

Abstract
Ligands from dying cells are a source of Toll-like receptor (TLR) activating agents. Although TLR3 is known to respond to RNA from necrotic cells, the relative importance of this response in vivo during acute inflammatory processes has not been fully explored. We observed the involvement of TLR3 activation during experimental polymicrobial septic peritonitis and ischemic gut injury in the absence of an exogenous viral stimulus. In TLR3-deficient mice, increased chemokine/cytokine levels and neutrophil recruitment characterized the initial inflammatory responses in both injury models. However, the levels of inflammatory chemokines and tumor necrosis factor α quickly returned to baseline in tlr3−/− mice, and these mice were protected from the lethal effects of sustained inflammation. Macrophages from tlr3−/− mice responded normally to other TLR ligands but did not respond to RNA from necrotic neutrophils. Importantly, an immunoneutralizing antibody directed against TLR3 attenuated the generation of inflammatory chemokines evoked by byproducts from necrotic neutrophils cultured with wild-type macrophages. In vivo, anti-TLR3 antibody attenuated the tissue injury associated with gut ischemia and significantly decreased sepsis-induced mortality. Collectively, these data show that TLR3 is a regulator of the amplification of immune response and serves an endogenous sensor of necrosis, independent of viral activation.
The innate immune system provides a first line of defense via the detection and elimination of pathogens. Pathogen-associated molecular patterns (PAMPs) are recognized by Toll-like receptors (TLRs), which activate inflammatory pathways leading to de novo cytokine and chemokine generation. When innate immunity is not appropriately regulated or the pathogen not contained, local and systemic inflammation can lead to severe pathophysiologic consequences (1, 2). Factors released from stressed or damaged tissues serve as “danger signals,” and these might provide the innate immune system with activation ligands (3). “Endogenous ligands” are potent activators of the innate immune response (4–7), and the TLRs appear to be the target of these factors. In this regard, TLR3 activation aggravates and/or potentiates preexisting inflammation associated with the kidney (8), rheumatic synovium (9), and gastrointestinal tract (10, 11). In addition, TLR3 ligand amplifies the systemic hyperinflammatory response observed during sepsis (12). Although TLR3 recognizes viral double-stranded RNA, recent studies have suggested that RNA released from either damaged tissue or contained within endocytosed cells might serve as a ligand for TLR3 (13). Whether TLR3 functions in this manner during inflammatory responses in vivo is presently unknown.
This study addressed the hypothesis that TLR3 regulates the inflammatory response during polymicrobial septic peritonitis and ischemic bowel injury. In the absence of an exogenous viral pathogen, TLR3 activation was important for the initiation and the amplification of inflammation via recognition of cellular by-products from necrotic, but not apoptotic, cells. Interestingly, tlr3−/− mice were protected from the lethal effects of total cecal ligation alone and cecal ligation and puncture (CLP)–induced experimental sepsis. In addition, antibody-mediated immunoneutralization of TLR3 activation suppressed the tissue injury mediated by necrosis of the gut and sepsis-induced mortality in WT mice. Collectively, these data demonstrate that TLR3 regulates amplification events during inflammation mediated by nonviral mechanisms.
RESULTS
Up-regulation of TLR3 in macrophages and neutrophils after sepsis
PAMPs activate TLR-induced signaling pathways, leading to the induction of innate immune responses. A mixed pathological picture is associated with CLP, which includes the elicitation of various leukocyte populations, vascular permeability changes, and significant necrosis of the gut tissue (14). This latter pathology may hold an important key in understanding endogenous signals that amplify the innate response, as growing evidence has suggested that host-derived RNA (9, 13) from necrotic cells serve as major stimuli for TLR3 activation. To assess the expression pattern of TLR3 during the development of CLP sepsis, we initially localized the expression of TLR3. Although peritoneal macrophages and neutrophils recovered from the sham WT mice did not express TLR3, the expression of TLR3, via immunohistochemistry, was detected in these cell populations from mice undergoing experimental sepsis induced by CLP (Fig. 1 A). As a control, no expression of this receptor was detected in thioglycollate-elicited peritoneal macrophages from tlr3−/− mice (Fig. 1 B), confirming the specificity of this antibody. At 24 h, CLP mice showed increased intracellular expression of TLR3 in peritoneal and splenic CD11b+ cells (Fig. 1, C and D, respectively) when compared with sham mice. Surprisingly, CLP also induced the extracellular expression of TLR3 in CD11b+ cells isolated from peritoneal lavage (Fig. 1 E). No statistical differences were observed at the extracellular expression of this molecule in spleen cells from CLP and sham surgery mice (Fig. 1 F).
Figure 1.

CLP surgery induced local and systemic expression of TLR3. (A) Representative immunochemistry analysis of TLR3 expression in peritoneal cells from sham and CLP 6 h after surgery. TLR3 expression is observed in brown. Bars, 20 μm. (B) Immunochemistry analysis of TLR3 expression in thioglycollate-elicited macrophages of tlr3−/− mice. Bar, 20 μm. (C and D) Histograms exhibit TLR3 expression in permeabilized peritoneal (C) and splenic (D) CD11b+ cells. (E and F) Nonpermeabilized peritoneal (E) and splenic (F) CD11b+ cells (gray line, isotype control antibody; shaded, sham mice; black line, CLP mice). Data are means ± SEM from three independent experiments. (G) Immunoreactive CCL3 and MIP-2 supernatant levels from thioglycollate-elicited WT and tlr3−/− peritoneal neutrophils exposed to poly(I:C). *, P < 0.05 compared with sham mice; §, P < 0.05 compared with WT mice.
Given that we observed the presence of TLR3+ neutrophils in cytospins of peritoneal cells from septic mice (Fig. 1 A), we further evaluated the effects of polyriboinosinic-polyribocytidylic acid (poly(I:C)) on chemokine generation by purified peritoneal neutrophils (see Materials and methods). As shown in Fig. 1 G, poly(I:C) promoted the release of CCL3 and macrophage inflammatory protein 2 (MIP-2) from WT neutrophils but not from tlr3−/− neutrophils. Thus, peritoneal inflammatory stimuli promoted the expression of functional TLR3 in macrophages and neutrophils.
The absence of TLR3 enhanced the influx of neutrophils, but suppressed necrosis and apoptosis, during experimental peritonitis
The contribution of TLR3 to septic peritonitis in WT and tlr3−/− mice after sham or CLP surgery is summarized in Fig. 2. A significantly greater number of peritoneal cells were present in the tlr3−/− group compared with the WT group at 24 h after CLP surgery (Fig. 2 A). Significantly higher peritoneal levels of myeloperoxidase (MPO; a specific marker of neutrophil recruitment) were also present in tlr3−/− mice compared with WT mice (Fig. 2 B). Moreover, peritoneal washes from septic mice were characterized by considerable necrotic cellular debris and bacterial contamination, whereas similar washings from septic tlr3−/− mice contained greater numbers of neutrophils, little necrotic cellular debris, and fewer bacteria (Fig. 2 C). It is worth noting that although peritoneal neutrophil numbers were significantly greater in the tlr3−/− group compared with the WT group after 24 h following CLP surgery, neutrophil numbers returned to equivalent levels in the peritoneal cavity in the surviving WT and knockout groups at 72 h after CLP (unpublished data). Consistent with the decreased presence of bacteria contamination in peritoneal washes from the tlr3−/− group at 24 h after CLP surgery, we observed that bacteria were not detected in the peritoneal fluid and blood in any of the tlr3−/− mice, whereas WT mice exhibited 104–106 log bacteria/CFU/μl of peritoneal wash and 105 log bacteria/CFU/μl of blood.
Figure 2.

Increased peritoneal neutrophil influx and a marked reduction in necrotic peritoneal cells during septic peritonitis in tlr3−/− mice. (A) WT and tlr3−/− peritoneal cells at 24 h after sham and CLP surgeries. (B) MPO levels in peritoneal lavage samples at 24 h after either sham or CLP surgery. Data are means ± SEM from two separate experiments. (C) Toluidine blue staining of peritoneal cells from WT and tlr3−/− mice at 24 h after CLP surgery. Bars, 50 μm. (D and E) Apoptotic and necrotic cells were identified by annexin V+/PI− and annexin V+/PI+ staining, respectively. The data represent means ± SEM (n = 5 mice per condition). *, P < 0.05; and **, P < 0.01 compared with C57BL/6 mice after CLP surgery.
The increased peritoneal neutrophilia in septic tlr3−/− mice at 24 h after surgery might be explained by an enhanced responsiveness of immune cells from these mice to other bacterial PAMPs, including TLR4 and 9 agonists. Both TLRs were increased in whole spleen from septic tlr3−/− mice relative to identical spleen samples from septic WT mice (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20081370/DC1). Consistent with this observation, isolated peritoneal neutrophils released significantly greater levels of CXCL10 after exposure to synthetic oligodeoxynucleotide (CpG-ODN) and the chemokine KC after exposure to LPS (Fig. S1 B). Also, the marked absence of bacteria in peritoneal washes from the knockout group might be explained by our observation that tlr3−/− neutrophils exhibited a marked increase in basal reactive oxygen intermediate generation compared with WT neutrophils (unpublished data).
FACS analysis confirmed the marked absence of necrotic cells in the peritoneal cavity of septic tlr3−/− mice. At 24 h, 46 ± 7% of peritoneal cells from septic mice were necrotic and ∼22 ± 1% of peritoneal cells in septic tlr3−/− mice were necrotic (Fig. 2 D), and this difference reached statistical significance. Between 4 and 7% of peritoneal cells in both septic groups were apoptotic (Fig. 2 E). Collectively, these findings demonstrate that TLR3 regulates the amplification of acute inflammatory response evoked by a septic peritoneal challenge.
TLR3 activation contributes to organ injury induced by septic peritonitis and gut ischemia
To evaluate the contribution of TLR3 activation on the mortality induced by septic peritonitis, we monitored survival rates in tlr3−/− and WT mice for 8 d after CLP. The tlr3−/− animals were significantly protected from sepsis-induced lethality (Fig. 3 A). The data show that ∼80% of WT mice were dead at 4 d after CLP surgery, whereas ∼80% of tlr3−/− mice were alive at day 8 after surgery. Mortality in the CLP model is caused by multiorgan failure (14), including ischemic injury to the bowel. In addition to the septic injury, we examined the role of TLR3 in ischemic injury to the gastrointestinal tract induced by complete ligation of the cecum. As shown in Fig. 3 B, complete ligation of the cecum without punctures for 24 h in WT mice caused severe necrotic injury to this portion of the bowel, characterized by the near total loss of tissue architecture and integrity. In contrast, the same procedure in tlr3−/− mice was associated with markedly less destruction to the architecture of the cecum and the cell infiltrate, and the contents of the cecum were still apparent in this organ, suggesting that reduced intralumen of its contents had leaked into the peritoneal cavity. Compared with WT mice, tlr3−/− mice showed significantly reduced levels of alanine aminotransferase, aspartate aminotransferase (AST), bilirubin, and lactic acid dehydrogenase (LDH) after 6 and 24 h following CLP surgery, and at 24 h after total cecal ligation (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20081370/DC1). Collectively, these findings are consistent with the concept that TLR3 activation contributes to the magnitude of the inflammatory response evoked by both septic and necrotic processes in the damaged gastrointestinal tract.
Figure 3.

tlr3−/− mice are resistant to lethality after CLP surgery and exhibit less tissue damage after gut ischemia. (A) Septic peritonitis was induced by CLP in WT (n = 20) and tlr3−/− (n = 17) mice. Mortality was monitored every 24 h for 8 d. ***, P < 0.0001 compared with the WT group. (B) Representative Masson's trichrome staining of cecum at 24 h after gut ischemia induced by total cecal ligation (n = 6). Bars, 100 μm.
TLR3 amplified local and systemic inflammatory responses
We next assessed whether the generation of inflammatory cytokines and chemokines differed between WT and tlr3−/− mice during septic inflammatory responses in the peritoneal cavity. At 6 h after CLP surgery, peritoneal levels of IFN-β were significant reduced in the tlr3−/− group compared with the WT group (Fig. 4 A). The peritoneal levels of CCL5 (Fig. 4 B) and CXCL10 (Fig. 4 C) were higher in the tlr3−/− group compared with the WT group at this time, but levels of TNF-α (Fig. 4 D), CCL3 (Fig. 4 E), and MIP-2 (Fig. 4 F) were similar in the two groups of mice at 6 h after CLP. At 24 h after CLP surgery, all peritoneal levels of the cytokines and chemokines were significantly reduced in the tlr3−/− CLP group versus the WT CLP group (Fig. 4, A–F).
Figure 4.

Acute but not sustained peritoneal inflammation in tlr3−/− mice after CLP or gut ischemia. (A–F) At 6 and 24 h after CLP surgery, WT and tlr3−/− peritoneal cytokine and chemokine concentrations were measured by ELISA. (G) WT and tlr3−/− peritoneal cytokine and chemokine concentrations were measured by ELISA. The data represent means ± SEM obtained from three independent experiments for CLP and two independent experiments for gut ischemia. *, P < 0.05 compared with WT groups. nd, not detected.
Ischemia of the cecum was also associated with significant increases in inflammatory chemokines and cytokines, including CCL3, CCL5, and TNF-α. Analysis of peritoneal lavage from cecal-ligated mice revealed that all three of these inflammatory mediators were significantly attenuated at 24 h after surgery in tlr3−/− mice when compared with WT mice (Fig. 4 G).
Multiorgan injury caused by septic inflammatory responses was significantly attenuated in tlr3−/− mice
Our data demonstrate that the absence of TLR3 had a dramatic regulatory effect on the magnitude and duration of the inflammatory response in the peritoneal cavity; therefore, we next assessed whether this modulatory effect extended to other organs. Most notably, whole lung and spleen levels of CXCL10, CCL3, and CCL2 were significantly lower in septic tlr3−/− mice when compared with septic WT mice (Fig. 5, A–C). Collectively, these data indicated that the inflammatory response in tlr3−/− mice after a septic inflammatory stimulus was restrained and regulated in a manner that markedly decreased the degree of inflammation in other organs in these mice.
Figure 5.

TLR3 amplified systemic inflammatory responses and secondary tissue injury after septic peritonitis. (A–C) At 24 h after sham and CLP surgeries, chemokines were measured in lung and spleen homogenates using ELISA. Data are means ± SEM from two independent experiments (n = 4–5 mice per experiment). *, P < 0.05; and **, P < 0.01 compared with WT groups.
Absence of signaling through TLR3 blocked the activation of macrophages by necrotic neutrophils
We addressed the possibility that the lack of amplification of the inflammatory response in tlr3−/− mice after injury was caused by their inability to respond to byproducts from necrotic cells. Accordingly, we examined the role of TLR3 in the generation of inflammatory chemokines by isolated peritoneal WT and tlr3−/− macrophages after their co-culture with necrotic polymorphonuclear neutrophils (PMNs) or apoptotic splenocytes. In cultures of WT macrophages, the addition of necrotic neutrophils (97% of cells were propidium iodide+ (PI+), as shown in Fig. 6 A) for 24 h significantly increased the levels of CCL5 (Fig. 6 B), MIP-2 (Fig. 6 C), CCL3 (Fig. 6 D), KC (Fig. 6 E), and CXCL10 (Fig. 6 F) above control (medium alone) levels. In marked contrast, the addition of necrotic neutrophils to cultures of tlr3−/− macrophages did not induce the expression of these chemokines (Fig. 6, B–F). However, chemokine responses evoked by the addition of apoptotic spleen cells (depicted) or neutrophils (not depicted) did not appear to be TLR3 dependent (Fig. 6, B–F).
Figure 6.

TLR3 was required for chemokine generation by peritoneal macrophages after co-culture with necrotic but not apoptotic cells. (A) The necrotic cells were identified by analyzing the side scatter (SSC) and PI+ staining. (B–F) Chemokine concentrations in the supernatants of WT or tlr3−/− peritoneal macrophages co-cultured with necrotic neutrophils, apoptotic splenocytes, poly(I:C), CpG-ODN, Pam3Cys, or LPS and quantified using ELISA and/or Bio-Plex. The data are means ± SEM from three to four combined experiments. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 compared with tlr3−/− mice. (G and H) KC (G) and MIP-2 (H) protein levels in 96-well tissue culture plates containing WT or tlr3−/− macrophages and one of untreated, RNase-, or Benzonase-treated necrotic PMNs. **, P < 0.01 when necrotic PMNs were compared with medium; #, P < 0.05 when enzyme-treated necrotic PMNs were compared with nontreated necrotic PMNs.
Further analysis of cytokine and chemokine generation by cultured peritoneal WT and tlr3−/− macrophages confirmed that the latter group of macrophages was significantly less responsive to poly(I:C) activation, but responses to this synthetic ligand were still present in TLR3-deficient cells, as has been shown previously (15). We also observed that tlr3−/− macrophages generated significantly greater levels of CCL3 after TLR9 activation via hypomethylated DNA (i.e., CpG-ODN) for 24 h compared with WT macrophages (Fig. 6 D). However, activators of macrophages derived from necrotic/apoptotic cells, namely vimentin (16) and high mobility group box 1 (HMGB1) protein (17), activated WT and tlr3−/− macrophages to the same extent (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20081370/DC1).
Because RNA released from damaged tissue might serve as a ligand for TLR3 (13), we next explored the possibility that RNA from necrotic PMNs was the signal from these cells that drove chemokine synthesis in peritoneal macrophages. To further explore this possibility, we added RNase or Benzonase to necrotic PMN samples, and RNA quantification showed that both of these enzyme treatments reduced the concentration of nucleic material within these samples by ∼50% or more compared with untreated PMN samples. As shown in Fig. 6 (G and H), the presence of RNase or Benzonase did not evoke the release of the chemokines KC (Fig. 6 G) or MIP-2 (Fig. 6 H) to the same extent as untreated necrotic PMN samples. Further, we again observed that necrotic PMNs failed to evoke KC or MIP-2 synthesis by tlr3−/− macrophages, and the addition of RNase- or Benzonase-treated PMN samples to these cells did not change chemokine levels in culture. Collectively, these data demonstrate that nucleic acid products from injured and/or necrotic cells contribute to the inflammatory response evoked in macrophages via TLR3 ligation.
Antibody-mediated immunoneutralization of TLR3 inhibited WT macrophage chemokine synthetic responses to the presence of necrotic neutrophils
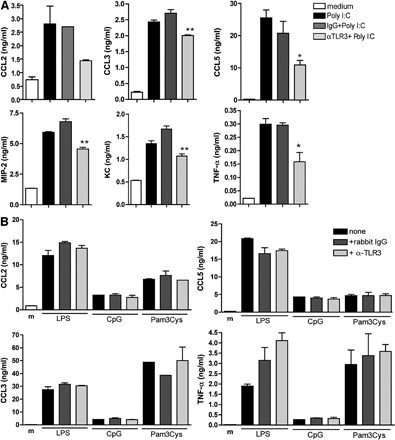
Using Western blot analysis, we examined the specificity of our polyclonal antibody directed against mouse TLR3. As shown in Fig. 7 A, this antibody detected recombinant TLR3 protein (a band at ∼140 kD), as well as in poly(I:C)-stimulated macrophages from WT (lane 1) and tlr7−/− (lane 2) mice, but not in tlr3−/− (lane 3) macrophages (Fig. 7 A). As would be expected given the polyclonal nature of this antibody, other bands were detected in the macrophage samples from the WT and tlr7−/− mice, but it appeared that these additional bands (∼60–80 kD) were specific for TLR3 because they were not detected by this antibody in tlr3−/− macrophages. Next, a functional analysis of TLR3 signaling was performed in purified WT peritoneal macrophage cultures exposed to necrotic neutrophils or stimulated by poly(I:C) (Fig. 7 B and Fig. S4 [available at http://www.jem.org/cgi/content/full/jem.20081370/DC1], respectively). After exposure to poly(I:C) alone or poly(I:C) and control IgG for 24 h, CCL2, CCL3, CCL5, MIP-2, KC, and TNF-α were prominently expressed in supernatants from these cultures. In contrast, the presence of anti-TLR3 antibody significantly reduced the chemokine generation by poly(I:C)-activated peritoneal macrophages (Fig. S4 A). However, the anti-TLR3 antibody did not have any effect on CCL2, CCL3, CCL5, and TNF-α production induced by LPS, CpG-DNA, and PamCys3 (Fig. S4 B).
Figure 7.

TLR3 was required for chemokine generation by WT peritoneal macrophages after co-culture with necrotic neutrophils. (A) The specificity of polyclonal rabbit anti-TLR3 was determined by Western blot analysis of TLR3 expression in the following samples: lane 1, WT macrophages; lane 2, tlr7−/− macrophages; lane 3, tlr3−/− macrophages; and lane 4, recombinant TLR3. The black line indicates that intervening lanes have been spliced out. (B-E) Chemokine concentrations in the supernatants of WT peritoneal macrophages co-cultured with necrotic neutrophils with IgG or anti-TLR3 polyclonal antibody were quantified using Bio-Plex. The data are means ± SEM of triplicate wells and are representative of three independent experiments. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 compared with IgG group.
We also examined the relative effect of endosomal acidification via chloroquine treatment on the neutralizing ability of this anti-TLR3 antibody. In cultures of peritoneal macrophages from thioglycollate-challenged mice stimulated with poly(I:C) in the presence of IgG or anti-TLR3 antibody, the presence of 10 μM chloroquine did not markedly impair the neutralizing activity of anti-TLR3 antibody when compared with IgG treatment (Fig. S5 A, available at http://www.jem.org/cgi/content/full/jem.20081370/DC1). These data suggested that activity of this antibody might not be restricted to TLR3 blockade in the endosome. In addition, the presence of chloroquine did not alter the effects of anti-TLR3 antibody on the synthesis of CCL5, CCL3, KC, and MIP-2 (Fig. S5 A). As a control, the production of chemokines by LPS-activated macrophages, which depends of TLR4, is neither affected by chloroquine or anti-TLR3 blockade (Fig. S5 B). These data confirmed that this TLR3-directed antibody specifically alters the activating effects of a known TLR3 ligand.
In additional in vitro studies, WT necrotic neutrophils were co-cultured with WT peritoneal macrophages in a 1:1 ratio with either control IgG, anti–IFN-β antibody, or anti-TLR3 antibody. The presence of necrotic neutrophils and IgG significantly increased the synthesis of MIP-2 (Fig. 7 B), CCL5 (Fig. 7 C), CCL3 (Fig. 7 D), and KC (Fig. 7 E) by WT macrophage. The inclusion of an antibody directed against IFN-β in this co-culture system resulted in chemokine generation that was similar to that observed in co-cultures with IgG (unpublished data). However, the addition of anti-TLR3 antibody to this cell co-culture system significantly decreased levels of all four chemokines (Fig. 7, B–E). Thus, these data showed that the activation of peritoneal macrophages by necrotic neutrophils was dependent on TLR3.
Antibody-mediated immunoneutralization of TLR3 attenuated the tissue damage mediated by gut ischemia and significantly reduced sepsis-induced mortality
Given that the genetic absence of TLR3 provided a dramatic protective and modulatory effect during septic and ischemic inflammatory responses in the peritoneal cavity, we next assessed whether this effect could be achieved through antibody-mediated immunoneutralization of this TLR. We therefore assessed the effect of this neutralizing anti-TLR3 antibody in WT mice subjected to total cecal ligation and CLP. IgG administration before total cecal ligation provided no protective effect, as indicated by severe tissue necrosis and marked disruption of the normal gastrointestinal architecture (Fig. 8 A, inset) observed at 24 h after surgery. In marked contrast, anti-TLR3 antibody administration preserved the histological appearance of cecal tissue analyzed in WT mice that received anti-TLR3 antibody (Fig. 8 B). The protective effect of neutralizing TLR3 receptors in this necrosis model was reflected by decreased levels of AST and LDH serum enzymes (Fig. 8 C). To examine the role of TLR3 during septic responses in WT mice, anti-TLR3 antibody was given to mice according to the following posttreatment regimens: (a) at 3 h before CLP surgery only; (b) 3 and 24 h after CLP surgery; and (c) at 6 and 24 h after CLP surgery. Although anti-TLR3 treatment regimens a and b did not affect the overall survival after CLP, anti-TLR3 treatment regimen c had a marked protective effect on the survival of WT mice after the induction of sepsis (Fig. 8 D). Collectively, these data demonstrate that the beneficial effects of TLR3 gene deficiency can be achieved via an anti-TLR3 antibody–directed approach.
Figure 8.

Anti-TLR3 antibody markedly reduced cecal damage induced by gut ischemia and enhanced survival after the induction of severe sepsis. (A and B) Representative histological sections from WT mice that received either IgG (A) or anti-TLR3 (B) antibody during gut ischemia. The inset in A is a representative histological cross section of cecum from a naive WT mouse. Bars, 100 μm. (C) At 24 h after surgery, serum from WT mice subjected to cecal ischemia and either IgG or anti-TLR3 antibody treatments (n = 4 mice per group) were analyzed for AST and LDH. The data are means ± SEM. *, P < 0.05 compared with the IgG group. (D) WT mice received 3 mg of anti-TLR3 antibody or isotype control at 6 and 24 h after CLP surgery, and the survival rates were monitored for 54 h (n = 7 for the anti-TLR3 antibody group; n = 8 for the IgG group).
DISCUSSION
Initiation and amplification events in the inflammatory response are complex, and the relative role of the TLR system in these events adds to this complexity. The recognition of PAMPs by TLRs has garnered much of the research attention in the elucidation of innate immunity in the context of pathogen exposure. However, growing evidence suggests that TLRs exert a prominent role in the response to endogenous signals provided by the host during injury events (7). In the present study, we examined the relative importance of TLR3, a TLR initially characterized for its responses to double-stranded RNA (15) but more recently for its response to endogenous nucleic acids from necrotic cells (9, 13). We observed that during septic peritonitis, intracellular as well as extracellular TLR3 expression was markedly induced in CD11b+ cells. The absence of TLR3 in mice was protective in the context of both CLP surgery and gut ischemia. Two key features were noted in these injury models. First, the initiation of the inflammatory response in tlr3−/− mice was enhanced compared with appropriate WT controls. Specifically, we observed that neutrophil influx into the peritoneal cavity was significantly increased, and this influx was associated with increased CXC chemokine levels. Second, the inflammatory response was quickly contained within tlr3−/− mice, in large part because of the absence of an amplifying effect of byproducts from necrotic but not apoptotic cells. Importantly, this latter feature in tlr3−/− mice was replicated in WT mice via the use of an immunoneutralizing antibody directed against TLR3. Thus, the absence or the modulation of TLR3 activity has major beneficial effects in mouse models of peritonitis characterized by infection and tissue necrosis.
TLR3 is expressed in DCs (18, 19), astrocytes (20), Schwann cells (21), macrophages (22), glomerular mesangial cells (8, 23), T cells (24), epithelial cells (25, 26), fibroblasts (27), and hepatocytes (11). Although TLR3 is present mainly within the endosomic compartment of these cell types, epithelial cells appear to uniquely express this TLR on their cell surface as well (26); however, we observed, like others (28), that during inflammation macrophages express this receptor on the surface. These results were confirmed in studies in which the presence of chloroquine did not alter the inhibitory effect of the anti-TLR3 antibody during poly(I:C)-induced chemokine synthesis by inflamed macrophages. Viral infection drives TLR3 expression in epithelial cells (29, 30), but TLR3 expression can be driven during chronic inflammation in the absence of virus or viral byproducts (31). Herein, we observed that TLR3 protein was expressed in macrophages and neutrophils after sepsis. The observation that TLR3 was induced in septic mouse neutrophils was novel because this TLR has only been reported in isolated human neutrophils (32). The presence of functional TLR3 in inflamed mouse neutrophils was confirmed by the addition of poly(I:C), which induced CCL3 and MIP-2 in WT but not tlr3−/− neutrophils. Thus, TLR3 expression was induced in macrophages and neutrophils in the absence of a viral stimulus.
Severe sepsis, a lethal syndrome after infection or injury, is the third leading cause of mortality in the United States (2), and it is characterized by multiorgan damage because of the fact that the excessive inflammation directed toward the infectious pathogen precipitates major tissue damage. Several initiation signals direct the migration of mononuclear cells and neutrophils during sepsis, but this response must be tightly regulated to avoid tissue injury (33). In the present study, the absence of TLR3 was associated with an increase in neutrophil recruitment into the peritoneal cavity in mice undergoing CLP and into the wall of the ischemic cecum at the 24-h time point. One explanation for the increased initial inflammatory response in tlr3−/− mice stems from our observation that neutrophils and macrophages generated significantly greater amounts of CXCL10 and KC, two potent neutrophil chemoattractants (34), after exposure to hypomethylated DNA, a ligand for TLR9. Consistent with this explanation, it was observed that significantly greater quantities of CXCL10 were detected in the peritoneal cavities of septic tlr3−/− mice compared with WT mice. Also, at 6 h after CLP both TLR4 and TLR9 were markedly increased in innate immune cells from tlr3−/− mice. The activation of these receptors might account for the increased neutrophil recruitment and effective bacterial killing initially observed in the knockout.
The amplified inflammatory response during peritonitis is characterized by the exuberant presence of cytokines and chemokines, and these mediators contribute directly and indirectly to tissue and organ destruction (33, 35, 36). Previous studies in tlr3−/− mice have noted that these mice exhibit markedly altered cytokine and chemokine generation in response to poly(I:C) (15) or a viral infection (11, 37–40). The intestinal tract appears to be highly susceptible to the damaging effects of double-stranded RNA, leading to a breakdown in the homeostasis in this organ system (10). Recently, Doughty et al. (12) reported that the activation of a TLR3 pathway with poly(I:C) in mice before septic peritonitis caused by CLP surgery significantly augmented the inflammatory response in this model, in part because of the up-regulation of IFN-α/β. The results from the present study expand these previous findings in that tlr3−/− mice were markedly resistant to the lethal effects of CLP. This decreased mortality in tlr3−/− mice was also associated with significantly lower levels of many proinflammatory cytokines (including IFN-β and TNF-α) and chemokines at 24 h after CLP surgery. Similarly, during total cecal ischemia in tlr3−/− mice, cytokine and chemokine levels were significantly reduced compared with WT mice, suggesting that the amplification of the inflammatory cascade was blunted in tlr3−/− mice. Thus, the inflammatory response in tlr3−/− mice was regulated in a manner consistent with tissue preservation.
The factors contributing to poorly regulated inflammatory responses such as those observed in sepsis and gut ischemia are not well understood, but tissue necrosis might provide a major “danger” signal that inappropriately amplifies inflammation (3). In the present study, significantly fewer necrotic cells were observed in tlr3−/− mice after sepsis. Further, the coincubation of necrotic neutrophils with WT macrophages promoted the release of many chemokines, most notably CXCL10. This effect was restricted to co-cultures of necrotic neutrophils and WT macrophages, because the co-culture of necrotic neutrophils and tlr3−/− macrophages failed to show similar elevations in proinflammatory cytokines and chemokines. Tissue necrosis leads to the release of several cellular components, including heat shock proteins, HMGB1, and nucleic acids. Heat shock proteins and HMGB1 have been shown or suggested to be agonists for TLR2 and TLR4 (41), whereas mRNA is an endogenous ligand for TLR3 (13). More recently, Brentano et al. (9) showed that RNA released from necrotic synovial cells activated fibroblasts in the arthritic joint in a TLR3-dependent manner. Also, necrotic neuronal cells were found to induce inflammatory Schwann cell activation via TLR3 (21). Although vimentin and HMGB1 are intracellular proteins, which are released during cell apoptosis and necrosis, respectively, neither factor appeared to activate peritoneal macrophages in a TLR3-dependent manner. However, the treatment of necrotic neutrophils with endonuclease or RNase significantly inhibited the ability of these cells to induce inflammatory chemokine secretion by WT “inflamed” macrophages. These findings lend credence to our hypothesis that RNA released from necrotic cells activates TLR3, leading to the amplification of the inflammatory response. Thus, TLR3 amplifies the inflammatory process in the peritoneal cavity, and its role in this process appears to be related to the detection of RNA from necrotic cells.
The development and characterization of an antibody directed against TLR3 allowed for the examination of TLR3 blockade in in vitro and in vivo experiments. We observed that this antibody recognized TLR3 in WT and tlr7−/− peritoneal macrophages but failed to detect any protein product in tlr3−/− macrophages. Moreover, the anti-TLR3 antibody significantly attenuated the cytokine- and chemokine-inducing effects of poly(I:C) and necrotic neutrophils by WT macrophages. Studies with chloroquine-treated “inflamed” peritoneal macrophages raised the possibility that this antibody worked via the blockade of TLR3 at the cell surface, but we cannot exclude the possibility that the neutralizing TLR3 antibody was internalized by macrophages via Fc γ receptor–mediated endocytosis. Importantly, the findings in tlr3−/− mice were duplicated in vivo in WT mice using a neutralizing antibody directed against TLR3. Specifically, tissue necrosis was significantly reduced after the administration of this antibody before the induction of gut ischemia. Interestingly, the administration of anti-TLR3 antibody at 6 and 24 h after CLP surgery, but not at earlier times after CLP surgery, had a significant protective effect, which is likely caused by the time interval needed to induce cell necrosis in this model. These findings are consistent with the role of TLR3 in the amplification of the inflammatory response after sepsis. Thus, TLR3 can now be included with the select group of molecules known to contribute to the amplification of the septic response. Other members on this list include HMGB1 (17) and TREM-1 (42).
In summary, tlr3−/− mice showed regulated peritoneal and systemic inflammation after tissue injury in the absence of a viral challenge. Thus, the use of antibody directed against TLR3 might serve as a therapeutic clinical option in the treatment of necrosis-induced inflammatory events.
MATERIALS AND METHODS
Mice.
Specific pathogen-free, male C57BL/6 (WT) mice (aged 6–8 wk; Taconic) were housed at the University of Michigan. tlr3−/− mice, backcrossed 10 generations onto a C57BL/6 background, were originally provided by R. Flavell (Yale University School of Medicine, New Haven, CT); a breeding colony of tlr3−/− mice was established at the University of Michigan. tlr7−/− mice were provided by G. Nuñez (University of Michigan Medical School, Ann Arbor, MI). The Animal Use Committee at the University of Michigan approved all in vivo protocols used in this study. Age- and sex-matched mice were used in these studies.
Models.
Thioglycollate-induced sterile peritonitis was induced by the i.p. injection of 2% thioglycollate (Sigma-Aldrich).
For polymicrobial sepsis, WT and tlr3−/− mice were subjected to sham or CLP surgery, as previously described in detail (43). CLP and sham mice were anesthetized, bled, and killed at 6 and 24 h after surgery; lung, spleen, and peritoneal cells were collected. Peritoneal lavage fluids were harvested, and these fluids and peripheral blood from each mouse were serially diluted and plated on brain heart infusion agar and incubated overnight at 37°C, and bacteria colonies were counted. Cell-free peritoneal fluids were used for chemokine, cytokine, and MPO detection by ELISA. Single-cell peritoneal suspensions were stained with Toluidine blue (Thermo Fisher Scientific). In separate experiments, WT and tlr3−/− CLP groups were monitored for survival over 8 d.
Total ischemic bowel injury was induced by the complete ligation of the cecum in WT and tlr3−/− mice. Cecums were exteriorized as described, but not punctured with a needle, and were returned to the peritoneal cavity, and the incision was closed with a surgical staple. At 24 h, the peritoneal cavity was flushed with saline, blood was drawn for clinical chemistry, and the cecum was collected for histology.
Generation of rabbit anti–mouse polyclonal TLR3-specific antibody.
TLR3 antibody was generated in New Zealand white rabbits with recombinant mouse TLR3 (carrier free; R&D Systems), as previously described in detail (44). This antibody was tittered by ELISA against TLR3 coated onto 96-well plates. Serum from nonimmunized rabbits was used for a control treatment group.
Flow cytometry.
Splenic and peritoneal cells from WT mice were analyzed for TLR3 at 24 h after surgery. For intracellular staining, Fc binding was blocked with anti–mouse CD16/32 antibody (eBioscience), and the cells were incubated with label PE–conjugated anti-CD11b. Cells were fixed, permeabilized with Cytofix/Cytoperm (BD Biosciences), incubated with 2 μg of biotinylated polyclonal rabbit anti–mouse TLR3 or biotinylated rabbit IgG, and stained with streptavidin-allophycocyanin. For extracellular staining of TLR3, the cells were not permeabilized. All stained cells were analyzed using a Cytomics FC 500 (Beckman Coulter). Peritoneal cells from both groups of mice were also stained with Alexa Fluor 488–labeled Annexin V and 1 μg/ml PI (both from Invitrogen) for 15 min. Flow cytometry analysis was performed immediately thereafter.
Immunocytochemistry.
Cytospin preparations of peritoneal cells were fixed in a 4% equal mixture of paraformaldehyde and sucrose, and incubated with purified rabbit polyclonal anti-TLR3 antibody or control IgG. The presence of TLR3 was revealed using a cell and tissue staining and horseradish peroxidase–DAB kit (R&D Systems).
Western blot analysis.
Macrophages from WT, tlr3−/−, and tlr7−/− mice stimulated with poly(I:C) for 24 h were lysed, kept on ice for 20 min, and centrifuged at 15,000 g for 15 min. Equal amounts of cell lysates (18 μg) and 10 ng of recombinant mouse TLR3 (R&D Systems) were fractionated by SDS-PAGE (NuPage; Invitrogen), followed by transfer onto nitrocellulose (Invitrogen). After overnight incubation with polyclonal rabbit anti–mouse TLR3 antibody (dilution = 1:1,000), membranes were counterstained with peroxidase-conjugated anti–rabbit IgG and visualized with enhanced chemiluminescence detection reagents (ECL; GE Healthcare). Membranes were stripped in Restore Buffer (Thermo Fisher Scientific) at 50°C for 45 min, blocked with 5% nonfat milk, and reprobed with anti-GAPDH (Abcam), followed by peroxidase-conjugated anti–rabbit IgG. The images were analyzed using ImageJ software (version 1.37; National Institutes of Health).
Proteomic analysis of cytokines, chemokines, and MPO.
Immunoreactive levels of chemokines and cytokines were measured in cultured cell supernatants, peritoneal lavage, lung, and spleen homogenates using a modified double-ligand ELISA assay, as previously described in detail (44, 45), or a cytokine assay (Bio-Plex; Bio-Rad Laboratories). Before each ELISA, snap-frozen lung and spleen samples were thawed on ice and homogenized in a solution containing 2 mg of protease inhibitor (Complete; Boehringer Mannheim) per milliliter of normal saline. Cell-free supernatants from the lung and spleen homogenates were analyzed. MPO activity in peritoneal lavage samples was determined by ELISA (Hycult biotechnology). All cytokine and chemokine levels were normalized to the total protein concentration from tissues and peritoneal lavage, as determined by the Bradford method (Bio-Rad Laboratories).
In vitro macrophage and neutrophil activation.
Macrophages and neutrophils were obtained from thioglycollate-challenged WT and tlr3−/− mice. One of the following stimuli was added to triplicate wells of isolated macrophages and neutrophils: 2.5 μg/ml tri-palmitoyl-S-glyceryl cysteine (Pam3Cys; EMC Microcollections), 10 μg/ml poly(I:C) (GE Healthcare), 1 μg/ml LPS from Escherichia coli 0111:B4 (Sigma-Aldrich), or 2 μM CpG-ODN (Hycult biotechnology). Inflammatory chemokine generation by cultured macrophages and neutrophils was determined by ELISA or Bio-Plex at 24 and 5 h, respectively. In separate experiments, cultures of peritoneal macrophages cultures were co-cultured with necrotic neutrophils or apoptotic splenocytes in the ratio 0.5–1:1 macrophage, vimentin (1, 2, or 5 μg/ml; Cytoskeleton), or recombinant HMGB1 (10 μg/ml; R&D Systems). Necrotic neutrophils were generated from peritoneal neutrophils (73.4% were Gr1highCD11cneg) harvested at 16 h after i.p. thioglycollate injection and incubated at 55°C for 25 min, as previously described (46). Apoptotic splenocytes or neutrophils were generated by the addition of 1 μM staurosporine (Sigma Aldrich) for 6 h. In additional experiments, samples of 106 necrotic PMNs were treated with either 20 μg RNase (DNase and protease free; Roche) or 10 U/106 cells of Benzonase Nuclease (EMD) for 2 h at 37°C. After incubation, the necrotic cells were thoroughly washed and co-cultured with WT macrophages at a 1:1 ratio. Nucleic acid content in all necrotic PMN samples was determined with a RediPlate 96 RiboGreen RNA Quantitation kit (Invitrogen) according to the manufacturer's directions.
Immunoneutralization of TLR3.
Cultures of peritoneal macrophages were exposed to 150 μg/ml anti-TLR3 of control IgG antibody for 1.5 h. Subsequently, TLR2, 3, 4, and 9 ligands or necrotic neutrophils were added. In separate experiments, macrophages were preincubated with 10 μM chloroquine. 2 h later, all cells were washed and incubated with neutralizing anti-TLR3 or IgG antibodies for 90 min. After this incubation, the cultures were stimulated with poly(I:C). In in vivo experiments, control IgG or 1 mg anti-TLR3 antibody per mouse were injected i.p. at 2 h before and 6 h after total cecal ligation. 24 h later, cecums and serum were collected for analysis. In the CLP model, control IgG or anti-TLR3 (3 mg per mouse) were injected i.p. according to one of the following treatment regimens: (a) at 3 h before CLP surgery only; (b) 3 and 24 h after CLP surgery; and (c) at 6 and 24 h after CLP surgery. Survival was monitored for 54 h after the CLP surgery.
Statistics.
Data are expressed as mean ± SEM. The Student's unpaired t test was used to evaluate the differences between WT and tlr3−/− groups. Survival studies were analyzed with the log-rank test. P ≤ 0.05 was considered significant.
Online supplemental material.
Fig. S1 A shows that the mRNA expression of TLR4 and 9 is increased in tlr3−/− mice after CLP. Fig. S1 B shows that the absence of TLR3 in neutrophils increased the responsiveness (i.e., chemokine production) of these cells to TLR4 (LPS). Fig. S2 shows that TLR3 activation increased the levels of AST, bilirubin, and LDH induced by septic peritonitis and gut ischemia. Fig. S3 shows that vimentin as well as HMGB1 proteins are not involved in the activation of TLR3 during acute inflammation. Fig. S4 shows that the polyclonal anti-TLR3 antibody used in the in vitro and in vivo studies attenuated the poly(I:C)-induced chemokine production and did not have any effect in the chemokine production induced by other TLR ligands. Fig. S5 shows that chemokine production mediated after TLR3 activation is not completely restricted by the endosomal acidification with chloroquine.
Supplementary Material
Acknowledgments
We thank Ms. Robin Kunkel for graphic assistance, and Ms. Holly Evanoff, Mr. Aaron Berlin, Dr. Ron Allen, and Ms. Lisa Riggs for technical assistance.
This work was supported in part by National Institutes of Health grants HL31237 and HL31963 (to S.L. Kunkel).
The authors have declared that no competing financial interests exist.
Abbreviations used: AST, aspartate aminotransferase; CLP, cecal ligation and puncture; HMGB1, high mobility group box 1; LDH, lactic acid dehydrogenase; MIP-2, macrophage inflammatory protein 2; MPO, myeloperoxidase; PAMP, pathogen-associated molecular pattern; PI, propidium iodide; PMN, polymorphonuclear neutrophil; poly(I:C), polyriboinosinic-polyribocytidylic acid; TLR, Toll-like receptor.
References
- 1.Pinsky, M.R. 2004. Dysregulation of the immune response in severe sepsis. Am. J. Med. Sci. 328:220–229. [DOI] [PubMed] [Google Scholar]
- 2.Cohen, J. 2002. The immunopathogenesis of sepsis. Nature. 420:885–891. [DOI] [PubMed] [Google Scholar]
- 3.Matzinger, P. 1994. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12:991–1045. [DOI] [PubMed] [Google Scholar]
- 4.Gallucci, S., M. Lolkema, and P. Matzinger. 1999. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5:1249–1255. [DOI] [PubMed] [Google Scholar]
- 5.Sauter, B., M.L. Albert, L. Francisco, M. Larsson, S. Somersan, and N. Bhardwaj. 2000. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 191:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi, Y., W. Zheng, and K.L. Rock. 2000. Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc. Natl. Acad. Sci. USA. 97:14590–14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marshak-Rothstein, A. 2006. Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 6:823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patole, P.S., H.J. Grone, S. Segerer, R. Ciubar, E. Belemezova, A. Henger, M. Kretzler, D. Schlondorff, and H.J. Anders. 2005. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J. Am. Soc. Nephrol. 16:1326–1338. [DOI] [PubMed] [Google Scholar]
- 9.Brentano, F., O. Schorr, R.E. Gay, S. Gay, and D. Kyburz. 2005. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum. 52:2656–2665. [DOI] [PubMed] [Google Scholar]
- 10.Zhou, R., H. Wei, R. Sun, and Z. Tian. 2007. Recognition of double-stranded RNA by TLR3 induces severe small intestinal injury in mice. J. Immunol. 178:4548–4556. [DOI] [PubMed] [Google Scholar]
- 11.Lang, K.S., P. Georgiev, M. Recher, A.A. Navarini, A. Bergthaler, M. Heikenwalder, N.L. Harris, T. Junt, B. Odermatt, P.A. Clavien, et al. 2006. Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J. Clin. Invest. 116:2456–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doughty, L.A., S. Carlton, B. Galen, I. Cooma-Ramberan, C.S. Chung, and A. Ayala. 2006. Activation of common antiviral pathways can potentiate inflammatory responses to septic shock. Shock. 26:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kariko, K., H. Ni, J. Capodici, M. Lamphier, and D. Weissman. 2004. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 279:12542–12550. [DOI] [PubMed] [Google Scholar]
- 14.Bone, R.C., W.J. Sibbald, and C.L. Sprung. 1992. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest. 101:1481–1483. [DOI] [PubMed] [Google Scholar]
- 15.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 16.Mor-Vaknin, N., A. Punturieri, K. Sitwala, and D.M. Markovitz. 2003. Vimentin is secreted by activated macrophages. Nat. Cell Biol. 5:59–63. [DOI] [PubMed] [Google Scholar]
- 17.Wang, H., O. Bloom, M. Zhang, J.M. Vishnubhakat, M. Ombrellino, J. Che, A. Frazier, H. Yang, S. Ivanova, L. Borovikova, et al. 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–251. [DOI] [PubMed] [Google Scholar]
- 18.Muzio, M., D. Bosisio, N. Polentarutti, G. D'Amico, A. Stoppacciaro, R. Mancinelli, C. van't Veer, G. Penton-Rol, L.P. Ruco, P. Allavena, and A. Mantovani. 2000. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J. Immunol. 164:5998–6004. [DOI] [PubMed] [Google Scholar]
- 19.Visintin, A., A. Mazzoni, J.H. Spitzer, D.H. Wyllie, S.K. Dower, and D.M. Segal. 2001. Regulation of Toll-like receptors in human monocytes and dendritic cells. J. Immunol. 166:249–255. [DOI] [PubMed] [Google Scholar]
- 20.Farina, C., M. Krumbholz, T. Giese, G. Hartmann, F. Aloisi, and E. Meinl. 2005. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J. Neuroimmunol. 159:12–19. [DOI] [PubMed] [Google Scholar]
- 21.Lee, H., E.K. Jo, S.Y. Choi, S.B. Oh, K. Park, J.S. Kim, and S.J. Lee. 2006. Necrotic neuronal cells induce inflammatory Schwann cell activation via TLR2 and TLR3: implication in Wallerian degeneration. Biochem. Biophys. Res. Commun. 350:742–747. [DOI] [PubMed] [Google Scholar]
- 22.Jiang, W., R. Sun, H. Wei, and Z. Tian. 2005. Toll-like receptor 3 ligand attenuates LPS-induced liver injury by down-regulation of toll-like receptor 4 expression on macrophages. Proc. Natl. Acad. Sci. USA. 102:17077–17082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wornle, M., H. Schmid, B. Banas, M. Merkle, A. Henger, M. Roeder, S. Blattner, E. Bock, M. Kretzler, H.J. Grone, and D. Schlondorff. 2006. Novel role of toll-like receptor 3 in hepatitis C-associated glomerulonephritis. Am. J. Pathol. 168:370–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tabiasco, J., E. Devevre, N. Rufer, B. Salaun, J.C. Cerottini, D. Speiser, and P. Romero. 2006. Human effector CD8+ T lymphocytes express TLR3 as a functional coreceptor. J. Immunol. 177:8708–8713. [DOI] [PubMed] [Google Scholar]
- 25.Sha, Q., A.Q. Truong-Tran, J.R. Plitt, L.A. Beck, and R.P. Schleimer. 2004. Activation of airway epithelial cells by toll-like receptor agonists. Am. J. Respir. Cell Mol. Biol. 31:358–364. [DOI] [PubMed] [Google Scholar]
- 26.Uehara, A., Y. Fujimoto, K. Fukase, and H. Takada. 2007. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol. Immunol. 44:3100–3111. [DOI] [PubMed] [Google Scholar]
- 27.Rudd, B.D., E. Burstein, C.S. Duckett, X. Li, and N.W. Lukacs. 2005. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J. Virol. 79:3350–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flandin, J.F., F. Chano, and A. Descoteaux. 2006. RNA interference reveals a role for TLR2 and TLR3 in the recognition of Leishmania donovani promastigotes by interferon-gamma-primed macrophages. Eur. J. Immunol. 36:411–420. [DOI] [PubMed] [Google Scholar]
- 29.Sajjan, U.S., Y. Jia, D.C. Newcomb, J.K. Bentley, N.W. Lukacs, J.J. LiPuma, and M.B. Hershenson. 2006. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 20:2121–2123. [DOI] [PubMed] [Google Scholar]
- 30.Le Goffic, R., V. Balloy, M. Lagranderie, L. Alexopoulou, N. Escriou, R. Flavell, M. Chignard, and M. Si-Tahar. 2006. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patole, P.S., R.D. Pawar, M. Lech, D. Zecher, H. Schmidt, S. Segerer, A. Ellwart, A. Henger, M. Kretzler, and H.J. Anders. 2006. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol. Dial. Transplant. 21:3062–3073. [DOI] [PubMed] [Google Scholar]
- 32.Bellocchio, S., S. Moretti, K. Perruccio, F. Fallarino, S. Bozza, C. Montagnoli, P. Mosci, G.B. Lipford, L. Pitzurra, and L. Romani. 2004. TLRs govern neutrophil activity in aspergillosis. J. Immunol. 173:7406–7415. [DOI] [PubMed] [Google Scholar]
- 33.Takala, A., I. Nupponen, M.L. Kylanpaa-Back, and H. Repo. 2002. Markers of inflammation in sepsis. Ann. Med. 34:614–623. [DOI] [PubMed] [Google Scholar]
- 34.Schuh, J.M., K. Blease, and C.M. Hogaboam. 2002. CXCR2 is necessary for the development and persistence of chronic fungal asthma in mice. J. Immunol. 168:1447–1456. [DOI] [PubMed] [Google Scholar]
- 35.Decker, T., M. Muller, and S. Stockinger. 2005. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 5:675–687. [DOI] [PubMed] [Google Scholar]
- 36.Matsukawa, A., C.M. Hogaboam, N.W. Lukacs, and S.L. Kunkel. 2000. Chemokines and innate immunity. Rev. Immunogenet. 2:339–358. [PubMed] [Google Scholar]
- 37.Wang, T., T. Town, L. Alexopoulou, J.F. Anderson, E. Fikrig, and R.A. Flavell. 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10:1366–1373. [DOI] [PubMed] [Google Scholar]
- 38.Rudd, B.D., J.J. Smit, R.A. Flavell, L. Alexopoulou, M.A. Schaller, A. Gruber, A.A. Berlin, and N.W. Lukacs. 2006. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J. Immunol. 176:1937–1942. [DOI] [PubMed] [Google Scholar]
- 39.Gowen, B.B., M.H. Wong, K.H. Jung, A.B. Sanders, W.M. Mitchell, L. Alexopoulou, R.A. Flavell, and R.W. Sidwell. 2007. TLR3 is essential for the induction of protective immunity against Punta Toro Virus infection by the double-stranded RNA (dsRNA), poly(I:C12U), but not Poly(I:C): differential recognition of synthetic dsRNA molecules. J. Immunol. 178:5200–5208. [DOI] [PubMed] [Google Scholar]
- 40.Hardarson, H.S., J.S. Baker, Z. Yang, E. Purevjav, C.H. Huang, L. Alexopoulou, N. Li, R.A. Flavell, N.E. Bowles, and J.G. Vallejo. 2007. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 292:H251–H258. [DOI] [PubMed] [Google Scholar]
- 41.Tsan, M.F., and B. Gao. 2004. Endogenous ligands of Toll-like receptors. J. Leukoc. Biol. 76:514–519. [DOI] [PubMed] [Google Scholar]
- 42.Bouchon, A., F. Facchetti, M.A. Weigand, and M. Colonna. 2001. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 410:1103–1107. [DOI] [PubMed] [Google Scholar]
- 43.Walley, K.R., N.W. Lukacs, T.J. Standiford, R.M. Strieter, and S.L. Kunkel. 1996. Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect. Immun. 64:4733–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evanoff, H.L., M.D. Burdick, S.A. Moore, S.L. Kunkel, and R.M. Strieter. 1992. A sensitive ELISA for the detection of human monocyte chemoattractant protein-1 (MCP-1). Immunol. Invest. 21:39–45. [DOI] [PubMed] [Google Scholar]
- 45.Matsukawa, A., M.H. Kaplanc, C.M. Hogaboam, N.W. Lukacs, and S.L. Kunkel. 2001. Pivotal role of signal transducer and activator of transcription (Stat)4 and Stat6 in the innate immune response during sepsis. J. Exp. Med. 193:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cocco, R.E., and D.S. Ucker. 2001. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol. Biol. Cell. 12:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}