model for which dCC ∼ 70 Hz and rCC ∼ 4.8 Å.
Strand arrangement models
Fig. 10 displays three models: (a) parallel β strand with all strands in-register; (b) parallel β strand with adjacent strands two residues out-of-register; and (c) antiparallel β strand with adjacent strand crossing between Phe-8 and Leu-9. Each model is shown with three strands although we do not have information about the numbers of strands in the LM3e-associated HFPtr oligomer. Table 1 displays Leu-7 CO/Phe-11 N and interstrand Leu-7 CO/Leu-7 CO distances from REDOR and fpCTDQBU experiments and from the models. The model distances are averages for parallel strand regions of cutinase and antiparallel strand regions of human gamma-D crystalline R58H mutant whose crystal structures have been refined to 1.0 Å and 1.15 Å, respectively (91, 92).
Figure 10.
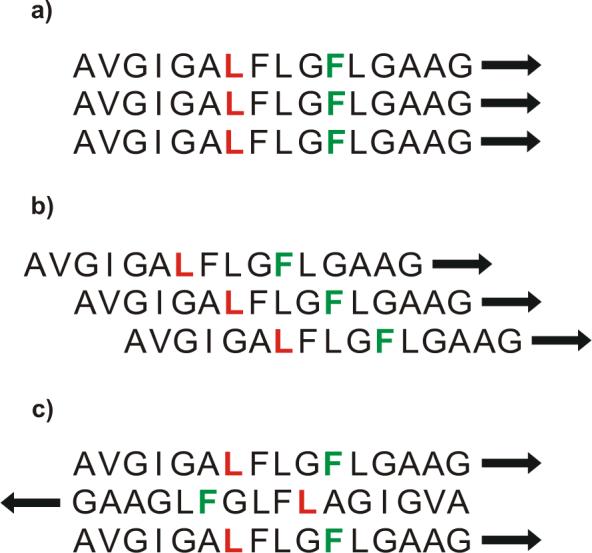
Structural models for strand arrangements of LM3e-associated HFPtr: (a) parallel in-register; (b) parallel with adjacent strands two-residues out-of-register; and (c) antiparallel with adjacent strand crossing between Phe-8 and Leu-9. The first sixteen residues of each strand are displayed, the 13CO labeled Leu-7 are colored red, and the 15N labeled Phe-11 are colored green.
Table 1.
Experimental and Model Distancesa
| rCN (Å)b | rCC (Å)c | |
|---|---|---|
| Experiment | 5.8(3) | 8.2(9) |
| Parallel in-register modeld | 11.0(3) | 4.8(2) |
| Parallel out-of-register modele | 6.5(3) | 8.2(3) |
| Antiparallel modelf | 6.1(3) | 8.5(3) |
Each model distance was determined from a region of a high-resolution crystal structure which has the same strand arrangement as the model. Several interatomic distances were measured in this region. An average distance is reported in the table as well as the standard deviation in units of 0.1 Å in parentheses.
Leu-7 CO/Phe-11 N distance.
Interstrand Leu-7 CO/Leu-7 CO distance.
Model distances were determined from the region of the cutinase protein corresponding to residues 34−39, 113−120, and 143−148.
Adjacent strands are two-residues out-of-register. Model distances were determined from the cutinase protein.
Adjacent strands cross between Phe-8 and Leu-9. Model distances were determined from the region of the human gamma-D crystalline R58H mutant protein corresponding to residues 2−7, 14−18, and 34−38.
There are significant differences between the experimental distances and in-register parallel strand model distances. The experimental Leu-7 13CO/Phe-11 15N distance is ∼6 Å which is much shorter than the ∼11 Å intrastrand or interstrand distance in the model. The lower limit on the experimental interstrand Leu-7 13CO/Leu-7 13CO distance is >7 Å and is significantly longer than the 4.8 Å distance in the model.
Distances in the out-of-register parallel strand and antiparallel strand models are more consistent with the experimentally-derived distances. There are ∼6 Å interstrand Leu-7 CO/Phe-11 N distances because of the strand registers in the models. In addition, the interstrand Leu-7 CO/Leu-7 CO distances are ∼8 Å. Calculations of REDOR and fpCTDQBU (ΔS/S0)isim for the two models agreed reasonably well with (ΔS/S0)icor. The data were more poorly fit by other strand arrangement models.
Further experiments are required to distinguish between the two models. For example, we recently observed Ala-6 13C/Gly-10 13C crosspeaks in 2D spectra of LM3-associated HFP which was uniformly 13C labeled at Ala-6 and Gly-10 (97). The crosspeaks were generated by magnetization exchange due to proton-driven spin diffusion which typically occurs between 13C nuclei separated by < 6 Å. For the out-of-register parallel and antiparallel models, the closest distance between Ala-6 and Gly-10 13Cs is ∼7.1 Å and ∼4.5 Å, respectively, and the spectra are more consistent with the antiparallel model. Future experiments could be based on the REDOR approaches used to recently elucidate strand arrangements in the PG-1 antimicrobial peptide (59).
A turn structure in the Leu-7 to Phe-11 region could also be consistent with the experimental Leu-7 CO/Phe-11 N distance. Although this possibility cannot be definitively ruled out, measurements of 13C chemical shifts for nuclei in this region of LM3-associated HFP are all consistent with β strand conformation (29, 97). We also note the possibility of populations of distinct strand arrangements which would necessitate fitting REDOR and fpCTDQBU signals with populations of distinct spin geometries.
In summary, the REDOR and fpCTDQBU data are consistent with a parallel strand arrangement with adjacent strands two-residues out-of-register and with an antiparallel arrangement whose adjacent strand crossing is between Phe-8 and Leu-9. The data are not consistent with an in-register parallel strand arrangement.
DISCUSSION
Conformational plasticity
The structural plasticity of membrane-associated viral fusion peptides has been observed by several groups using a variety of experimental probes, and it is known that they can exist in helical or nonhelical forms (7). Other investigators have developed structural models for the helical form of the peptides based principally on liquid-state NMR, ESR, and IR data (33, 38, 39, 73, 74). IR and solid-state NMR studies of the β strand form of HFP and its oligomeric constructs have shown parallel strand arrangement, anti-parallel strand arrangement and a mixture of parallel and antiparallel arrangements (26, 43, 44). In the present study, the structure of membrane-associated HFPtr was explored with solid-state NMR methods including chemical shift, heteronuclear distance, and homonuclear distance measurements. The data suggest that the region of HFPtr between Leu-7 and Phe-11 is α helical when associated with membranes without cholesterol and is β strand when associated with membranes which have a lipid headgroup and cholesterol composition similar to that of host cells of the virus. These results are consistent with our observations that for a single FP:lipid mol ratio, cholesterol favors formation of the β strand conformation of fusion peptides (27, 76, W. Qiang and D. P. Weliky, unpublished experiments). This conformational difference has been seen both at room temperature as well as at the −50 °C temperature of the experiments in this paper (29). The observed fusion peptide conformations appear to be equilibrium rather than kinetically-trapped structures (98).
The presence of cholesterol in membranes is known to increase the lateral molecular packing density and membrane tensile strength, decrease in-plane elasticity and permeability of water through the membrane, and promote formation of the “liquid-ordered phase” (99-104). This phase is characterized by a rapid lateral molecular translational diffusion coefficient similar to that of the “liquid-disordered” phase and high configurational order of the lipid acyl chains similar to that of the “solid-ordered” phase (104, 105). These latter phases exist for non-cholesterol containing membranes at higher and lower temperature, respectively. Fluorescence data suggest that HFPmn in its helical form inserts more deeply into the membrane than HFPmn in its β strand form (106). The higher molecular packing density in the cholesterol-containing membranes may make HFPtr insertion more difficult and thus favor a more surface-associated β strand form.
Because the rapid fusion rate of HFPtr is observed both for vesicles which contain cholesterol and for vesicles which do not contain cholesterol, one reasonable hypothesis is that both the helical and β strand structures of HFPtr are highly fusogenic (46). Other investigators have proposed that the peptide structure responsible for fusion is irregular and may be transient (107, 108). Although the present paper does not directly address these issues, it is noted: (1) cholesterol-associated structural variation is also observed for the influenza virus fusion peptide (27, 76); and (2) pH-triggered fusion can be observed both for helical influenza fusion peptide bound to non-cholesterol containing membranes and for β strand influenza fusion peptide bound to cholesterol-containing membranes (109). These results argue in favor of fusogenic activity of both helical and β strand structures. Lentz and coworkers have proposed that fusion peptides catalyze fusion by filling void spaces in nonbilayer fusion intermediates and that this function can be done by peptides in either helical or β strand conformation (110). Our results are consistent with this model and two fusogenic conformations may allow the virus to fuse with a wider variety of membrane compositions. Membranes of uninfected host cells of the virus contain ∼30 mol% cholesterol and there is some depletion of cholesterol in these membranes after HIV infection (77, 78). The HIV membrane contains ∼45 mol% cholesterol and there is some evidence that HIV fuses with cholesterol-rich regions of host cell membranes (111).
The 15−40 fold higher rate of HFPtr-induced lipid mixing relative to HFPmn is observed both with PC-PG vesicles for which the final HFPtr conformation is helical and with LM3 vesicles for which the final conformation is β strand. For either conformation, there is a higher local concentration of peptide strands at the membrane surface with HFPtr than with HFPmn and this larger concentration could cause greater perturbation of the membrane and provide a general model for the increased fusion rate of HFPtr. Other possible differences between HFPtr and HFPmn are their depths and angles of membrane insertion as well as the arrangement and interactions of individual helices or strands.
Strand arrangement
Previous solid-state NMR REDOR data for HFPmn associated with cholesterol-containing membranes were consistent with a mixture of parallel and antiparallel strand arrangements (43). A predominant parallel strand arrangement is an appealing structural model to explain the increased fusion rate of β strand HFPtr because it places the apolar N-terminal regions of the strands close to one another and thereby provides a larger apolar volume to perturb the membrane. The solid-state NMR data of this paper are consistent with a parallel model with adjacent strands two-residues out of register.
The data are also consistent with antiparallel strands with adjacent strands crossing between Phe-8 and Leu-9. For this antiparallel arrangement, the sixteen N-terminal apolar residues (A1 to G16) could form a hydrogen-bonded β strand oligomer and the more polar C-terminal residues would be outside the hydrogen-bonded oligomer. If residues A1-G16 form a single β strand without turns, then a structural building block could be based on two HFPtr molecules “A” and “B”. The A1, A2, and A3 strands would run in the same direction (as enforced by the cross-linking) and the B1, B2, and B3 strands would run in the opposite direction. An antiparallel interleaved arrangement could be A1B3A2B2A3B1; i.e. residues in strand B3 are hydrogen bonded to residues in strands A1 and A2. This antiparallel structure would place the bulky C-terminal regions of the HFPtr molecules on either side of the oligomer and could allow more efficient assembly of multiple HFPtr trimers at the membrane surface with consequent greater membrane perturbation and fusion rate. The total number of strands in a hydrogen-bonded oligomer may not be large as evidenced by poor room-temperature cross-polarization and the inferred high molecular mobility (29).
For membrane-associated HFPmn, IR data from several groups support an antiparallel strand arrangement (26, 30, 110, 112). However, IR data on constructs which contain the first 34 or 70 residues of gp41 support an in-register parallel strand arrangement (44). These latter constructs contain the 23-residues of HFPmn as well as an additional 11 or 47 C-terminal residues. The variation in strand arrangements among the constructs may be due to sequence or cross-linking differences (113).
Accurate measurement of weak couplings
Model compound results in this paper provide insight into the accuracy with which solid-state NMR determines weak couplings and long internuclear distances in peptides and proteins with selective isotopic labeling. This is a significant issue because most structurally interesting distances are long. After consideration of fractional labeling, natural abundance nuclei, and the population of molecules with random coil structures, the I4 REDOR data were fit well to dCN of 44.8 Hz and rCN of 4.11 Å. The latter value agrees quantitatively with rCN of a α helix which is the predominant conformation of the I4 peptide.
After consideration of fractional labeling and natural abundance nuclei, the GFF fpCTDQBU data were fit well to dCC of 42.3 Hz and rCC of 5.67 Å. These values are respectively ∼15% smaller and ∼5% larger than the crystallographic values. Possible contributions to this discrepancy include: (1) vibrational averaging of dCC through the large numbers of bonds between the two labeled nuclei; and (2) inaccuracy in the D-GFF:unlabeled GFF ratio. Overall, the experiment was a robust measure of dCC and rCC.
Incorporation of finite π pulses in the fpCTDQBU experiment resulted in higher ΔS/S0 buildup rate and signal-to-noise. To our knowledge, previous application of fpRFDR sequences for distance determination has typically been done at high (≥ 20 kHz) MAS frequencies with the exception of work by Chan et al. on 31P-31P distances in phosphate compounds (114). The present work demonstrates application of fpRFDR at moderate spinning frequencies to measurement of structurally interesting and weak (<50 Hz) 13C-13C dipolar couplings. This is significant for membrane-associated peptides and proteins because it is often desirable to work at smaller protein:lipid ratio and therefore larger volume rotors with concomitant slower MAS frequencies. It was also found that constant-time was required to obtain interpretable RFDR data. Constant-time reduces signal intensity so reasonably high decoupling and judicious choice of the constant-time value were critical to obtain satisfactory data for the HFPtr sample. The fpCTDQBU sequence of the present study relies on the intensity difference of two spectra as a function of dephasing time while the related fpRFDR-CT method relies on the intensity of a single spectrum as a function of dephasing time (51, 65). The fpRFDR-CT technique may therefore yield data with higher signal-to-noise.
Supplementary Material
ACKNOWLEDGEMENT
We acknowledge the Michigan State mass spectrometry facility, the Michigan State Max T. Rogers NMR facility and the Michigan Center for Biological Information for use of mass spectrometers, NMR spectrometers and compute nodes, respectively. We acknowledge Dr. David L. DeWitt for use of his ultracentrifuge, Dr. Robert I. Cukier and Paul A. Reed for their assistance in installation of SIMPSON, and the SIMPSON/SIMMOL Discussion Forum for helpful discussion.
ABBREVIATIONS
- CD
circular dichroism
- CP
cross-polarization
- CSA
chemical shift anisotropy
- dCC
13C-13C dipolar coupling
- dCN
13C-15N dipolar coupling
- DCM
dichloromethane
- D-GFF
doubly labeled GFF
- D-NAL, 1-13C
N-13C doubly labeled NAL
- DTPC
1,2-di-O-tetradecyl-sn-glycero-3-phosphocholine
- DTPG
1,2-di-O-tetradecyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt
- DPhPE
1,2-di-O-phytanyl-sn-glycero-3-phosphoethanolamine
- ESR
electron spin resonance
- FMOC
9-fluorenylmethoxycarbonyl
- fpCTDQBU
constant-time double-quantum build-up with finite pulses
- fpRFDR-CT
constant-time finite-pulse rf-driven recoupling
- GFF
glycyl-L-phenylalanyl-L-phenylalanine
- HEPES
N-(2-hydroxyethyl)piperazine-N'-2-ethanesulfonic acid
- HIV
human immunodeficiency virus
- HFP
HIV fusion peptide
- HFPdm
HIV fusion peptide dimer
- HFPmn
HIV fusion peptide monomer
- HFPtr
HIV fusion peptide trimer
- HPLC
high-performance liquid chromatography
- IR
infrared
- LM3
lipid mixture 3
- LM3e
ether-linked lipid mixture 3
- LUV
large unilamellar vesicle
- MAS
magic angle spinning
- NAL
N-acetyl-L-leucine
- NMP
N-methylpyrrolidone
- NMR
nuclear magnetic resonance
- PC-PG
4:1 POPC:POPG mixture
- PDB
Protein Data Bank
- PI
phosphatidylinositol
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-(1-glycerol)
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine
- rf
radio frequency
- rCC
13C-13C distance
- rCN
13C-15N distance
- REDOR
rotational-echo double resonance
- RFDR
radio frequency-driven recoupling
- SIMPSON
simulation program for solid-state NMR spectroscopy
- TBME
t-butyl methyl ether
- TFA
trifluoroacetic acid
- TPPM
two-pulse phase-modulation
- 2D
two-dimensional
Footnotes
This work was supported by NIH award AI47153 to D. P. W.
SUPPORTING INFORMATION AVAILABLE
Equations are derived for determination of (ΔS/S0)cor for REDOR and fpCTDQBU analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Hernandez LD, Hoffman LR, Wolfsberg TG, White JM. Virus-cell and cell-cell fusion. Annu. Rev. Cell. Dev. Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 2.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 3.Colman PM, Lawrence MC. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 2003;4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 4.Freed EO, Myers DJ, Risser R. Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc. Natl. Acad. Sci. U.S.A. 1990;87:4650–4654. doi: 10.1073/pnas.87.12.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pereira FB, Goni FM, Muga A, Nieva JL. Permeabilization and fusion of uncharged lipid vesicles induced by the HIV-1 fusion peptide adopting an extended conformation: dose and sequence effects. Biophys. J. 1997;73:1977–1986. doi: 10.1016/S0006-3495(97)78228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mobley PW, Lee HF, Curtain CC, Kirkpatrick A, Waring AJ, Gordon LM. The amino-terminal peptide of HIV-1 glycoprotein 41 fuses human erythrocytes. Biochim. Biophys. Acta. 1995;1271:304–314. doi: 10.1016/0925-4439(95)00048-9. [DOI] [PubMed] [Google Scholar]
- 7.Durell SR, Martin I, Ruysschaert JM, Shai Y, Blumenthal R. What studies of fusion peptides tell us about viral envelope glycoprotein-mediated membrane fusion. Mol. Membr. Biol. 1997;14:97–112. doi: 10.3109/09687689709048170. [DOI] [PubMed] [Google Scholar]
- 8.Freed EO, Delwart EL, Buchschacher GL, Jr., Panganiban AT. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc. Natl. Acad. Sci. U.S.A. 1992;89:70–74. doi: 10.1073/pnas.89.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pereira FB, Goni FM, Nieva JL. Liposome destabilization induced by the HIV-1 fusion peptide effect of a single amino acid substitution. FEBS Lett. 1995;362:243–246. doi: 10.1016/0014-5793(95)00257-a. [DOI] [PubMed] [Google Scholar]
- 10.Kliger Y, Aharoni A, Rapaport D, Jones P, Blumenthal R, Shai Y. Fusion peptides derived from the HIV type 1 glycoprotein 41 associate within phospholipid membranes and inhibit cell-cell Fusion. Structure- function study. J. Biol. Chem. 1997;272:13496–13505. doi: 10.1074/jbc.272.21.13496. [DOI] [PubMed] [Google Scholar]
- 11.Suarez T, Gallaher WR, Agirre A, Goni FM, Nieva JL. Membrane interface-interacting sequences within the ectodomain of the human immunodeficiency virus type 1 envelope glycoprotein: putative role during viral fusion. J. Virol. 2000;74:8038–47. doi: 10.1128/jvi.74.17.8038-8047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimitrov AS, Rawat SS, Jiang S, Blumenthal R. Role of the fusion peptide and membrane-proximal dornain in HIV-I envelope glycoprotein-mediated membrane fusion. Biochemistry. 2003;42:14150–14158. doi: 10.1021/bi035154g. [DOI] [PubMed] [Google Scholar]
- 13.Shnaper S, Sackett K, Gallo SA, Blumenthal R, Shai Y. The C- and the N-terminal regions of glycoprotein 41 ectodomain fuse membranes enriched and not enriched with cholesterol, respectively. J. Biol. Chem. 2004;279:18526–18534. doi: 10.1074/jbc.M304950200. [DOI] [PubMed] [Google Scholar]
- 14.Pascual R, Moreno MR, Villalain J. A peptide pertaining to the loop segment of human immunodeficiency virus gp41 binds and interacts with model biomembranes: Implications for the fusion mechanism. J. Virol. 2005;79:5142–5152. doi: 10.1128/JVI.79.8.5142-5152.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 16.Tan K, Liu J, Wang J, Shen S, Lu M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. U.S.A. 1997;94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 18.Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang ZN, Mueser TC, Kaufman J, Stahl SJ, Wingfield PT, Hyde CC. The crystal structure of the SIV gp41 ectodomain at 1.47 A resolution. J. Struct. Biol. 1999;126:131–144. doi: 10.1006/jsbi.1999.4116. [DOI] [PubMed] [Google Scholar]
- 20.Markosyan RM, Cohen FS, Melikyan GB. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell. 2003;14:926–938. doi: 10.1091/mbc.E02-09-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimitrov AS, Louis JM, Bewley CA, Clore GM, Blumenthal R. Conformational changes in HIV-1 gp41 in the course of HIV-1 envelope glycoprotein-mediated fusion and inactivation. Biochemistry. 2005;44:12471–12479. doi: 10.1021/bi051092d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rafalski M, Lear JD, DeGrado WF. Phospholipid interactions of synthetic peptides representing the N-terminus of HIV gp41. Biochemistry. 1990;29:7917–7922. doi: 10.1021/bi00486a020. [DOI] [PubMed] [Google Scholar]
- 23.Martin I, Schaal H, Scheid A, Ruysschaert JM. Lipid membrane fusion induced by the human immunodeficiency virus type 1 gp41 N-terminal extremity is determined by its orientation in the lipid bilayer. J. Virol. 1996;70:298–304. doi: 10.1128/jvi.70.1.298-304.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtain C, Separovic F, Nielsen K, Craik D, Zhong Y, Kirkpatrick A. The interactions of the N-terminal fusogenic peptide of HIV-1 gp41 with neutral phospholipids. Eur. Biophys. J. 1999;28:427–436. doi: 10.1007/s002490050225. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Gabrys CM, Weliky DP. Solid-state nuclear magnetic resonance evidence for an extended beta strand conformation of the membrane-bound HIV-1 fusion peptide. Biochemistry. 2001;40:8126–8137. doi: 10.1021/bi0100283. [DOI] [PubMed] [Google Scholar]
- 26.Gordon LM, Mobley PW, Pilpa R, Sherman MA, Waring AJ. Conformational mapping of the N-terminal peptide of HIV-1 gp41 in membrane environments using 13C-enhanced Fourier transform infrared spectroscopy. Biochim. Biophys. Acta-Biomembr. 2002;1559:96–120. doi: 10.1016/s0005-2736(01)00443-6. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Parkanzky PD, Bodner ML, Duskin CG, Weliky DP. Application of REDOR subtraction for filtered MAS observation of labeled backbone carbons of membrane-bound fusion peptides. J. Magn. Reson. 2002;159:101–110. doi: 10.1016/s1090-7807(02)00033-2. [DOI] [PubMed] [Google Scholar]
- 28.Yang R, Yang J, Weliky DP. Synthesis, enhanced fusogenicity, and solid state NMR measurements of cross-linked HIV-1 fusion peptides. Biochemistry. 2003;42:3527–3535. doi: 10.1021/bi027052g. [DOI] [PubMed] [Google Scholar]
- 29.Bodner ML, Gabrys CM, Parkanzky PD, Yang J, Duskin CA, Weliky DP. Temperature dependence and resonance assignment of 13C NMR spectra of selectively and uniformly labeled fusion peptides associated with membranes. Magn. Reson. Chem. 2004;42:187–194. doi: 10.1002/mrc.1331. [DOI] [PubMed] [Google Scholar]
- 30.Castano S, Desbat B. Structure and orientation study of fusion peptide FP23 of gp41 from HIV-1 alone or inserted into various lipid membrane models (mono-, bi- and multibi-layers) by FT-IR spectroscopies and Brewster angle microscopy. Biochim. Biophys. Acta-Biomembr. 2005;1715:81–95. doi: 10.1016/j.bbamem.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 31.Nieva JL, Agirre A. Are fusion peptides a good model to study viral cell fusion? Biochim. Biophys. Acta-Biomembr. 2003;1614:104–115. doi: 10.1016/s0005-2736(03)00168-8. [DOI] [PubMed] [Google Scholar]
- 32.Buzon V, Padros E, Cladera J. Interaction of fusion peptides from HIV gp41 with membranes: A time-resolved membrane binding, lipid mixing, and structural study. Biochemistry. 2005;44:13354–13364. doi: 10.1021/bi050382r. [DOI] [PubMed] [Google Scholar]
- 33.Chang DK, Cheng SF, Chien WJ. The amino-terminal fusion domain peptide of human immunodeficiency virus type 1 gp41 inserts into the sodium dodecyl sulfate micelle primarily as a helix with a conserved glycine at the micelle-water interface. J. Virol. 1997;71:6593–6602. doi: 10.1128/jvi.71.9.6593-6602.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamath S, Wong TC. Membrane structure of the human immunodeficiency virus gp41 fusion domain by molecular dynamics simulation. Biophys. J. 2002;83:135–143. doi: 10.1016/S0006-3495(02)75155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maddox MW, Longo ML. Conformational partitioning of the fusion peptide of HIV-1 gp41 and its structural analogs in bilayer membranes. Biophys. J. 2002;83:3088–3096. doi: 10.1016/S0006-3495(02)75313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langham A, Kaznessis Y. Simulation of the N-terminus of HIV-1 glycoprotein 41000 fusion peptide in micelles. J. Pept. Sci. 2005;11:215–224. doi: 10.1002/psc.623. [DOI] [PubMed] [Google Scholar]
- 37.Morris KF, Gao XF, Wong TC. The interactions of the HIV gp41 fusion peptides with zwitterionic membrane mimics determined by NMR spectroscopy. Biochim. Biophys. Acta-Biomembr. 2004;1667:67–81. doi: 10.1016/j.bbamem.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 38.Gordon LM, Mobley PW, Lee W, Eskandari S, Kaznessis YN, Sherman MA, Waring AJ. Conformational mapping of the N-terminal peptide of HIV-1 gp41 in lipid detergent and aqueous environments using 13C-enhanced Fourier transform infrared spectroscopy. Protein Sci. 2004;13:1012–1030. doi: 10.1110/ps.03407704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaroniec CP, Kaufman JD, Stahl SJ, Viard M, Blumenthal R, Wingfield PT, Bax A. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 2005;44:16167–16180. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- 40.Taylor SE, Desbat B, Blaudez D, Jacobi S, Chi LF, Fuchs H, Schwarz G. Structure of a fusion peptide analogue at the air-water interface, determined from surface activity, infrared spectroscopy and scanning force microscopy. Biophys. Chem. 2000;87:63–72. doi: 10.1016/s0301-4622(00)00182-4. [DOI] [PubMed] [Google Scholar]
- 41.Pritsker M, Rucker J, Hoffman TL, Doms RW, Shai Y. Effect of nonpolar substitutions of the conserved Phe11 in the fusion peptide of HIV-1 gp41 on its function, structure, and organization in membranes. Biochemistry. 1999;38:11359–11371. doi: 10.1021/bi990232e. [DOI] [PubMed] [Google Scholar]
- 42.Gordon LM, Curtain CC, Zhong YC, Kirkpatrick A, Mobley PW, Waring AJ. The amino-terminal peptide of HIV-1 glycoprotein 41 interacts with human erythrocyte membranes: peptide conformation, orientation and aggregation. Biochim. Biophys. Acta. 1992;1139:257–274. doi: 10.1016/0925-4439(92)90099-9. [DOI] [PubMed] [Google Scholar]
- 43.Yang J, Weliky DP. Solid state nuclear magnetic resonance evidence for parallel and antiparallel strand arrangements in the membrane-associated HIV-1 fusion peptide. Biochemistry. 2003;42:11879–11890. doi: 10.1021/bi0348157. [DOI] [PubMed] [Google Scholar]
- 44.Sackett K, Shai Y. The HIV fusion peptide adopts intermolecular parallel β-sheet structure in membranes when stabilized by the adjacent N-terminal heptad repeat: A 13C FTIR study. J. Mol. Biol. 2005;350:790–805. doi: 10.1016/j.jmb.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 45.Munoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J. Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang R, Prorok M, Castellino FJ, Weliky DP. A trimeric HIV-1 fusion peptide construct which does not self-associate in aqueous solution and which has 15-fold higher membrane fusion rate. J. Am. Chem. Soc. 2004;126:14722–14723. doi: 10.1021/ja045612o. [DOI] [PubMed] [Google Scholar]
- 47.Epand RF, Macosko JC, Russell CJ, Shin YK, Epand RM. The ectodomain of HA2 of influenza virus promotes rapid pH dependent membrane fusion. J Mol Biol. 1999;286:489–503. doi: 10.1006/jmbi.1998.2500. [DOI] [PubMed] [Google Scholar]
- 48.Lau WL, Ege DS, Lear JD, Hammer DA, DeGrado WF. Oligomerization of fusogenic peptides promotes membrane fusion by enhancing membrane destabilization. Biophys. J. 2004;86:272–284. doi: 10.1016/S0006-3495(04)74103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wexler-Cohen Y, Sackett K, Shai Y. The role of the N-terminal heptad repeat of HIV-1 in the actual lipid mixing step as revealed by its substitution with distant coiled coils. Biochemistry. 2005;44:5853–5861. doi: 10.1021/bi047666g. [DOI] [PubMed] [Google Scholar]
- 50.Benzinger TL, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Propagating structure of Alzheimer's β-amyloid(10−35) is parallel β-sheet with residues in exact register. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Supramolecular structure in full-length Alzheimer's β-amyloid fibrils: Evidence for a parallel β-sheet organization from solid-state nuclear magnetic resonance. Biophys. J. 2002;83:1205–1216. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 53.Anderson RC, Gullion T, Joers JM, Shapiro M, Villhauer EB, Weber HP. Conformation of [1-13C,15N]acetyl-L-carnitine - rotational-echo, double-resonance nuclear-magnetic-resonance spectroscopy. J. Am. Chem. Soc. 1995;117:10546–10550. [Google Scholar]
- 54.Kimura S, Naito A, Saito H, Ogawa K, Shoji A. Characterization of α-helix structures in polypeptides, revealed by 13C=O...H-15N hydrogen bond lengths determined by 13C REDOR NMR. J. Mol. Struct. 2001;562:197–203. [Google Scholar]
- 55.Murphy OJ, 3rd, Kovacs FA, Sicard EL, Thompson LK. Site-directed solid-state NMR measurement of a ligand-induced conformational change in the serine bacterial chemoreceptor. Biochemistry. 2001;40:1358–1366. doi: 10.1021/bi0015109. [DOI] [PubMed] [Google Scholar]
- 56.Smith SO, Eilers M, Song D, Crocker E, Ying WW, Groesbeek M, Metz G, Ziliox M, Aimoto S. Implications of threonine hydrogen bonding in the glycophorin A transmembrane helix dimer. Biophys. J. 2002;82:2476–2486. doi: 10.1016/S0006-3495(02)75590-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishimura K, Kim SG, Zhang L, Cross TA. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 58.Toke O, Maloy WL, Kim SJ, Blazyk J, Schaefer J. Secondary structure and lipid contact of a peptide antibiotic in phospholipid Bilayers by REDOR. Biophys. J. 2004;87:662–674. doi: 10.1529/biophysj.103.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mani R, Tang M, Wu X, Buffy JJ, Waring AJ, Sherman MA, Hong M. Membrane-bound dimer structure of a beta-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry. 2006;45:8341–8349. doi: 10.1021/bi060305b. [DOI] [PubMed] [Google Scholar]
- 60.Long JR, Shaw WJ, Stayton PS, Drobny GP. Structure and dynamics of hydrated statherin on hydroxyapatite as determined by solid-state NMR. Biochemistry. 2001;40:15451–15455. doi: 10.1021/bi010864c. [DOI] [PubMed] [Google Scholar]
- 61.Bennett AE, Ok JH, Griffin RG, Vega S. Chemical-shift correlation spectroscopy in rotating solids - radio frequency-driven dipolar recoupling and longitudinal exchange. J. Chem. Phys. 1992;96:8624–8627. [Google Scholar]
- 62.Gullion T, Vega S. A simple magic angle spinning NMR experiment for the dephasing of rotational echoes of dipolar coupled homonuclear spin pairs. Chem. Phys. Lett. 1992;194:423–428. [Google Scholar]
- 63.Bennett AE, Rienstra CM, Griffiths JM, Zhen WG, Lansbury PT, Griffin RG. Homonuclear radio frequency-driven recoupling in rotating solids. J. Chem. Phys. 1998;108:9463–9479. [Google Scholar]
- 64.Bennett AE, Weliky DP, Tycko R. Quantitative conformational measurements in solid state NMR by constant-time homonuclear dipolar recoupling. J. Am. Chem. Soc. 1998;120:4897–4898. [Google Scholar]
- 65.Ishii Y. 13C-13C dipolar recoupling under very fast magic angle spinning in solid-state nuclear magnetic resonance: Applications to distance measurements, spectral assignments, and high-throughput secondary-structure determination. J. Chem. Phys. 2001;114:8473–8483. [Google Scholar]
- 66.Bak M, Rasmussen JT, Nielsen NC. SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000;147:296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 67.Chang CD, Waki M, Ahmad M, Meienhofer J, Lundell EO, Haug JD. Preparation and properties of N-α-9-fluorenylmethyloxycarbonylamino acids bearing tert-butyl side chain protection. Int. J. Pept. Protein Res. 1980;15:59–66. doi: 10.1111/j.1399-3011.1980.tb02550.x. [DOI] [PubMed] [Google Scholar]
- 68.Lapatsanis L, Milias G, Froussios K, Kolovos M. Synthesis of N-2,2,2-(trichloroethoxycarbonyl)-L-amino acids and N-(9-fluorenylmethoxycarbonyl)-L-amino acids involving succinimidoxy anion as a leaving group in amino-acid protection. Synthesis-Stuttgart. 1983;8:671–673. [Google Scholar]
- 69.Williamson KL. Macroscale and Microscale Organic Experiments. 2nd ed. Heath and Company; Lexington, Massachusetts, Toronto: 1994. [Google Scholar]
- 70.Waskowska A, Lukaszewicz K, Kuzmina LG, Strutshkov YT. The Crystal Structure of N-acetyl-L-leucine. Bull. Pol. Acad. Sci., Ser. Sci. Chim. 1975;23:149–153. [Google Scholar]
- 71.Precigoux G, Cotrait M, Geoffre S. Structure of glycyl-l-phenylalanyl-L-phenylalanine hemihydrate. Acta Crystallogr. Sect. C. 1986;42:315–317. [Google Scholar]
- 72.Long HW, Tycko R. Biopolymer conformational distributions from solid-state NMR: α-helix and 310-helix contents of a helical peptide. J. Am. Chem. Soc. 1998;120:7039–7048. [Google Scholar]
- 73.Macosko JC, Kim CH, Shin YK. The membrane topology of the fusion peptide region of influenza hemagglutinin determined by spin-labeling EPR. J. Mol. Biol. 1997;267:1139–1148. doi: 10.1006/jmbi.1997.0931. [DOI] [PubMed] [Google Scholar]
- 74.Han X, Bushweller JH, Cafiso DS, Tamm LK. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 2001;8:715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- 75.Afonin S, Dur UHN, Glaser RW, Ulrich AS. ‘Boomerang’-like insertion of a fusogenic peptide in a lipid membrane revealed by solid-state 19F NMR. Magn. Reson. Chem. 2004;42:195–203. doi: 10.1002/mrc.1340. [DOI] [PubMed] [Google Scholar]
- 76.Wasniewski CM, Parkanzky PD, Bodner ML, Weliky DP. Solid-state nuclear magnetic resonance studies of HIV and influenza fusion peptide orientations in membrane bilayers using stacked glass plate samples. Chem. Phys. Lipids. 2004;132:89–100. doi: 10.1016/j.chemphyslip.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 77.Aloia RC, Tian H, Jensen FC. Lipid composition and fluidity of the human immunodeficiency virus envelope and host cell plasma membranes. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5181–5185. doi: 10.1073/pnas.90.11.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krasslich HG. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hope MJ, Bally MB, Webb G, Cullis PR. Production of large unilamellar vesicles by a rapid extrusion procedure - characterization of size distribution, trapped volume and ability to maintain a membrane-potential. Biochim. Biophys. Acta-Biomembr. 1985;812:55–65. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- 80.Morcombe CR, Zilm KW. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 81.Zhang HY, Neal S, Wishart DS. RefDB: A database of uniformly referenced protein chemical shifts. J. Biomol. NMR. 2003;25:173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 82.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 1995;103:6951–6958. [Google Scholar]
- 83.Gullion T. Introduction to rotational-echo, double-resonance NMR. Concepts Magn. Reson. 1998;10:277–289. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 84.McDowell LM, Holl SM, Qian SJ, Li E, Schaefer J. Intertryptophan distances in rat cellular retinol-binding Protein Ii by solid-state NMR. Biochemistry. 1993;32:4560–4563. doi: 10.1021/bi00068a011. [DOI] [PubMed] [Google Scholar]
- 85.Balazs YS, Thompson LK. Practical methods for solid-state NMR distance measurements on large biomolecules: Constant-time rotational resonance. J. Magn. Reson. 1999;139:371–376. doi: 10.1006/jmre.1999.1791. [DOI] [PubMed] [Google Scholar]
- 86.Bevington PR, Robinson DK. Data Reduction and Error Analysis for the Physical Sciences. 2nd ed. McGraw-Hill; Boston: 1992. [Google Scholar]
- 87.Oas TG, Hartzell CJ, McMahon TJ, Drobny GP, Dahlquist FW. The carbonyl 13C chemical shift tensors of 5 peptides determined from 15N dipole-coupled chemical shift powder patterns. J. Am. Chem. Soc. 1987;109:5956–5962. [Google Scholar]
- 88.Herzfeld J, Berger AE. Sideband intensities in NMR spectra of samples spinning at the magic angle. J. Chem. Phys. 1980;73:6021–6030. [Google Scholar]
- 89.Naito A, Ganapathy S, Akasaka K, McDowell CA. Chemical shielding tensor and 13C-14N dipolar splitting in single crystals of l-alanine. J. Chem. Phys. 1981;74:3190–3197. [Google Scholar]
- 90.Facelli JC, Gu ZT, McDermott A. Carbon-13 chemical-shift tensors of carboxylic-acids: GIAO calculations in acetic-acid + methylamine dimer. Mol. Phys. 1995;86:865–872. [Google Scholar]
- 91.Longhi S, Czjzek M, Lamzin V, Nicolas A, Cambillau C. Atomic resolution (1.0 Å) crystal structure of Fusarium solani cutinase: stereochemical analysis. J. Mol. Biol. 1997;268:779–799. doi: 10.1006/jmbi.1997.1000. [DOI] [PubMed] [Google Scholar]
- 92.Basak A, Bateman O, Slingsby C, Pande A, Asherie N, Ogun O, Benedek GB, Pande J. High-resolution X-ray crystal structures of human γD crystallin (1.25 Å) and the R58H mutant (1.15 Å) associated with aculeiform cataract. J. Mol. Biol. 2003;328:1137–1147. doi: 10.1016/s0022-2836(03)00375-9. [DOI] [PubMed] [Google Scholar]
- 93.Schmidt-Rohr K, Spiess HW. Multidimensional Solid-State NMR and Polymers. 2nd ed. Academic Press; San Diego: 1994. [Google Scholar]
- 94.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical Recipes in FORTRAN 77: The Art of Scientific Computing. 2nd ed. Fortran Numerical Recipes. Cambridge, New York: 1996. [Google Scholar]
- 95.Kricheldorf HR, Muller D. Secondary structure of peptides. 3. 13C NMR cross polarization/magic angle spinning spectroscopic characterization of solid polypeptides. Macromolecules. 1983;16:615–623. [Google Scholar]
- 96.Saito H. Conformation-dependent 13C chemical shifts - a new means of conformational characterization as obtained by high-resolution solid-state 13C NMR. Magn. Reson. Chem. 1986;24:835–852. [Google Scholar]
- 97.Bodner ML. Ph. D. thesis. Michigan State University; East Lansing: 2006. Solid state nuclear magnetic resonance of the HIV-1 and influenza fusion peptides associated with membranes. [Google Scholar]
- 98.Yang J, Prorok M, Castellino FJ, Weliky DP. Oligomeric beta structure of the membrane-bound HIV-1 fusion peptide formed from soluble monomers. Biophys. J. 2004;87:1951–1963. doi: 10.1529/biophysj.103.028530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bloom M, Evans E, Mouritsen OG. Physical properties of the fluid lipid-bilayer component of cell membranes: a perspective. Quat. Rev. Biophys. 1991;24:293–397. doi: 10.1017/s0033583500003735. [DOI] [PubMed] [Google Scholar]
- 100.Smaby JM, Momsen MM, Brockman HL, Brown RE. Phosphatidylcholine acyl unsaturation modulates the decrease in interfacial elasticity induced by cholesterol. Biophys. J. 1997;73:1492–1505. doi: 10.1016/S0006-3495(97)78181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li XM, Momsen MM, Smaby JM, Brockman HL, Brown RE. Cholesterol decreases the interfacial elasticity and detergent solubility of sphingomyelins. Biochemistry. 2001;40:5954–5963. doi: 10.1021/bi002791n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Silvius JR. Role of cholesterol in lipid raft formation: lessons from lipid model systems. Biochim. Biophys. Acta-Biomembr. 2003;1610:174–183. doi: 10.1016/s0005-2736(03)00016-6. [DOI] [PubMed] [Google Scholar]
- 103.Binder WH, Barragan V, Menger FM. Domains and rafts in lipid membranes. Ange. Chem.-Int. Ed. Engl. 2003;42:5802–5827. doi: 10.1002/anie.200300586. [DOI] [PubMed] [Google Scholar]
- 104.Simons K, Vaz WLC. Model systems, lipid rafts, and cell membranes. Ann. Rev. Biophys. Biomol. Struct. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 105.McMullen TPW, Lewis R, McElhaney RN. Cholesterol-phospholipid interactions, the liquid-ordered phase and lipid rafts in model and biological membranes. Curr. Opin. Colloid Interface Sci. 2004;8:459–468. [Google Scholar]
- 106.Saez-Cirion A, Nieva JL. Conformational transitions of membrane-bound HIV-1 fusion peptide. Biochim. Biophys. Acta-Biomembr. 2002;1564:57–65. doi: 10.1016/s0005-2736(02)00400-5. [DOI] [PubMed] [Google Scholar]
- 107.Afonin S, Glaser RW, Berditchevskaia M, Wadhwani P, Guhrs KH, Mollmann U, Perner A, Ulrich AS. 4-fluorophenylglycine as a label for 19F NMR structure analysis of membrane-associated peptides. Chembiochem. 2003;4:1151–1163. doi: 10.1002/cbic.200300568. [DOI] [PubMed] [Google Scholar]
- 108.Hofmann MW, Weise K, Ollesch J, Agrawal P, Stalz H, Stelzer W, Hulsbergen F, de Groot H, Gerwert K, Reed J, Langosch D. De novo design of conformationally flexible transmembrane peptides driving membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14776–14781. doi: 10.1073/pnas.0405175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Parkanzky PD. Ph. D. thesis. Michigan State University; East Lansing: 2006. Solid state nuclear magnetic resonance studies of the influenza fusion peptide associated with membrane bilayers. [Google Scholar]
- 110.Haque ME, Koppaka V, Axelsen PH, Lentz BR. Properties and structures of the influenza and HIV fusion peptides on lipid membranes: Implications for a role in fusion. Biophys. J. 2005;89:3183–3194. doi: 10.1529/biophysj.105.063032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ono A, Freed EO. Role of lipid rafts in virus replication. In: Roy P, editor. Virus Structure And Assembly. Elsevier Academic Press; San Diego: 2005. pp. 311–358. [DOI] [PubMed] [Google Scholar]
- 112.Nieva JL, Nir S, Muga A, Goni FM, Wilschut J. Interaction of the HIV-1 fusion peptide with phospholipid vesicles: different structural requirements for fusion and leakage. Biochemistry. 1994;33:3201–3209. doi: 10.1021/bi00177a009. [DOI] [PubMed] [Google Scholar]
- 113.Gordon DJ, Balbach JJ, Tycko R, Meredith SC. Increasing the amphiphilicity of an amyloidogenic peptide changes the beta-sheet structure in the fibrils from antiparallel to parallel. Biophys. J. 2004;86:428–434. doi: 10.1016/S0006-3495(04)74119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tseng YH, Mou Y, Mou CY, Chan JCC. Double-quantum NMR spectroscopy based on finite pulse RFDR. Solid State Nucl. Magn. Reson. 2005;27:266–270. doi: 10.1016/j.ssnmr.2005.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.