

Abstract
The neuropeptide Phe-Met-Arg-Phe-NH2 (FMRFa) can induce transcription-dependent long-term synaptic depression (LTD) in Aplysia sensorimotor synapses. We investigated the role of the ubiquitin–proteasome system and the regulation of one of its components, ubiquitin C-terminal hydrolase (ap-uch), in LTD. LTD was sensitive to presynaptic inhibition of the proteasome and was associated with upregulation of ap-uch mRNA and protein. This upregulation appeared to be mediated by CREB2, which is generally regarded as a transcription repressor. Binding of CREB2 to the promoter region of ap-uch was accompanied by histone hyperacetylation, suggesting that CREB2 cannot only inhibit but also promote gene expression. CREB2 was phosphorylated after FMRFa, and blocking phospho-CREB2 blocked LTD. In addition to changes in the expression of ap-uch, the synaptic vesicle-associated protein synapsin was downregulated in LTD in a proteasome-dependent manner. These results suggest that proteasome-mediated protein degradation is engaged in LTD and that CREB2 may act as a transcription activator under certain conditions.
Keywords: p38 kinase, lactacystin, ubiquitin C-terminal hydrolase, CREB2, histone acetylation, synapsin
Introduction
Long-term synaptic plasticity, which is a mechanism for long-term memory, requires synthesis of new RNA and protein (Pittenger and Kandel, 2003). This requirement holds true regardless of the direction of the plasticity, potentiation or depression (Stanton and Sarvey, 1984; Frey et al., 1988; Montarolo et al., 1988; Bailey et al., 1992; Nguyen et al., 1994; Linden, 1996; Ahn et al., 1999). In Aplysia, long-term depression (LTD), a transcription- and translation-dependent form of synaptic plasticity, can be induced by application of the endogenous tetrapeptide Phe-Met-Arg-Phe-NH2 (FMRFa) (Montarolo et al., 1988; Bailey et al., 1992). Knowledge about the molecular targets of FMRFa during LTD is limited (Guan et al., 2002). It is known, however, that FMRFa leads to activation of p38 mitogen-activated protein kinase (MAPK), which is necessary for LTD (Guan et al., 2003; Fioravante et al., 2006). It is also known that FMRFa promotes structural changes, including loss of presynaptic varicosities and retraction of neurites (Schacher and Montarolo, 1991). The structural rearrangements could depend on selective protein degradation; however, this hypothesis has not been previously investigated.
Recently, a role for protein degradation in long-term plasticity has emerged (Bingol and Schuman, 2005). A central pathway through which selective proteins are degraded involves the ubiquitin–proteasome system (UPS) (Hegde and DiAntonio, 2002). In this system, target proteins are tagged by the attachment of ubiquitin molecules, which serve as a recognition signal for degradation by the proteasome system. This process is highly regulated by many enzymes, including deubiquitinating enzymes. By removing ubiquitin from proteins before their degradation, deubiquitinating enzymes could promote recycling of ubiquitin and facilitate processing through the UPS (Wing, 2003). Early evidence for the importance of the UPS in synaptic plasticity came from studies of long-term facilitation (LTF) in Aplysia (Chain et al., 1999a), in which injection of UPS inhibitors blocked LTF. Subsequent studies in mammalian systems corroborated the requirement of the UPS in plasticity and memory (Lopez-Salon et al., 2001; Colledge et al., 2003; Wood et al., 2005). However, little is known about the role of UPS in LTD in Aplysia.
A key component of the UPS that is involved in the regulation of LTF in Aplysia is ubiquitin C-terminal hydrolase (ap-uch) (Hegde et al., 1997). Synthesis of ap-uch, a deubiquitinating enzyme, is increased during LTF and is necessary for consolidation of LTF (Hegde et al., 1997). The promoter region of ap-uch was recently cloned and a cAMP response element (CRE) was identified (Mohamed et al., 2005). The presence of a CRE raised the possibility that transcription factors of the CRE-binding protein (CREB) family may regulate expression of ap-uch.
The gene family of CREB transcription factors is important for long-term synaptic plasticity in both vertebrates and invertebrates (Lonze and Ginty, 2002). In Aplysia, two CREB genes have been identified: creb1 and creb2. The creb1 gene encodes two alternatively spliced mRNAs that are translated into three proteins, CREB1a, CREB1b, and CREB1c (Bartsch et al., 1998). CREB1a, often referred to simply as CREB1, is homologous to mammalian CREB and is a transcription activator necessary for LTF (Dash et al., 1990). The creb2 gene gives rise to CREB2 protein, which is homologous to vertebrate CREB2 and ATF4 (Bartsch et al., 1995) and is regarded as a transcriptional repressor that may pose inhibitory constraints on memory formation (Abel et al., 1998; Guan et al., 2002). This constraint can be regulated by phosphorylation (Guan et al., 2002, 2003).
In this study, we investigated mechanisms underling FMRFa-induced LTD. We report that LTD was sensitive to presynaptic disruption of proteasomal function and correlated with increased levels of protein ubiquitination. We also identified ap-uch as a target gene that is upregulated during LTD at least partly in a p38 MAPK-dependent manner. This upregulation was probably mediated by CREB2, which is traditionally regarded as a transcription repressor. CREB2 was phosphorylated during LTD at a putative MAPK site and this phosphorylation of CREB2 was necessary for LTD. Finally, we found that synapsin, a synaptic vesicle protein implicated in synapse formation and stability, was downregulated during LTD in a proteasome-dependent manner.
Materials and Methods
Treatments.
Ganglia and/or cocultures were treated with FMRFa (American Peptide; 10 μm) or artificial seawater (ASW) (for composition, see Antzoulatos et al., 2003) using two protocols, as indicated: five 5 min pulses at an interpulse interval of 20 min (∼85 min in total); or 90 min continuous application. The p38 MAPK inhibitor 4-(4-fluorophenyl)-2-(4-methylsulfonylphenyl)-5-(4-pyridyl)-1H-imidazole (SB203580) (EMD Chemicals; 20 μm) was applied 30 min before FMRFa treatment and was present throughout FMFRa exposure.
Electrophysiology.
Sensory-motor cocultures of pleural sensory neurons and L7 motor neurons were prepared as described previously (Angers et al., 2002). EPSPs were recorded from motor neurons with 10–15 MΩ sharp electrodes filled with 3 m potassium acetate. Stimulation of presynaptic sensory neurons was performed extracellularly using a blunt, patch-type electrode filled with 50% modified L15 medium–50% ASW. Data acquisition was performed using pClamp (Molecular Devices). For statistical analysis, the amplitude of the EPSP in millivolts was measured during the pretest and 24 h after exposure to FMRFa or vehicle (posttest) and the post/pre ratio was formed.
Aplysia CREB2 antibodies.
The polyclonal anti-phospho-CREB2 and anti-CREB2 antibodies used in this study have been described previously (Mohamed et al., 2005). Briefly, both antibodies were raised by a commercial vendor (Genemed Synthesis) against a hybrid Aplysia CREB2 peptide with sequence SPPDSPEQGPSSPET. This peptide was constructed to juxtapose the sequences immediately surrounding two putative MAPK phosphorylation sites on Aplysia CREB2 (underlined). The phosphorylated version of the peptide, with phosphate groups chemically added on Ser152 (first underline) and Ser237 (second underline), was used to raise the anti-phospho-CREB2 antibody, whereas the unphosphorylated version of the peptide was used to raise the anti-CREB2 antibody. Both antibodies were purified through columns with the corresponding antigen. The anti-phospho-CREB2 antibody was further purified through a second affinity column with the unphosphorylated peptide.
Antibody injections.
Anti-phospho-CREB2 antibody or purified rabbit IgG (both at 0.43 mg/ml) in injection buffer (100 mm KCl, 0.1% 10 kDa dextran-Alexa 488) was pressure-injected in the cytoplasm of sensory neurons. The efficiency of injections was monitored with a fluorescence microscope. Purified IgG, as opposed to preimmune serum, was used in this experiment because the anti-phospho-CREB2 antibody was affinity-purified. Purified IgG has been used as control for affinity-purified antibodies in various systems (Pepperkok et al., 1994; Buffo et al., 2000; Iriyama et al., 2007).
Quantitative reverse transcription-PCR analysis of ap-uch mRNA.
Naive animals were anesthetized by injection of isotonic MgCl2 (0.5 ml/g), and the two pairs of pleural–pedal ganglia were surgically removed. Each pair of ganglia was randomly assigned to either control or FMRFa group. After trimming of the connective sheath tissue in isotonic MgCl2:ASW (1:1 v/v), ganglia in both groups were rinsed with L15:ASW (1:1 v/v) and rested at 18°C for 1–2 h. Subsequently, groups were treated with either vehicle (L15:ASW) or 10 μm FMRFa for 90 min. At the end of the treatment, ganglia were rinsed with L15:ASW, rapidly frozen on dry ice, and stored at −80°C until additional processing. Total RNA was isolated from frozen pleural ganglia by addition of 750 μl of Trizol (Invitrogen) and treated with RNase-free DNase I to remove any contaminating genomic DNA. The quantification of the mRNAs was done by quantitative reverse transcription-PCR (QRT-PCR), conducted in the Quantitative Genomics Core Laboratory of the Department of Integrative Biology and Pharmacology at The University of Texas Medical School at Houston, using a 7700 Sequence Detector (Applied Biosystems) and following published procedures (Wall et al., 2003). Specific assays for ap-uch mRNAs, and for 18S rRNA, were developed with Primer Express software (Applied Biosystems). FAM (6-carboxyfluorescein), TAMRA (6-carboxy-tetramethyrhodamine), and the following primers and synthetic DNAs (sDNAs) were used: 18S rRNA, GenBank accession num-ber X94268 (Winnepenninckx et al., 1998), [1047]-CGATGCCAA-CTAGCGATCC-[1065], [1115]-CACTTTGGTTTCCCGGAAG-[1097]; sDNA 18S rRNA, CGATGCCAACTAGCGATCCGCAGGAGTTGCTT-TGATGACTCTGCGGGCAGCTTCCGGGAAACCAAAGTG; ap-uch, GenBank accession number U90177 (Hegde et al., 1997), [587]-GAA-AGGAAGCACCTGTTGTCC-[607], [656]-ACAACTTCAGCAGCGTCC-TC-[637]; sDNA-ap-uch, GGATGGGAGAAAGGAAGCACCTGTTGTC-CATGGAACGACCTCAGCAGACACATTTCTTGAGGACGCTGCTGA-AGTTGT.
Western blot analysis.
Proteins were extracted in lysis buffer [25 mm Tris, pH 7.5, 1 mm EDTA, 1% Triton X-100, 1% protease inhibitor mixture (Sigma-Aldrich), 1% phosphatase inhibitor mixture 1 and 2 (also from Sigma-Aldrich)] at the times indicated after FMRFa treatment. Equal protein amount from each sample was resolved by SDS-PAGE and transferred to nitrocellulose membrane. For analysis of ubiquitination levels in pleural–pedal ganglia, membranes were incubated with an anti-ubiquitin monoclonal antibody (P4D1; Cell Signaling Technology) and immunoreactive bands were visualized by enhanced chemiluminescence (AP Biotech). Signals were analyzed with ImageQuant (GE Healthcare). For analysis of ap-uch levels, membranes were incubated with anti-ap-uch antibody (kindly provided by Dr. Ashok Hegde, Wake Forest University, Winston-Salem, NC). A polyclonal antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Abcam) was also used for normalization purposes. For the two experiments that examined the role of p38 MAPK in the upregulation of ap-uch, protein was isolated from Trizol-extracted samples used for mRNA quantification, as follows: after removal of the RNA-containing top phase and the interphase, 150 μl of 100% ethanol was added to the phenol layer. After centrifugation at 12,000 × g for 20 min, 750 μl of isopropanol was added to the supernatant and incubated at room temperature for 15 min. Subsequent centrifugation resulted in formation of a protein-containing pellet. The pellet was washed twice with ethanol, air-dried, and solubilized in 100 μl of 0.1 m NaOH. pH was neutralized with 5 μl of 20% HCl and 10 μl of 1 m Tris, pH 6.8. After addition of loading buffer, 40 μl of each sample was subjected to gel electrophoresis. Subsequent analysis followed procedures described above.
Immunofluorescence analysis.
Sensory neurons from pleural ganglia were cultured for 4–5 d as described previously (Angers et al., 2002). Neurons were fixed with 4% paraformaldehyde in PBS containing 30% sucrose, rinsed with PBS, and blocked for 30 min at room temperature in Superblock blocking buffer (Pierce) supplemented with 0.2% Triton X-100 and 3% normal goat serum. Overnight incubation with rabbit polyclonal anti-phospho-CREB2 antibody (primary) at 4°C was followed by PBS rinses and 1 h incubation with secondary antibody (goat anti-rabbit secondary antibody conjugated to Cy-3; Jackson ImmunoResearch Laboratories) at room temperature. For the experiments examining the stability of injected anti-phospho-CREB2 antibody, no primary antibody was added. Neurons were mounted on slides using Prolong anti-fade medium (Invitrogen). Images were obtained with a Bio-Rad 1024 MP confocal microscope using a 60× oil-immersion lens. A z-series of optical sections through the cell body (0.5 μm increments) were taken, and the section through the middle of the nucleus was used for analysis of mean fluorescence intensity with the MetaMorph off-line software (Molecular Devices).
Synaptosomal preparation.
Synaptosomes from six Aplysia pleural–pedal ganglia per group were prepared as described previously (Chin et al., 1989; Levenson et al., 2000). The synaptosomal pellet (P3) was resuspended in 1% SDS–10 mm Tris, pH 7.5, boiled, and centrifuged at 14,000 rpm. The cellular fractions (∼1 ml each) were concentrated to ∼100 μl using Centricon centrifugal filter units (Millipore). After addition of SDS and Tris, pH 7.5, to a final concentration of 1% SDS and 10 mm Tris, samples were boiled and centrifuged at 14,000 rpm. Proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and probed with anti-synapsin, anti-synaptobrevin/vesicle-associated membrane protein (VAMP) (kindly provided by Dr. K. C. Martin, University of California, Los Angeles, Los Angeles, CA), and anti-GAPDH antibodies, as described by Angers et al. (2002) and above. For the experiments with the proteasome inhibitor, clasto-lactacystin β-lactone (50 μm; Calbiochem) was added to the ganglia 2 h before and during FMRFa treatment. After FRMFa washout, the inhibitor was readded to the bath until lysis. β-Lactone was used in this experiment because it has previously been proven effective in blocking the proteasome in Aplysia ganglia (Upadhya et al., 2004). The effectiveness of lactacystin to block the proteasome has been documented in Aplysia cultures (Zhao et al., 2003) but not in ganglia.
Chromatin immunoprecipitation assay.
The technique of chromatin immunoprecipitation (ChIP) is used to detect the interaction of a protein to a region of DNA, through a two-step process as described previously (Weinmann et al., 2001; Mohamed et al., 2005). Briefly, pleural–pedal ganglia were isolated and treated with FMRFa or vehicle for 90 min. Immediately after the treatment, the ganglia were treated with 1% formaldehyde to cross-link proteins to DNA. The reaction was quenched by the addition of glycine (0.125 m). After cell lysis in the presence of protease inhibitors, nuclei were recovered by low-speed centrifugation, resuspended, and sonicated to shear the genomic DNA to lengths of 0.3–1.3 kb. The lysates were then diluted with ChIP dilution buffer and a portion of the lysates was kept for input control. Subsequently, the lysate was precleared with salmon sperm DNA/protein A-agarose beads, followed by brief centrifugation to pull down the beads. Antibodies were added to the resulting supernatant fraction and incubated overnight at 4°C with rotation. The following antibodies were used: anti-CREB1 (2 μg); anti-CREB2 (2 μg); anti-acetyl-histone H3 (Millipore; catalog #06-599; 5 μg); anti-acetyl-histone H4 (Millipore; catalog #06-866; 5 μg); anti-histone deacetylase 5 (HDAC5) (catalog #SC-5250; Santa Cruz Biotechnology; 5 μg). Some samples were incubated with preimmune sera (or no antibody at all for the experiments in Fig. 4) as negative controls. Immune complexes were recovered by the addition of 60 μl of salmon sperm DNA/protein A-agarose slurry for 1 h at 4°C with rotation. The beads were washed with low- and high-salt washing buffers and 1× TE buffer. After washing, the immune complexes were eluted and cross-linking was reversed by addition of NaCl (0.3 m) followed by overnight incubation at 65°C. After treatment with proteinase K, DNA was recovered by phenol/chloroform extraction and ethanol precipitation. DNA was resuspended in H2O and a small aliquot was analyzed by PCR. The input samples were not submitted to immunoprecipitation; rather, they were used directly in PCR to control for the amount of chromatin. The primer sequences (5′ to 3′ direction) used for detection of the ap-uch promoter region were the following: forward primer, [−785]-GCATTAAAGACTAGACTCTAGACTC; reverse primer, [−568]-AGATTCTAGATCTA-CCGGCAAGTAC.
Figure 4.

Binding of CREB2, but not CREB1, to the promoter region of ap-uch coincides with histone hyperacetylation during LTD induction. After treatment with FMRFa or vehicle (ASW; control), chromatin was prepared from ganglia and immunoprecipitated with the indicated antibodies. The immunoprecipitated chromatin was analyzed by PCR as described in Materials and Methods. Chromatin samples before immunoprecipitation (Input) were also analyzed to control for amount of starting material. A, Under basal conditions, no association of CREB2 or CREB1 with the ap-uch promoter region was detected (A1, A2, left lane). FMRFa did not induce detectable binding of CREB1 to the promoter region (A1, right lane), but it did stimulate binding of CREB2 (A2, right lane), indicating that CREB2 is recruited to the ap-uch promoter region during LTD induction. No change in input was observed. Shown are representative examples of three independent experiments. B, The acetylation state of histones in a promoter region correlates with transcription competence: increased acetylation correlates with transcription initiation, whereas deacetylation by HDATs may lead to transcription repression. Using anti-acetyl-H3 or anti-acetyl-H4 antibodies, basal acetylation of H3 and H4 histones was detected in the ap-uch promoter region (B1, B2, left lane). Basal binding of HDAC5 to the promoter region was also observed (B3, left lane). FMRFa caused increased acetylation of both histones H3 and H4 (B1, B2, right lane) as well as reduced recruitment of HDAC5 at the ap-uch promoter region (B3, right lane), indicating that the promoter region of ap-uch becomes hyperacetylated, and therefore permissive to transcription, after FMRFa. No change in input was observed. Shown are representative examples of three independent experiments.
Statistical analysis.
Statistical tests were performed using MegaStat 9.1 and Jandel SigmaStat 2.0. Student's t tests were used for two-sample comparisons (see Figs. 1, 3, 5A,B). ANOVA followed by post hoc comparisons were used whenever more than two groups were compared (see Figs. 5C, 6). Values of p < 0.05 were considered statistically significant.
Figure 1.

Lactacystin blocked FMRFa-induced LTD of sensorimotor synapse. A1, B1, Bright-field images of sensorimotor cocultures. MN, Motor neuron; SN, sensory neuron. A2, B2, Sensory neurons filled with Alexa 488-dextran, indicating successful injection of DMSO (A2) or lactacystin (B2). A3, B3, Examples of EPSPs measured before the presynaptic injection (pre) and 24 h after treatment with FRMFa (post). EPSPs were elicited by extracellular stimulation of the sensory neuron. C, For statistical analysis, the ratio of post over pre EPSP amplitude was formed for each coculture. *p < 0.01. D, E, Examples of EPSPs measured before (pre) and ∼24 h after (post) presynaptic injection of DMSO (vehicle) or lactacystin. F, Lactacystin did not significantly affect synaptic strength over a 24 h period (p = 0.54). Error bars indicate SEM.
Figure 3.
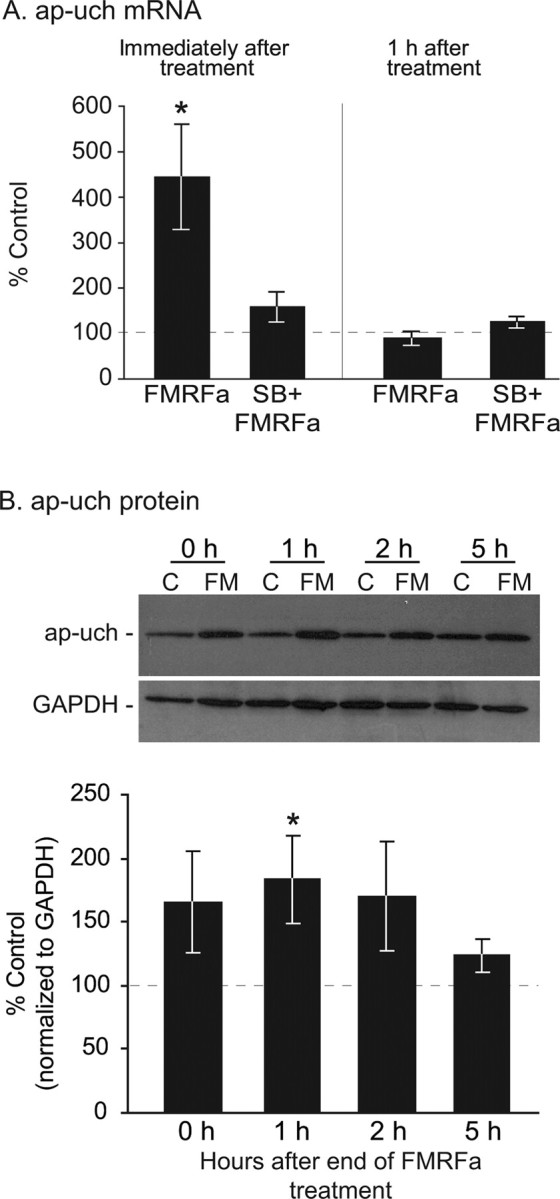
FMRFa upregulates ap-uch mRNA and protein in pleural ganglia. A, QRT-PCR analysis of mRNA extracted from ganglia either immediately or 1 h after the end of 90 min treatment with FMRFa or vehicle (control). Immediately after the end of treatment, a significant increase in ap-uch mRNA was observed compared with time-matched control (*p < 0.05). In the presence of the p38 kinase inhibitor SB203580, FMRFa did not induce an increase in ap-uch mRNA. The effect of FMRFa on ap-uch was transient because 1 h after the end of treatment the mRNA was at control levels. B, Using an antibody specific for ap-uch, Western blot analysis was conducted on extracts from ganglia treated with FMRFa (FM) or vehicle [artificial seawater; control (C)] and lysed immediately, 1, 2, or 5 h after the end of treatment. Membranes were also probed with an anti-GAPDH antibody for normalization purposes. Compared with time-matched controls, ap-uch protein significantly increased only at the 1 h time point, although a tendency for increase was observed at all time points examined (*p < 0.05). These results indicate that the upregulation of ap-uch mRNA is accompanied by increased ap-uch protein synthesis. Error bars indicate SEM.
Figure 5.
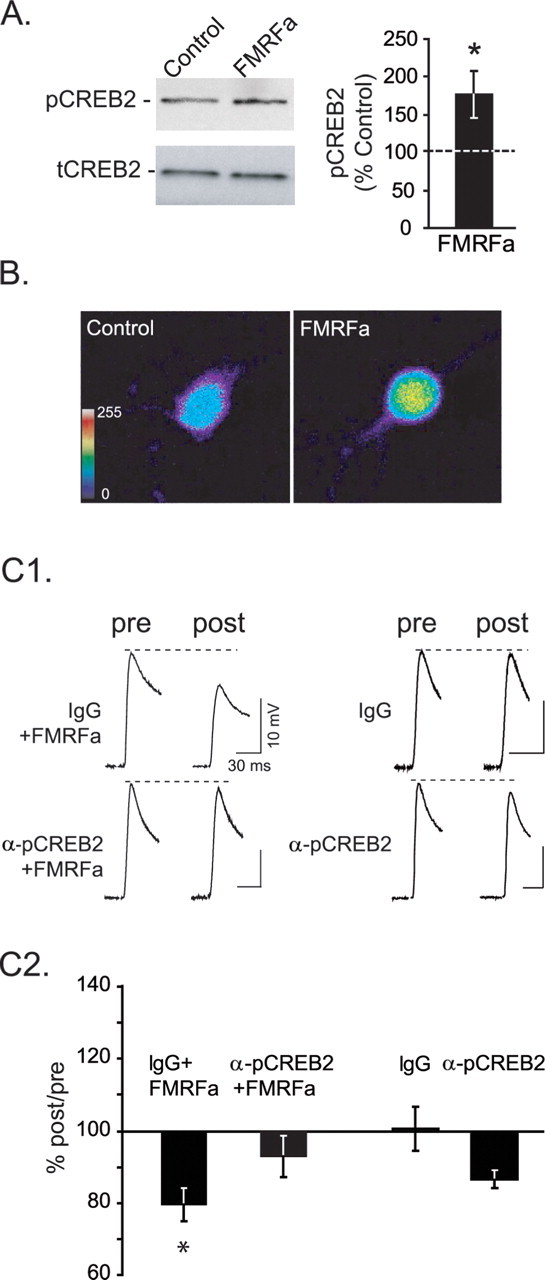
CREB2 is phosphorylated after FMRFa treatment and is necessary for FMRFa-induced LTD. A, Immunoblot analysis using the anti-phospho-CREB2 antibody showed that immediately after the end of treatment with FMRFa, levels of pCREB2 were significantly increased compared with control. Exposure to FMRFa did not affect levels of total CREB2 (*p < 0.05). B, Under control conditions, pCREB2 was observed primarily in the nucleus of cultured isolated sensory neurons and to a lesser extend in the cytoplasm, displaying a distribution pattern that is similar to that of mammalian CREB2 in rat cultured cortical neurons (White et al., 2000). After FMRFa exposure, an increase in pCREB2 was observed in both the cytoplasm and nucleus, suggesting that the presence of a postsynaptic target is not required for CREB2 to be phosphorylated. C1, Representative EPSPs recorded from IgG-injected (top traces) or phosphospecific anti-CREB2-injected (bottom traces) cocultures, before and after treatment with FMRFa or vehicle. C2, For statistical analysis, the peak amplitude of EPSP at the 24 h posttest was normalized to pretest EPSP. Injection of anti-phospho-CREB2 antibody blocked LTD (*p < 0.05) without significantly affecting basal transmission. Error bars indicate SEM.
Figure 6.

Synapsin is downregulated in LTD in a proteasome-dependent manner. A, Western blot analysis of synaptosomes prepared from FMRFa-treated or control ganglia. Membranes were probed with anti-synapsin, anti-VAMP, and anti-GAPDH antibodies. GAPDH signal was used for normalization purposes. Results indicated that, compared with untreated ganglia, synapsin was significantly decreased 24 h after FMRFa (*p < 0.05). This decrease was blocked by preincubation with the proteasome inhibitor clasto-lactacystin β-lactone. Levels of the synaptic vesicle-associated membrane protein synaptobrevin/VAMP were unaffected by the treatment. B, Western blot analysis of the cellular fractions from the same cell fractionation experiment as in A. FMRFa treatment had no significant effect on levels of synapsin or VAMP. Note enrichment of synapsin and VAMP in the synaptosomal fraction (A) compared with the cellular fraction (B). Error bars indicate SEM.
Results
An inhibitor of the proteasome blocks LTD
As a first step toward investigating the role of the proteasome in long-term depression in Aplysia, we examined the effect of lactacystin, an irreversible inhibitor of the 20S component of the proteasome (Myung et al., 2001), on FMRFa-induced depression of the sensorimotor synapse (Fig. 1). Lactacystin is commonly used in Aplysia and rodents to block the proteasome, and its actions are comparable with those of other proteasome inhibitors (Chain et al., 1999a; Lopez-Salon et al., 2001; Zhao et al., 2003; Khoutorsky and Spira, 2005). To distinguish between a presynaptic versus postsynaptic requirement for the proteasome, we injected lactacystin in the presynaptic sensory neuron and monitored the effects on synaptic strength after treatment with FMRFa. Briefly, after electrophysiological assessment of basal synaptic strength (Fig. 1A3,B3, pre), we pressure-injected lactacystin (10 μm) or DMSO (0.1%; vehicle for lactacystin) into sensory neurons paired with L7 motor neurons. The efficiency of the injection was monitored under fluorescence through the addition of Alexa 488-dextran in the injection buffer (Fig. 1A2,B2). One to 2 h later, cultures were treated with five 5 min pulses of FMRFa to induce LTD. This time point was chosen based on the protocol of Zhao et al. (2003). The extent of LTD and the effect of lactacystin on LTD were assessed 24 h later during the posttest (Fig. 1A3,B3, post). The DMSO plus FMRFa group (mean ± SEM, 66.31 ± 4.5%; n = 4) was significantly different from the lactacystin plus FMRFa group (90.40 ± 3.11%; n = 3; t(5) = 4.06; p < 0.01), indicating that lactacystin blocked FMRFa-induced depression (Fig. 1C). In a separate set of experiments, we examined whether lactacystin affected basal transmission over 24 h. The lactacystin-injected group (96.24 ± 9.07%; n = 4) did not differ significantly from the DMSO-injected group (87.81 ± 8.51%; n = 3; t(5) = 0.65; p = 0.54), suggesting that lactacystin did not affect synaptic strength over a 24 h period (Fig. 1F). These results provide the first evidence that presynaptic proteasomal function is necessary for LTD in Aplysia.
Blockage of LTD by the inhibitor might have resulted from interfering with the initial signaling cascade activated by FMRFa (e.g., by affecting the basal turnover of proteins involved in this cascade, rather than LTD mechanisms). We think this possibility is unlikely because in our experiments the inhibitor was applied only 1–2 h before FMRFa; therefore, the time window to affect the transduction cascade through a protein turnover mechanism was limited. Moreover, we found no effect of the inhibitor on basal synaptic transmission over 24 h (Fig. 1). Finally, at present there is no evidence implicating the ubiquitin–proteasome system in short-term FMRFa-induced depression. Therefore, the block of LTD after injection of the proteasome inhibitor probably reflects a requirement of the proteasome in LTD.
FMRFa exposure increased protein ubiquitination
An important step in the proteasome-mediated degradation of proteins is the attachment of ubiquitin, which serves as a tag for subsequent degradation (but see Conaway et al., 2002, for a review of proteasome-independent functions of ubiquitin). If the ubiquitin–proteasome system is necessary for LTD, then it should be possible to detect increased incorporation of ubiquitin to proteins. One method of detecting protein ubiquitination is through immunoblots using an anti-ubiquitin antibody (Ehlers, 2003). After treatment of pleural–pedal ganglia with FMRFa or vehicle (ASW; control), we monitored ubiquitination of proteins at various time points: immediately, 1, 2, 3, and 5 h after treatment. In four independent experiments, we observed a reproducible increase in protein ubiquitination 1–2 h after the end of FMRFa exposure (Fig. 2A). This increase was not an artifact of unequal protein loading, as indicated by Ponceau staining of total protein (Fig. 2B). The time course of protein ubiquitination suggested that recruitment of the ubiquitin pathway by FMRFa is rapid but transient (Fig. 2A).
Figure 2.
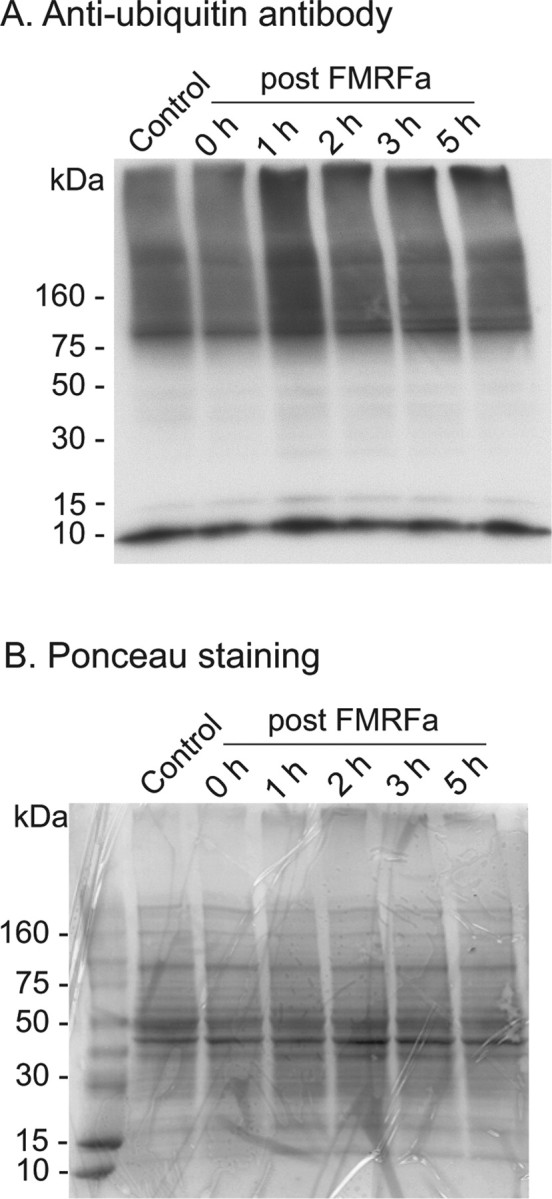
Protein ubiquitination is increased 1–2 h after treatment with FMRFa. After exposure to FMRFa or vehicle (ASW; control), pleural–pedal ganglia were lysed at various time points as indicated and processed with an anti-ubiquitin antibody to monitor levels of protein ubiquitination. Note that the 0 h time point corresponds to ∼85 min of FMRFa exposure. A, Increased protein ubiquitination was observed 1–2 h after the end of FMRFa exposure. The 10 kDa band is most probably free ubiquitin. B, Ponceau staining of the nitrocellulose membrane confirmed equal loading of samples. Representative examples of four independent experiments.
Ubiquitin hydrolase mRNA and protein are regulated in LTD
The transient nature of the increase in ubiquitination levels after treatment with FMRFa (Fig. 2) could be explained by either the transient regulation of a ubiquitinating enzyme or the engagement of deubiquitinating processes. Because one deubiquitinating enzyme, ap-uch, is known to be necessary for and upregulated during serotonin-induced long-term synaptic plasticity (Hegde et al., 1997), we examined whether ap-uch is also regulated during FMRFa-induced long-term plasticity. Pleural–pedal ganglia were exposed to FMRFa (10 μm) or vehicle (ASW; control) for 85 min, followed by excision of the pleural ganglia either immediately or 1 h after treatment (Fig. 3A). QRT-PCR analysis of isolated RNA indicated that compared with time-matched controls, FMRFa induced a fourfold increase in ap-uch mRNA immediately after the end of the 90 min treatment (mean ± SEM, normalized to time-matched control: FMRFa, 444.73 ± 114.74%; n = 5; t(4) = 3.01; p < 0.05) (Fig. 3A). This upregulation was transient, with ap-uch mRNA returning to baseline 1 h after the end of treatment (normalized to time-matched control: FMRFa, 89.19 ± 17.12%; n = 5; t(4) = 0.63; p = 0.56). These results indicate that ap-uch mRNA is regulated by FMRFa.
Previous studies showed that p38 MAPK is necessary for FMRFa-induced synaptic plasticity (Guan et al., 2003). To test whether this kinase is also involved in the FMRFa-induced regulation of ap-uch, we preincubated ganglia for 30 min with SB203580, a p38 MAPK inhibitor. This treatment blocked the increase in ap-uch mRNA immediately after FMRFa (compared with time-matched SB group: SB plus FMRFa, 159% ± 32.91; n = 5; t(4) = 1.8; p = 0.15). As expected, SB203580 had no effect 1 h after the end of FMRFa treatment (compared with time-matched SB group: SB plus FMRFa, 124.98 ± 13.94%; n = 5; t(4) = 1.79; p = 0.15). These results suggest that the upregulation of ap-uch mRNA by FMRFa requires the p38 MAPK signaling pathway.
We next examined whether the increased ap-uch mRNA was associated with an increase in ap-uch protein (Fig. 3B) using a polyclonal antibody against ap-uch in Western blots. This antibody was raised against the Aplysia uch and was previously characterized and used in the study by Hegde et al. (1997). Membranes were reblotted for GAPDH, which was used for normalization purposes. Compared with time-matched controls, FMRFa significantly increased ap-uch levels 1 h after the end of treatment (183.8 ± 34.76%; n = 5; t = −2.9; p < 0.05), but not immediately (165 ± 39.53%; n = 5; t = −2.17; p = 0.1), 2 h (170 ± 42.47%; n = 5; t = −1.41; p = 0.23), or 5 h (123 ± 12.37%; n = 5; t = −1.44; p = 0.22) after the end of treatment, although a moderate increase is evident at these time points. In two independent experiments, inhibiting p38 MAPK with SB203580 blocked the ap-uch increase 1 h after FMRFa (normalized to SB group: SB plus FMRFa, 81.2 ± 21.8%). These findings identify ap-uch as a target for regulation by FMRFa and suggest that proteolytic degradation through the ubiquitin pathway may play a role in LTD. Moreover, these results support the role of p38 MAPK as an important mediator of the actions of FMRFa.
CREB2, but not CREB1, binds to the promoter region of ap-uch after LTD induction
The ap-uch promoter region was recently cloned and a variant CRE was identified (Mohamed et al., 2005), raising the possibility that members of the CREB family of transcription factors may be involved in the regulation of ap-uch. Mohamed et al. (2005) investigated the interaction of CREB1 and CREB2 with the ap-uch promoter region under basal conditions and after treatment with 5-HT. Neither transcription factor bound to the promoter region. However, it remains possible that CREB1 or CREB2 may bind to the ap-uch promoter region under different conditions (e.g., after treatment with FMRFa). To explore this possibility, we performed ChIP assays on samples from vehicle- and FMRFa-treated ganglia using anti-CREB1 and anti-CREB2 antibodies. In three independent experiments, no basal binding of CREB1 or CREB2 to the ap-uch promoter region was detected, as indicated by the lack of PCR product in control groups (Fig. 4A1,A2, left lane). Moreover, treatment with FMRFa did not induce detectable binding of CREB1 (Fig. 4A1, right lane), suggesting that binding of CREB1 to the promoter region of ap-uch is not regulated by FMRFa. In contrast, binding of CREB2 to the promoter region was detected immediately after treatment with FMRFa (Fig. 4A2, right lane), indicating that CREB2 is recruited to the ap-uch promoter region during LTD induction. No changes were observed in input controls. The temporal dynamics of CREB2 association with the promoter region of ap-uch are not known. A transient association could potentially explain the transient change in ap-uch mRNA after FMRFa (Fig. 3). The antibodies used for the ChIP experiments are specific for CREB1 and CREB2 (Mohamed et al., 2005). Moreover, the anti-CREB1 antibodies successfully detect the association of CREB1 with the promoter region of other genes (e.g., creb1) (Mohamed et al., 2005), suggesting that the observed lack of CREB1 association with the ap-uch promoter region is not a false-negative result. These findings suggest that CREB2, but not CREB1, may participate in the regulation of ap-uch by FMRFa.
CREB2 binding to the ap-uch promoter region correlates with increased histone acetylation in the ap-uch promoter region
The observation that CREB2 is recruited to the ap-uch promoter region in response to FMRFa raises the interesting possibility that CREB2 may be involved in the FMRFa-induced regulation of ap-uch. To further investigate this hypothesis, we examined the acetylation state of histones H3 and H4 in the promoter region of ap-uch. Binding of transcription factors to the promoter region of target genes recruits histone acetylases (HATs) or deacetylases (HDATs), which modify lysine residues on core histones, thus regulating the transcriptional competence of a gene. Increased acetylation by HATs correlates with transcription initiation, whereas deacetylation by HDATs may lead to transcription repression (for review, see Davie and Spencer, 1999; Eberharter and Becker, 2002) (but see Shahbazian and Grunstein, 2007). The acetylation state of chromatin is finely regulated during long-term synaptic plasticity and memory formation (Guan et al., 2002; Alarcón et al., 2004; Korzus et al., 2004; Levenson et al., 2004).
Using anti-acetyl-H3 or anti-acetyl-H4 antibodies in three independent ChIP experiments, some basal acetylation of H3 and H4 histones in the ap-uch promoter region was detected, as indicated by the presence of a PCR product in the control groups (Fig. 4B1,B2, left lane). This observation is in agreement with the constitutive expression of ap-uch (Hegde et al., 1997). Basal binding of HDAC5 to the promoter region was also observed (Fig. 4B3, left lane), suggesting that the acetylation level of the promoter histones is dynamically regulated at rest. After treatment with FMRFa, an increase in acetyl-H3 and acetyl-H4 signals was observed compared with controls (Fig. 4B1,B2, right lane), suggesting that both histones H3 and H4 in the promoter region of ap-uch are hyperacetylated in response to FMRFa.
We further asked whether FMRFa signaling modulates the activity of histone deacetylases that may, in turn, alter acetylation of histones associated with the ap-uch promoter. HDAC5 in particular is known to be modulated by FMRFa (Guan et al., 2002). We found that recruitment of HDAC5 to the ap-uch promoter region was impaired (Fig. 4B3, right lane), indicating that the deacetylation of the promoter region is reduced. No changes were observed in input controls. These FMRFa-induced changes in acetylation levels are consistent with the observation that ap-uch is actively transcribed during LTD, as indicated by the increase in mRNA levels (Fig. 3A). More importantly, the hyperacetylation of the promoter region occurs at the same time and in response to the same treatment that induces CREB2 binding to the promoter region, raising the possibility that CREB2 is recruited by FMRFa to activate transcription of ap-uch.
CREB2 is phosphorylated at a putative MAPK site during LTD induction and is necessary for LTD
The results from the ChIP experiments point to CREB2 as an important mediator of the effects of FMRFa on gene expression. If CREB2 is a key player in regulating gene expression during LTD, then CREB2 should be phosphorylated, and activated, in response to FMRFa. In vitro kinase assays suggested that CREB2 can be phosphorylated by Erk and p38 MAPK (Bartsch et al., 1995; Guan et al., 2002). However, the phosphorylation of CREB2 in vivo has not been previously demonstrated.
To examine the potential of FMRFa to induce phosphorylation of CREB2 at a putative MAPK site, we used a phosphospecific anti-CREB2 antibody, which detects CREB2 phosphorylated at either one of two putative MAPK sites (Mohamed et al., 2005). The specificity of the antibody has been demonstrated in Western blots of membranes treated with lambda protein phosphatase (Mohamed et al., 2005), as well as in fixed cultured sensory neurons through antigen preabsorption experiments or intracellular injection of lambda protein phosphatase (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) (see also Materials and Methods). Using this antibody, we found that immediately after the end of treatment with FMRFa, levels of phosphorylated CREB2 (pCREB2) were significantly increased compared with control samples (FMRFa, 174.97 ± 31.58; n = 4; t = −3.44; p < 0.05) (Fig. 5A). Exposure to FMRFa did not affect levels of total CREB2 (tCREB2). The increase in pCREB2 after FMRFa exposure was further observed in cultured isolated sensory neurons (Fig. 5B). Compared with controls, FMRFa treatment increased levels of pCREB2 in both the nucleus (in arbitrary units: control, 46.42 ± 1.52; FMRFa, 90.38 ± 5.85; t(13) = −6.83; p < 0.001) and the cytoplasm (control, 48.79 ± 1.57; FMRFa, 100.26 ± 4.96; t(13) = −9.32; p < 0.001) of sensory neurons, suggesting that a postsynaptic target is not required for CREB2 to be phosphorylated in response to FMRFa. Furthermore, these results show that FMRFa can have the same long-term biochemical effects on cells in culture and in ganglia.
We further hypothesized that blocking phosphorylated CREB2 would impair LTD. To investigate the role of phosphorylated CREB2 in LTD, we injected presynaptic sensory neurons cocultured with motor neurons with the same anti-phospho-CREB2 antibody that we used for our Western blots and immunofluorescence experiments, and we monitored the effects on LTD expression (Fig. 5C). The anti-phospho-CREB2 antibody was expected to functionally sequester phospho-CREB2 in the cytoplasm, preventing its nuclear translocation and blocking pCREB2-dependent functions. Indeed, our immunofluorescence experiments showed that phosphorylated CREB2 localizes in both the nucleus and the cell body (Fig. 5B), as observed in rat cultured cortical neurons (White et al., 2000). Moreover, Lai et al. (2006) showed that, after treatment with FMRFa, CREB2 translocates to the nucleus in an importin-dependent manner. The antibody was not expected to enter the nucleus because of size exclusion (Ohno et al., 1998).
The specificity of the anti-phospho-CREB2 antibody, which was also used in Western blots and immunofluorescence experiments (Fig. 5A,B), is demonstrated by the following: (1) BLAST searches indicated that the peptide sequence of Aplysia CREB2 against which the antibody was raised, is not contained in any other known Aplysia protein; (2) the antibody was affinity-purified twice by passing through a phospho-CREB2 column and a total CREB2 column (Mohamed et al., 2005); (3) in Western blots, the antibody specifically immunoreacted with a single band of the expected molecular mass (50 kDa) (Fig. 5A); (4) preabsorption of the antibody with a CREB2 phosphopeptide blocked immunoreactivity in cultured sensory neurons (supplemental Fig. S1A1,A2, available at www.jneurosci.org as supplemental material), whereas preabsorption with the unphosphorylated version of the same peptide did not (supplemental Fig. S1A3,A4, available at www.jneurosci.org as supplemental material); (5) antibody immunoreactivity was dramatically reduced in sensory neurons after dephosphorylation of endogenous CREB2 by injecting a protein phosphatase (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material); (6) brief treatment of membranes with protein phosphatase impaired immunoreactivity of the anti-phospho-CREB2 antibody in Western blots of Aplysia protein extracts (Mohamed et al., 2005).
After assessment of synaptic strength (Fig. 5C1, pre), we injected anti-phospho-CREB2 (anti-pCREB2) antibody or purified rabbit IgG into sensory neurons. After a rest period of 1 h, cocultures were exposed to five, 5 min pulses of FMRFa or vehicle (ASW) at an interpulse interval of 20 min. Synaptic strength was reassessed 24 h later (Fig. 5C1, post) to evaluate the extent of LTD. Two-way ANOVA indicated that there was a significant interaction between factors [the injected antibodies (anti-pCREB2 or rabbit IgG) and the treatment (FMRFa or vehicle)] (F(1,28) = 8.013; p < 0.05). Post hoc comparisons revealed that in the IgG-injected group, FMRFa induced significant depression compared with vehicle-treated controls (IgG plus FMRFa, 79.43 ± 4.58%; IgG, 100.5 ± 6.2%; q = 4.00; p < 0.05). However, in the anti-pCREB2 group, FMRFa failed to induce significant depression (pCREB2, 86.5 ± 7.3%; pCREB2 plus FMRFa, 92.97 ± 5.63%; q = 1.464; p > 0.05). Importantly, a significant difference was found between the anti-pCREB2 plus FMRFa group and the IgG plus FMRFa group (q = 3.04; p < 0.05), indicating that injection of anti-pCREB2 antibody blocked FMRFa-induced LTD (Fig. 5C2). Injection of IgG or anti-pCREB2 antibody alone did not significantly impair transmission over the 24 h period (q = 2.67; p > 0.05). Collectively, these results suggest that CREB2 is phosphorylated in LTD and that pCREB2 is necessary for LTD.
One concern about the antibody injection experiment was whether the anti-CREB2 antibody would be stable in the cellular milieu so as to functionally block endogenous CREB2. Our finding that anti-pCREB2 injection blocked LTD would argue for the stability of the antibody. Nonetheless, we performed a control experiment to examine the presence of the antibody within the cell over time. To this end, we injected the anti-pCREB2 antibody in sensory neurons and then fixed them either 2 or 24 h after injection. Cells were incubated only with secondary antibody (to detect levels of injected primary antibody) and processed for immunofluorescence. pCREB2 immunofluorescence could be detected both at 2 and 24 h after injection (pCREB2 signal normalized to injection dye: 2 h, 0.97 ± 0.06; 24 h, 0.92 ± 0.04). The small decrease in signal intensity that was observed at 24 h compared with 2 h was not statistically significant (t(17) = 0.69; p = 0.50). These results indicate that the pCREB2 antibody is stable in vivo for at least 24 h.
Levels of synapsin are decreased in synaptosomes during LTD
Results from the experiments discussed above point to the ubiquitin–proteasome system as a necessary mechanism for LTD, supporting a role of regulated protein degradation in the induction and/or consolidation of LTD. Previous work also showed that FMRFa-induced LTD of sensorimotor synapses is accompanied by a significant loss of sensory neuron varicosities and neurites (Schacher and Montarolo, 1991), raising the possibility that synaptic proteins are targeted for degradation during LTD. Synapsin is a protein that has been implicated not only in the regulation of synaptic strength (Llinas et al., 1985; Hilfiker et al., 1998) and plasticity (Humeau et al., 2001; Chi et al., 2003; Fioravante et al., 2007) but also in synapse formation and maintenance as well as neurite elongation (Ferreira and Rapoport, 2002). The Aplysia homolog of synapsin was cloned and found to localize primarily at varicosities (Angers et al., 2002). To begin to investigate mechanisms of LTD expression, we tested whether synapsin is degraded during LTD. Twenty-four hours after treatment with FMRFa or vehicle (ASW; control), we prepared synaptosomes from pleural–pedal ganglia and examined levels of synapsin using Western blot analysis. Results from four independent experiments indicated that significantly less synapsin was present in synaptosomes from ganglia treated with FMRFa compared with controls (synapsin, 64% ± 9.19; t(3) = 3.33; p < 0.05).
To begin to examine the mechanisms underlying synapsin degradation, we repeated the above experiment with or without the proteasome inhibitor clasto-lactacystin β-lactone (Fig. 6A). This irreversible inhibitor of the proteasome is a derivative of lactacystin that effectively blocks protein degradation in Aplysia ganglia (Upadhya et al., 2004) and cultures (Zhao et al., 2003; Khoutorsky and Spira, 2005). In three independent experiments, we replicated our previous finding that FMRFa decreased levels of synaptosomal synapsin 24 h after treatment (mean ± SEM, expressed as percentage control: FMRFa, 21.17 ± 6.94) (Fig. 6A). Preincubation with β-lactone at 50 μm, a concentration used by Upadhya et al. (2004), blocked this decrease (β-lactone plus FMRFa, 96.08 ± 2.37). One-way ANOVA showed a significant difference among the four groups [DMSO (vehicle for β-lactone), DMSO plus FMRFa, β-lactone, β-lactone plus FMRFa: F(3,11) = 7.79, p < 0.01]. Subsequent post hoc comparisons indicated that the β-lactone plus FMRFa group differed significantly from the FMRFa group (q = 2.65; p < 0.05) but not from the β-lactone group (q = 0.16; p > 0.05), or the DMSO group (q = 2.16; p > 0.05), suggesting that the inhibitor blocked the FMRFa-induced decrease in synapsin. This effect of FMRFa targeted synapsin specifically and probably did not reflect loss of synaptic vesicles because levels of the synaptic vesicle marker synaptobrevin/VAMP were not affected by FMRFa treatment (FMRFa, 92.68 ± 19.4%) (Fig. 6A). Finally, the decrease in synapsin was not observed in the cellular fraction (FMRFa, 108.11 ± 9.88%) (Fig. 6B), suggesting that the synaptosomal decrease in synapsin levels results from protein degradation and not subcellular redistribution. A decrease in synapsin may or may not be evident in total lysates, depending on whether synapsin degradation is a predominantly synaptic event. The 8% increase in the cellular fraction is within the margin of statistical error. Our interpretation of these results is that, during the first 24 h after FMRFa treatment, synapsin is downregulated at synaptic sites, probably through the action of synaptically localized proteasomes (Bingol and Schuman, 2005; Upadhya et al., 2006), but not in other cellular compartments.
Discussion
Using the Aplysia model system, we sought to identify mechanisms mediating a form of long-term heterosynaptic depression induced by the neuropeptide FMRFa. For our studies, we used both the Aplysia sensorimotor synapse, which is a well studied model for the investigation of synaptic plasticity (Kandel, 2001), as well as whole pleural–pedal ganglia. The approach of using both whole ganglia and cultured sensory neurons in a complementary way to investigate mechanisms of synaptic plasticity has been used successfully in the work of others (Guan et al., 2002, 2003; Si et al., 2003; Udo et al., 2005; Lee et al., 2007), yielding significant findings.
Since its discovery in bivalves (Price and Greenberg, 1977), an extensive family of FMRFa-related peptides (FaRPs) has been described in Drosophila (for review, see Taghert, 1999), Caenorhabditis elegans (for review, see Li et al., 1999), and rodents (for review, see Raffa, 1990). However, the function of FaRPs in these systems is mostly unknown. In Aplysia, FMRFa-containing neurons are widespread (Brown et al., 1985; Lloyd et al., 1987; Xu et al., 1994) and activation of some of them produces presynaptic inhibition of sensory neuron synapses (Mackey et al., 1987; Small et al., 1992). FMRFa induces both short- and long-term synaptic depression. Short-term depression, which has been observed in both cultures and ganglia (Pieroni and Byrne, 1992; Guan et al., 2003), emerges at least in part from modulation of ionic conductances and the release machinery (Belardetti et al., 1987; Piomelli et al., 1987; Dale and Kandel, 1990; Critz et al., 1991). Long-term depression, which at present has only been examined in cultures, requires gene expression (Montarolo et al., 1988; Bailey et al., 1992). However, the identities of LTD-related genes and of the transcription factors that regulate them are mostly unexplored. The present study provides the first evidence that a proteasome-associated ubiquitin hydrolase is upregulated in LTD and identifies CREB2 as a potential mediator of this regulation.
Using a pharmacological inhibitor, we found that the ubiquitin–proteasome system is necessary for LTD. Other evidence from Aplysia and mammals also points to the role of this system in synaptic plasticity and learning. Specifically, the proteasome is involved in long-term synaptic facilitation (LTF) (Chain et al., 1999a; Zhao et al., 2003). In LTF, the regulatory subunit of protein kinase A (PKA) is degraded, giving rise to persistently active catalytic subunits. Long-term PKA activation could prolong phosphorylation of transcription activators, such as CREB1 (Chain et al., 1999a; Liu et al., 2008). Moreover, the transcription inhibitor CREB1b is targeted for degradation during LTF (Upadhya et al., 2004). Together, these molecular events could lead to increased expression of genes essential for consolidation of LTF. Also, in rodents the ubiquitin pathway seems to be important in memory formation because bilateral infusion of a proteasome inhibitor in the hippocampus impaired inhibitory avoidance learning (Lopez-Salon et al., 2001) and extinction of fear conditioning (Lee et al., 2008). Furthermore, long-term potentiation (LTP) and context-dependent learning were impaired in mice carrying a mutation in E6-AP ubiquitin ligase, an enzyme involved in the ubiquitin pathway (Jiang et al., 1998). Recently, LTP induction was reported to accelerate protein degradation via the proteasome system (Karpova et al., 2006) (but see Dong et al., 2008). Finally, the proteasome and protein ubiquitination are important for AMPA receptor internalization and LTD in the hippocampus (Colledge et al., 2003).
After exposure to FMRFa, we observed a rapid, yet transient, increase in protein ubiquitination. Even if ubiquitination persisted for 24 h, a causal link between ubiquitination and LTD could not be claimed without additional analysis. Nonetheless, it seems likely that the proteasome is involved in LTD based on the following findings: (1) ubiquitination is increased in response to FMRFa treatment; (2) blocking the proteasome by injecting lactacystin blocks LTD; and (3) lactacystin blocks the FMRFa-induced downregulation of synapsin. Increased protein ubiquitination has also been observed in cortical neurons after pharmacologically induced changes in synaptic activity (Ehlers, 2003).
The duration and extent of protein ubiquitination is partly controlled by a large family of deubiquitinating enzymes (DUBs), which are involved in removing and recycling ubiquitin from ubiquitin-tagged proteins (Glickman and Ciechanover, 2002). Therefore, we examined the potential involvement of DUBs in LTD. In Aplysia, one DUB is ubiquitin hydrolase (ap-uch), a neuron-specific hydrolase that is necessary for LTF and is upregulated by 5-HT (Hegde et al., 1997). This upregulation appears to be independent of CREB1 and CREB2 (Mohamed et al., 2005). In our study, we found that ap-uch is upregulated early during LTD in a p38 MAPK-dependent manner, suggesting that ap-uch may participate in regulating the duration of ubiquitination. If ap-uch is involved in both LTF and LTD, how is the direction of synaptic plasticity (facilitation vs depression) attained? One possible explanation is that increased protein degradation is important for both forms of synaptic plasticity, and that the direction of plasticity at least partly derives from specific combinations of ubiquitin conjugating and ubiquitin ligase enzymes, which would target particular proteins for degradation (Cline, 2003; Yi and Ehlers, 2005). Such target proteins may include the regulatory subunit of PKA, whose dissociation from the catalytic subunit is triggered by 5-HT and seems to be required for its degradation by the proteasome (Chain et al., 1999b).
In our experiments, CREB2 bound to the ap-uch promoter in response to FMRFa, but not under basal conditions, and this binding correlated with histone hyperacetylation, suggesting that the ap-uch promoter becomes transcriptionally active under the control of CREB2. Indeed, CREB2 is known to induce gene expression in F9 cells (Bartsch et al., 1995). Moreover, mammalian CREB2, which shows significant homology to Aplysia CREB2, can function as an activator of transcription (Siu et al., 2002; Luo et al., 2003), although it is traditionally regarded as a repressor (Karpinski et al., 1992). Furthermore, mammalian CREB2 possesses a strong transcription activator domain but no repressor domain (Schoch et al., 2001; Thiel et al., 2005), suggesting that CREB2 may act as an activator of transcription under certain conditions.
We also found that FMRFa induced phosphorylation of CREB2 at a putative MAPK site. Furthermore, in agreement with previous observations that blocking total CREB2 impaired LTD (Guan et al., 2002), intracellular injections of anti-phospho-CREB2 antibody blocked LTD. However, one possibility is that blockage of LTD arises from interfering with the initial signaling cascade activated by FMRFa. We believe that this possibility is unlikely because in contrast to long-term depression, short-term depression does not depend on protein synthesis (Montarolo et al., 1988; Bailey et al., 1992). Moreover, in our hands (Fig. 5) as well as in experiments of others (Bartsch et al., 1995; Guan et al., 2002), injection of anti-CREB2 antibodies does not significantly affect basal synaptic transmission. Therefore, the block of LTD after injection of anti-phospho-CREB2 antibodies probably reflects a requirement of phospho-CREB2 in LTD.
In previous studies, CREB2 was assigned the role of transcription repressor because binding of CREB2 to the promoter of the gene CCAAT enhancer binding protein (c/ebp), which is upregulated by 5-HT and is necessary for LTF (Alberini et al., 1994), recruited HDAT5 and correlated with reduced C/EBP mRNA levels. Our results confirm the essential role of CREB2 in LTD and provide additional information on its phosphorylation state after FMRFa exposure. p38 MAPK probably mediates the FMRFa-induced phosphorylation of CREB2 in vivo because p38 MAPK is activated by FMRFa, is necessary for LTD, and can phosphorylate CREB2 in vitro (Guan et al., 2003). Moreover, our results showed that pretreatment with a p38 MAPK inhibitor blocked the FMRFa-induced regulation of ap-uch. Finally, based on our ChIP assays, we propose a role for CREB2 as transcription activator and we identify ap-uch as one candidate gene whose regulation may be mediated by CREB2.
Recent studies revealed that, in parallel to long-term potentiation/facilitation (Bailey and Kandel, 1993; Yuste and Bonhoeffer, 2001), LTD protocols produce persistent morphological changes. In hippocampal slices, low-frequency stimulation, which gives rise to LTD, induces spine retraction (Nägerl et al., 2004) and shrinkage (Zhou et al., 2004). In Aplysia, LTD in sensorimotor cocultures is accompanied by reduction in the number of presynaptic varicosities and retraction of processes (Schacher and Montarolo, 1991). Nonetheless, specific target proteins associated with these structural changes have not been identified. The synaptic vesicle protein synapsin, apart from its role in vesicle regulation (Hilfiker et al., 1999), is known to play a role in synapse formation and neurite elongation (Chin et al., 1995; Ferreira et al., 1995; Kao et al., 2002). We now provide evidence that synapsin is a target for regulated protein degradation during LTD. Levels of synapsin are downregulated in synaptosomes 24 h after LTD induction. This downregulation depends on the proteasome and does not seem to reflect loss of synaptic vesicles because levels of another vesicle protein, VAMP, are not affected by FMRFa at least during the first 24 h after treatment. Because our synaptosomal preparations were obtained from pleural–pedal ganglia, which include sensory neurons as well as other types of cells, an alternative explanation for the differential effect of FMRFa on synapsin and VAMP could be that the two proteins are differentially regulated in various cell types. Another possibility is that the time course of varicosity elimination differs in cultures and ganglia. In cultures, FMRFa-induced varicosity elimination and retraction of neurites occurs within 24 h (Schacher and Montarolo, 1991), whereas these phenomena have not been characterized in ganglia. We propose that regulation of synapsin may be important for LTD, in a manner that remains to be described. Also, the temporal dynamics of synapsin regulation during LTD remain unknown. Based on our observation that total protein ubiquitination is increased early after FMRFa (Fig. 2), we would predict that synapsin downregulation should also be detectable at time points earlier than 24 h. Presynaptic proteins including syntaxin and RIM1 are known to be regulated through ubiquitination (Chin et al., 2002; Chen et al., 2003; Yao et al., 2007), but a link between proteasome-mediated degradation of presynaptic proteins and long-term plasticity has not been previously reported.
We propose a conceptual model that begins to explain some of the mechanisms involved in long-term depression. On FMRFa stimulation, p38 MAPK becomes activated and phosphorylates CREB2. This phosphorylation of CREB2 triggers both repression and enhancement of transcription, depending on the target genes. For example, binding of CREB2 to the promoter regions of genes such as c/ebp would repress transcription. In contrast, binding of phosphorylated CREB2 to the promoter regions of a different set of genes (probably with different response elements than c/ebp) including ap-uch, would enhance transcription. Protein ubiquitination is increased in response to FMRFa, tagging proteins for degradation. Increased levels of ap-uch would help enhance degradation of ubiquitinated proteins, which is an essential step in the induction of LTD. One such protein could be synapsin, the degradation of which may be important for downregulating synaptic strength and structures.
Footnotes
This work was supported by National Institutes of Health Grant NS019895. We thank W. Yao for performing several experiments in this study. We also thank A. Eskin and J. Levenson (The University of Houston, Houston, TX) for help with the technique of synaptosome isolation, J. Liu for preparing the cultures, A. Hegde (Wake Forest University, Winston-Salem, NC) for providing the ap-uch antibody, K. C. Martin (University of California, Los Angeles, Los Angeles, CA) for the anti-VAMP antibody, and P. Smolen and E. Antzoulatos for helpful comments on a previous version of this manuscript.
References
- Abel T, Martin KC, Bartsch D, Kandel ER. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science. 1998;279:338–341. doi: 10.1126/science.279.5349.338. [DOI] [PubMed] [Google Scholar]
- Ahn S, Ginty DD, Linden DJ. A late phase of cerebellar long-term depression requires activation of CaMKIV and CREB. Neuron. 1999;23:559–568. doi: 10.1016/s0896-6273(00)80808-9. [DOI] [PubMed] [Google Scholar]
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Alberini CM, Ghirardi M, Metz R, Kandel ER. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 1994;76:1099–1114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Angers A, Fioravante D, Chin J, Cleary LJ, Bean AJ, Byrne JH. Serotonin stimulates phosphorylation of Aplysia synapsin and alters its subcellular distribution in sensory neurons. J Neurosci. 2002;22:5412–5422. doi: 10.1523/JNEUROSCI.22-13-05412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzoulatos EG, Cleary LJ, Eskin A, Baxter DA, Byrne JH. Desensitization of postsynaptic glutamate receptors contributes to high-frequency homosynaptic depression of Aplysia sensorimotor connections. Learn Mem. 2003;10:309–313. doi: 10.1101/lm.61403. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER. Structural changes accompanying memory storage. Annu Rev Physiol. 1993;55:397–426. doi: 10.1146/annurev.ph.55.030193.002145. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Montarolo P, Chen M, Kandel ER, Schacher S. Inhibitors of protein and RNA synthesis block structural changes that accompany long-term heterosynaptic plasticity in Aplysia. Neuron. 1992;9:749–758. doi: 10.1016/0896-6273(92)90037-e. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Casadio A, Karl KA, Serodio P, Kandel ER. CREB1 encodes a nuclear activator, a repressor, and a cytoplasmic modulator that form a regulatory unit critical for long-term facilitation. Cell. 1998;95:211–223. doi: 10.1016/s0092-8674(00)81752-3. [DOI] [PubMed] [Google Scholar]
- Belardetti F, Kandel ER, Siegelbaum SA. Neuronal inhibition by the peptide FMRFamide involves opening of S K+ channels. Nature. 1987;325:153–156. doi: 10.1038/325153a0. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–541. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Brown RO, Gusman D, Basbaum AI, Mayeri E. Identification of Aplysia neurons containing immunoreactive FMRFamide. Neuropeptides. 1985;6:517–526. doi: 10.1016/0143-4179(85)90113-1. [DOI] [PubMed] [Google Scholar]
- Buffo A, Zagrebelsky M, Huber AB, Skerra A, Schwab ME, Strata P, Rossi F. Application of neutralizing antibodies against NI-35/250 myelin-associated neurite growth inhibitory proteins to the adult rat cerebellum induces sprouting of uninjured purkinje cell axons. J Neurosci. 2000;20:2275–2286. doi: 10.1523/JNEUROSCI.20-06-02275.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999a;22:147–156. doi: 10.1016/s0896-6273(00)80686-8. [DOI] [PubMed] [Google Scholar]
- Chain DG, Schwartz JH, Hegde AN. Ubiquitin-mediated proteolysis in learning and memory. Mol Neurobiol. 1999b;20:125–142. doi: 10.1007/BF02742438. [DOI] [PubMed] [Google Scholar]
- Chen H, Polo S, Di Fiore PP, De Camilli PV. Rapid Ca2+-dependent decrease of protein ubiquitination at synapses. Proc Natl Acad Sci U S A. 2003;100:14908–14913. doi: 10.1073/pnas.2136625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Chin GJ, Shapiro E, Vogel SS, Schwartz JH. Aplysia synaptosomes. I. Preparation and biochemical and morphological characterization of subcellular membrane fractions. J Neurosci. 1989;9:38–48. doi: 10.1523/JNEUROSCI.09-01-00038.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Liu RY, Cleary LJ, Eskin A, Byrne JH. TGF-beta1-induced long-term changes in neuronal excitability in Aplysia sensory neurons depend on MAPK. J Neurophysiol. 2006;95:3286–3290. doi: 10.1152/jn.00770.2005. [DOI] [PubMed] [Google Scholar]
- Chin LS, Li L, Ferreira A, Kosik KS, Greengard P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9230–9234. doi: 10.1073/pnas.92.20.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin LS, Vavalle JP, Li L. Staring, a novel E3 ubiquitin-protein ligase that targets syntaxin 1 for degradation. J Biol Chem. 2002;277:35071–35079. doi: 10.1074/jbc.M203300200. [DOI] [PubMed] [Google Scholar]
- Cline H. Synaptic plasticity: importance of proteasome-mediated protein turnover. Curr Biol. 2003;13:R514–R516. doi: 10.1016/s0960-9822(03)00443-3. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway RC, Brower CS, Conaway JW. Emerging roles of ubiquitin in transcription regulation. Science. 2002;296:1254–1258. doi: 10.1126/science.1067466. [DOI] [PubMed] [Google Scholar]
- Critz SD, Baxter DA, Byrne JH. Modulatory effects of serotonin, FMRFamide, and myomodulin on the duration of action potentials, excitability, and membrane currents in tail sensory neurons of Aplysia. J Neurophysiol. 1991;66:1912–1926. doi: 10.1152/jn.1991.66.6.1912. [DOI] [PubMed] [Google Scholar]
- Dale N, Kandel ER. Facilitatory and inhibitory transmitters modulate spontaneous transmitter release at cultured Aplysia sensorimotor synapses. J Physiol. 1990;421:203–222. doi: 10.1113/jphysiol.1990.sp017941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–721. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- Davie J, Spencer V. Control of histone modifications. J Cell Biochem. 1999;(Suppl 32-33):141–148. doi: 10.1002/(sici)1097-4644(1999)75:32+<141::aid-jcb17>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Dong C, Upadhya SC, Ding L, Smith TK, Hegde AN. Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn Mem. 2008;15:335–347. doi: 10.1101/lm.984508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberharter A, Becker P. Histone acetylation: a switch between repressive and permissive chromatin. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Rapoport M. The synapsins: beyond the regulation of neurotransmitter release. Cell Mol Life Sci. 2002;59:589–595. doi: 10.1007/s00018-002-8451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Han HQ, Greengard P, Kosik KS. Suppression of synapsin II inhibits the formation and maintenance of synapses in hippocampal culture. Proc Natl Acad Sci U S A. 1995;92:9225–9229. doi: 10.1073/pnas.92.20.9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Smolen PD, Byrne JH. The 5-HT- and FMRFa-activated signaling pathways interact at the level of the Erk MAPK cascade: potential inhibitory constraints on memory formation. Neurosci Lett. 2006;396:235–240. doi: 10.1016/j.neulet.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Liu RY, Netek AK, Cleary LJ, Byrne JH. Synapsin regulates basal synaptic strength, synaptic depression and serotonin-induced facilitation of sensorimotor synapses in Aplysia. J Neurophysiol. 2007;98:3568–3580. doi: 10.1152/jn.00604.2007. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Guan Z, Kim JH, Lomvardas S, Holick K, Xu S, Kandel ER, Schwartz JH. p38 MAP kinase mediates both short-term and long-term synaptic depression in Aplysia. J Neurosci. 2003;23:7317–7325. doi: 10.1523/JNEUROSCI.23-19-07317.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, DiAntonio A. Ubiquitin and the synapse. Nat Rev Neurosci. 2002;3:854–861. doi: 10.1038/nrn961. [DOI] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell. 1997;89:115–126. doi: 10.1016/s0092-8674(00)80188-9. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Doussau F, Vitiello F, Greengard P, Benfenati F, Poulain B. Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short-term plasticity in Aplysia. J Neurosci. 2001;21:4195–4206. doi: 10.1523/JNEUROSCI.21-12-04195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriyama A, Chen YN, Tamaki Y, Yanagi Y. Effect of anti-VEGF antibody on retinal ganglion cells in rats. Br J Ophthalmol. 2007;91:1230–1233. doi: 10.1136/bjo.2007.117309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kao HT, Song HJ, Porton B, Ming GL, Hoh J, Abraham M, Czernik AJ, Pieribone VA, Poo MM, Greengard P. A protein kinase A-dependent molecular switch in synapsins regulates neurite outgrowth. Nat Neurosci. 2002;5:431–437. doi: 10.1038/nn840. [DOI] [PubMed] [Google Scholar]
- Karpinski BA, Morle GD, Huggenvik J, Uhler MD, Leiden JM. Molecular cloning of human CREB-2: an ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc Natl Acad Sci U S A. 1992;89:4820–4824. doi: 10.1073/pnas.89.11.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Thomas U, Knöpfel T, Behnisch T. Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J Neurosci. 2006;26:4949–4955. doi: 10.1523/JNEUROSCI.4573-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoutorsky A, Spira ME. Calcium-activated proteases are critical for refilling depleted vesicle stores in cultured sensory-motor synapses of Aplysia. Learn Mem. 2005;12:414–422. doi: 10.1101/lm.92105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Zhao Y, Martin KC. Nuclear translocation of CREB2, a potential cargo of importin, in Aplysia neurons in response to synaptic plasticity. Soc Neurosci Abstr. 2006;32:632–632. [Google Scholar]
- Lee SH, Lim CS, Park H, Lee JA, Han JH, Kim H, Cheang YH, Lee SH, Lee YS, Ko HG, Jang DH, Kim H, Miniaci MC, Bartsch D, Kim E, Bailey CH, Kandel ER, Kaang BK. Nuclear translocation of CAM-associated protein activates transcription for long-term facilitation in Aplysia. Cell. 2007;129:801–812. doi: 10.1016/j.cell.2007.03.041. [DOI] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Levenson J, Endo S, Kategaya LS, Fernandez RI, Brabham DG, Chin J, Byrne JH, Eskin A. Long-term regulation of neuronal high-affinity glutamate and glutamine uptake in Aplysia. Proc Natl Acad Sci U S A. 2000;97:12858–12863. doi: 10.1073/pnas.220256497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Li C, Kim K, Nelson LS. FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. Brain Res. 1999;848:26–34. doi: 10.1016/s0006-8993(99)01972-1. [DOI] [PubMed] [Google Scholar]
- Linden DJ. A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron. 1996;17:483–490. doi: 10.1016/s0896-6273(00)80180-4. [DOI] [PubMed] [Google Scholar]
- Liu RY, Fioravante D, Shah S, Byrne JH. cAMP response element-binding protein 1 feedback loop is necessary for consolidation of long-term synaptic facilitation in Aplysia. J Neurosci. 2008;28:1970–1976. doi: 10.1523/JNEUROSCI.3848-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, McGuinness T, Leonard C, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci U S A. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd PE, Frankfurt M, Stevens P, Kupfermann I, Weiss KR. Biochemical and immunocytological localization of the neuropeptides FMRFamide, SCPA, SCPB, to neurons involved in the regulation of feeding in Aplysia. J Neurosci. 1987;7:1123–1132. doi: 10.1523/JNEUROSCI.07-04-01123.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lopez-Salon M, Alonso M, Vianna MR, Viola H, Mello e Souza T, Izquierdo I, Pasquini JM, Medina JH. The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur J Neurosci. 2001;14:1820–1826. doi: 10.1046/j.0953-816x.2001.01806.x. [DOI] [PubMed] [Google Scholar]
- Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem. 2003;278:37375–37385. doi: 10.1074/jbc.M303619200. [DOI] [PubMed] [Google Scholar]
- Mackey SL, Glanzman DL, Small SA, Dyke AM, Kandel ER, Hawkins RD. Tail shock produces inhibition as well as sensitization of the siphon-withdrawal reflex of Aplysia: possible behavioral role for presynaptic inhibition mediated by the peptide Phe-Met-Arg-Phe-NH2. Proc Natl Acad Sci U S A. 1987;84:8730–8734. doi: 10.1073/pnas.84.23.8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed HA, Yao W, Fioravante D, Smolen PD, Byrne JH. cAMP-response elements in Aplysia creb1, creb2, and Ap-uch promoters: implications for feedback loops modulating long-term memory. J Biol Chem. 2005;280:27035–27043. doi: 10.1074/jbc.M502541200. [DOI] [PubMed] [Google Scholar]
- Montarolo PG, Kandel ER, Schacher S. Long-term heterosynaptic inhibition in Aplysia. Nature. 1988;333:171–174. doi: 10.1038/333171a0. [DOI] [PubMed] [Google Scholar]
- Myung J, Kim KB, Crews CM. The ubiquitin-proteasome pathway and proteasome inhibitors. Med Res Rev. 2001;21:245–273. doi: 10.1002/med.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Ohno M, Fornerod M, Mattaj IW. Nucleocytoplasmic transport: the last 200 nanometers. Cell. 1998;92:327–336. doi: 10.1016/s0092-8674(00)80926-5. [DOI] [PubMed] [Google Scholar]
- Pepperkok R, Lorenz P, Ansorge W, Pyerin W. Casein kinase II is required for transition of G0/G1, early G1, and G1/S phases of the cell cycle. J Biol Chem. 1994;269:6986–6991. [PubMed] [Google Scholar]
- Pieroni JP, Byrne JH. Differential effects of serotonin, FMRFamide, and small cardioactive peptide on multiple, distributed processes modulating sensorimotor synaptic transmission in Aplysia. J Neurosci. 1992;12:2633–2647. doi: 10.1523/JNEUROSCI.12-07-02633.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D, Volterra A, Dale N, Siegelbaum SA, Kandel ER, Schwartz JH, Belardetti F. Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature. 1987;328:38–43. doi: 10.1038/328038a0. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Kandel ER. In search of general mechanisms for long-lasting plasticity: Aplysia and the hippocampus. Philos Trans R Soc Lond B Biol Sci. 2003;358:757–763. doi: 10.1098/rstb.2002.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DA, Greenberg MJ. Structure of a molluscan cardioexcitatory neuropeptide. Science. 1977;197:670–671. doi: 10.1126/science.877582. [DOI] [PubMed] [Google Scholar]
- Raffa RB. The actions of FMRF-NH2 and FMRF-NH2 related peptides on mammals. NIDA Res Monogr. 1990;105:243–249. [PubMed] [Google Scholar]
- Schacher S, Montarolo PG. Target-dependent structural changes in sensory neurons of Aplysia accompany long-term heterosynaptic inhibition. Neuron. 1991;6:679–690. doi: 10.1016/0896-6273(91)90166-w. [DOI] [PubMed] [Google Scholar]
- Schoch S, Cibelli G, Magin A, Steinmüller L, Thiel G. Modular structure of cAMP response element binding protein 2 (CREB2) Neurochem Int. 2001;38:601–608. doi: 10.1016/s0197-0186(00)00127-3. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in Aplysia. Cell. 2003;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- Siu F, Bain PJ, LeBlanc-Chaffin R, Chen H, Kilberg MS. ATF4 is a mediator of the nutrient-sensing response pathway that activates the human asparagine synthetase gene. J Biol Chem. 2002;277:24120–24127. doi: 10.1074/jbc.M201959200. [DOI] [PubMed] [Google Scholar]
- Small SA, Cohen TE, Kandel ER, Hawkins RD. Identified FMRFamide-immunoreactive neuron LPL16 in the left pleural ganglion of Aplysia produces presynaptic inhibition of siphon sensory neurons. J Neurosci. 1992;12:1616–1627. doi: 10.1523/JNEUROSCI.12-05-01616.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci. 1984;4:3080–3088. doi: 10.1523/JNEUROSCI.04-12-03080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghert PH. FMRFamide neuropeptides and neuropeptide-associated enzymes in Drosophila. Microsc Res Tech. 1999;45:80–95. doi: 10.1002/(SICI)1097-0029(19990415)45:2<80::AID-JEMT3>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Thiel G, Al Sarraj J, Vinson C, Stefano L, Bach K. Role of basic region leucine zipper transcription factors cyclic AMP response element binding protein (CREB), CREB2, activating transcription factor 2 and CAAT/enhancer binding protein alpha in cyclic AMP response element-mediated transcription. J Neurochem. 2005;92:321–336. doi: 10.1111/j.1471-4159.2004.02882.x. [DOI] [PubMed] [Google Scholar]
- Udo H, Jin I, Kim JH, Li HL, Youn T, Hawkins RD, Kandel ER, Bailey CH. Serotonin-induced regulation of the actin network for learning-related synaptic growth requires Cdc42, N-WASP, and PAK in Aplysia sensory neurons. Neuron. 2005;45:887–901. doi: 10.1016/j.neuron.2005.01.044. [DOI] [PubMed] [Google Scholar]
- Upadhya SC, Smith TK, Hegde AN. Ubiquitin-proteasome-mediated CREB repressor degradation during induction of long-term facilitation. J Neurochem. 2004;91:210–219. doi: 10.1111/j.1471-4159.2004.02707.x. [DOI] [PubMed] [Google Scholar]
- Upadhya SC, Ding L, Smith TK, Hegde AN. Differential regulation of proteasome activity in the nucleus and the synaptic terminals. Neurochem Int. 2006;48:296–305. doi: 10.1016/j.neuint.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW. Localization of pendrin in mouse kidney. Am J Physiol Renal Physiol. 2003;284:F229–F241. doi: 10.1152/ajprenal.00147.2002. [DOI] [PubMed] [Google Scholar]
- Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol Cell Biol. 2001;21:6820–6832. doi: 10.1128/MCB.21.20.6820-6832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JH, McIllhinney RA, Wise A, Ciruela F, Chan WY, Emson PC, Billinton A, Marshall FH. The GABAB receptor interacts directly with the related transcription factors CREB2 and ATFx. Proc Natl Acad Sci U S A. 2000;97:13967–13972. doi: 10.1073/pnas.240452197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing SS. Deubiquitinating enzymes—the importance of driving in reverse along the ubiquitin-proteasome pathway. Int J Biochem Cell Biol. 2003;35:590–605. doi: 10.1016/s1357-2725(02)00392-8. [DOI] [PubMed] [Google Scholar]
- Winnepenninckx B, Steiner G, Backeljau T, De Wachter R. Details of gastropod phylogeny inferred from 18S rRNA sequences. Mol Phylogenet Evol. 1998;9:55–63. doi: 10.1006/mpev.1997.0439. [DOI] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Brensinger CM, Guo W, Abel T. Ubiquitin C-terminal hydrolase L3 (Uchl3) is involved in working memory. Hippocampus. 2005;15:610–621. doi: 10.1002/hipo.20082. [DOI] [PubMed] [Google Scholar]
- Xu Y, Cleary LJ, Byrne JH. Identification and characterization of pleural neurons that inhibit tail sensory neurons and motor neurons in Aplysia: correlation with FMRFamide immunoreactivity. J Neurosci. 1994;14:3565–3577. doi: 10.1523/JNEUROSCI.14-06-03565.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, Macgregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Ubiquitin and protein turnover in synapse function. Neuron. 2005;47:629–632. doi: 10.1016/j.neuron.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Hegde AN, Martin KC. The ubiquitin proteasome system functions as an inhibitory constraint on synaptic strengthening. Curr Biol. 2003;13:887–898. doi: 10.1016/s0960-9822(03)00332-4. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]