

Abstract
Overexpression of the epidermal growth factor receptor (EGFR) in human papillomavirus type 16-immortalized human keratinocytes (HKc) is caused by the viral oncoprotein E6, which targets p53 for degradation. We have previously observed that expression of p53 RNAi in normal HKc is associated with an increase in EGFR mRNA and protein. We now report that p53 RNAi induces EGFR promoter activity up to approximately 10-fold in normal HKc, and this effect does not require intact p53 binding sites on the EGFR promoter. Exogenous wild-type p53 inhibits the EGFR promoter at low levels, and activates it at higher concentrations. Yin Yang 1 (YY1), which negatively regulates p53, induces EGFR promoter activity, and this effect is augmented by p53 RNAi. Intact p53 binding sites on the EGFR promoter are not required for activation by YY1. In addition, Sp1 and YY1 synergistically induce the EGFR promoter in normal HKc, indicating that Sp1 may recruit YY1 as a co-activator. Wild-type p53 suppressed Sp1- and YY1-mediated induction of the EGFR promoter. We conclude that acute loss of p53 in normal HKc induces EGFR expression bya mechanism that involves YY1 and Sp1 and does not require p53 binding to the EGFR promoter.
Keywords: p53, EGFR promoter, HPV16, human keratinocytes, YY1, Sp1
Introduction
In our model system for human papillomavirus type 16 (HPV16)-mediated human cell carcinogenesis, normal human keratinocytes (HKc) immortalized by transfection with HPV16 DNA (HKc/HPV16) progress toward malignancy in a step-wise manner characterized by growth factor independent (HKc/GFI) and differentiation-resistant (HKc/DR) stages (Pirisi et al., 1987, 1988). We have previously shown that epidermal growth factor receptor (EGFR) mRNA and protein levels increase dramatically during in vitro progression of HKc/HPV16, and constitutive tyrosine phosphorylation of the EGFR contributes to the autonomous growth of HKc/GFI (Zyzak et al., 1994). The EGFR plays an important role in cell growth and development (Yarden and Sliwkowski, 2001; Arteaga, 2002) and is overexpressed in many types of cancer, including cervical cancer (Hu et al., 1997; Mathur et al., 2001). The HPV16 E6 oncoprotein is primarily responsible for the increase in EGFR mRNA and protein observed in HKc/HPV16 (Akerman et al., 2001). Recent work in our laboratory demonstrated that E6-mediated p53 degradation is necessary for induction of the EGFR promoter (Bheda et al., manuscript in preparation), and that expression of a p53 RNAi causes an increase in EGFR mRNA and protein comparable to that produced by E6 (Emmel et al., manuscript in preparation). Therefore, we set out to investigate in detail the effects of loss of p53 on EGFR promoter activity in normal HKc.
p53 and the multifunctional transcription regulator Yin Yang 1 (YY1) (Shi et al., 1997; Gordon et al., 2006) exist in a functional balance with each other: YY1 promotes p53 degradation via the proteosomal pathway and, in turn, loss of p53 results in increased levels of YY1 (Gronroos et al., 2004; Sui et al., 2004). YY1 can bind to p53 responsive elements and modulate p53 activation or repression of gene expression (Yakovleva et al., 2004). Using the Patch software (www.generegulation.com), we identified 12 putative YY1 binding sites on the EGFR promoter. Some of these sites overlap the binding sites of other transcription factors such as specificity protein 1 (Sp1) (Xu et al., 1993), activator protein (Ap)2α (Wang et al., 2006), Ap1 (Johnson et al., 2000) and p53 (Deb et al., 1994; Ludes-Meyers et al., 1996).
Sp1 is an important activator of the EGFR promoter (Kageyama et al., 1988). Sp1 recruits YY1 as a co-activator (Lee et al., 1993). Therefore, we asked whether Sp1 and YY1 regulate the EGFR promoter and modulate the EGFR promoter’s response to p53 RNAi. In addition, as p53 can repress Sp1-mediated transcription of some promoters (Innocente and Lee, 2005), we explored the role of exogenous wild-type p53 on Sp1-mediated transactivation of the EGFR promoter.
Our results show that loss of p53 by p53 RNAi induces EGFR promoter activity in normal HKc. This effect does not require intact p53 binding sites on the promoter, and is mediated by YY1 and Sp1, which can also activate the EGFR promoter in a p53-independent manner. We conclude that E6-mediated p53 degradation is the likely cause of EGFR overexpression in HPV16-transformed cells.
Results
RNAi-mediated loss of p53 induces EGFR promoter activity in normal HKc, but not in C33A cells
Four different individual normal HKc strains and C33A cervical carcinoma cells were co-transfected with a pGL3-EGFR promoter construct (containing nucleotides -1100 to -19 of the EGFR sequence) along with increasing amounts of the pSuper p53RNAi-1 construct, a commercially available p53 RNAi targeting position 1026-1045 in p53 mRNA, or the control pSuper vector. C33A are HPV-negative and express a mutant p53 devoid of DNA binding activity (Kilic et al., 1999). p53RNAi-1 produced a dose-dependent increase in luciferase activity driven by the EGFR promoter in normal HKc, but not in C33A (Figure 1a).
Figure 1.
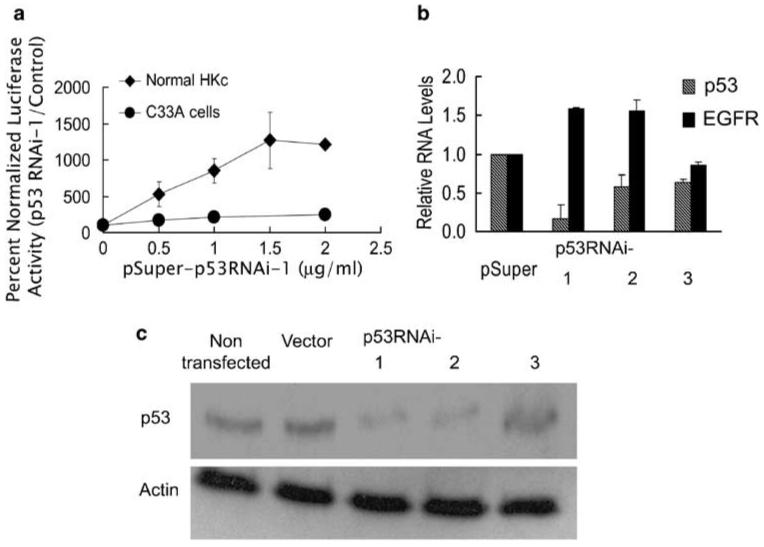
Expression of a p53 RNAi results in induction of EGFR promoter activity in normal HKc. (a) EGFR promoter activity in normal HKc (◆) and C33A (●) in the presence of p53 RNAi. Normal HKc and C33A were transiently transfected with the pGL3-EGFR promoter construct (0.5 μg per well), pRL-TK (50 ng per well) and increasing concentrations of p53RNAi-1 or control pSuper plasmid. The total amount of DNA in the transfection mix was brought to 2.5 μg per well by addition of pSuper plasmid. Cells were harvested 48 h post-transfection for dual-luciferase assays. Data represent the averages±s.d. of four separate determinations using different normal HKc strains. (b) p53 and EGFR mRNA levels in normal HKc expressing p53 RNAi. Normal HKc were transfected with 5 μg per 60-mm dish of p53RNAi-1, p53RNAi-2, p53RNAi-3 or pSuper control plasmid and 1 μg per dish of pLXSN plasmid. Cells were selected with G418 (100 μgml-1) for a week, RNA was extracted and real-time RT-PCR was performed. The p53 and EGFR mRNA levels in the p53RNAi-transfected cells were normalized to RPLPO and expressed as a fold change compared with control (pSuper). Data represent averages±s.d. of triplicate determinations. (c) p53 protein levels in normal HKc expressing p53 RNAi. p53 or actin protein levels were detected by western blot analysis in extracts from normal HKc transfected with the various p53 RNAi constructs as described in (b). EGFR, epidermal growth factor receptor; HKc, human keratinocytes; RNAi, RNA interference; RPLPO, ribosomal protein large protein O; RT-PCR, reverse transcription-PCR.
To confirm that the RNAi caused specific inhibition of p53, we constructed two additional p53 RNAi constructs targeting two different positions on the p53 mRNA: position 220-239 (p53RNAi-2) and position 1588-1607 (p53RNAi-3). We co-transfected each p53 RNAi construct into normal HKc, along with a neomycin-selectable plasmid (pLXSN), selected with G418 for 7 days, and then determined p53 and EGFR mRNA levels by real-time RT-PCR (reverse transcription-PCR) (Figure 1b) and p53 protein levels by western blot analysis (Figure 1c). p53RNAi-1 produced a significant decrease in p53 mRNA, whereas p53RNAi-2 only slightly decreased p53 mRNA levels (Figure 1b). However, both p53RNAi-1 and p53RNAi-2 caused a decrease in p53 protein levels (Figure 1c). p53RNAi-3 decreased p53 RNA to an extent but did not have much of an effect on p53 protein levels. The two p53 RNAi constructs that caused a decrease in p53 protein levels (p53RNAi-1 and p53RNAi-2) also produced an increase in EGFR mRNA, whereas p53RNAi-3, unable to decrease p53 protein levels, did not induce EGFR mRNA (Figure 1b). Similar to p53RNAi-1 (Figure 1a), p53RNAi-2 activated the EGFR promoter in transient transfection assays of normal HKc (data not shown). These experiments suggest that a loss of endogenous p53 activates the EGFR promoter and increases EGFR mRNA levels in normal HKc.
Mutations in the p53 binding sites of the EGFR promoter differentially alter promoter basal activity, but do not abolish induction by p53 RNAi
The EGFR proximal promoter region contains three p53 binding elements. The p53 binding site contained between nucleotides -265 and -239 is particularly well characterized (Deb et al., 1994; Ludes-Meyers et al., 1996). We will refer to this site as p53 binding site 1. The two additional p53 binding sites reside in the region -167/-105 (Sheikh et al., 1997). We will refer to these as p53 binding sites 2 (distal) and 3 (proximal to the translation start site). Figure 2a shows the exact location of the three p53 binding sites on the human EGFR promoter. Double-stranded oligonucleotides spanning each of these three p53 binding sites were constructed, along with oligonucleotides carrying mutations in key positions within the p53 responsive elements (Figure 2a; Materials and methods). To confirm that the mutations in the p53 binding sites of the EGFR promoter abolished p53 binding, we conducted electrophoretic mobility shift assays using nuclear extracts from UV-treated normal HKc or C33A (negative control). The two-nucleotide change in the p53 binding site 1 completely abolished p53 binding (Figure 2b). The two oligonucleotides containing mutated p53 binding sites 2 and 3 produced similar results (data not shown). We then performed site-directed mutagenesis in the parental pGL3-EGFR promoter plasmid, producing pGL3-EGFR promoter plasmids carrying one, two or all three mutated p53 binding sites. Mutations of the p53 binding sites on the EGFR promoter affected EGFR promoter basal activity in normal HKc, with different effects depending on the specific site mutated and the number of sites mutated. Individual site mutations reduced basal promoter activity to some extent, with the p53 binding site 2 mutant causing the greatest decrease, about 50% (Figure 2c). Of the double mutants, the mutant 2, 3 had no effect, 1, 3 slightly increased EGFR promoter activity and 1, 2 caused a 50% reduction. Only the 1, 2, 3 triple mutant showed a dramatic reduction (over 80%) in promoter basal activity. Next, we asked whether the different mutations of the p53 binding sites on the EGFR promoter would affect induction of the promoter by p53 RNAi. We transfected normal HKc with the p53RNAi-1 construct or control pSuper vector, along with the wild type or mutant pGL3-EGFR promoter constructs. None of the single or double p53 binding site mutants abolished promoter induction by p53RNAi-1 (Figure 2d). Induction by p53RNAi-1 of the double mutant 1, 2 was about twice that of the wild type, while induction of the triple p53 binding site pGL3-EGFR mutant was about four times greater than that of the wild-type pGL3-EGFR promoter (Figure 2d). Hence, despite the fact that the basal activity of the triple mutant was about fourfold lower than that of the wild-type pGL3-EGFR promoter (Figure 2c), p53 RNAi induced the triple mutant and the wild-type promoters to comparable absolute levels of activity. These results demonstrate that intact p53 binding sites are not necessary for induction of the pGL3-EGFR promoter construct by p53RNAi-1. These experiments were repeated using the p53RNAi-2 construct, with similar results (data not shown).
Figure 2.
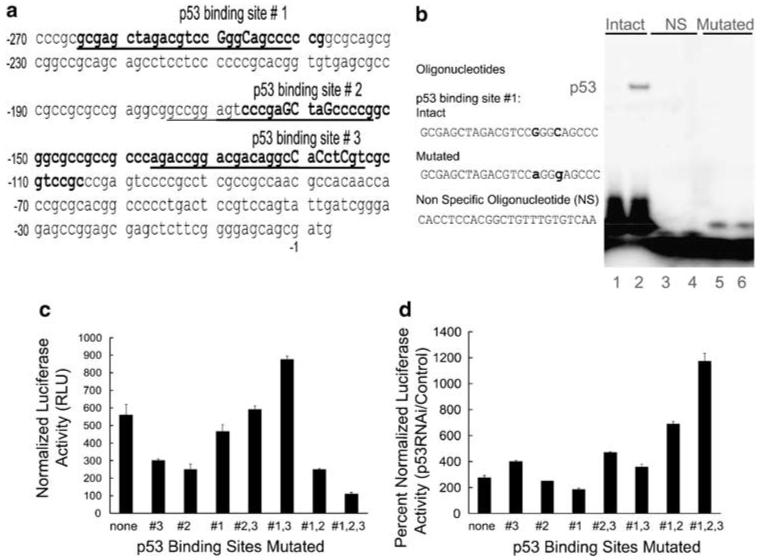
Site directed mutations in p53 binding sites do not abolish induction of EGFR promoter activity by p53 RNAi. (a) Partial EGFR promoter sequence (nucleotides -270 to +3) containing the three regions where p53 binding sites have been previously identified, shown here in bold. The sequences we used in EMSAs are underlined. The specific nucleotides we mutated are shown in uppercase. (b) EMSAs were performed on synthetic oligonucleotides containing the p53 binding site 1, intact or mutated (M), and a non-specific control oligonucleotide (NS). These oligonucleotides were labeled with [γ- 32P] ATP and were incubated with nuclear extracts from C33A (lanes 1, 3 and 5) or UV-treated normal HKc (lanes 2, 4 and 6) and EMSA were performed as described in Materials and methods. (c) Basal activity and (d) p53RNAi-induced activity of the EGFR promoter p53 binding site mutants (single, double and triple) in transient transfections of normal HKc. Normal HKc were co-transfected with the indicated wild-type or mutated pGL3-EGFR promoter constructs (1.5 μg per well), along with pRL-Null (10 ng per well) (c) or with p53RNAi-1 or control pSuper plasmid (1 μg per well) and pRL-Null (10 ng per well) (d). Cells were harvested 48 h post-transfection for dual-luciferase assays. EGFR, epidermal growth factor receptor; EMSA, electrophoretic mobility shift assays; HKc, human keratinocytes; RNAi, RNA interference.
Exogenous wild-type p53 both inhibits and activates the EGFR promoter in normal HKc
Our observation of increased EGFR promoter activity with suppression of endogenous p53 expression by p53 RNAi was in contrast with the published data, which showed that the p53 binding sites in the EGFR promoter were activating, not inhibitory, when wild-type p53 was overexpressed in p53-null cells (Deb et al., 1994; Ludes-Meyers et al., 1996). To explore this apparent contradiction, we determined pGL3-EGFR promoter activity in normal HKc transfected with increasing amounts of a wild-type p53 expression plasmid. As shown in Figure 3a, wild-type p53 inhibited EGFR promoter activity at low concentrations, with nearly complete suppression with 500 ng per well of the p53 expression plasmid. However, 3 μg per well of the p53 expression vector activated the EGFR promoter about twofold. We also explored in normal HKc the effects of exogenous wild-type p53 on the activity of two of our mutant EGFR promoter constructs (the no.2 single mutant and the no.1, 2, 3 triple mutant). Repression of EGFR promoter activity by 500 ng per well of the wild-type p53 expression vector was partially relieved by mutation of either a single p53 binding site (2) or all three (Figure 3b). Similarly, induction of EGFR promoter activity by 3 μg per well of wild-type p53 was reduced by over 60% by mutation of all three p53 binding sites (data not shown).
Figure 3.

Expression of exogenous wild-type p53 produces a dual response on EGFR promoter activity in normal HKc, and mutations of p53 binding sites on the EGFR promoter inhibit these responses. (a) Normal HKc were co-transfected with the pGL3-EGFR promoter construct (1.5 μg per well) along with increasing amounts of a p53 expression vector or its control plasmid (pcDNA 3.1) and with pRL-Null (10 ng per well). (b) Normal HKc were co-transfected with wild type or mutant pGL3-EGFR promoter constructs (1.5 μg per well) along with the wild-type p53 expression vector (500 ng per well) or pcDNA 3.1 control plasmid (500 ng per well) and pRL-Null (10 ng per well). Cells were harvested 48 h post-transfection for dual-luciferase assays. EGFR, epidermal growth factor receptor; HKc, human keratinocytes.
YY1 protein levels increase with loss of p53, and exogenous YY1 induces EGFR promoter activity
YY1 levels inversely correlate with p53 levels (Sui et al., 2004). We compared the levels of YY1 by western blot analysis in normal HKc stably transfected with p53RNAi-1 or an expression vector for HPV16 E6 (pLXSN-16E6) to their respective vector-transfected controls. The decrease in p53 and the expression of E6 RNA in the stable transfectants were confirmed by western blotting and real-time PCR, respectively (data not shown). YY1 was undetectable in normal HKc transfected with control plasmids (pSuper and pLXSN) but robust expression of YY1 was observed in normal HKc transfected with p53RNAi-1, and to a lesser extent in those transfected with pLXSN-16E6 (Figure 4a). Immunofluorescent staining and confocal microscopy showed that YY1 protein increases in early passage HKc/HPV16 in comparison with normal HKc, and increases further in high passage HKc/HPV16 and HKc/DR (Figure 4b). To determine the effects of YY1 on EGFR promoter activity, we co-transfected normal HKc with increasing concentrations of a YY1 expression plasmid along with the pGL3-EGFR promoter construct: exogenous YY1 induced EGFR promoter activity in a dose-dependent fashion (Figure 4c).
Figure 4.
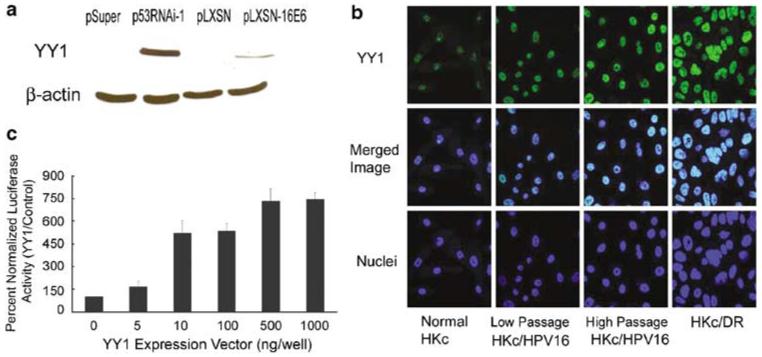
Loss of p53 induces YY1 protein levels in normal HKc and YY1 levels increase during in vitro progression of HKc/HPV16. Exogenous YY1 induces the EGFR promoter. (a) Western blot analysis for YY1 and β-actin (as a loading control) of cell lysates of normal HKc stably transfected with the p53RNAi-1 expression plasmid, or an HPV16 E6 expression vector (pLXSN-16E6) or their respective controls (pSuper or pLXSN). (b) Immunofluorescence staining for YY1 (green) in normal HKc, HKc/HPV16 at passage 15 (HKc/HPV16 low passage), 127 (HKc/HPV16 high passage) and HKc/DR. Cell nuclei (blue) were stained with DAPI and confocal microscopy was performed. (c) Normal HKc were transiently co-transfected with the pGL3-EGFR promoter construct (1.5 μg per well) and increasing amounts of a YY1 expression vector or its control plasmid (pcDNA 3.1) and with pRL-Null (10 ng per well). Cells were harvested 48 h post-transfection for dual-luciferase assays. EGFR, epidermal growth factor receptor; HKc, human keratinocytes; YY1, Yin Yang 1.
YY1’s induction of the EGFR promoter is augmented by loss of endogenous p53, and YY1-mediated induction does not require intact p53 binding sites
Next, we sought to determine the combined effect of loss of endogenous p53 and overexpression of YY1 on the EGFR promoter. We co-transfected normal HKc with pGL3-EGFR promoter and p53RNAi-1, in the presence or absence of the YY1 expression vector. Loss of endogenous p53 potentiates YY1 induction of EGFR promoter activity (Figure 5a). We then determined if p53 binding sites were necessary for YY1-mediated induction of the EGFR promoter: EGFR promoter constructs containing intact or mutated p53 binding sites were all activated by exogenous YY1 (Figure 5b).
Figure 5.

YY1 induction of the EGFR promoter is potentiated by p53 RNAi, and mutations in the p53 binding sites on the EGFR promoter do not alter YY1-mediated induction. (a) Normal HKc were transiently co-transfected with the pGL3-EGFR promoter construct (1 μg per well) and the YY1 expression vector or its control pcDNA 3.1 plasmid (500 ng per well), along with p53RNAi-1 or its control, the pSuper plasmid (1 μg per well) and pRL-Null (10 ng per well). Cells were harvested 48 h post-transfection for dual-luciferase assays. (b) Normal HKc were transiently co-transfected with wild-type pGL3-EGFR promoter construct or EGFR promoter constructs containing mutations in the p53 binding sites (1.5 μg per well), the YY1 expression plasmid or its pcDNA 3.1 control vector (10 ng per well) and pRL-Null (10 ng per well). Cells were harvested 48 h post-transfection for dual-luciferase assays. EGFR, epidermal growth factor receptor; HKc, human keratinocytes; YY1, Yin Yang 1.
YY1 and Sp1 synergize to activate the EGFR promoter
Sp1 is a key transcription factor in the regulation of the EGFR promoter (Kageyama et al., 1988) and can work synergistically with YY1 to activate promoters (Kawada et al., 2005). Therefore, we investigated whether YY1 and Sp1 synergized to activate the EGFR promoter. When expressed individually, exogenous Sp1 or YY1 induced EGFR promoter activity six- to sevenfold. However, when expressed together, exogenous YY1 and Sp1 induced EGFR promoter activity up to 40-fold (Figure 6a).
Figure 6.

Exogenous Sp1 induces the EGFR promoter and potentiates YY1 induction; expression of exogenous wild-type p53 suppresses EGFR promoter induction by YY1 and Sp1 in normal HKc. (a) Normal HKc were transiently co-transfected with the pGL3-EGFR promoter construct (1.5 μg per well) and the pcDNA 3.1 YY1 expression plasmid or the pcDNA 3.1 control plasmid (10 ng per well), along with the pPacSp1 expression vector or the pPac control plasmid (100 ng per well) and pRL-Null (10 ng per well). Cells were harvested 48 h post-transfection for dual-luciferase assays. (b) Normal HKc were transiently co-transfected with the pGL3-EGFR promoter construct (1.5 μg per well), and the pcDNA 3.1 wild-type p53 expression plasmid, or the pcDNA 3.1 control plasmid (500 ng per well), along with the pPacSp1 expression vector or the pPac control plasmid (100 ng per well) and pRL-Null (10 ng per well). Cells were harvested 48 h post-transfection for dual-luciferase assays. EGFR, epidermal growth factor receptor; HKc, human keratinocytes; YY1, Yin Yang 1.
Exogenous wild-type p53 prevents the induction of the EGFR promoter by Sp1 and YY1
p53 has been shown to inhibit Sp1-mediated promoter activation (Innocente and Lee, 2005). Therefore, we determined the effect of exogenous p53 overexpression on Sp1-mediated activation of the EGFR promoter. Exogenous wild-type p53 abrogated Sp1-mediated activation of the EGFR promoter (Figure 6b) and also counteracted induction of the EGFR promoter by YY1 (data not shown).
Discussion
These studies demonstrate that a loss of endogenous p53 results in enhanced EGFR promoter activity in normal HKc. This effect does not require intact p53 binding sites on the EGFR promoter, and is mediated by YY1 and Sp1, which synergistically activate the EGFR promoter. In addition, expression of exogenous wild-type p53 produces inhibition of the EGFR promoter at low doses, and induction at high doses, and both these effects require intact p53 binding sites on the EGFR promoter. These results point to at least two distinct activities of p53 in the control of the EGFR promoter: one that is direct, possibly by-phasic, and mediated by p53 binding elements; the other that is indirect, independent of p53 binding sites, and mediated at least in part by YY1 and Sp1.
Loss of p53 resulted in EGFR promoter induction in all normal individual HKc strains studied. However, the extent of activation of the EGFR promoter elicited by p53 RNAi varied from individual to individual. Given the short life span of these cells, different normal individual HKc isolates were used for each experiment. The observed interindividual variability in the magnitude of the EGFR promoter response to p53 RNAi parallels a marked variability of normal HKc responses to HPV16 E6 that we have described previously (Akerman et al., 2001). The molecular basis for these variable responses remain to be defined, and are probably due to more than one genetically-determined characteristic of each individual normal HKc strain. Intrinsic host cell factors that modulate a cell’s response to HPV, loss of p53 and other factors that mediate transformation are likely to be important co-determinants of the risk for transformation and progression of HPV-infected cells. Hence, more work is warranted in this area. The magnitude of the p53 RNAi effects on the promoter was always greater than the magnitude of induction of endogenous EGFR mRNA, from both p53 RNAi and E6 (Akerman et al., 2001; Bheda et al., manuscript in preparation). We interpret this observation based on the fact that dramatic increases in EGFR expression are not well tolerated by normal HKc (Akerman et al., 2001).
Different p53 RNAi constructs elicited different effects on p53 expression, confirming that RNAi effects can be elicited at different levels (Dykxhoorn and Lieberman, 2005). However, both p53RNAi-1 and -2, which reduced p53 protein levels, increased EGFR mRNA levels and promoter activity.
We determined that exogenous wild-type p53 inhibited the EGFR promoter at low concentrations, and activated it at high concentrations. This dual response resolves the apparent discrepancy between our results and published observations that overexpression of p53 in p53-null cells activated the EGFR promoter (Deb et al., 1994; Ludes-Meyers et al., 1996). In addition, this observation is in line with reports showing that different levels of p53 activate different sets of genes (Zhao et al., 2000) and that transcriptional responses to p53 are heterogeneous (Yu et al., 1999).
Our results show that intact p53 binding sites in the EGFR promoter were not required for induction by p53 RNAi, YY1 or Sp1, yet were required for the promoter’s responses to exogenous wild-type p53. This observation indicates that mutation of all three p53 binding sites (which completely abolishes p53 binding to the promoter) is not functionally equivalent to removal of p53 from the intracellular milieu by p53 RNAi. We presume that the balance of transcription factors interacting with the intact EGFR promoter may be quite different in cells treated with p53 RNAi, compared to the transcription factor composition on the triple mutant EGFR promoter. More information on how YY1, Sp1 and possibly other transcription factors, interact with the EGFR promoter (wild-type and triple mutant) under various levels of p53 expression, and also information on how p53 affects EGFR promoter activity in cells devoid of YY1 is needed before we can separate direct effects of p53 binding on the EGFR promoter from indirect effects associated with the raise and fall of YY1, for example, as a consequence of loss or gain of p53. Clearly, p53 controls EGFR expression both directly and indirectly, as our results indicate. In addition, p53 might also affect the choice of a transcription start site, as the region including the three p53 binding sites (nucleotides -265 to -105) also contains the six transcription start sites for this gene (Ishii et al., 1985; Haley et al., 1987). p53 RNAi induction of EGFR promoter activity in the triple p53 binding sites mutant is likely due to increased YY1 and facilitated binding of YY1 and Sp1 to their respective sites, in the absence of p53 binding.
Exogenous YY1 induced EGFR promoter activity in normal HKc. This activation was augmented by loss of endogenous p53, even on promoters with mutated p53 binding sites, again indicating that YY1 activates this promoter independent of p53 binding. However, we still need to determine exactly how YY1 mediates its effect on the EGFR promoter, whether by directly binding to the promoter, or indirectly via interactions with co-activators. We found synergism of YY1 and Sp1 in the activation of the EGFR promoter. In addition, YY1 interacts with the co-activators p300 and P/CAF, which facilitate YY1 activation of transcription by acetylating YY1 and the core histones (Yao et al., 2001). Accordingly, YY1-mediated induction of the EGFR promoter was increased up to fivefold by p300 and to a lesser extent by P/CAF, strongly indicating that YY1 does not act alone to induce EGFR promoter activity (Bheda et al., unpublished).
To illustrate our findings, we propose a simplified model of the principal mechanisms of EGFR promoter activation/repression by p53 RNAi, YY1 and p53 (Figure 7). p53 represses and YY1 activates the EGFR promoter; under normal homeostatic conditions, YY1 and p53 levels are in balance (Figure 7a). When p53 levels decrease (that is by p53 RNAi, E6) YY1 increases causing degradation of p53 protein, and therefore removing repression of the promoter by p53, while at the same time activating the promoter (Figure 7b). These results link the EGFR overexpression we have previously reported in HPV16-transformed cells (Zyzak et al., 1994; Akerman et al., 2001) specifically to the p53 degradation caused by the HPV16 oncoprotein E6, and identify at least some of the molecular mediators of EGFR overexpression in HPV-transformed cells.
Figure 7.
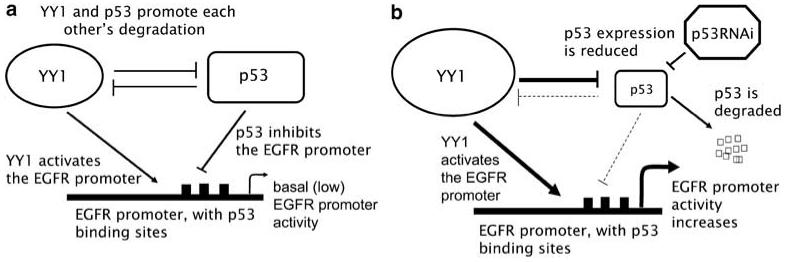
Proposed model for the interplay of p53 and YY1 in the regulation of the EGFR promoter under normal homeostatic conditions (a) or in the presence of p53 RNAi (b). EGFR, epidermal growth factor receptor; RNAi, RNA interference; YY1, Yin Yang 1.
Materials and methods
Cell culture
Normal HKc isolated from neonatal foreskins were cultured in serum free MCDB 153-LB basal media supplemented with epidermal growth factor (5 ng ml-1), bovine pituitary extract (35-50 μg protein per milliliter), hydrocortisone (0.2 μm), calcium chloride (0.1 nm), triiodothyronine (10 nm), transferrin (10 μg ml-1), insulin (5 μg ml-1) and gentamycin (50 mg ml-1) (complete medium) (Pirisi et al., 1987). C33a cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and penicillin-streptomycin (100 units ml-1). All cells were maintained at 37 °C in 5% CO2 in air.
Plasmid constructs
The YY1 expression plasmid and control plasmid YY1 (Ai et al., 2000) were a generous gift from Dr Ann Roman. The YY1 cDNA was recloned into the BglII and XbaI site of the expression plasmid pcDNA 3.1. The EGFR promoter—luciferase plasmid (-1100 to -19 bp)—a gift from Dr Gordon Gill) was re-cloned into the HindIII site of the pGL3 basic plasmid (Promega, Madison, WI, USA), referred to as the pGL3-EGFR promoter construct. The pcDNA 3.1 p53 expression plasmid was a gift from Dr Curtis Harris. The pPac Sp1 expression plasmid was from Dr Robert Tijan.
A commercially available p53 RNAi pSuper vector was obtained from OligoEngine and is referred to as p53RNAi-1. Two additional siRNAs against p53 were designed using the OligoEngine design software. Their sequences are as follows: p53 RNAi targeting position 220-239: TTGGCAGCCA GACTGCCTT referred to as p53RNAi-2. p53 RNAi targeting position 1588-1607: ACAAGTTGGCCTGCACTGG referred to as p53RNAi-3. These sequences were then cloned into the BglII and HindIII sites of the pSuper retro vector (Oligoengine, Seattle, WA, USA). The nucleotide sequence of all of the plasmids obtained or re-cloned was confirmed by direct DNA sequencing of the cDNAs.
Quantitative real-time PCR
Total RNA was extracted using Qiagen RNeasy Total RNA isolation kit (Qiagen, Valencia, CA, USA). The reverse transcriptase reaction (1 μg of total RNA) was performed using the IScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). Real-time PCR (1:25 dilution of the cDNA synthesis reaction) was performed using the ICycler IQ detection system (Bio-Rad) using the following program: one cycle, 95 °C for 8.5 min; 50 cycles, 30 s at 95 °C, 30 s at 55 °C, and 30 s at 72 °C, plus a melting curve of 55 °C at 0.5 °C intervals for 10 s for 80 cycles; and 58 °C for 30 s for one cycle, ending with a hold at 10 °C. All samples were assayed in triplicate. The differences in expression of p53 were evaluated using a relative quantification method where the expression of p53 was normalized to the reference gene—RPLPO (ribosomal protein, large protein O). The primers used in the study were: p53 forward: 5′-CGTGTGGAGTATTTGGATGAC, p53 reverse: 5′-AGTCTTCCAGTGTGATGATGG, RPLPO forward: 5′-CCACATTCCCCCGGATATGA, RPLPO reverse: 5′-TTA AACCCCCTCGTGGCAATC.
Transient transfections and dual-luciferase assays
Only first or second passage individual normal HKc strains at 70% confluency were used for transient transfections. Normal HKc were co-transfected using TransFast (Promega) with equal amounts of experimental plasmid or control and the pGL3-EGFR promoter construct along with pRL-Null or pRL-TK Renilla luciferase constructs (Promega) as a control for transfection efficiency. The cells were incubated with the transfection mix for 1 h in basal MCDB 153-LB medium at 37 °C. Similar experimental protocols were followed to transfect C33A cells, in Dulbecco’s modified Eagle’s medium. After transfection, cells were washed once with Dulbecco’s phosphate buffered saline (PBS) and were then fed with complete medium with no antibiotics. Cells were harvested 48 h post-transfection and luciferase assays were performed using the Dual-Luciferase Assay System (Promega). Relative light units were determined using a luminometer (Berthold Lumat LB9501) for both firefly and Renilla luciferase. Firefly luciferase activity was normalized for Renilla luciferase activity. In some cases in which extremely high levels of Renilla luciferase activity made data interpretation problematic, we confirmed the results by two independent methods: repeated transfection assays without Renilla luciferase, and by determining the levels of firefly and Renilla luciferase RNA by RT-PCR.
Nuclear extracts
Nuclei were isolated using modifications of a previously described procedure (Baldwin et al., 2004). Briefly, cells were washed twice with ice-cold PBS, scraped from the dish, transferred to a pre-chilled microfuge tube, and collected by centrifugation at 12 000 × g for 1 min at 4 °C. The cell pellet was re-suspended in 100 μl of hypotonic buffer (10 mm HEPES, pH 7.9, 2 mm MgCl2, 10mm KCl, 0.1 mm EDTA, 0.5% NP40, 1mm DTT, 0.5 mm PMSF and protease inhibitors including 0.1% aprotinin, 0.1% leptin and 0.1% pepstatin A were added fresh), incubated on ice for 10 min, and then centrifuged at 15 000 × g for 1 min at 4 °C. The supernatant was removed and the pellet was re-suspended in 100 μl of high salt buffer (50 mm HEPES, pH 7.9, 300 mm NaCl, 50 mm KCL, 0.1 mm EDTA, 10% glycerol and 1 mm DTT, 0.5 mm PMSF; protease inhibitors including 0.1% aprotinin, 0.1% leptin and 0.1% pepstatin A were added fresh), incubated with rotation at 4 °C for 30 min, and centrifuged at 15 000 × g for 30 min to remove the nuclear remnants. The nuclear protein was transferred to a fresh tube and stored at -80 °C.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays were performed using the following double-stranded oligonucleotides containing p53 responsive elements and surrounding sequences from the human EGFR promoter, and the corresponding mutant oligonucleotides (Figure 2a):
For p53 binding site 1:GGCGAGCTAGACGTCCG(G/A)GGC(C/G)AGCCC,
For p53 binding site 2:gccggagtcccgag(G/T)C(C/G)TAG(G/T)CCCCG, and
For p53 binding site 3: AGACCGGACGACAGGCC(C/G)AC(C/G)CTC(C/A)GT,
where the nucleotides bolded and in brackets represent the site and nature of the mutations introduced. Wild type and mutant oligonucleotides were 5′ end labeled with [γ-32P] ATP (Amersham, Piscataway, NJ, USA) using T4 polynucleotide kinase (New England Biolabs, Ipswich, MA, USA). Labeled probes (100 000 c.p.m.) were incubated with nuclear extracts (4 μg protein) from normal HKc or C33A in a reaction mixture containing 10 mm Tris HCl, pH 7.5, 1 mm DTT, 50 mm NaCl, 5 mm MgCl2, 20 mm EDTA, 6% glycerol, 0.1 mg ml-1 bovine serum albumin, 1 μg of poly (dI)-poly (dC), 0.5 μg of herring sperm DNA and 0.05% NP-40 for 20 min at room temperature. As a control, the nuclear extracts were also incubated with 100-fold excess unlabeled specific or non-specific oligonucleotides. The entire reaction mixture was loaded without dye and resolved on a 4% non-denaturing Tris-boric polyacrylamide gel (3.5 h at 120 V). Gels were dried and visualized using a Bio-Rad K-screen and phosphoimager.
Western blot analysis
Samples (22 μg protein) were separated by electrophoresis in a 12% denaturing polyacrylamide gel at 120 V for 1 h and transferred to a nitrocellulose membrane by electroblotting (Bio-Rad) at 75 mA overnight. The blot was blocked in 5% non-fat dry milk for 1 h at room temperature. The blots were probed with mouse anti-p53 antibody (Oncogene, Cambridge, MA, USA, 1:1000 dilution) or mouse anti-YY1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:1000 dilution) at 4 °C overnight followed with horseradish peroxidase-conjugated goat anti-mouse (Boehringer Mannheim, Gaithersburg, MD, USA, 1:5000 dilution) secondary antibody. Proteins were detected using the Super Signal West Pico Chemiluminescence Detection Kit (Pierce, Rockford, IL, USA). The blots were also probed for actin, as a protein loading control, with rabbit anti-actin antibodies (Sigma, St Louis, MO, USA, 1:5000 dilution) at 4 °C overnight and a secondary goat anti-rabbit antibody (Vector Laboratories, Burlingame, CA, USA, 1:10 000 dilution).
Site directed mutagenesis
The EGFR promoter was mutated at different p53 binding sites using the Quick-Change Site Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). Double or triple nucleotide changes were introduced into the parental plasmid by oligonucleotide primers carrying the same specific mutations in the p53 binding sites that we had confirmed by electrophoretic mobility shift assays to abolish p53 binding to the promoter. The promoter was mutated at a single p53 binding site, two p53 binding sites at a time, or all three p53 binding sites. The parental DNA template was then digested with DpnI. All mutations and the integrity of the remainder of the promoter were confirmed by DNA sequencing.
Immunofluorescence staining
Normal HKc, HKc/HPV16 or HKc/DR (Pirisi et al., 1987, 1988) were grown on cover slips coated with poly-lysine until 75% confluent. The cells were then washed twice with ice-cold PBS, fixed with 4% paraformaldehyde for 20 min at room temperature, permeablized with 0.5% Triton and 1% glycine and then blocked using 0.5% bovine serum albumin and 5% goat serum for 30 min at room temperature. Cells were next incubated with mouse anti-YY1 antibody (Santa Cruz Biotechnology, 1:250) in blocking solution overnight at 4 °C and then washed three times with PBS for 5 min at room temperature. Next, the cells were incubated with Cy3 conjugated goat-anti-mouse secondary antibody (Vector Laboratories, 1:400) for 1 h at 37 °C, washed once with PBS, mounted on to the slides with DABCO mounting media, sealed and visualized using a confocal microscope.
Acknowledgements
We thank Dr Omar Bagasra for discussion and advice in the construction and use of siRNA. This work was supported by grants from the National Institutes of Health: R01CA89502 and P20CA096427 to KEC, and EXPORT Center P20MD001770.
References
- Ai W, Narahari J, Roman A. Yin yang 1 negatively regulates the differentiation-specific E1 promoter of human papillomavirus type 6. J Virol. 2000;74:5198–5205. doi: 10.1128/jvi.74.11.5198-5205.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman GS, Tolleson WH, Brown KL, Zyzak LL, Mourateva E, Engin TS, et al. Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signaling shortens the life span of normal human keratinocytes. Cancer Res. 2001;61:3837–3843. [PubMed] [Google Scholar]
- Arteaga CL. Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncologist. 2002;7(Suppl 4):31–39. doi: 10.1634/theoncologist.7-suppl_4-31. [DOI] [PubMed] [Google Scholar]
- Baldwin A, Pirisi L, Creek KE. NFI-Ski interactions mediate transforming growth factor beta modulation of human papilloma-virus type 16 early gene expression. J Virol. 2004;78:3953–3964. doi: 10.1128/JVI.78.8.3953-3964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb SP, Munoz RM, Brown DR, Subler MA, Deb S. Wild-type human p53 activates the human epidermal growth factor receptor promoter. Oncogene. 1994;9:1341–1349. [PubMed] [Google Scholar]
- Dykxhoorn DM, Lieberman J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu Rev Med. 2005;56:401–423. doi: 10.1146/annurev.med.56.082103.104606. [DOI] [PubMed] [Google Scholar]
- Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- Gronroos E, Terentiev AA, Punga T, Ericsson J. YY1 inhibits the activation of the p53 tumor suppressor in response to genotoxic stress. Proc Natl Acad Sci USA. 2004;101:12165–12170. doi: 10.1073/pnas.0402283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley J, Whittle N, Bennet P, Kinchington D, Ullrich A, Waterfield M. The human EGF receptor gene: structure of the 110 kb locus and identification of sequences regulating its transcription. Oncogene Res. 1987;1:375–396. [PubMed] [Google Scholar]
- Hu G, Liu W, Mendelsohn J, Ellis LM, Radinsky R, Andreeff M, et al. Expression of epidermal growth factor receptor and human papillomavirus E6/E7 proteins in cervical carcinoma cells. J Natl Cancer Inst. 1997;89:1271–1276. doi: 10.1093/jnci/89.17.1271. [DOI] [PubMed] [Google Scholar]
- Innocente SA, Lee JM. p53 is a NF-Y- and p21-independent, Sp1-dependent repressor of cyclin B1 transcription. FEBS Lett. 2005;579:1001–1007. doi: 10.1016/j.febslet.2004.12.073. [DOI] [PubMed] [Google Scholar]
- Ishii S, Xu YH, Stratton RH, Roe BA, Merlino GT, Pastan I. Characterization and sequence of the promoter region of the human epidermal growth factor receptor gene. Proc Natl Acad Sci USA. 1985;82:4920–4924. doi: 10.1073/pnas.82.15.4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AC, Murphy BA, Matelis CM, Rubinstein Y, Piebenga EC, Akers LM, et al. Activator protein-1 mediates induced but not basal epidermal growth factor receptor gene expression. Mol Med. 2000;6:17–27. [PMC free article] [PubMed] [Google Scholar]
- Kageyama R, Merlino GT, Pastan I. Epidermal growth factor (EGF) receptor gene transcription. Requirement for Sp1 and an EGF receptor-specific factor. J Biol Chem. 1988;263:6329–6336. [PubMed] [Google Scholar]
- Kawada H, Nishiyama C, Takagi A, Tokura T, Nakano N, Maeda K, et al. Transcriptional regulation of ATP2C1 gene by Sp1 and YY1 and reduced function of its promoter in Hailey-Hailey disease keratinocytes. J Invest Dermatol. 2005;124:1206–1214. doi: 10.1111/j.0022-202X.2005.23748.x. [DOI] [PubMed] [Google Scholar]
- Kilic G, Cardillo M, Ozdemirli M, Arun B. Human papillomavirus 18 oncoproteins E6 and E7 enhance irradiation- and chemotherapeutic agent-induced apoptosis in p53 and Rb mutated cervical cancer cell lines. Eur J Gynaecol Oncol. 1999;20:167–171. [PubMed] [Google Scholar]
- Lee JS, Galvin KM, Shi Y. Evidence for physical interaction between the zinc-finger transcription factors YY1 and Sp1. Proc Natl Acad Sci USA. 1993;90:6145–6149. doi: 10.1073/pnas.90.13.6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludes-Meyers JH, Subler MA, Shivakumar CV, Munoz RM, Jiang P, Bigger JE, et al. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol Cell Biol. 1996;16:6009–6019. doi: 10.1128/mcb.16.11.6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur SP, Mathur RS, Rust PF, Young RC. Human papilloma virus (HPV)-E6/E7 and epidermal growth factor receptor (EGF-R) protein levels in cervical cancer and cervical intraepithelial neoplasia (CIN) Am J Reprod Immunol. 2001;46:280–287. doi: 10.1034/j.1600-0897.2001.d01-14.x. [DOI] [PubMed] [Google Scholar]
- Pirisi L, Creek KE, Doniger J, DiPaolo JA. Continuous cell lines with altered growth and differentiation properties originate after transfection of human keratinocytes with human papilloma-virus type 16 DNA. Carcinogenesis. 1988;9:1573–1579. doi: 10.1093/carcin/9.9.1573. [DOI] [PubMed] [Google Scholar]
- Pirisi L, Yasumoto S, Feller M, Doniger J, DiPaolo JA. Transformation of human fibroblasts and keratinocytes with human papillomavirus type 16 DNA. J Virol. 1987;61:1061–1066. doi: 10.1128/jvi.61.4.1061-1066.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh MS, Carrier F, Johnson AC, Ogdon SE, Fornace AJ., Jr Identification of an additional p53-responsive site in the human epidermal growth factor receptor gene promoter. Oncogene. 1997;15:1095–1101. doi: 10.1038/sj.onc.1201264. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lee JS, Galvin KM. Everything you have ever wanted to know about Yin Yang 1. Biochim Biophys Acta. 1997;1332:F49–F66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- Sui G, Affar el B, Shi Y, Brignone C, Wall NR, Yin P, et al. Yin Yang 1 is a negative regulator of p53. Cell. 2004;117:859–872. doi: 10.1016/j.cell.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Wang X, Bolotin D, Chu DH, Polak L, Williams T, Fuchs E. AP-2alpha: a regulator of EGF receptor signaling and proliferation in skin epidermis. J Cell Biol. 2006;172:409–421. doi: 10.1083/jcb.200510002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Thompson KL, Shephard LB, Hudson LG, Gill GN. T3 receptor suppression of Sp1-dependent transcription from the epidermal growth factor receptor promoter via overlapping DNA-binding sites. J Biol Chem. 1993;268:16065–16073. [PubMed] [Google Scholar]
- Yakovleva T, Kolesnikova L, Vukojevic V, Gileva I, Tan-No K, Austen M, et al. YY1 binding to a subset of p53 DNA-target sites regulates p53-dependent transcription. Biochem Biophys Res Commun. 2004;318:615–624. doi: 10.1016/j.bbrc.2004.04.065. [DOI] [PubMed] [Google Scholar]
- Yao YL, Yang WM, Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol. 2001;21:5979–5991. doi: 10.1128/MCB.21.17.5979-5991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Rago C, Kinzler KW, Vogelstein B. Identification and classification of p53-regulated genes. Proc Natl Acad Sci USA. 1999;96:14517–14522. doi: 10.1073/pnas.96.25.14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, et al. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–993. [PMC free article] [PubMed] [Google Scholar]
- Zyzak LL, MacDonald LM, Batova A, Forand R, Creek KE, Pirisi L. Increased levels and constitutive tyrosine phosphorylation of the epidermal growth factor receptor contribute to autonomous growth of human papillomavirus type 16 immortalized human keratinocytes. Cell Growth Differ. 1994;5:537–547. [PubMed] [Google Scholar]