

Abstract
An effective AIDS vaccine will need to protect against globally diverse isolates of HIV. To address this issue in macaques, we administered a live-attenuated simian immunodeficiency virus (SIV) vaccine and challenged with a highly pathogenic heterologous isolate. Vaccinees reduced viral replication by ∼2 logs between weeks 2–32 (P ≤ 0.049) postchallenge. Remarkably, vaccinees expressing MHC-I (MHC class I) alleles previously associated with viral control completely suppressed acute phase replication of the challenge virus, implicating CD8+ T cells in this control. Furthermore, transient depletion of peripheral CD8+ lymphocytes in four vaccinees during the chronic phase resulted in an increase in virus replication. In two of these animals, the recrudescent virus population contained only the vaccine strain and not the challenge virus. Alarmingly, however, we found evidence of recombinant viruses emerging in some of the vaccinated animals. This finding argues strongly against an attenuated virus vaccine as a solution to the AIDS epidemic. On a more positive note, our results suggest that MHC-I–restricted CD8+ T cells contribute to the protection induced by the live-attenuated SIV vaccine and demonstrate that vaccine-induced CD8+ T cell responses can control replication of heterologous challenge viruses.
The enormous variability of HIV is one of the major hurdles that must be overcome in the development of a successful AIDS vaccine. Accumulated nucleotide changes within the highly mutable env gene form the basis for classifying HIV-1 into different groups (M, N, and O) and subtypes (clades A, B, C, D, E, F, G, and K). Evolutionary analysis of nucleotide sequences shows that env can vary by up to 35% among different clades (1). Even within clades, this diversity can reach 20% (1). This variability is driven by several factors, including recombination between different strains of HIV and the high rate of mutation associated with HIV RT (2–4). For this reason, many HIV-1 vaccine designs focused on inducing cell-mediated immunity have abandoned using Env as an immunogen in favor of concentrating on more conserved regions of the virus. However, even relatively minor variations in these proteins may have grave implications for vaccine efficacy, as single-aa differences can negatively affect recognition by vaccine-induced antibodies and CD8+ T cells (5, 6). To address this issue, several strategies have been proposed to improve vaccine design by including polyvalent formulations or immunogens based on ancestral, center of tree, or consensus HIV-1 sequences (1, 7, 8). Each of these methods seeks to minimize differences between the vaccine sequence and circulating viruses while maximizing cross-reactive immune responses.
Currently, most AIDS vaccine strategies include a prime/boost component to induce antiviral immune responses. Recent results from the HIV Vaccine Trial Network and Merck's STEP trial (http://www.hvtn.org/media/pr/step111307.html) have clearly shown that current versions of these approaches fail to either protect against pathogenic infections or reduce viral replication. In contrast, immunization of rhesus macaques with live-attenuated simian immunodeficiency virus (SIV) has consistently induced protective immunity against homologous pathogenic SIV challenge (9–11). However, characterization of the vaccine-induced immune responses accounting for this protection has proven difficult. Antibodies (12, 13), CD4+ and CD8+ T cells (11, 13–15), NK cells (16, 17) and even viral interference (18–20) have been implicated in mediating live-attenuated SIV-induced protection. Understanding the underlying mechanisms for this protection should facilitate the design of improved HIV vaccines (21).
Despite the effectiveness of live-attenuated SIV vaccination against homologous virus challenge, only a few small-scale studies have addressed the ability of live-attenuated SIV to control heterologous SIV challenge and have had mixed results with regard to vaccine efficacy (9, 22, 23). We therefore sought to determine whether macaques vaccinated with SIVmac239Δnef could effectively control heterologous virus replication in a large-scale study designed to achieve statistical significance. We included MHC-I (MHC class I)–defined macaques to facilitate careful monitoring of CD8+ T cell responses. Vaccinated animals, along with ten naive controls matched for the MHC-I alleles of interest, were challenged i.v. with the highly pathogenic heterologous “swarm” virus SIVsmE660, and their mean plasma virus concentrations were compared at different time intervals.
RESULTS
SIVmac239Δnef vaccination
To address the issue of whether an HIV vaccine can ameliorate the pathogenic effects of a heterologous challenge, we used a well-described macaque AIDS model of protective immunity. We induced antiviral immune responses by inoculating ten MHC-I–defined rhesus macaques with the attenuated SIV strain SIVmac239Δnef (24). We included animals expressing Mamu-A*01, -A*02, -A*11, -B*08, or -B*17 (n = 2 for each) because all of the SIVmac239 epitopes bound by these MHC-I molecules had previously been defined (25–31). Most animals had a peak of virus replication at 2 wk postinoculation (p.i.), with SIVmac239Δnef ranging between 3.2 × 103 and 9.4 × 105 viral RNA (vRNA) copy equivalents (Eq)/ml of plasma (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20081524/DC1). 8 of the 10 vaccinated animals controlled SIVmac239Δnef plasma virus replication below 500 vRNA copy Eq/ml between weeks 12 and 26 p.i. (Fig. S1).
All vaccinated macaques developed broad, but low frequency, cellular immune responses. We monitored the development of SIV-specific cellular immune responses using a combination of IFN-γ ELISPOT and MHC-I tetramer staining. We did not conduct such an extensive analysis in the Mamu-A*11+ animals because MHC-I tetramers were unavailable. Throughout the vaccine phase of the study, the frequency of tetramer binding cells in PBMC varied from animal to animal and within animals from epitope to epitope (Table S1, available at http://www.jem.org/cgi/content/full/jem.20081524/DC1). In most animals, at least one tetramer-specific response attained a frequency of >0.3% of the circulating CD8+ T cells. At the time of challenge, 26 wk p.i., the frequency of tetramer binding cells in the PBMC for each specificity ranged from undetectable to 0.2% of the CD8+ T cells. In IFN-γ ELISPOT assays, most animals had detectable cellular immune responses to multiple peptide pools from several SIV proteins before challenge (Table S2), ranging between 50 and 850 spot-forming cells (SFC) per million PBMC (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20081524/DC1). To differentiate CD4+ and CD8+ T cell responses, we performed IFN-γ ELISPOT assays using total PBMC and PBMC depleted of CD8+ cells. Most animals made CD4+ T cell responses against multiple peptide pools derived from several proteins (Fig. S2). We only measured responses directed against Gag, Tat, Rev, Vif, and the intact portion of Nef using PBMC depleted of CD8+ cells because of the limited number of available cells. The CD4+ T cell responses that we did detect were primarily directed against Gag (Fig. S2).
Pathogenic heterologous SIV challenge
To assess the protective capacity of SIVmac239Δnef-induced immune responses against heterologous SIV infection, we challenged the 10 vaccinated animals, along with 10 naive controls matched for expression of the MHC-I of interest, i.v. with the highly pathogenic heterologous swarm virus SIVsmE660 at 6 mo p.c. with SIVmac239Δnef. SIVsmE660 differs from SIVmac239 by ∼15% of its amino acids (Table I). This difference approximates the variation between single clade–based HIV vaccines and circulating HIV isolates within that clade (1).
Table I.
Percentage difference in amino acid sequence among different strains of immunodeficiency viruses
| Protein | Consensus Clade Ba versus Clade B isolatesb |
SIVmac239a versus SIVsmE660b |
Consensus Clade Ba versus Clade C isolatesb |
|---|---|---|---|
| % | % | % | |
| Tat | 12.5 | 26.0 | 30.5 |
| Rev | 12.1 | 25.0 | 35.3 |
| Nef | 17.7 | 21.3 | 35.2 |
| Vif | 6.8 | 17 | 18.1 |
| Env | 27.4 | 15.1 | 53.1 |
| Vpr | 9.8 | 12.0 | 17.3 |
| Pol | 3.2 | 8.3 | 10.0 |
| Gag | 4.4 | 8.0 | 16.4 |
The mean percentage difference per protein of five clade B and five clade C isolates in comparison to the consensus clade B amino acid sequence. The clade B and C isolates were obtained from the Los Alamos National Laboratory HIV sequence database.
Vaccine strain.
Challenge strain.
During acute infection, the SIVmac239Δnef-vaccinated animals reduced plasma virus concentrations by 2–3 logs in comparison to the naive controls (Fig. 1, a and b). At 2 (P = 0.011), 3 (P < 0.001), and 4 (P = 0.005) wk postchallenge (p.c.), the mean plasma virus concentrations in the vaccinated animals were significantly lower than that of the naive controls. The vaccinated animals maintained a reduction in mean plasma virus concentrations through 8 mo p.c. (the point at which some animals began experiencing simian AIDS). Only at week 24 p.c. did the difference in viral loads fall below the level of significance (P = 0.052). Despite gradually increasing plasma virus levels in some of the vaccinees, the majority of the time points showed significant difference between vaccines and controls (weeks p.c.: 6, P < 0.001; 8, P = 0.001; 12, P = 005; 16, P = 0.002; 20, P = 0.049; 28, P = 0.046; and 32, P = 0.008).
Figure 1.

Plasma virus concentrations after SIVsmE660 challenge. (a) Plasma virus concentrations of the ten SIVmac239Δnef-vaccinated macaques and their ten MHC-I–matched naive controls. (b) Geometric means for the ten vaccinated and ten naive controls. (c) Plasma virus concentrations of the vaccinated and naive Mamu-B*08+ and -B*17+ macaques. (d) Geometric means for the vaccinated and naive Mamu-B*08+ and -B*17+ macaques. (e) Plasma virus concentrations of the vaccinated and naive Mamu-A*01+, -A*02+, and -A*11+ macaques. (f) Geometric means for the vaccinated and naive Mamu-A*01+, -A*02+, and -A*11+ macaques.
Vaccinated macaques expressing the MHC-I alleles Mamu-B*08 or Mamu-B*17, which have previously been associated with spontaneous control of pathogenic SIVmac239 replication in naive macaques (25, 32), manifested highly effective control of acute infection after the heterologous challenge (Fig. 1, c and d). The four animals expressing either of these two protective alleles had plasma virus concentrations of <30, 40, 240, and 14,000 vRNA copy Eq/ml at 2 wk p.c., which is normally the peak of virus replication. By comparison, the MHC-I–matched naive controls expressing these same protective alleles had more than one million copy Eq/ml plasma. The high level of acute viral replication in Mamu-B*08+ and -B*17+ naive macaques was expected because these two alleles only exert their protective effect in the chronic phase in naive animals (25, 32). Differences in mean plasma virus replication between the vaccinated and naive Mamu-B*08+ or -B*17+ macaques were significant at 1 (P = 0.002; n = 4), 2 (P = 0.014), 3 (P = 0.002), 4 (P < 0.001), 6 (P = 0.002), and 8 (P = 0.011) wk p.c. The two Mamu-B*08+ vaccinated macaques maintained control of SIV replication below 5,000 vRNA copy Eq/ml plasma out to 11 mo p.c. However, despite low virus replication in the acute phase peak, virus replication in the vaccinated Mamu-B*17+ macaques progressively increased during the chronic phase of infection (Fig. 1 c). The Mamu-B*08+ and -B*17+ vaccinated animals no longer had significantly different (P > 0.055) plasma virus levels in comparison to the naive Mamu-B*08+or -B*17+ animals by week 12 p.c. (Fig. 1, c and d).
Significant reduction in plasma virus concentrations observed in SIVmac239Δnef-vaccinated animals was not solely caused by the effect of Mamu-B*08+ and -B*17+ on SIVsmE660 replication. Vaccinated Mamu-A*01+, -A*02+, and -A*11+ macaques also showed significantly reduced plasma replication at 3 (P = 0.004; n = 6), 4 (P = 0.031), 8 (P = 0.044), 16 (P = 0.036), and 32 (P = 0.037) wk p.c. in comparison with their MHC-I–matched naive control counterparts (Fig. 1, e and f). However, only two of these animals (one Mamu-A*01+ and one Mamu-A*02+) maintained replication below 500 copy Eq/ml plasma at 11 mo p.c.
Interestingly, we found no evidence that vaccine-induced neutralizing antibodies contributed to the control of viral replication. We assessed the level of preexisting neutralizing antibodies in the plasma of two animals that controlled acute or chronic virus replication (01022 and 01003) and two animals that did not control virus replication p.c. (rhAO84 and rh2000). None of the animals had any neutralizing antibody activity against SIVsmE660 (Fig. 2). This indicates that cellular immune responses likely play the main role for controlling virus replication after heterologous virus challenge.
Figure 2.

No in vitro neutralization of SIVsmE660 by vaccine-induced antibodies. Percent neutralization of SIVsmE660 replication in rhesus macaque PBMC after 7 d in culture by plasma collected before challenge in SIVmac239Δnef vaccinees. The chimeric molecule CD4-IgG2 was used as a positive control for neutralization and plasma collected from an unvaccinated animal, 84070, before challenge was used as a negative control. Percent neutralization is presented as the reduction in the amount of virus at the end of the assay in comparison to cells infected with SIVsmE660 alone. Similar negative plasma neutralization results were observed in a TZM-bl cell neutralization assay (65). Error bars represent SEM of experiments done in triplicate.
Cellular immune responses p.c.
Cross-reactive vaccine-primed immune responses recognizing the infecting virus will likely determine the efficacy of HIV-1 vaccines. We therefore monitored cellular immune responses elicited by SIVmac239Δnef vaccination to determine which, if any, expanded after challenge with SIVsmE660. In IFN-γ ELISPOT assays using pools of peptides spanning the entire SIVmac239 proteome, we observed varying degrees of expansion in cellular immune responses above prechallenge levels at 2 wk p.c. (Fig. 3). Expanded responses were primarily directed against Gag and Pol, which are the most conserved proteins between SIVmac239Δnef and SIVsmE660 (Table I). To more closely examine the effect that amino acid changes within CD8+ T cell epitopes have on p.c. expansion, we used MHC-I–restricted peptides corresponding to the minimal optimal epitopes in SIVmac239Δnef. We found that some of the epitopes with 1 or 2 aa differences between SIVmac239Δnef and SIVsmE660 stimulated responses above prechallenge levels (Table II and Fig. S3 a, available at http://www.jem.org/cgi/content/full/jem.20081524/DC1). Correlating with the IFN-γ ELISPOT assays, some of the vaccinated animals displayed a twofold or more expansion of at least one tetramer binding population during acute infection (Fig. 4). Interestingly, the animals that controlled SIVsmE660 replication at 2 wk p.c. generally had the lowest frequency of p.c. cellular immune responses in the PBMC.
Figure 3.

Expansion of virus-specific cells in SIVmac239Δnef-vaccinated macaques at 2 wk p.c. with SIVsmE660. Expansion in vaccinated macaques of the number of SFC above prechallenge levels in IFN-γ ELISPOT assays at 2 wk p.c. with SIVsmE660. To determine the expansion of SFC p.c., we subtracted the number of SFC per pool before challenge from the number of SFC detected p.c. The sum of the expanded cells per protein is displayed in the graph.
Table II.
Comparison of amino acid sequences of known CD8+ T cell epitopes between SIVmac239Δnef, SIVsmE660, and circulating virus during the chronic phase of infection in the vaccinated animals
| Sequence origin | Epitope sequence | |||||||
|---|---|---|---|---|---|---|---|---|
| Mamu-A*01 | Gag149-157LW9 | Gag181-189CM9 | Gag254-262QI9 | Gag372-379LF8 | Pol147-156LV10 | Pol363-372GM10 | Pol592-600QV9 | Pol625-633SV9 |
| SIVmac239Δnef | LSPRTLNAW | CTPYDINQM | QNPIPVGNI | LAPVPIPF | LGPHYTPKIV | GSPAIFQYTM | QVPKFHLPV | STPPLVRLV |
| SIVsmE660 | ......... | ......... | ......... | .R.DQL.. | ...N....V. | .......... | E........ | ......... |
| 02092 | ......... | .A....... | ......... | .R.DQL.. | ...N....V. | .......... | E.....V.. | ......... |
| 88085 | ......... | ......... | ......... | ........ | .......... | .......... | ......... | ......... |
| Mamu-A*01 | Env233-241CL9 | Env622-630TL9 | Env729-738ST10 | Tat28-35SL8 | Vif144-152QA9 | Vpx8-18II11 | ||
| SIVmac239Δnef | CAPPGYALL | TVPWPNASL | SSPPSYFQQT | STPESANL | QVPSLQYLA | IPPGNSGEETI | ||
| SIVsmE660 | ......... | ......ET. | ....A.V..I | P....... | ......... | ........... | ||
| 02092 | ......... | ......GT. | ....A.V..I | P....... | ......... | ........... | ||
| 88085 | ......... | ......ET. | ....A.V..I | ....L... | ......... | |||
| Mamu-A*02 | Gag71-79GY9 | Pol324-332FF9 | Pol518-526LY9 | Env296-304RY9 | Env317-325KM9 | Env359-367QY9 | Env519-528GF10 | Env760-768SY9 |
| SIVmac239Δnef | GSENLKSLY | FSIPLDEEF | LSQEQEGCY | RTIISLNKY | KTVLPVTIM | QTIVKHPRY | GTSRNKRGVF | SSWPWQIEY |
| SIVsmE660 | ......... | ......... | ......... | ......... | ......... | E.L...... | .A........ | R........ |
| 01022 | ......... | ......... | ||||||
| rhAO84 | ......... | ......... | ......... | ......... | ...f..... | E.L...... | .A........ | R........ |
| Mamu-A*02 | Env788-795RY8 | Nef20-28LY9 | Vif89-97IW9 | Vif97-104WY8 | Vpr63-71RM9 | |||
| SIVmac239Δnef | RTLLSRVY | LLRARGETY | ITWYSKNFW | WTDVTPNY | RILQRALFM | |||
| SIVsmE660 | .DW.L.T. | ..Q...... | .....R... | ......D. | ........I | |||
| 01022 | ......... | |||||||
| rhAO84 | .DW.L.T. | ..Q...... | .....R... | ......D. | K.......I | |||
| Mamu-A*11 | Gag178-186SI9 | Pol92-100AL9 | Pol457-465RI9 | Pol507-517AI11 | Env495-502GI8 | Vpr13-21RV9 | ||
| SIVmac239Δnef | SEGCTPYDI | AERKQREAL | RETWTVNDI | AEAEYEENKII | GDYKLVEI | REPWDEWVV | ||
| SIVsmE660 | ......... | G.–...T. | ......... | ........... | ........ | ......... | ||
| 01009 | ......... | RK–...T. | ......... | ........... | ........ | ......... | ||
| rh2000 | ......... | G.–...T. | ......... | ........... | ........ | |||
| Mamu-B*08 | Gag263-272YL9 | Env524-532KF9 | Env573-581KL9 | Env717-725LF9 | Env868-876RL9 | Rev12-20KL9 | Rev44-51RL8 | Nef8-16RL9 |
| SIVmac239Δnef | YRRWIQLGL | KRGVFVLGF | KRQQELLRL | LRQGYRPVF | RRIRQGLEL | KRLRLIHLL | RRRWQQLL | RRSRPSGDL |
| SIVsmE660 | ......... | ......... | ...H..... | ......... | ......... | .......F. | .Q....I. | KQC.RG.N. |
| 02132 | ......... | ...G..R.. | ...H..... | ......... | ..V...... | .......F. | .Q....I. | KQCRRR.N. |
| 01048 | ......... | ......... | .S....... | ......... | ........ | ......... | ||
| Mamu-B*08 | Vif123-131RL9 | Vif172-179RL8 | ||||||
| SIVmac239Δnef | RRAIRGEQL | RRDNRRGL | ||||||
| SIVsmE660 | .......K. | G.N...s. | ||||||
| 02132 | ||||||||
| 01048 | ......... | .....G.. | ||||||
| Mamu-B*17 | Pol372-379MF8 | Pol435-443FW9 | Pol604-613VW10 | Env241-251LF11 | Env816-825LY10 | Env830-838FW9 | Vif44-52HW9 | Vif66-73HW8 |
| SIVmac239Δnef | MRHVLEPF | FQWMGYELW | VWEQWWTDYW | LRCNDTNYSGF | LRTELTYLQY | FHEAVQAVW | HFKVGWAWW | HLEVQGYW |
| SIVsmE660 | ..N..... | ......... | I......... | .....S..... | I..GIA.... | .Q.....W. | .H....... | ........ |
| 01003 | ........ | ......... | .......... | .....S..... | .......... | .Y....... | .H....... | Q....... |
| 01079 | ..N..... | ......... | I......... | .....S..... | .......... | .Y....... | .H....... | ........ |
| Mamu-B*17 | Vif135-143CY9 | cryptic RW9 | ||||||
| SIVmac239Δnef | CRFPRAHKY | RHLAFKCLW | ||||||
| SIVsmE660 | .K..K...N | .A..S..FR | ||||||
| 01003 | .K..K...N | .A..S..FR | ||||||
| 01079 | .K..K...N | .A..S..FR | ||||||
Periods represent conserved amino acids in comparison to SIVmac239Δnef. The aa sequence for each epitope corresponds to the vaccine strain SIVmac237Δnef. Any aa substitutions in these epitopes present in our stock of SIVsmE660 or in the plasma virus of our vaccinated animals during the chronic phase of infection are identified with capital letters or lowercase letters for mixed populations. Periods represent conserved aa in comparison to SIVmac239Δnef.
Figure 4.

Frequency of tetramer binding cells in the PBMC of SIVmac239Δnef-vaccinated macaques p.c. with SIVsmE660. (a) The frequency of MHC-I tetramer binding cells in the PBMC after challenge with SIVsmE660. Frequencies of tetramer positive cells are reported as the percentage of CD3+/CD8+/tetramer+ lymphocytes. The plasma virus concentrations for each animal are displayed with the red line and symbols. * distinguishes Nef221-229YY9 from Nef159-167YY9. (b) Comparison of amino acid sequences between SIVmac239-derived peptides and SIVsmE660. The SIVmac239 sequence is shown above SIVsmE660 sequence for comparison of the two viruses. Periods represent conserved amino acids, capital letters indicate amino acid substitutions, and lowercase letters represent mixed populations.
The Mamu-B*08+ animal 02132 experienced a massive expansion of Mamu-B*08–restricted CD8+ T cells after SIVsmE660 challenge during acute infection. CD8+ T cells against peptide pools Nef A, Vif C and D (both Vif pools containing the Mamu-B*08–restricted epitope Vif123-131RL9), and Env O (containing the Mamu-B*08–restricted epitope Env573-581KL9) were the only vaccine-induced T cell responses to expand after heterologous challenge (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20081524/DC1). This animal had a peak concentration of plasma virus (14,000 vRNA Eq/ml) at 2 wk p.c. (Fig. 1 and Fig. 4), which was subsequently brought under control. CD4+ T cells against two epitopes in Gag (pools B and C) also expanded in this animal (Fig. S4). Thus, in this Mamu-B*08+ macaque, vaccine-induced Nef-, Vif-, and Env-specific CD8+ T cells expanded during the time in which control of virus replication was established. Remarkably, three of the four vaccine-induced CD8+ T cell responses were restricted by Mamu-B*08.
Transient depletion of circulating CD8+ cells
Because neutralizing antibodies did not appear to be responsible for control of viral replication, we next investigated whether CD8+ cells were important in vaccine-mediated control of replication. Therefore, we depleted CD8+ cells in vivo at 11 mo p.c. in the four SIVmac239Δnef-vaccinated animals controlling virus replication below 5,000 vRNA copy Eq/ml plasma. We administered a monoclonal antibody specific for CD8α to transiently deplete CD8+ cells, which includes both CD8+ T cells and NK cells, from the periphery. 1 d after administering the antibody, CD8+ T cells were almost completely absent from the PBMC in three of the animals and reduced by >50% in the fourth. During the period of CD8+ cell depletion, plasma virus concentrations increased by 1–3 logs (Fig. 5 a). Plasma virus replication peaked 7–10 d after in vivo depletion of CD8+ cells and ranged between 2.3 × 104 and 5.5 × 105 vRNA copy Eq/ml. CD8+ cells reemerged in the periphery 17–28 d after depletion, concomitant with a decrease in plasma virus concentrations (Fig. 5, b and c). Only animal 01048, however, controlled virus replication at or below predepletion levels by 5 mo after CD8+ cell depletion. Conversely, animal 02132 never regained control of virus replication below 5,000 vRNA Eq/ml.
Figure 5.

Plasma virus concentrations and CD8+ T cell and NK cell counts after in vivo depletion of CD8+ cells. (a) Plasma virus concentrations of the four SIVmac239Δnef-vaccinated macaques were transiently depleted of their peripheral CD8+ cells. (b and c) The number of CD8+ T cells (CD3+/CD8+ lymphocytes; b) and the number of NK cells (CD3−/CD8+/CD16+; c) in the PBMC after in vivo CD8 depletion.
Because these animals controlled virus replication to extremely low levels before CD8+ cell depletion, we wanted to determine the predominant species of virus replicating after depletion. Using a sequence-specific quantitative RT-PCR (QRT-PCR) assay, we were surprised to detect only SIVmac239Δnef replicating in two of the animals (01022 and 01048), whereas SIVsmE660 was the primary virus replicating in the remaining two animals. These results prompted us to reexamine samples spanning the entire study for all of the vaccinated animals. After this retrospective analysis, we only detected the vaccine strain and not the challenge strain at any point p.c. in 01022 and 01048. In the remaining vaccinated animals, SIVsmE660 was the principal virus replicating at all time points measured after challenge.
For the animals transiently depleted of their CD8+ cells, we examined the cellular immune responses by IFN-γ ELISPOT after the CD8+ cells returned to the periphery, reasoning that these cells might be involved in reestablishing control of viral replication. We assessed both CD8+ and CD4+ T cell responses using whole PBMC and PBMC depleted of CD8+ cells. We detected broad responses in two of the four animals using freshly isolated PBMC. Animals 01022 and 01048 made responses against 27 and 20 peptide pools, respectively. These responses were directed against multiple viral proteins (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20081524/DC1). Conversely, animals 02132 (11 pools) and 88085 (4 pools) had more focused responses. Of note, 88085 consistently had high background levels of IFN-γ in the PBMC, which resulted in an increased threshold for positive responses and probably accounted for the generally low number of responses detected in this animal.
We also examined CD4+ T cell responses in the CD8+ cell–depleted animals using a combination of fresh and frozen cells. Because of limitations on cell availability, we restricted our initial analysis of CD4+ T cell responses with fresh PBMC depleted of CD8+ cells to pools of peptides spanning Gag, Tat, Rev, and Vif at 4 or 5 wk after CD8+ cell depletion. We used frozen cells to determine how many peptide pool–specific responses outside of Gag, Tat, Rev, and Vif that were identified using whole PBMC were associated, at least in part, with CD4+ cells. Remarkably, animals 88085 (10 pools), 01048 (17 pools), and 01022 (19 pools) made broad CD4+ T cell responses against multiple proteins (Fig. S5). The largest magnitude response at 4 wk after depletion in animal 01022 was a CD4+ T cell response directed against a peptide pool spanning the first 51 aa of Rev (1,965 SFC per million PBMC and 2,083 SFC per million CD8+ cell–depleted PBMC). This region of Rev is known to be the target of potent CD4+ T cell responses (33–35). From animal 02132, however, we only detected three CD4+ T cell responses. Although it is likely that some peptide pools stimulated both CD4+ and CD8+ T cell responses, we were able to associate over half of the detectable peptide pool–specific responses in three of the four animals with CD4+ T cells. It is difficult to determine whether these CD4+ T cell responses directly contributed to controlling virus replication or their presence is merely the result of low virus replication. These results do underscore, however, the potential importance of SIV-specific CD4+ T cell responses in generating successful antiimmunodeficiency virus immunity. Results also indicate the likelihood that SIV-specific CD4+ T cell responses are not necessarily deleterious, though some have speculated they would be (36).
Escape in SIVsmE660?
Most of the vaccinated macaques in our study controlled virus replication below 10,000 vRNA copy Eq/ml for at least 4 mo p.c. However, the majority of them experienced gradually increasing virus replication as the study progressed. We therefore set out to determine whether increased plasma virus replication was caused by escape in known CD8+ T cell epitopes. We sequenced the virus from the plasma of six of the animals during the chronic phase of infection. Because of the low number of virions in the plasma of the animals controlling virus replication during the chronic phase of infection, we used post–CD8+ cell depletion samples for the CD8+ T cell escape analysis. We found evidence for amino acid substitutions in known CD8+ T cell epitopes in the circulating viruses of these animals. Animal rhAO84, the only vaccinee not to control virus replication at any point p.c. and the fastest to progress to simian AIDS, had circulating virus at its time of death with changes in only one of the 13 Mamu-A*02–restricted epitopes, with respect to the SIVsmE660 sequence, and a mixed population in a second (Table II). This corresponds with the limited expansion of epitope-specific responses detected in this animal p.c. (Fig. 4 and Fig. S3 e). Conversely, animal 02092 made vigorous responses against several Mamu-A*01–restricted epitopes during acute infection, particularly the immunodominant epitope Gag181-189CM9 (Gag CM9; Fig. 4 and Fig. S3). During the chronic phase of infection, however, we detected mutations in several of the Mamu-A*01–restricted epitopes recognized during acute infection, including Gag CM9. Escape from Gag CM9–specific CD8+ T cells has previously been associated with a loss of viral control (37, 38).
We also investigated viral evolution in the two vaccinated Mamu-B*17+ animals. These animals controlled virus replication at two weeks p.c. below 250 vRNA copy Eq/ml plasma but subsequently experienced near-identical increases in plasma virus replication with respect to time and magnitude (Fig. 6). We detected the same amino acid changes in the two Mamu-B*17–restricted Env epitopes in both animals, Env816-825LY10 (Env LY10) and Env830-838FW9 (Env FW9; Table II). Curiously, the mutations observed in these two epitopes were nearly identical to the sequence of SIVmac239Δnef, with only a histidine-to-tyrosine substitution in Env FW9 differing from the SIVmac239Δnef sequence in these animals.
Figure 6.

Virus replication in Mamu-B*17+ macaques. Plasma virus concentrations in SIVmac239Δnef vaccinated (blue) and unvaccinated (red) Mamu-B*17+ macaques for 60 wk p.c.
Recombination between SIVmac239Δnef and SIVsmE660
One possible explanation for the emergence of SIVmac239-like sequences in these two epitopes was that recombination had occurred between the vaccine and challenge viruses. Indeed, bulk sequencing of the entire viral genome at 8 mo p.c. showed that multiple recombination events had occurred in the two Mamu-B*17+ animals. Further analysis of all of the vaccinated animals revealed recombination occurring in several other animals (Fig. 7). Additionally, sequence analysis of animals 01022 and 01048 confirmed that only SIVmac239Δnef was replicating in these animals. Interestingly, the virus in the two vaccinated Mamu-B*17+ macaques had similar patterns of recombination and replicated to a level ∼1 log higher at 1 yr p.c. than the unvaccinated Mamu-B*17+ animals infected with SIVsmE660 alone (Figs. 6 and 7). This suggests that recombination provided a fitness advantage for replication in Mamu-B*17+ animals.
Figure 7.

Recombination events between SIVmac239Δnef and SIVsmE660. During the chronic phase of infection or after in vivo CD8 depletion we used bulk sequencing to detect recombination between SIVmac239Δnef and SIVsmE660. Represented are the regions that most closely align to either SIVmac239Δnef (red) or SIVsmE660 (blue). Positioning of putative break points and size of either SIVmac239Δnef or SIVsmE660 regions are approximations based on sequencing data. Open boxes represent regions of the virus where sequences could not be obtained.
A recent study found that bulk sequencing of plasma vRNA might result in recombination because of PCR artifacts (39). Therefore, to confirm recombination between the vaccine and challenge strains, we used limiting dilution PCR to achieve single-genome amplification (SGA). We focused our analysis on a putative break point at the end of env and the beginning of nef in the vaccinated Mamu-B*17+ animals. SGA analysis corroborated the bulk sequencing results indicating that recombination had indeed occurred in the two Mamu-B*17+ animals, as all 10 clones from 01003 and all 11 clones from 01079 showed evidence of recombination (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20081524/DC1). The presence of recombination in the vaccinees, particularly the Mamu-B*17+ animals, indicates another mechanism by which the virus can evade protective immunity in live-attenuated SIV-vaccinated macaques.
DISCUSSION
In this study, we examined the ability of live-attenuated SIV to protect macaques against heterologous virus challenge. The vaccinated animals significantly reduced plasma virus replication at most time points between 2 and 32 wk p.c. (P ≤ 0.049) in comparison to the unvaccinated controls. Surprisingly, we found no evidence of SIVsmE660 replication in two of the vaccinated animals. We also explored the nature of the antiviral immunity that provided protection in the vaccinated animals. Several lines of evidence emerged strongly implicating vaccine-induced MHC-I–restricted CD8+ cell responses as the primary force involved in protecting SIVmac239Δnef-vaccinated macaques from heterologous challenge. We discovered, however, evidence of recombination between the vaccine and challenge strains, which likely contributed to a loss of immune-mediated control of virus replication.
We have known that live-attenuated SIV vaccines can protect macaques against pathogenic SIV challenges for almost 20 yr. However, the underlying mechanism of this protective immunity has remained elusive. CD4+ and CD8+ T cells (10, 12–14, 16), antibodies (9, 10), NK cells (11, 15), and viral interference (16–18) have been implicated in vaccine-induced control. In this study, we provide several lines of evidence showing that MHC-I–restricted CD8+ responses are playing the key role in reducing virus replication after heterologous virus challenge in SIVmac239Δnef-vaccinated macaques. First, there is no evidence that vaccine-induced neutralizing antibodies contributed to controlling virus replication. Second, we found an association between two MHC-I alleles and near complete control of acute virus replication after heterologous challenge. Finally, transient depletion of CD8+ cells from the periphery of macaques controlling SIV replication quickly led to a 3–4 log increase in plasma virus concentrations. Our results eliminate viral interference, CD4+ T cells, and antibodies as sole providers of protective immunity in live-attenuated SIV vaccinated macaques because these responses are not influenced by protective MHC-I alleles and virus replication in CD8+ cell–depleted animals in which SIV-specific antibodies were still present. Our results do not eliminate the possibility that helper CD4+ T cells or binding antibodies, either through the development of immune complexes or removal of infected cells via antibody-dependent cell-mediated cytotoxicity, play an important supportive role in controlling virus replication. We also cannot rule out the possibility that NK cells contributed to the control of virus replication. However, a recent study demonstrated that NK cells have a limited role in controlling primary SIV replication (40). But the chronic low level of replication by live-attenuated SIV may provide persistent stimulation and activation to NK cells to allow them to help eliminate pathogenic SIV replication. Additionally, depletion of peripheral CD8+ cells during the chronic phase of infection indicates that these cells are responsible for controlling virus replication during this phase of infection but leaves open the possibility that additional factors contribute to the reduction in acute virus replication, particularly in the Mamu-B*08+ and -B*17+ animals. To more accurately assess the role of CD8+ T cells in controlling acute virus replication in Mamu-B*08+ and -B*17+ macaques, additional animals will need to be vaccinated with SIVmac239Δnef and depleted of CD8+ cells before SIVsmE660 challenge.
Throughout the course of the study, we used peptides corresponding to the SIVmac239 proteome to detect virus-specific cellular immune responses. We feel that these peptides effectively detected cross-reactive CD8+ and CD4+ T cell responses between the vaccine and challenge strains because these responses are likely responsible for controlling virus replication in the vaccinated animals. Nevertheless, there may be additional CD4+ and CD8+ T cell responses that contribute to controlling SIV replication but cannot be detected using SIVmac239-specific peptides. The number of these unaccounted for responses may vary from animal to animal.
Our inability to detect SIVsmE660 replication by QRT-PCR or bulk sequencing in two of the vaccinated animals suggested that they achieved sterilizing immunity against a heterologous virus. These results give hope that CD8+ T cell–based HIV vaccines can realize a similar level of efficacy. However, it is unclear why these two animals completely suppressed SIVsmE660 replication, whereas the remaining eight vaccinees did not. Also, we cannot rule out the possibility that these two animals harbor a small number of cells latently infected with SIVsmE660.
The protective capacity of vaccine-induced CD8+ T cells elicited by live-attenuated SIV vaccines differs from current prime-boost regimens, which provide only limited reduction in viral replication after homologous virus challenge (41–44). There may be several reasons for this discrepancy in protective efficacy. CD8+ T cell responses induced by SIVmac239Δnef during the vaccine phase of the study were roughly equivalent in frequency to those produced by prime-boost vaccine regimens (41, 45–47). The magnitude of vaccine-induced CD8+ T cell responses, therefore, is likely not the reason why live-attenuated SIV vaccines succeed at controlling virus replication and recombinant virus–based vaccines largely fail. Current HIV vaccine modalities focus the immune responses on only a few viral proteins. This may not generate the breadth of responses needed to control replication after a heterologous virus challenge. SIVmac239Δnef vaccination generated broad CD8+ T cell responses against several viral proteins, even those which are often considered too variable to be included in HIV vaccines. Cellular immune responses directed against these relatively variable proteins might have contributed to protective immunity in our study. Animal 02132, for example, made a very strong CD8+ T cell response to an immunodominant Mamu-B*08–restricted epitope in Vif, likely contributing to its control of virus replication after heterologous virus challenge. SIVmac239Δnef vaccination also provided consistent low-level antigen stimulation throughout the vaccine phase of the study. This contrasts with prime-boost strategies and single cycle SIV viruses where only limited replication of the virus/vector occurs (48). Under these circumstances anamnestic responses induced by prime-boost or single-cycle SIV may be incapable of responding fast enough to limit acute viral replication after exposure to pathogenic viruses. The constant antigen exposure provided by SIVmac239Δnef, alternatively, may induce effector responses that rapidly react after pathogenic immunodeficiency virus infection and limit virus replication. Finally, SIVmac239Δnef may have additional advantages over most current HIV/SIV vaccines in that it primes immune responses in the same tissues where the pathogenic viruses are likely to replicate. The discovery of recombinant viruses indicates that the two virus strains indeed replicated in the same cells, let alone the same tissues. Although we did not specifically sample mucosal tissues in our study, live-attenuated SIV vaccines are known to generate CD8+ T cell responses in these tissues (49, 50). Trafficking of antiviral immune responses to the mucosa maximizes the chances that these responses will encounter virally infected cells during the earliest stages of infection, even after i.v. inoculation. Therefore, to mimic live-attenuated SIV vaccines as closely as possible, improved HIV vaccines may need to use replicating vectors to provide constant antigenic stimulation to antiviral immune responses. Ideally these vectors will target responses to mucosal tissues and include as many viral proteins as possible to maximize the number of cross-reactive immune responses with infecting HIV viruses.
In spite of early control of virus replication after SIVsmE660 challenge, particularly those animals expressing Mamu-B*17, we observed a progressive increase in plasma virus replication in most vaccinees. This differs from the durable control observed in animals vaccinated with live-attenuated SIV and challenged with homologous viruses. We found evidence of viral escape from CD8+ T cell responses that likely contributed to the loss of viral containment. Although this finding further supports CD8+ T cells as playing an important role in controlling virus replication after heterologous virus challenge, it also demonstrates the fragility of this control. The already reduced number of epitopes recognized in the heterologous challenge virus may focus selective pressure on a few regions of the virus and provide an avenue by which only a small number of key mutations can result in a loss of viral control. These results, therefore, also emphasize the need to limit virus replication to as low a level as possible.
We discovered that recombination had occurred between the vaccine and the challenge strains. This may have potentially produced a more pathogenic recombinant strain. In vaccinated Mamu-B*17+ animals in particular we detected numerous recombination events. Surprisingly, most of these events were shared between the two animals. These recombination events appear to contribute to the loss of containment of virus replication even after control of acute virus replication was achieved. Startlingly, the recombinant viruses eventually replicated to around 5 × 106 copy Eq/ml plasma, approximately one log higher than the unvaccinated Mamu-B*17+ controls infected with SIVsmE660 alone. Although the number of Mamu-B*17+ animals in our study is relatively small, our results suggest that the recombinant virus had a fitness advantage in Mamu-B*17+ animals.
In retrospect, this recombination was hardly surprising. Recombination between different viral strains is well documented in HIV and has been noted before in the setting of live-attenuated SIV vaccines (3, 4, 51–54). However, the prospect of incoming HIV virions recombining with a live-attenuated strain to produce a more pathogenic strain should eliminate any remaining possibility of using live-attenuated HIV vaccines in humans.
We challenged our cohort of animals i.v. because previous studies had shown effective control of homologous virus by live-attenuated SIV-vaccinated macaques challenged via this route. This challenge is comparable to transmission of HIV-1 through contaminated needles or blood products. It is also the most difficult route of transmission for vaccines to protect against because all physical barriers are bypassed and viruses can enter freely into the body. Worldwide, however, sexual contact, and not direct blood-to-blood contact, is the primary route of HIV-1 transmission. Studies in macaques and recent studies in humans suggest that only a small population of virus crosses the mucosal barriers to establish infection after heterosexual transmission (39, 55, 56). Subsequent localized virus replication during the earliest stages of infection may allow vaccine-induced immune responses to prevent systemic infection. Thus, it is possible that the reduction in plasma virus replication observed in our study after i.v. challenge may be even greater in a model of sexual transmission.
In conclusion, our study demonstrates that SIVmac239Δnef-induced CD8+ T cell responses can significantly reduce heterologous AIDS virus replication. Most of the vaccinated animals controlled virus replication below 10,000 vRNA copy Eq/ml through 4 mo p.c. However, only four of them maintained low levels of virus replication out to 11 mo p.c. These four animals have various MHC-I backgrounds, suggesting that a properly designed HIV vaccine that induces CD8+ T cell responses can be effective in diverse populations. The combination of the association of the Mamu-B*08+ and -B*17+ MHC-I alleles with near complete control of acute virus replication, the rapid increase in virus replication after in vivo depletion of CD8+ cells in the periphery, and the inability of neutralizing antibodies to affect ongoing virus replication strongly implicate MHC-I–restricted CD8+ T cells as the major contributors to live-attenuated SIV-induced protective immunity. The discovery of recombination between the vaccine and challenge strains, however, militates strongly against the development of live-attenuated HIV vaccines.
MATERIALS AND METHODS
Animals and viruses.
Indian rhesus macaques (Macaca mulatta) from the Wisconsin National Primate Research Center were chosen based on expression of particular MHC-I alleles as determined by sequence-specific PCR (26, 57). All animals were housed and cared for according to the regulations set forth in the Guide for the Care and Use of Laboratory Animals of the National Research Council (National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington DC), as approved by the University of Wisconsin Institutional Animal Care and Use Committee. 10 macaques were inoculated i.v. with 10 ng p27 of SIVmac239Δnef, provided by R. Desrosiers (New England Primate Research Center, Southborough, MA). Vaccinees and control animals were challenged i.v. with 100 50% tissue culture infective doses of SIVsmE660.
In vivo CD8 depletion.
We transiently depleted peripheral CD8+ cells by administering monoclonal antibody cM-T807 (produced by Centocor and provided via Keith Reimann [Harvard Medical School, Boston, MA] and the National Institutes of Health Nonhuman Primate Reagent Resource) i.v. in a single dose at 50 mg/kg of body weight.
Viral load determination.
Levels of circulating plasma virus were determined using a previously described QRT-PCR assay (58). The challenge strain SIVsmE660 was differentiated from SIVmac239Δnef via a second QRT-PCR assay run on the same isolation of vRNA used for quantitating virus concentrations. The E660 primers Nefdel.f2 5′-CRCTCTCTTGTGAGCCTCAGAA-3′ and Nefdel.r2 5′-TCTTGGGTGCACTGAGACAC-3′ produce a small amplicon in Nef. The Nefdel.r2 primer and the probe 5′-(6-Fam)CAAGTCATCATCTTCATCATCCACATCATCCAT(BHQ1)-3′ bind entirely within the deleted portion of nef and detect full-length virus exclusively. These primers and probe are used under the same cycling conditions that were described previously (58). The concentration of full-length virus was determined by interpolation onto a standard curve of in vitro–transcribed nef RNA standards in serial 10-fold dilutions using the LightCycler 480 software (version 1.2.0; Roche).
IFN-γ ELISPOT assays.
Fresh PBMC isolated from EDTA anticoagulated blood were used in ELISPOT assays for the detection of IFN-γ–secreting cells as previously described (59). Peptides used in these assays were obtained through the AIDS Research and Reference reagent program, Division of AIDS, National Institute of Allergy and Infectious Disease, and National Institutes of Health. Additionally, we examined responses by CD8− cells by depleting PBMC of CD8+ cells using a CD8 Microbead kit for nonhuman primates (Miltenyi Biotec) according to the manufacturer's instructions.
Tetramer staining.
SIV-specific cells were enumerated by staining PBMC with MHC-I (Mamu-A*01, -A*02, or -B*17) tetrameric complexes loaded with synthetic peptides corresponding to previously identified epitopes (26, 28, 29, 60, 61). The Mamu-B*08 tetramers were refolded with either the synthetic peptides corresponding to Env573-581KL9 (62) or the recently identified Vif123-131RL9 or Vif172-179RL8 epitopes (30). All Mamu-B*17 tetramers and some Mamu-B*08 tetramers were refolded as previously outlined (63). The assay was performed using one million PBMC as previously described (41).
Neutralization assays.
Neutralization assays were performed as previously described (64) with the following exceptions. Rhesus PBMC was stimulated for 3 d with PHA and IL-2 before the addition of virus and plasma. After addition of virus and plasma, plates were incubated at 37°C overnight, and the cells were then washed four times with fresh culture medium. Half of the media was replaced with fresh media 3 d later. Culture supernatants were harvested for p27 analysis 7 d after infection.
Intracellular cytokine staining.
Intracellular cytokine staining was performed using thawed PBMC as previously described (42). In brief, 5 × 105–106 PBMC were incubated in the presence of Brefeldin A (Sigma-Aldrich) and pools of 10 peptides, 15 aa in length, for 5 h. The cells were then stained with anti-CD8α PerCP (Becton Dickinson) and anti-CD4 APC (Becton Dickinson) for 30 min. After the incubation, the cells were washed twice with PBS, fixed with 2% paraformaldehyde, and stored overnight at 4°C. The next day, the cells were washed twice with permeabilization buffer (PBS + 10% FBS + 0.1% saponin) and stained with anti–IL-2 PE (BD Biosciences) and anti–IFN-γ FITC (BD Biosciences) for 50 min. After washing the cells twice with permeabilization buffer, the cells were fixed with paraformaldehyde and stored at 4°C until being acquired on a FACSCalibur Flow Cytometer (Becton Dickinson). The data were analyzed using FlowJo software (Tree Star, Inc.).
CD8+ T cell and NK cell quantification.
We monitored the CD8+ cell depletion kinetics by staining PBMC with fluorescently labeled antibodies specific for CD8-PE (Dako), CD16–Pacific Blue (BD Biosciences), and CD3–Alexa 700 (BD Biosciences). In brief, 500,000 PBMC were incubated with these antibodies for 30 min at room temperature. The samples were then washed twice, fixed with paraformaldehyde, and run on a BD-LSR-II flow cytometer (Becton Dickinson) using FACSDiva software (BD Biosciences). Data were analyzed using FlowJo software. Absolute counts were calculated by multiplying the frequency of CD8+ T cells or CD3+/CD8+/CD16+ NK cells within the lymphocyte gate by the lymphocyte counts per microliter of blood obtained from matching complete blood counts.
Sequencing of plasma vRNA.
vRNA was extracted from plasma samples containing >1,000 vRNA copy Eq/ml as previously described (58). Samples were sequenced from 8 mo p.c., the time of death, for animals rhAO84 and rh2000, from 10 mo p.c. for animals 02092, 01009, 01003, and 01079, and 2 wk after depletion for animals 88085, 01022, 02132, and 01048. For each vRNA sample, several overlapping amplicons spanning the open reading frames of SIVmac239 or SIVsmE660 were amplified using the One-Step RT-PCR kit (QIAGEN) and pairs of sequence specific primers. The RT-PCR conditions for all amplicons were as follows: 50°C for 30 min, 95°C for 15 min, 45 cycles of 94°C for 30 s, 53°C for 1 min, 72°C for 150 s, and 68°C for 20 min. The amplified cDNAs were purified using the QIAquick 8 PCR Purification kit (QIAGEN). Sequencing reactions for each amplicon were set up using the DYEnamic ET Terminator Cycle Sequencing kit (GE Healthcare). The sequencing cycling conditions for all amplicons were as follows: 30 cycles of 95°C for 20 s, 50°C for 15 s, and 60°C for 1 min. Both strands of each amplicon were sequenced on a 3730 DNA Analyzer (Applied Biosystems). Sequences were assembled using Aligner version 2.0.6 (CodonCode Corporation). DNA sequences were conceptually translated and aligned to SIVsmE660 in MacVector 9.0 trial version (Accelrys).
SGA PCR.
cDNA was created from vRNA extracted from plasma using the MinElute kit (QIAGEN) per the manufacturer's instructions. cDNAs were created using the Superscript III cDNA synthesis kit (Invitrogen) using oligo dT primers per the manufacturer's instructions.
To determine the optimal cDNA concentration resulting in <30% positive reactions in PCR we used Phusion high-fidelity polymerase (Finnzymes) per the manufacturer's instructions. PCR reactions were set up using first round amplifications as template as follows: each reaction contained 4 μl of 5× HF buffer, 0.4 μl of 10 mM dNTP, 0.5 μl of 20 μM sense oligo, 0.5 μl of 20 μM antisense oligo, 2 μl of first-round PCR product, 0.2 μl Phusion high-fidelity polymerase, and DEPC water to 20 μl. Thermocycling was performed with the following parameters: initial denaturation at 98°C for 30 s, and then denature at 98°C for 5 s, anneal at 65°C for 10 s, and extend at 72°C for 10 s for 45 cycles. A final extension was done at 72°C for 5 min. The oligos used in this reaction were E543-8858F (ACCAAAGAAGGAGAAGAAGGAGA) and E453-9449R (TATTGCCAATTTGTATGTCATGG). When nested PCRs are complete, all reaction products were electrophoresed on a 1% agarose gel stained with SYBR safe DNA gel stain (Invitrogen). Positive reactions contained a band at ∼592 bp which was cut out and from which DNA was isolated using the QIAquick Gel Extraction kit (QIAGEN) as per the manufacturer's instructions.
Isolated DNA was sequenced as described in Sequencing of plasma vRNAs using the following oligos: E543-8858F (ACCAAAGAAGGAGAAGAAGGAGA), E453-9449R (TATTGCCAATTTGTATGTCATGG), E543_Frag14_F1 (GGCCTTGGCAGATAGAATATATTC), and E543_Frag14_R (CTGAKACMCCTACYAAGTCATCATC).
Statistical analysis.
Statistical analyses consisted of two sample Student's t tests of the variables studied, namely number of virions per milliliter of blood and viral load at different time points p.c. We performed these tests using the TTEST procedure in SAS (version 9.1) to do the following: compare the plasma virus concentrations of the SIVmac239Δnef-vaccinated macaques (n = 10) with their MHC-I–matched naive controls (n = 10); compare Mamu-B*08+/-B*17+ macaques (n = 4) with their MHC-I–matched naive controls (n = 4); and compare Mamu-A*01+/-A*02+/-A*11+ macaques (n = 6) with their MHC-I–matched controls (n = 6).
Before performing inferential testing, we checked key underlying assumptions of the Student's t tests (normality of residuals and homoscedasticity). The failure of the data to support these assumptions led us to transform the data via the natural logarithm, which improved the conformity to the assumptions. Student's t test were performed under equal and unequal group variance assumptions via the Welch correction, in which we used the folded form of F test (F′) to decide the test statistic (pooled or Satterthwaite). This form of F test gives the probability of a greater F value under the null hypothesis that group variances are equal. We also used one way nonparametric analysis using an exact Wilcoxon Rank-Sum test with the continuity correction, which does not assume normality of residuals, to further verify our results. Conclusions about statistical significance and direction of effects were the same across these three analytic approaches, so herein we report only the Student's t test results.
Online supplemental material.
Fig. S1 shows that most SIVmac239Δnef replication was controlled below 500 copy Eq/ml plasma at the time of the SIVsmE660 challenge. Fig. S2 shows the prechallenge IFN-γ ELISPOT responses for each animal using whole PBMC and CD8+-depleted PBMC. Fig. S3. shows the epitope-specific expansion of CD8+ T cell responses before challenge and p.c. for all of the animals. Fig. S4 shows prechallenge and p.c. peptide pool–specific responses from animal 02132 in IFN-γ ELISPOT assays. It also shows, through intracellular cytokine staining assays, that we can associate two of these responses with CD4+ T cells. Fig. S5 shows peptide-specific responses in whole PBMC and PBMC depleted of CD8+ cells after the return of CD8+ cells to the periphery. Fig. S6 shows recombination by single-genome analysis of virus circulating in the vaccinated Mamu-B*17+ macaques during the chronic phase of infection. Table S1 displays the frequency of tetramer binding cells in the PBMC during the vaccine phase of the study. Table S2 displays the peptide pools to which SIVmac239Δnef-vaccinated animals made responses in IFN-γ ELISPOT assays using whole PBMC or PBMC depleted of CD8+ cells. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20081524/DC1.
Supplementary Material
Acknowledgments
The following reagents were obtained through the AIDS Research and Reference reagent program, Division of AIDS, NIAID, National Institutes of Health: SIVmac239 Vif Peptides, complete set (6205); SIVmac239 Tat Peptides, complete set (6207); SIVmac239 Rev (15-mer) Peptides, complete set (6448); SIVmac239 Pol Peptides, complete set (6443); SIVmac239 Gag Peptides, complete set (6204); SIVmac239 Vpr Peptides, complete set (6449); SIVmac239 Vpx Peptides, complete set (6450); SIVmac239 Env Peptides, complete set (6883); and SIVmac239 full-length Nef Peptides, complete set (8762). We would like to thank the veterinary staff at the Wisconsin National Primate Research Center for their assistance and Ron Desrosiers for providing the SIVmac239Δnef. We would also like to thank Keith Reimann and Centocor for supplying the monoclonal antibody cM-T807.
This research was supported by funds from the International AIDS Vaccine Initiative, National Institutes of Health grants R01 AI049120 and R01 AI052056 to D.I. Watkins and P51 RR000167 to the Wisconsin National Primate Research Center from the National Center for Research Resources, a component of the National Institutes of Health. Additionally, this research was conducted at a facility constructed with support from Research Facilities Improvement Program grant numbers RR15459-01 and RR020141-01. D.A. Price is a Medical Research Council (UK) Senior Clinical Fellow. This publication's contents are solely the responsibility of the authors and do not necessarily represent the official views of National Center for Research Resources or National Institutes of Health.
The authors have no conflicting financial interests.
Abbreviations used: Eq, equivalents; p.c., postchallenge; p.i., postinoculation; QRT-PCR, quantitative RT-PCR; SFC, spot-forming cells; SGA, single genome amplification; SIV, simian immunodeficiency virus; vRNA, viral RNA.
References
- 1.Gaschen, B., J. Taylor, K. Yusim, B. Foley, F. Gao, D. Lang, V. Novitsky, B. Haynes, B.H. Hahn, T. Bhattacharya, and B. Korber. 2002. Diversity considerations in HIV-1 vaccine selection. Science. 296:2354–2360. [DOI] [PubMed] [Google Scholar]
- 2.Korber, B., M. Muldoon, J. Theiler, F. Gao, R. Gupta, A. Lapedes, B.H. Hahn, S. Wolinsky, and T. Bhattacharya. 2000. Timing the ancestor of the HIV-1 pandemic strains. Science. 288:1789–1796. [DOI] [PubMed] [Google Scholar]
- 3.Robertson, D.L., P.M. Sharp, F.E. McCutchan, and B.H. Hahn. 1995. Recombination in HIV-1. Nature. 374:124–126. [DOI] [PubMed] [Google Scholar]
- 4.Heeney, J.L., A.G. Dalgleish, and R.A. Weiss. 2006. Origins of HIV and the evolution of resistance to AIDS. Science. 313:462–466. [DOI] [PubMed] [Google Scholar]
- 5.Mo, H., L. Stamatatos, J.E. Ip, C.F. Barbas, P.W. Parren, D.R. Burton, J.P. Moore, and D.D. Ho. 1997. Human immunodeficiency virus type 1 mutants that escape neutralization by human monoclonal antibody IgG1b12. off. J. Virol. 71:6869–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goulder, P.J., and D.I. Watkins. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat. Rev. Immunol. 4:630–640. [DOI] [PubMed] [Google Scholar]
- 7.Nickle, D.C., M.A. Jensen, G.S. Gottlieb, D. Shriner, G.H. Learn, A.G. Rodrigo, and J.I. Mullins. 2003. Consensus and ancestral state HIV vaccines. Science. 299:1515–1518. [DOI] [PubMed] [Google Scholar]
- 8.Fischer, W., S. Perkins, J. Theiler, T. Bhattacharya, K. Yusim, R. Funkhouser, C. Kuiken, B. Haynes, N.L. Letvin, B.D. Walker, et al. 2007. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat. Med. 13:100–106. [DOI] [PubMed] [Google Scholar]
- 9.Wyand, M.S., K. Manson, D.C. Montefiori, J.D. Lifson, R.P. Johnson, and R.C. Desrosiers. 1999. Protection by live, attenuated simian immunodeficiency virus against heterologous challenge. J. Virol. 73:8356–8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wyand, M.S., K.H. Manson, M. Garcia-Moll, D. Montefiori, and R.C. Desrosiers. 1996. Vaccine protection by a triple deletion mutant of simian immunodeficiency virus. J. Virol. 70:3724–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson, R.P., R.L. Glickman, J.Q. Yang, A. Kaur, J.T. Dion, M.J. Mulligan, and R.C. Desrosiers. 1997. Induction of vigorous cytotoxic T-lymphocyte responses by live attenuated simian immunodeficiency virus. J. Virol. 71:7711–7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daniel, M.D., F. Kirchhoff, S.C. Czajak, P.K. Sehgal, and R.C. Desrosiers. 1992. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science. 258:1938–1941. [DOI] [PubMed] [Google Scholar]
- 13.Schmitz, J.E., R.P. Johnson, H.M. McClure, K.H. Manson, M.S. Wyand, M.J. Kuroda, M.A. Lifton, R.S. Khunkhun, K.J. McEvers, J. Gillis, et al. 2005. Effect of CD8+ lymphocyte depletion on virus containment after simian immunodeficiency virus SIVmac251 challenge of live attenuated SIVmac239delta3-vaccinated rhesus macaques. J. Virol. 79:8131–8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gauduin, M.C., R.L. Glickman, S. Ahmad, T. Yilma, and R.P. Johnson. 1999. Immunization with live attenuated simian immunodeficiency virus induces strong type 1 T helper responses and beta-chemokine production. Proc. Natl. Acad. Sci. USA. 96:14031–14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gauduin, M.C., Y. Yu, A. Barabasz, A. Carville, M. Piatak, J.D. Lifson, R.C. Desrosiers, and R.P. Johnson. 2006. Induction of a virus-specific effector–memory CD4+ T cell response by attenuated SIV infection. J. Exp. Med. 203:2661–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Igarashi, T., Y. Ami, H. Yamamoto, R. Shibata, T. Kuwata, R. Mukai, K. Shinohara, T. Komatsu, A. Adachi, and M. Hayami. 1997. Protection of monkeys vaccinated with vpr- and/or nef-defective simian immunodeficiency virus strain mac/human immunodeficiency virus type 1 chimeric viruses: a potential candidate live-attenuated human AIDS vaccine. J. Gen. Virol. 78:985–989. [DOI] [PubMed] [Google Scholar]
- 17.Stahl-Hennig, C., R.M. Steinman, P. Ten Haaft, K. Uberla, N. Stolte, S. Saeland, K. Tenner-Racz, and P. Racz. 2002. The simian immunodeficiency virus deltaNef vaccine, after application to the tonsils of Rhesus macaques, replicates primarily within CD4(+) T cells and elicits a local perforin-positive CD8(+) T-cell response. J. Virol. 76:688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silvera, P., A. Wade-Evans, E. Rud, R. Hull, K. Silvera, R. Sangster, N. Almond, and J. Stott. 2001. Mechanisms of protection induced by live attenuated simian immunodeficiency virus: III. Viral interference and the role of CD8+ T-cells and beta-chemokines in the inhibition of virus infection of PBMCs in vitro. J. Med. Primatol. 30:1–13. [DOI] [PubMed] [Google Scholar]
- 19.Stebbings, R.J., N.M. Almond, E.J. Stott, N. Berry, A.M. Wade-Evans, R. Hull, J. Lines, P. Silvera, R. Sangster, T. Corcoran, et al. 2002. Mechanisms of protection induced by attenuated simian immunodeficiency virus. Virology. 296:338–353. [DOI] [PubMed] [Google Scholar]
- 20.Rud, E.W., L. Oglivie, B.E. Clarke, N. Almond, K. Kent, L. Chan, M. Page, P. Kitchin, J. Stott, N. Cook, et al. 1994. A naturally attenuated SIVmac32H vaccine or viral interference? In Vaccines ‘94: Modern Approaches to New Vaccines Including Prevention of AIDS. E. Norrby, F. Brown, R.M. Chanock, and H.S. Ginsberg, editors. Cold Spring Harbor Laboratory Press, Plainview, N.Y. 217-223.
- 21.Koff, W.C., P.R. Johnson, D.I. Watkins, D.R. Burton, J.D. Lifson, K.J. Hasenkrug, A.B. McDermott, A. Schultz, T.J. Zamb, R. Boyle, and R.C. Desrosiers. 2006. HIV vaccine design: insights from live attenuated SIV vaccines. Nat. Immunol. 7:19–23. [DOI] [PubMed] [Google Scholar]
- 22.Abdel-Motal, U.M., J. Gillis, K. Manson, M. Wyand, D. Montefiori, K. Stefano-Cole, R.C. Montelaro, J.D. Altman, and R.P. Johnson. 2005. Kinetics of expansion of SIV Gag-specific CD8+ T lymphocytes following challenge of vaccinated macaques. Virology. 333:226–238. [DOI] [PubMed] [Google Scholar]
- 23.Nilsson, C., B. Makitalo, R. Thorstensson, S. Norley, D. Binninger-Schinzel, M. Cranage, E. Rud, G. Biberfeld, and P. Putkonen. 1998. Live attenuated simian immunodeficiency virus (SIV)mac in macaques can induce protection against mucosal infection with SIVsm. AIDS. 12:2261–2270. [DOI] [PubMed] [Google Scholar]
- 24.Kestier, H.W. III, D.J. Ringler, K. Mori, D.L. Panicali, P.K. Sehgal, M.D. Daniel, and R.C. Desrosiers. 1991. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 65:651–662. [DOI] [PubMed] [Google Scholar]
- 25.Loffredo, J.T., J. Maxwell, Y. Qi, C.E. Glidden, G.J. Borchardt, T. Soma, A.T. Bean, D.R. Beal, N.A. Wilson, W.M. Rehrauer, et al. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J. Virol. 81:8827–8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogel, T.U., T.C. Friedrich, D.H. O'Connor, W. Rehrauer, E.J. Dodds, H. Hickman, W. Hildebrand, J. Sidney, A. Sette, A. Hughes, et al. 2002. Escape in one of two cytotoxic T-lymphocyte epitopes bound by a high-frequency major histocompatibility complex class I molecule, Mamu-A*02: a paradigm for virus evolution and persistence? J. Virol. 76:11623–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans, D.T., P. Jing, T.M. Allen, D.H. O'Connor, H. Horton, J.E. Venham, M. Piekarczyk, J. Dzuris, M. Dykhuzen, J. Mitchen, et al. 2000. Definition of five new simian immunodeficiency virus cytotoxic T-lymphocyte epitopes and their restricting major histocompatibility complex class I molecules: evidence for an influence on disease progression. J. Virol. 74:7400–7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen, T.M., B.R. Mothe, J. Sidney, P. Jing, J.L. Dzuris, M.E. Liebl, T.U. Vogel, D.H. O'Connor, X. Wang, M.C. Wussow, et al. 2001. CD8(+) lymphocytes from simian immunodeficiency virus-infected rhesus macaques recognize 14 different epitopes bound by the major histocompatibility complex class I molecule mamu-A*01: implications for vaccine design and testing. J. Virol. 75:738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mothe, B.R., J. Sidney, J.L. Dzuris, M.E. Liebl, S. Fuenger, D.I. Watkins, and A. Sette. 2002. Characterization of the peptide-binding specificity of Mamu-B*17 and identification of Mamu-B*17-restricted epitopes derived from simian immunodeficiency virus proteins. J. Immunol. 169:210–219. [DOI] [PubMed] [Google Scholar]
- 30.Loffredo, J.T., T.C. Friedrich, E.J. Leon, J.J. Stephany, D.S. Rodrigues, S.P. Spencer, A.T. Bean, D.R. Beal, B.J. Burwitz, R.A. Rudersdorf, et al. 2007. CD8 T Cells from SIV Elite Controller Macaques Recognize Mamu-B*08-Bound Epitopes and Select for Widespread Viral Variation. PLoS ONE. 2:e1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maness, N.J., L.E. Valentine, G.E. May, J. Reed, S.M. Piaskowski, T. Soma, J. Furlott, E.G. Rakasz, T.C. Friedrich, D.A. Price, et al. 2007. AIDS virus–specific CD8+ T lymphocytes against an immunodominant cryptic epitope select for viral escape. J. Exp. Med. 204:2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yant, L.J., T.C. Friedrich, R.C. Johnson, G.E. May, N.J. Maness, A.M. Enz, J.D. Lifson, D.H. O'Connor, M. Carrington, and D.I. Watkins. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 80:5074–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel, T.U., H. Horton, D.H. Fuller, D.K. Carter, K. Vielhuber, D.H. O'Connor, T. Shipley, J. Fuller, G. Sutter, V. Erfle, et al. 2002. Differences between T cell epitopes recognized after immunization and after infection. J. Immunol. 169:4511–4521. [DOI] [PubMed] [Google Scholar]
- 34.Giraldo-Vela, J.P., R. Rudersdorf, C. Chung, Y. Qi, L.T. Wallace, B. Bimber, G.J. Borchardt, D.L. Fisk, C.E. Glidden, J.T. Loffredo, et al. 2008. The major histocompatibility complex class II alleles Mamu-DRB1*1003 and -DRB1*0306 are enriched in a cohort of simian immunodeficiency virus-infected rhesus macaque elite controllers. J. Virol. 82:859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dzuris, J.L., J. Sidney, H. Horton, R. Correa, D. Carter, R.W. Chesnut, D.I. Watkins, and A. Sette. 2001. Molecular determinants of peptide binding to two common rhesus macaque major histocompatibility complex class II molecules. J. Virol. 75:10958–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pantaleo, G., and R.A. Koup. 2004. Correlates of immune protection in HIV-1 infection: what we know, what we don't know, what we should know. Nat. Med. 10:806–810. [DOI] [PubMed] [Google Scholar]
- 37.Barouch, D.H., J. Kunstman, J. Glowczwskie, K.J. Kunstman, M.A. Egan, F.W. Peyerl, S. Santra, M.J. Kuroda, J.E. Schmitz, K. Beaudry, et al. 2003. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J. Virol. 77:7367–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barouch, D.H., J. Kunstman, M.J. Kuroda, J.E. Schmitz, S. Santra, F.W. Peyerl, G.R. Krivulka, K. Beaudry, M.A. Lifton, D.A. Gorgone, et al. 2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature. 415:335–339. [DOI] [PubMed] [Google Scholar]
- 39.Salazar-Gonzalez, J.F., E. Bailes, K.T. Pham, M.G. Salazar, M.B. Guffey, B.F. Keele, C.A. Derdeyn, P. Farmer, E. Hunter, S. Allen, et al. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82:3952–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi, E.I., K.A. Reimann, and N.L. Letvin. 2008. In vivo natural killer cell depletion during primary simian immunodeficiency virus infection in rhesus monkeys. J. Virol. 82:6758–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson, N.A., J. Reed, G.S. Napoe, S. Piaskowski, A. Szymanski, J. Furlott, E.J. Gonzalez, L.J. Yant, N.J. Maness, G.E. May, et al. 2006. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J. Virol. 80:5875–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vogel, T.U., M.R. Reynolds, D.H. Fuller, K. Vielhuber, T. Shipley, J.T. Fuller, K.J. Kunstman, G. Sutter, M.L. Marthas, V. Erfle, et al. 2003. Multispecific vaccine-induced mucosal cytotoxic T lymphocytes reduce acute-phase viral replication but fail in long-term control of simian immunodeficiency virus SIVmac239. J. Virol. 77:13348–13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horton, H., T.U. Vogel, D.K. Carter, K. Vielhuber, D.H. Fuller, T. Shipley, J.T. Fuller, K.J. Kunstman, G. Sutter, D.C. Montefiori, et al. 2002. Immunization of rhesus macaques with a DNA prime/modified vaccinia virus Ankara boost regimen induces broad simian immunodeficiency virus (SIV)-specific T-cell responses and reduces initial viral replication but does not prevent disease progression following challenge with pathogenic SIVmac239. J. Virol. 76:7187–7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown, C.R., M. Czapiga, J. Kabat, Q. Dang, I. Ourmanov, Y. Nishimura, M.A. Martin, and V.M. Hirsch. 2007. Unique pathology in simian immunodeficiency virus-infected rapid progressor macaques is consistent with a pathogenesis distinct from that of classical AIDS. J. Virol. 81:5594–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santra, S., D.H. Barouch, B. Korioth-Schmitz, C.I. Lord, G.R. Krivulka, F. Yu, M.H. Beddall, D.A. Gorgone, M.A. Lifton, A. Miura, et al. 2004. Recombinant poxvirus boosting of DNA-primed rhesus monkeys augments peak but not memory T lymphocyte responses. Proc. Natl. Acad. Sci. USA. 101:11088–11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Demberg, T., R.H. Florese, M.J. Heath, K. Larsen, I. Kalisz, V.S. Kalyanaraman, E.M. Lee, R. Pal, D. Venzon, R. Grant, et al. 2007. A replication-competent adenovirus-human immunodeficiency virus (Ad-HIV) tat and Ad-HIV env priming/Tat and envelope protein boosting regimen elicits enhanced protective efficacy against simian/human immunodeficiency virus SHIV89.6P challenge in rhesus macaques. J. Virol. 81:3414–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu, J., B.A. Ewald, D.M. Lynch, M. Denholtz, P. Abbink, A.A. Lemckert, A. Carville, K.G. Mansfield, M.J. Havenga, J. Goudsmit, and D.H. Barouch. 2008. Magnitude and phenotype of cellular immune responses elicited by recombinant adenovirus vectors and heterologous prime-boost regimens in rhesus monkeys. J. Virol. 82:4844–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans, D.T., J.E. Bricker, H.B. Sanford, S. Lang, A. Carville, B.A. Richardson, M. Piatak Jr., J.D. Lifson, K.G. Mansfield, and R.C. Desrosiers. 2005. Immunization of macaques with single-cycle simian immunodeficiency virus (SIV) stimulates diverse virus-specific immune responses and reduces viral loads after challenge with SIVmac239. J. Virol. 79:7707–7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cranage, M.P., A.M. Whatmore, S.A. Sharpe, N. Cook, N. Polyanskaya, S. Leech, J.D. Smith, E.W. Rud, M.J. Dennis, and G.A. Hall. 1997. Macaques infected with live attenuated SIVmac are protected against superinfection via the rectal mucosa. Virology. 229:143–154. [DOI] [PubMed] [Google Scholar]
- 50.Cromwell, M.A., R.S. Veazey, J.D. Altman, K.G. Mansfield, R. Glickman, T.M. Allen, D.I. Watkins, A.A. Lackner, and R.P. Johnson. 2000. Induction of mucosal homing virus-specific CD8(+) T lymphocytes by attenuated simian immunodeficiency virus. J. Virol. 74:8762–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khatissian, E., V. Monceaux, M.C. Cumont, M.P. Kieny, A.M. Aubertin, and B. Hurtrel. 2001. Persistence of pathogenic challenge virus in macaques protected by simian immunodeficiency virus SIVmacDeltanef. J. Virol. 75:1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gundlach, B.R., M.G. Lewis, S. Sopper, T. Schnell, J. Sodroski, C. Stahl-Hennig, and K. Uberla. 2000. Evidence for recombination of live, attenuated immunodeficiency virus vaccine with challenge virus to a more virulent strain. J. Virol. 74:3537–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim, E.Y., M. Busch, K. Abel, L. Fritts, P. Bustamante, J. Stanton, D. Lu, S. Wu, J. Glowczwskie, T. Rourke, et al. 2005. Retroviral recombination in vivo: viral replication patterns and genetic structure of simian immunodeficiency virus (SIV) populations in rhesus macaques after simultaneous or sequential intravaginal inoculation with SIVmac239Deltavpx/Deltavpr and SIVmac239Deltanef. J. Virol. 79:4886–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wooley, D.P., R.A. Smith, S. Czajak, and R.C. Desrosiers. 1997. Direct demonstration of retroviral recombination in a rhesus monkey. J. Virol. 71:9650–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller, C.J., Q. Li, K. Abel, E.Y. Kim, Z.M. Ma, S. Wietgrefe, L. La Franco-Scheuch, L. Compton, L. Duan, M.D. Shore, et al. 2005. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J. Virol. 79:9217–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haase, A.T. 2005. Perils at mucosal front lines for HIV and SIV and their hosts. Nat. Rev. Immunol. 5:783–792. [DOI] [PubMed] [Google Scholar]
- 57.Knapp, L.A., E. Lehmann, M.S. Piekarczyk, J.A. Urvater, and D.I. Watkins. 1997. A high frequency of Mamu-A*01 in the rhesus macaque detected by polymerase chain reaction with sequence-specific primers and direct sequencing. Tissue Antigens. 50:657–661. [DOI] [PubMed] [Google Scholar]
- 58.Friedrich, T.C., L.E. Valentine, L.J. Yant, E.G. Rakasz, S.M. Piaskowski, J.R. Furlott, K.L. Weisgrau, B. Burwitz, G.E. May, E.J. Leon, et al. 2007. Subdominant CD8+ T-cell responses are involved in durable control of AIDS virus replication. J. Virol. 81:3465–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loffredo, J.T., B.J. Burwitz, E.G. Rakasz, S.P. Spencer, J.J. Stephany, J.P. Vela, S.R. Martin, J. Reed, S.M. Piaskowski, J. Furlott, et al. 2007. The antiviral efficacy of simian immunodeficiency virus-specific CD8+ T cells is unrelated to epitope specificity and is abrogated by viral escape. J. Virol. 81:2624–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loffredo, J.T., J. Sidney, C. Wojewoda, E. Dodds, M.R. Reynolds, G. Napoe, B.R. Mothe, D.H. O'Connor, N.A. Wilson, D.I. Watkins, and A. Sette. 2004. Identification of seventeen new simian immunodeficiency virus-derived CD8+ T cell epitopes restricted by the high frequency molecule, Mamu-A*02, and potential escape from CTL recognition. J. Immunol. 173:5064–5076. [DOI] [PubMed] [Google Scholar]
- 61.Allen, T.M., J. Sidney, M.F. del Guercio, R.L. Glickman, G.L. Lensmeyer, D.A. Wiebe, R. DeMars, C.D. Pauza, R.P. Johnson, A. Sette, and D.I. Watkins. 1998. Characterization of the peptide binding motif of a rhesus MHC class I molecule (Mamu-A*01) that binds an immunodominant CTL epitope from simian immunodeficiency virus. J. Immunol. 160:6062–6071. [PubMed] [Google Scholar]
- 62.Loffredo, J.T., A.T. Bean, D.R. Beal, E.J. Leon, G.E. May, S.M. Piaskowski, J.R. Furlott, J. Reed, S.K. Musani, E.G. Rakasz, et al. 2008. Patterns of CD8+ immunodominance may influence the ability of Mamu-B*08-positive macaques to naturally control simian immunodeficiency virus SIVmac239 replication. J. Virol. 82:1723–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hutchinson, S.L., L. Wooldridge, S. Tafuro, B. Laugel, M. Glick, J.M. Boulter, B.K. Jakobsen, D.A. Price, and A.K. Sewell. 2003. The CD8 T cell coreceptor exhibits disproportionate biological activity at extremely low binding affinities. J. Biol. Chem. 278:24285–24293. [DOI] [PubMed] [Google Scholar]
- 64.Parren, P.W., P.A. Marx, A.J. Hessell, A. Luckay, J. Harouse, C. Cheng-Mayer, J.P. Moore, and D.R. Burton. 2001. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J. Virol. 75:8340–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei, X., J.M. Decker, H. Liu, Z. Zhang, R.B. Arani, J.M. Kilby, M.S. Saag, X. Wu, G.M. Shaw, and J.C. Kappes. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
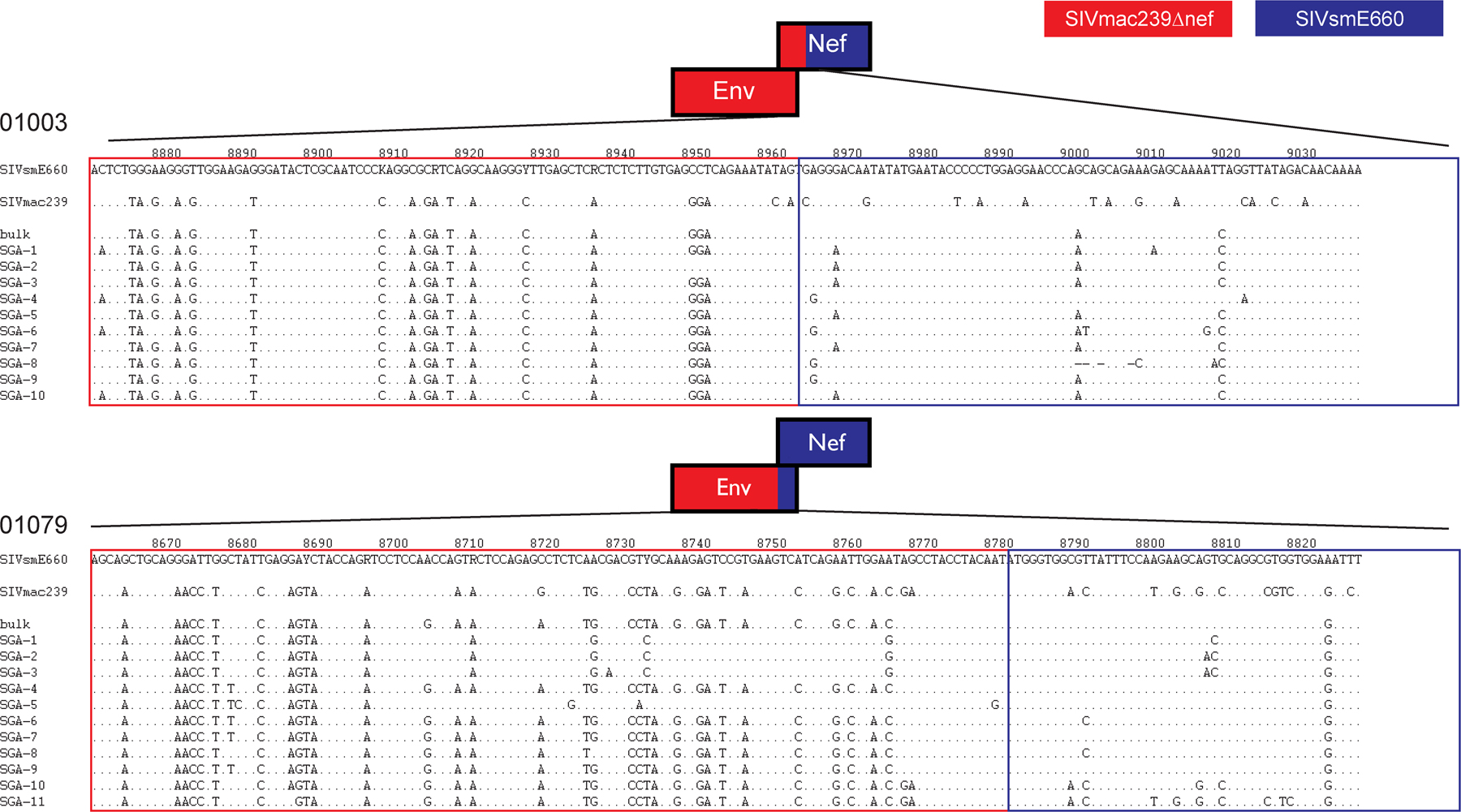{kind=link}
{kind=link}