

Abstract
The amyloid precursor protein (APP) is subject to alternative pathways of proteolytic processing, leading either to production of the amyloid-β (Aβ) peptides or to non-amyloidogenic fragments. Here, we report the first structural study of C99, the 99-residue transmembrane C-terminal domain of APP liberated by β-secretase cleavage. We also show that cholesterol, an agent that promotes the amyloidogenic pathway, specifically binds to this protein. C99 was purified into model membranes where it was observed to homodimerize. NMR data show that the transmembrane domain of C99 is an α-helix that is flanked on both sides by mostly disordered extramembrane domains, with two exceptions. First, there is a short extracellular surface-associated helix located just after the site of α-secretase cleavage that helps to organize the connecting loop to the transmembrane domain, which is known to be essential for Aβ production. Second, there is a surface-associated helix located at the cytosolic C-terminus, adjacent to the YENPTY motif that plays critical roles in APP trafficking and protein–protein interactions. Cholesterol was seen to participate in saturable interactions with C99 that are centered at the critical loop connecting the extracellular helix to the transmembrane domain. Binding of cholesterol to C99 and, most likely, to APP may be critical for the trafficking of these proteins to cholesterol-rich membrane domains, which leads to cleavage by β- and γ-secretase and resulting amyloid-β production. It is proposed that APP may serve as a cellular cholesterol sensor that is linked to mechanisms for suppressing cellular cholesterol uptake.
The human amyloid precursor protein (APP)1 is a single-span membrane protein that is alternatively processed by either α- or β-secretase to release its large ectodomain from the cell surface, a process referred to as “shedding”. β-Secretase (β-site APP cleaving enzyme 1, BACE1) cleaves APP after Met671, leading to production of the C-terminal 99-residue domain of APP, C99, a single-span membrane protein. Subsequent cleavage of C99 at membrane-disposed sites by γ-secretase leads to release of both the amyloid-β (Aβ) peptides and the water-soluble APP intracellular domain (AICD), the latter of which plays roles in transcriptional regulation (1). The Aβ peptides have heterogeneous C-termini owing to a somewhat imprecise γ-secretase-mediated intramembranous cleavage, and these peptides have varying tendencies to aggregate and then to seed amyloid deposits in neural tissue (2–4). Thus, BACE1 cleavage is closely linked to the etiology of Alzheimer's disease. Alternative processing of APP by α-secretase results in cleavage after Lys687 and release of an 83-residue C-terminal fragment, C83 (5). While C83 (also known as p10) is also a transmembrane protein and is also subject to γ-secretase cleavage, the resulting polypeptide products are not amyloidogenic. Pharmacological elevation of α-secretase cleavage of APP at the expense of β-secretase cleavage has attracted interest as a strategy for reducing predisposition to Alzheimer's disease (4, 6, 7).
The partitioning of APP between amyloidogenic and non-amyloidogenic pathways is closely linked to the intracellular trafficking of APP and C99, a process in which cholesterol is also intimately involved. α-Secretase cleavage occurs primarily at the cell surface in the cholesterol-poor bulk plasma membrane (8) whereas β- and γ-secretase proteolysis occurs primarily in cholesterol-rich lipid rafts (also known as detergent-resistant membranes, DRMs), most often following internalization of the protein into cholesterol-rich endosomes (9–12). Cholesterol was identified more than a decade ago as a factor that could modulate β-amyloid production (13, 14), and subsequent investigations have confirmed the dependence of amyloidogenic processing on cholesterol (15, 16) and the association of APP or BACE1 with lipid rafts (10, 17). These observations have laid the foundation for a model of APP processing in which the proteolytic fate of APP is governed by its dynamic equilibrium between lipid rafts and the bulk phase of the membrane. According to this increasingly well-substantiated model, raft-associated APP is processed in an amyloidogenic fashion, often in association with the endocytic pathway, whereas APP in the bulk phase of the membrane is cleaved primarily by α-secretase at the cell surface. Further support for this model has been provided by the observations that cholesterol depletion and filipin-mediated caveolar destruction augment α-secretase cleavage (18).
Factors that induce the association of APP with raft-like membrane domains remain uncharacterized. Various proteins have been suggested to regulate the membrane trafficking of APP, including the low-density lipoprotein (LDL) receptor-related protein 1 (LRP1), LRP1b, SorLA/SorL1/LR11, and ApoER2/LR8 (see review in ref 19). These proteins, all of which are members of the LDLR family, influence the trafficking kinetics of APP and serve to retain it in particular organelles or induce its endocytosis. SorLA, for instance, is thought to engage in retrograde trafficking from the endosomal system to the Golgi, thereby protecting APP from BACE1 (20). LRP1 promotes the endocytosis of APP (21), whereas LRP1b (22) and ApoER2 (23) retain APP at the cell surface. While competition among these proteins for APP dictates its intracellular trafficking, they are not likely to be the proximate factors that induce association of APP with lipid rafts or promote raft formation around APP clusters. The raft-associated protein flotillin was shown to interact with APP and promote its clustering and subsequent endocytosis in a cholesterol-dependent fashion. Clustering of membrane proteins is known to promote raft association (24), and APP clustering is evidently dependent on cholesterol (25). Therefore, the possibility exists that direct interactions of APP with cholesterol contribute to translocation of APP from the bulk phase of the membrane to cholesterol-enriched lipid rafts. This latter possibility would require APP/C99 to have a specific affinity for cholesterol.
In this work we examine the possibility that the critical C99 domain of APP can directly recognize and bind cholesterol. We also present the first detailed structural characterization of C99, the product of β-secretase cleavage and a substrate for cleavage by γ-secretase. Although the amyloid-β peptides and the extramembrane domains of APP have been the subject of numerous structural studies (25–42), analysis of C99 and shorter transmembrane domain- (TMD-) containing fragments of APP have been limited to elucidation of the putative homodimeric state of the TMD (43–46) and to some limited CD studies (43, 47). The studies of C99 in this paper reveal surprising structural and cholesterol-binding properties that appear to be critically related to the proteolytic processing and membrane trafficking of C99 and APP.
Methods
Overexpression and Purification of the C-Terminal Transmembrane Domain of APP (C99)
The β-secretase cleavage product of the full-length human amyloid precursor protein (residues 672–770, C99) was cloned into a pET-21a vector, with an added methionine encoded at the N-terminus and an added spacer/purification tag (QGRILQISITLAAALEHHHHHH) at the C-terminus. This vector was then used to transform the BL21(DE3) strain of Escherichia coli.
The transformed cells were plated on Luria broth (LB)/ampicillin plates and then cultured in 5–10 mL of LB/ampicillin at 37 °C. These cultures were then used to inoculate 1 L of M9 minimal medium (40 mM Na2HPO4, 20 mM KH2PO4, 10 mM NaCl, 20 mM NH4Cl, pH 7.0) supplemented with 0.1 mM CaCl2, 1 mM MgSO4, 0.4% glucose, vitamins (150 mg of a pulverized CVS Spectravite vitamin), and 0.1 mg/mL ampicillin. The cultures were then incubated with vigorous shaking at 37 °C until their OD600 reached ca. 0.8, at which point the cultures were induced by adding isopropyl thiogalactoside to 1 mM. Expression was allowed to proceed for 5 h at 37 °C, and cells were then harvested by centrifugation.
To each gram of harvested cell paste was added 20 mL of lysis buffer (75 mM Tris, 300 mM NaCl, 0.2 mM EDTA, pH 7.8), followed by lysozyme (0.2 mg/mL), DNase (0.02 mg/mL), RNase (0.02 mg/mL), and magnesium acetate (5 mM). The lysate was agitated for 1 h at room temperature followed by sonication for 5 min (alternating 5 s on and 5 s off). The solution was then centrifuged at 15000g for 20 min, and the supernatant was discarded. The pellet, which contained C99-containing inclusion bodies, was resuspended in lysis buffer (20 mL for each original gram of cells), dispersed with a hand-held homogenizer, and recentrifuged (15000g for 20 min), following which the supernatant was discarded. The inclusion bodies were washed in this manner until the supernatant became clear, which typically required three iterations. The washed inclusion bodies were solubilized by tumbling overnight at room temperature in a urea/SDS buffer (20 mM Tris, 150 mM NaCl, 8 M urea, and 0.2% SDS, pH 7.8). The solution was then centrifuged at 18 °C for 20 min at 15000g, and the pellet was discarded. The supernatant could be frozen and stored prior to subsequent steps. The supernatant was then mixed with Qiagen (Valencia, CA) Ni-NTA-Superflow resin (2.5 mL of resin for each gram of cells represented by the supernatant) and tumbled for 20 min at room temperature. The resin was transferred to a column, which was then washed with 4 bed volumes of urea/SDS buffer, followed by 4 bed volumes of SDS rinse buffer (20 mM Tris, 150 mM NaCl, no urea, 0.2% SDS, pH 7.8). The protein was refolded by exchanging detergent from SDS to lysomyristoylphosphatidylglycerol (LMPG; Avanti, Alabaster, AL) by pulsing the column with a series of 8 × 1 bed volumes of Tris-buffered saline (20 mM Tris HCl, 200 mM NaCl, pH 7.8) containing 0.05% LMPG. Finally, C99 was eluted from the resin using a solution containing 0.05% LMPG and 250 mM imidazole (Calbiochem, UL-TROL grade) at pH 7.8. The purity of C99 was confirmed by SDS–PAGE using Coomassie staining. The concentration of C99 was determined by measuring the A280 and using an extinction coefficient of 5960 M−1 cm−1 (0.43 (mg/mL)−1 cm−1). The protein could then be flash-frozen in liquid nitrogen and stored at −80 °C. The typical yield of purified C99 was 20 mg/L of culture.
To prepare uniformly 13C- and/or 15N-labeled C99, 13C6-glucose and/or 15NH4Cl was used in the minimal medium. Perdeuterated C99 samples (70–85%) were prepared using the same method as above but replacing the H2O with 99% D2O in the minimal medium in which the cells were grown (final labeling was only 70–85% because the carbon source used was fully protonated glucose). In order to adapt the cells to growth in D2O, a 5 mL LB/ampicillin starter culture was inoculated, followed 10–14 h later by addition of 1 mL of this culture to 30 mL of minimal media with 70% D2O. The culture was allowed to grow until OD600 reached a value of 0.5, at which point 1 mL of this culture was used to inoculate 30 mL of minimal media in 99.9% D2O. This culture was grown until OD600 reached a value of 0.5, and 6 mL of this culture was then used to inoculate 1 L of minimal medium in 100% D2O. The culture was then grown until OD600 reached a value of 0.8, which required about 16 h, and induced with 1 mM IPTG. After induction, protein expression continued for 24 h, at which point the cells were harvested by centrifugation.
NMR Sample Preparation
To prepare C99 samples for NMR, EDTA was added to 1 mM and D2O to a concentration of 10%. Additional LMPG was added to adjust the final concentration to 9%. The pH was then adjusted to 6.5 using acetic acid and ammonium hydroxide. The solution was then concentrated to 1–3 mM C99 using an Amicon Ultra-15 (MWCO = 5000 Da) centrifugal ultrafiltration cartridge. For titration experiments, U-15N-labeled C99 was used, while for the backbone assignment, U-15N,13C,2H(70%)-C99 was employed. Relaxation measurements were carried out with U-15N,2H(70%)-C99. Between NMR experiments, NMR samples were stored at 4 °C where they remained stable almost indefinitely. Samples were tested for stability at 45 °C, and the kinetics of C99 precipitation was found to be inversely related to the concentration of LMPG. At 45 °C, C99 was observed to remain stable for 6 weeks in the presence of 9% LMPG, hence the use of high detergent concentrations in NMR samples.
Backbone NMR Resonance Assignments and Secondary Structural Analysis
3-D NMR data were recorded at 45 °C on a Varian 900 NMR spectrometer equipped with a conventional probe at the University of Georgia. The sequential backbone resonance assignments for U-2H(70%), 15N,13C-labeled C99 were carried out using TROSY-based triple resonance experiments (48): HNCA, HNCACB, HN(CO)CA, HN(COCA)CB, and HNCO with deuterium decoupling. NMR data were processed using NMRPipe (49) and analyzed using NMRView software (50) or Felix (FelixNMR, Inc.). Sequential connectivities were established by [15N-1H]-TROSY-HNCA and [15N-1H]-TROSY-HNCACB experiments, which provided intraresidue and sequential cross-peaks of Cα and Cα/Cβ, respectively.
Backbone chemical shifts were assessed in terms of secondary structure using chemical shift index (51) analysis in NMRView.
Paramagnetic Probe-Induced NMR Line Broadening Experiments
The 800 MHz 1H,15N-TROSY (52)spectrum of U-15N-C99 was monitored as C99 was titrated with either 16-doxylstearic acid (16-DSA) or Gd(III)-diethylenetriaminepentaacetic acid (Gd-DTPA, Aldrich, Milwaukee, WI). 16-DSA was titrated over a concentration range of 0–0.8 mM to a 1 mM U-15N-C99 sample from a stock solution consisting of 8 mM LMPG, 31 mM 16-DSA, 10% D2O, and 240 mM imidazole, pH 6.5. Peak intensities were measured at each titration point to assess the amount of paramagnet-induced line broadening.
Preliminary titrations with Gd-DTPA led to TROSY spectra of C99 that exhibited not only line broadening but also small peak shifts, the latter most likely resulting from transient coordination of Gd(III) ligand sites with Asp or Glu side chains on the protein. It was possible to suppress these unwanted interactions between ion and protein by simultaneously titrating the protein sample with Gd-DTPA and EDTA. The presence of additional chelating agent serves to effectively cap available Gd(III) coordination sites. Gd-DTPA was added over a concentration range of 0–11 mM to a 1 mM U-15N-C99 sample from a stock solution consisting of 150 mM Gd-DTPA, 250 mM free EDTA, and 150 mM imidazole, pH 6.5. During the course of this titration, the C99 concentration was diluted by 7%.
NOE and Relaxation Analysis
All experiments were carried out on a 900 MHz spectrometer equipped with a cryoprobe. Relaxation and steady-state NOE measurements were collected using U-15N,2H(70%)-C99.
The gNhsqc sequence in Biopack (Varian, Palo Alto, CA) was modified for the measurements of 15N T1 and T2 relaxation times in TROSY spectra. For T1, τ values of 0.01, 0.1, 0.25, 0.5, 0.8, 1.1, 1.4, 2.5, and 4.5 s were employed. The acquisition time was 116.5 ms and the delay between scans was 2 s. T2 relaxation times were determined using τ values of 0.01, 0.03, 0.05, 0.07, 0.09, and 0.11 s. The acquisition time was 116.5 ms and the delay between scans was 2.8 s. For resonances with T2 shorter than 30 ms, T2 values were determined from two time points: one at 10 ms and the other at 30 ms. Steady-state 1H-15N NOEs were measured by acquiring a pair of spectra (with and without amide proton presaturation) using the gNNOE pulse program in Biopack. Saturation of the amide protons was achieved by a train of 27.4 μs π pulses for 3 s. The total delay between scans was 7 s. Heteronuclear NOEs were obtained from the ratio of the resonance volume from the spectrum with presaturation to the volume from the spectrum without presaturation.
Amide–amide NOEs were measured using the 3D gnoesyNhsqc experiment in Biopack and a conventional probe. A mixing time of 120 ms was employed. Exchange cross-peaks to water were classified as strong, weak, or no exchange. Ideally, classification would be based on a percentage of the diagonal peak intensity. However, diagonal peaks often varied in intensity and overlapped extensively. Instead, the exchange cross-peaks between amide proton and water were classified as strong or weak based on their intensity relative to other NOE cross-peaks to the same amide proton. This is a reasonable based on the fact that most amide protons have at least one cross-peak to a non-water proton at a distance of 3.0–3.5 Å. If the exchange peak had the largest intensity, the amide proton resonance was classified as being strongly exchanging. Smaller exchange peaks were labeled as weak, reflecting slower exchange rates. Many amide protons exhibited no exchange cross-peak and were classified as nonexchanging.
Titration of C99 with Cholesterol Analogues
Cholesterol has a very low solubility in LMPG micelles and in most other types of detergent micelles. We therefore employed a water-soluble cholesterol analogue, β-CHOLBIMALT: β-cholesteryl-(maltosyl-β-(1,6)-maltoside) (Anatrace, Maumee, OH). 1H,15N-TROSY spectra of U-15N-C99 were recorded in LMPG micelles as β-CHOLBIMALT was titrated over a concentration range of 0–64 mol % (moles of β-CHOLBIMALT/(moles of β-CHOLBIMALT + moles of LMPG) × 100). The starting C99 solution was 550 μL and contained 2 mM protein. β-CHOLBIMALT was added from a 15% stock solution (in 10% D2O).
In order to carry out a limited titration using actual cholesterol, a stock 5% LMPG solution that contained 5 mol % cholesterol was prepared by codissolving LMPG and cholesterol in a 95:5 mixture of benzene:ethanol. This solution was then lyophilized, and the resulting powder was redissolved in 250 mM imidazole, pH 6.5, and then aliquoted into purified C99 solutions. Following subsequent concentration of the sample to 600 μL for NMR, the total LMPG concentration was 9%, while the cholesterol concentration was 5 mol %. 1H,15N-TROSY spectra were acquired for each sample. Efforts to find conditions that allowed higher concentrations of cholesterol to be attained in C99/LMPG samples were not successful.
Reported chemical shift changes represent a composite of the β-CHOLBIMALT-induced changes in the proton and nitrogen-15 chemical shifts according to the equation (53):
| (1) |
Preparation of Nitroxide Spin-Labeled C99
Wild-type C99 has no cysteine residues. To facilitate site-directed spin labeling of C99, a series of single-cysteine mutant forms of the protein (F675C, Q686C, E693C, T729C, H732C, and S750C) were generated using QuickChange site-directed mutagenesis (Stratagene). Each mutant was purified as described above for WT, except that 2 mM β-mercaptoethanol was added to the lysis buffer and 0.5 mM dithiothreitol (DTT) was added to all column chromatography buffers. Following purification, C99 in elution buffer was concentrated to 10 mg/mL, and DTT was increased to 1 mM to reduce intermolecular disulfide bonds. To these concentrated C99 samples, 4 mM 2,2,5,5-tetramethyl-3-pyrroline-3-methyl)methanethiosulfonate (MTSL; Toronto Research Chemicals) was added from a 75 mM stock solution in methanol (stored at −80 °C between use). The solution was gently agitated for 1 h at room temperature and then for 2 h at 37 °C, being preserved under argon throughout. MTSL-labeled DTT and excess MTSL were then removed from the sample by repeatedly diluting the sample with 15 mL of a 250 mM imidazole buffer (pH 6.5) and then concentrating with an Amicon Ultra-15 centrifugal filter device. Four to five iterations of this process were sufficient to eliminate undesired contaminants. D2O was then added to 10%, and EDTA was added to 1 mM. The sample was then concentrated to a level suitable for NMR (ca. 1 mM).
Titration of NMR-Labeled C99 with Spin-Labeled C99
The TROSY NMR spectrum of wild-type U-15N-C99 in LMPG micelles was monitored as it was titrated with a 15N-unlabeled Thr729Cys mutant form of C99 that had been modified at its lone cysteine with a nitroxide spin label. The starting sample, which contained only the U-15N-labeled protein at a concentration of 1.2 mM, was titrated over a range of 0–3.2 (molar ratio of spin-labeled C99 to 15N-labeled C99) using a stock solution containing 3.0 mM MTSL-labeled Thr729Cys in LMPG. TROSY spectra were acquired at each point, with the number of scans being adjusted so that the inherent sensitivity of all spectra was equal (accounting for the dilution of NMR-labeled C99 that occurred during this titration).
NMR peak line widths at half-maximal height (Δν1/2) for ten selected resonances were averaged and plotted as a function of the ratio of the spin-labeled subunit to the NMR-labeled subunit. If the assumption is made that the inclusion of the spin label does not bias the formation of dimers and that C99 dimers can exchange subunits at a rate that is rapid on the NMR time scale, then the population of each dimer (homodimeric NMR-labeled [A], homodimeric spin-labeled [B], and the NMR/spin-labeled heterodimer [C]) can be related to the molar ratio (x) of spin-labeled C99 to NMR-labeled C99 by the relations:
| (2) |
| (3) |
| (4) |
Only A and C (which contain 15N-labeled protein) will exhibit NMR resonances; therefore, only the levels of these two species need be considered in predicting spectral parameters produced by this collection of molecules. Based on this, the following equation predicts the resonance line widths:
| (5) |
The starting average line width is Δν1/2,A (the observed line width for the point at which χ = 0). The preceding equation can be adjusted for the line width of species C (Δν1/2,C) in order to optimally fit the model to the data. NLB is a nonspecific paramagnetic line broadening factor. Simulations were carried out to find the best match between the actual data and simulated data.
Glutaraldehyde Cross-Linking of C99
C99 samples in 1% LMPG and 50 mM PIPES (pH 6.5) were cross-linked by the addition of glutaraldehyde from a fresh 25% aqueous solution to a concentration of 16 mM. The solution was gently agitated for 12 h at room temperature, and aliquots were then analyzed by SDS–PAGE.
Results
Assignment of NMR Resonances for C99 in LMPG Micelles
Human C99 was expressed in E. coli and purified into micelles composed of LMPG, a phospholipid-derived detergent that generally maintains membrane proteins in native-like structural and functional states (54–56) (Figure 1). Backbone 15N, 13CO, 13Cα, and 13Cβ NMR resonances were then assigned (Figure 2) using a series of TROSY-based 3-D experiments. Assignment benefited greatly from collection of key data sets at 900 MHz. 3-D spectra acquired at 800 MHz had insufficient spectral dispersion to permit resonance assignments. Even at 900 MHz, a number of assignments proved elusive because of severe resonance overlap or extreme line broadening arising from intermediate time scale exchange. Nevertheless, 85% of the backbone and Cβ resonances could be successfully assigned, and these have been deposited in BioMagResBank (entry 15775). The assignments enabled a series of experiments to elucidate key features of C99's three-dimensional structure and dynamics, as described below.
Figure 1.
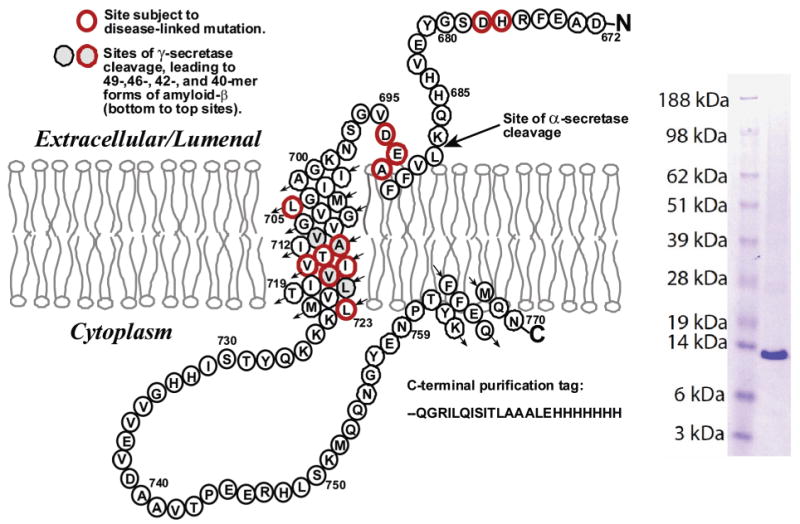
Sequence and topology of C99 (left) and SDS–PAGE gel with Coomassie staining of the purified protein (right). The locations of the transmembrane helix, the surface-associated extracellular helix at sites 690–693, and the surface-associated C-terminal helix at sites 761–770 reflect results from this work.
Figure 2.
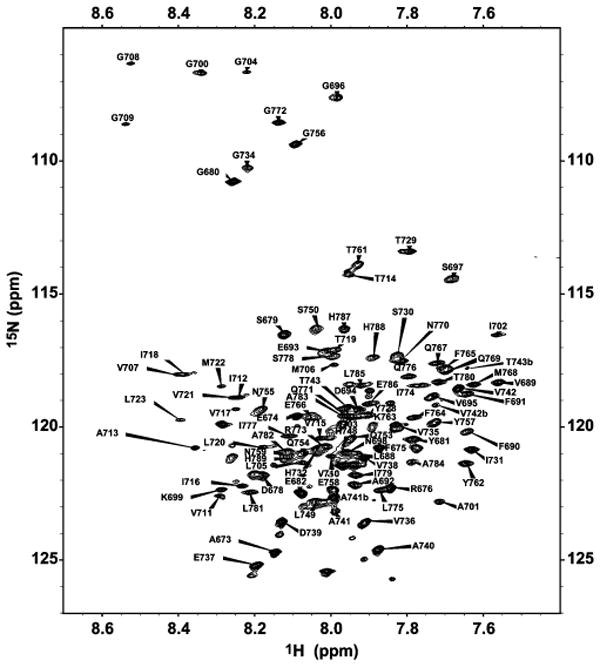
900 MHz 1H,15N-TROSY spectrum of U-2H(70%),13C,15N-C99 in LMPG micelles showing resonance assignments. The pH was 6.5 and the temperature was 45 °C.
C99 Is a Homodimer in LMPG Micelles
Results from other laboratories have suggested that C99 or its derived TMD is homodimeric under both membrane and model membrane conditions (43–46). We therefore examined C99's oligomeric state in LMPG micelles. Chemical cross-linking with glutaraldehyde led to formation of covalently linked dimers with moderate efficiency (Figure 3A). Moreover, unlike certain monomeric single-span membrane proteins such as KCNE1 (56), single-cysteine mutant forms of C99 were observed to be highly susceptible to disulfide-mediated dimerization in LMPG micelles, even in the presence of high concentrations of the thiol reducing agent, dithiothreitol (Figure 3B–D). C99 dimerization was also supported by titration of U-15N-labeled wild-type C99 with a T729C mutant form of C99 that was not NMR-labeled but was thiol-derivatized with a nitroxide spin label. This titration resulted in line broadening in the 1H,15N-TROSY spectra of the NMR-labeled subunit that clearly exhibits a dependence on the molar ratio consistent with the protein forming a statistically ideally mixed distribution of homo- and heterodimers (Figure 3E).
Figure 3.

C99 dimerizes in LMPG. (A) Glutaraldehyde cross-linking of C99 in LMPG micelles as followed by SDS–PAGE/Coomassie. The first lane shows C99 WT in the absence of a cross-linking reagent; the second lane shows C99 WT following cross-linking with glutaraldehyde. (B) SDS–PAGE of single-cysteine mutant forms of C99 following incubation in LMPG in the presence of DTT. The concentration of protein in LMPG was 10 mg/mL (0.73 mM), and the concentration of DTT was varied between 0 and 4 mM, as indicated on the figure. (C) SDS–PAGE of C99 F675C in the presence of varying concentrations of DTT. The C99 concentration in LMPG micellar solutions was 1.5 mg/mL (0.11 mM), and the concentration of DTT was varied between 0 and 10 mM, as indicated on the figure. (D) SDS–PAGE of C99 F675C in the presence of increasing quantities of DTT. The concentration of protein was 10 mg/mL (0.73 mM), and the concentration of DTT was varied between 0 and 10 mM, as indicated on the figure. Significant disulfide-mediated dimerization was observed even in the presence of ca. 30 equiv of reducing agent per cysteine. (E) Nitroxide spin-labeled C99 T729C was titrated into a solution of U-15N-labeled C99 WT in LMPG micelles. The average of the 1H line widths for ten 1H,15N-TROSY NMR peaks (V689, F691, G696, G700, A701, I702, G704, G708, G709, M722) is plotted as a function of the T729C:WT molar ratio (open circles). Also plotted are simulated data for the ideal case where C99 (15N-labeled and nitroxide-labeled) is assumed to form homo- and heterodimers with freely exchanging subunits (closed circles).
Secondary Structure of C99 in LMPG Micelles
Inspection of the backbone chemical shifts of C99 using chemical shift index (CSI) analysis reveals three helical segments (Figure 4). Other parts of the protein appear to lack a regular secondary structure. An α-helix that includes the transmembrane domain (TMD) extends from Asn698 through Leu723, where it is terminated by three consecutive lysines. A second short α-helical segment (Phe690 through Glu693) is located in the extracellular domain between the site of α-secretase cleavage (after Lys687) and the start of the TMD. The third helix is observed at the extreme C-terminus of C99, starting at Thr761 and extending through N770. This latter observation is reminiscent of results from previous NMR studies of the isolated cytosolic domain of C99 under micelle-free conditions showing this segment to have transient helical character (38, 39). It is acknowledged, however, that our studies of C99 were carried out in the presence of a 22-residue C-terminal purification tag, which could promote the formation of this third helix, although it is much more likely that formation of a stable C-terminal helix results from its capacity to form an amphipathic helix that binds with modest avidity to the micelle surface (see below). The retention of the tag in these studies was justified by the observation that tags at the C-termini of APP or C99 do not interfere with their proteolytic processing (57–60).
Figure 4.
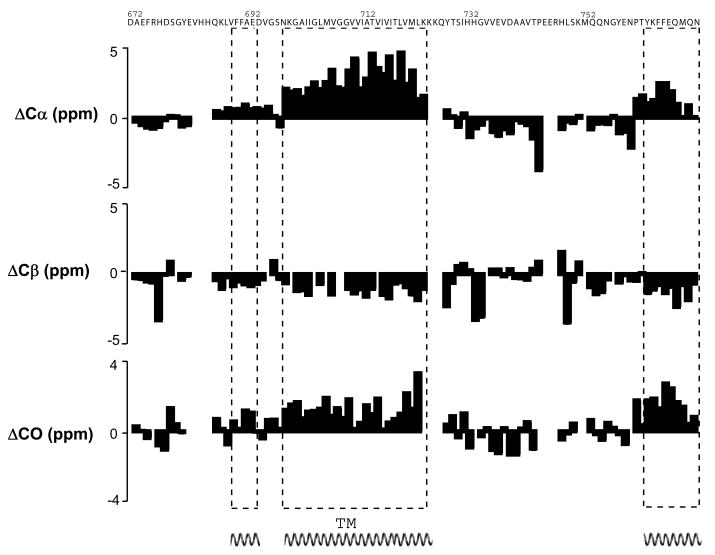
Chemical shift index (CSI) data and analysis for C99 in LMPG micelles. The ΔC values represent the differences between the observed 13C chemical shifts and standard residue-specific random coil 13C shift values for the backbone and Cβ carbons. As shown, these data are consistent with three α-helical segments.
Dynamics of C99 in LMPG Micelles
NMR relaxation measurements (Figure 5) are consistent with the results of CSI analysis and offer additional insight by showing that the nonhelical domains of C99 are flexible, suggesting that they are predominantly unstructured. These domains include both the extreme N-terminus (residues 672–682) and cytosolic residues 724 to 760, which connect the TMD and the C-terminal helix. The TMD, on the other hand, is seen to be uniformly immobile, indicating that its relaxation properties reflect the overall correlation time of the C99/LMPG micellar assemblies. A correlation time of 26 ns was calculated from the average T1/T2 ratio for amide 15N sites in the TMD and is consistent with the homodimeric oligomeric state of C99. We recently determined a correlation time of 21 ns for the single-span membrane protein, KCNE1, in LMPG micelles (56). The tagged form of KCNE1 has 137 residues, slightly longer than the tagged form of C99 examined in this work (121 residues). Assuming the KCNE1/LMPG micellar aggregate is ca. 60 kDa, the 5 ns increase in correlation time observed for C99/LMPG would correspond to an additional 14 kDa in mass, a value in agreement with C99's putative homodimeric state.
Figure 5.
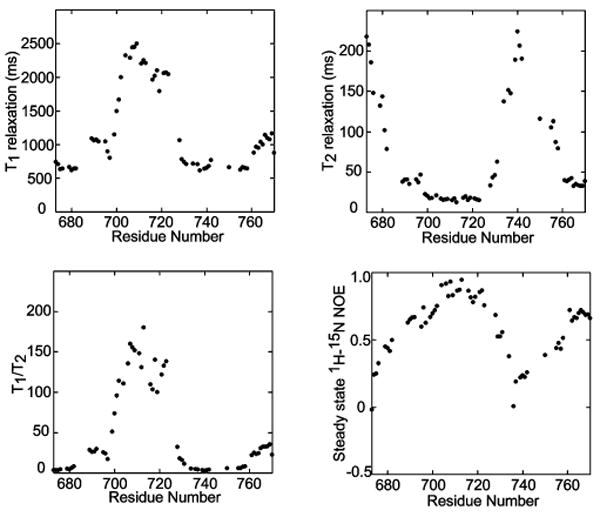
900 MHz backbone amide 15N T1 and T2 relaxation times and steady-state 1H–15N NOEs for U-15N,2H(70%)-C99 in LMPG micelles at pH 6.5 and 45 °C.
For the two extramembrane helices (residues 690–693 and 761–770), T2 values were observed to be much lower (and the T1/T2 ratios much higher) than for the adjacent disordered segments. This difference indicates that these helices are immobilized, although not to the extent of the TMD. As supported by additional data below, this appears to reflect avidity of these segments for the micellar surface.
Amide Exchange and Paramagnetic Probes of C99's Topology
The topology of C99 with respect to the micellar model membrane was probed by examining backbone amide proton exchange with water. Figure 6 summarizes the degree of water accessibility to amide sites in C99 as observed in NOESY spectra. While extensive resonance overlap and missing assignments precluded the evaluation of exchange for all sites, the data reveal that the backbone amide protons within the transmembrane domain undergo only slow exchange with protons from water, consistent both with their location in the apolar phase of the membrane and also with a lack of high amplitude dynamics of this domain, which would allow sites to make frequent excursions outside the micellar interior to the soluble phase where helices would be destabilized and exchange could occur. As expected, all sites observed to be disordered in the relaxation measurements were also seen to undergo relatively rapid proton exchange with water. For the two extramembrane helices associated with membrane surface (residues 690–693 and 761–770), some of the backbone amide protons exhibited a modest reduction in exchange with water relative to the disordered segments. In the case of the C-terminal helix, other sites showed strong exchange with water. These data suggest that both helices are at least partially water-exposed and that the stability of the C-terminal helix is modest.
Figure 6.
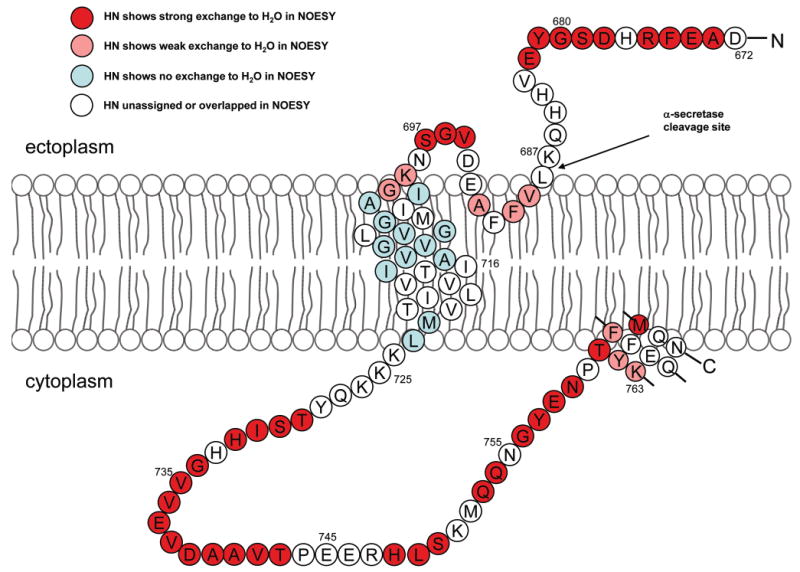
Site-specific backbone amide proton/water exchange as detected in 900 MHz NOESY spectra of U-15N-C99 in LMPG micelles at pH 6.5 and 45 °C.
A more detailed map of protein topology with respect to the membrane was obtained by assessing backbone amide proton accessibility to the polar and nonpolar paramagnetic probes Gd-DTPA and 16-doxylstearate (16-DSA). Paramagnet-induced reductions in peak intensities were recorded from TROSY spectra acquired in both the absence and presence of these probes (cf. Figure 7). These measurements are reported in Figure 8 for those peaks that are both assigned and sufficiently well resolved to allow unambiguous measurement of peak intensities. As expected, the disordered N-terminal residues 673–682 were fully exposed to the water-soluble Gd-DTPA and experienced relatively little line broadening in the presence of the lipophilic 16-DSA. The largely disordered membrane-proximal C-terminus that extends to site 761 is somewhat more complex. While many sites are fully exposed to Gd-DTPA and are largely inaccessible to 16-DSA, several sites (especially in segment containing residues 729–735) exhibit some degree of protection from Gd-DTPA and also some exposure to 16-DSA. This complexity may reflect transient interactions of this segment with the micelle surface as a result of the presence of apolar amino acids and nonrandom conformational preferences (as also reflected by deviations of the backbone chemical shifts from their random coil values; see Figure 4). Given that C99 appears to form dimers in LMPG, there also exists the possibility that there might be some inter-subunit contacts that also impact site accessibility to Gd-DTPA.
Figure 7.
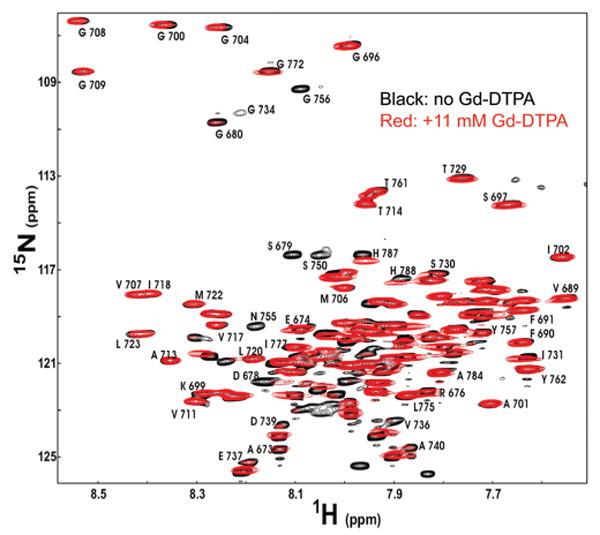
Effects of Gd-DTPA on 800 MHz 1H,15N-TROSY peak intensities for 1 mM U–15N-C99 in LMPG micelles at pH 6.5 and 45 °C. The black peaks represent the 1H,15N-TROSY spectrum of C99 in the absence of a paramagnetic probe. The superimposed red spectrum was acquired after addition of Gd-DTPA to a concentration of 11 mM.
Figure 8.
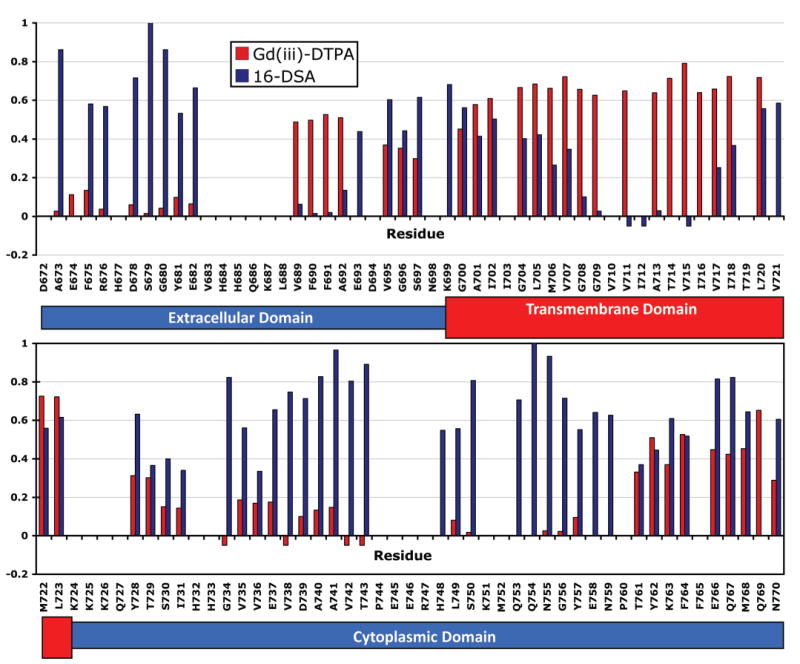
Site-specific reductions in 800 MHz 1H,15N-TROSY peak intensities for C99 as a result of adding either a water-soluble paramagnetic probe (Gd-DTPA) to 11 mM or a lipophilic probe (16-DSA) to 0.8 mM (2 mol %) to samples of U-15N-C99 in LMPG micelles at pH 6.5 and 45 °C. These experiments were performed twice, and the plotted values represent an average of the two measurements. A negative bar indicates that the peak was broadened by the paramagnet to the extent that it became effectively undetectable. The absence of a bar indicates that data were unavailable because of extensive peak overlap or lack of residue assignment. Peak intensity ratios were not corrected to account for the modest degree of sample dilution that occurred during the course of the titration (3% for 16-DSA and 7% for Gd-DTPA).
The C-terminal helix extending from Thr761 to N770 is observed to be largely inaccessible to both 16-DSA and Gd-DTPA (Figure 8). This is almost certainly due to association of this helix with the micelle surface. At the surface this helix is not well-accessed by the nitroxide group near the end of the stearate chain of the lipophilic 16-DSA. This trend is also observed for sites near both ends of the TMD. These interfacial sites are also seen to be largely inaccessible to Gd-DTPA, which may be surprising. The most likely explanation is that the water-exposed face of the C-terminal helix sits roughly at the same level as the negatively charged head groups of LMPG such that Gd-DTPA, which has a net charge of −2, is repelled from proximity to the negatively charged micelle surface.
The central residues of the TMD (708–715) are almost completely inaccessible to the Gd-DTPA and, conversely, are completely accessible to the nitroxide of 16-DSA (Figure 8). This suggests that the intramembrane subunit-subunit interface for homodimeric C99 is located N-terminal to the Gly708Gly709 segment such that the C-termini of the twin TMDs extend away from each other in a scissor-like fashion from the dimer interface. Closer to the micelle surface within the TMD (on both sides), residues remain inaccessible to Gd-DTPA and become gradually more inaccessible to 16-DSA as the ends of the TMD are approached.
A surprising result from the paramagnetic probe experiments concerns the juxtamembrane extracellular domain extending from the site of α-secretase cleavage to the start of the transmembrane helix at Asn698. Phe690 and Phe691 in the extracellular helix are observed to be fully exposed to the nitroxide probe of 16-DSA (Figure 8), which indicates that these sites must extend fairly deeply into the model membrane surface. Val689, which precedes this helix, also exhibits considerable accessibility to 16-DSA. On the other hand, Ala692 is at least partially protected from interactions with 16-DSA, whereas the peak from E693 undergoes only modest broadening from 16-DSA. These results suggest that the extracellular helix sits at the membrane surface with its N-terminal end more deeply buried than its C-terminal end.
Residues 694–697 that link the extracellular and transmembrane helices must form a loop or turn, as supported by the chemical shift and NMR relaxation data (Figures 4 and 5). At least three of these four residues also undergo facile proton exchange with water (Figure 6). However, this segment is largely inaccessible to both 16-DSA and Gd-DTPA (Figure 8). While it is possible that the observed inaccessibility to Gd-DTPA may arise partly from the proximity of this segment to the negatively charged micelle surface (that appears to repel the negatively charged Gd-DTPA, as noted above), it is also possible that these data reflect a relatively crowded tertiary/quaternary environment for these sites, an environment that may encompass both the adjacent homodimer interface and the extracellular helix.
Cholesterol Induces Specific and Saturable Changes in the NMR Spectrum of C99
Because cholesterol is thought to play an important role in the trafficking and proteolytic processing of APP and C99, the consequences of cholesterol addition on the structural properties of C99 were examined. Cholesterol is notoriously difficult to solubilize in detergent micelles; nevertheless, a cholesterol content of 5 mol % was attained in C99-containing LMPG micelles. The TROSY spectrum of C99 in this sample exhibited small but significant changes in many resonance chemical shifts as a result of the presence of cholesterol (Supporting Information Figure 1). The solubility limit of cholesterol in micelles precluded a full titration. Therefore, we turned to the use of novel cholesterol analogues that have enhanced cosolubility with detergent micelles: specifically, the α and β anomers of a cholesterol derivative glycosylated by a tetrose (β-(maltosyl-β-1,6-maltoside)) group, an addition that confers aqueous solubility to these compounds.2 These compounds are referred to as α- and β-CHOLBIMALT (the chemical structure of the latter is depicted in Figure 9). The changes in the NMR spectrum of C99 that were induced by cholesterol were generally very similar to those induced by a comparable level of β-CHOLBIMALT (Supporting Information Figure 1). This prompted a titration of C99 in LMPG with β-CHOLBIMALT over a concentration range of 0–64 mol %, which spans the range of cholesterol concentrations present in various mammalian membranes. The superimposed TROSY series for this titration is shown in Figure 9. For those peaks exhibiting the greatest chemical shift perturbation, the induced changes in peak position as a function of β-CHOLBIMALT concentration were found to be saturable, with half-maximal changes occurring at about 15 mol % (Figure 10A, inset). Figure 10A shows the changes in chemical shift that were induced by 32 mol % β-CHOLBIMALT. While some of the smaller (<0.1 ppm) changes could arise from glycoside-specific interactions, the large changes in the positions of the TROSY peaks of Gly696, Ser697, and Lys699 as a result β-CHOLBIMALT addition are striking. That the observed perturbations at these sites were induced by the cholesterol moiety of β-CHOLBIMALT is supported by the fact that these peaks also exhibited the greatest chemical shift perturbation in response to the addition of bona fide cholesterol at 5 mol % (Supporting Information Figure 1).
Figure 9.
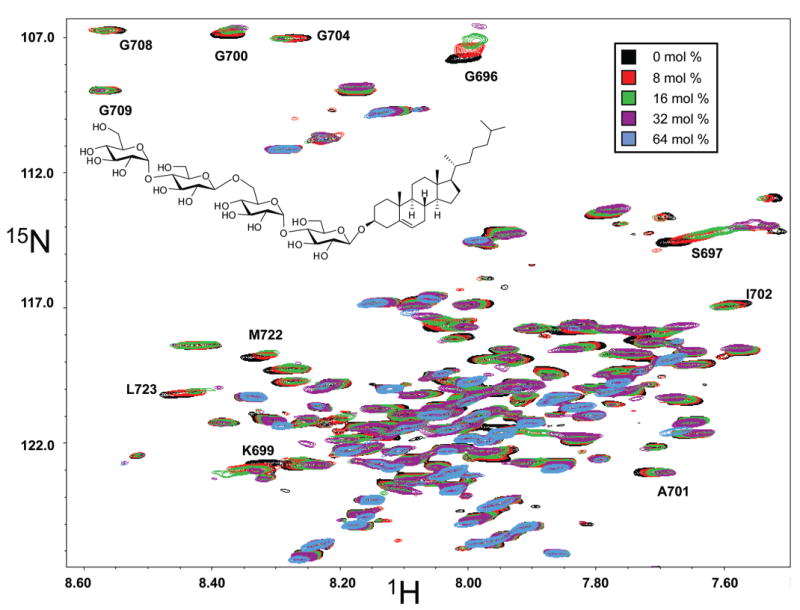
Titration of U-15N-C99 in LMPG micelles at pH 6.5 and 45 °C with the cholesterol analogue β-CHOLBIMALT, as monitored by 800 MHz 1H,15N-TROSY NMR spectroscopy. TROSY spectra for C99 (initial concentration of 2 mM) are superimposed and correspond to β-CHOLBIMALT concentrations of 0 mol % (black), 8 mol % (red), 16 mol % (green), 32 mol % (purple), and 64 mol % (blue). A number of peaks broaden and disappear by the end of the titration. Resonance assignments are shown for peaks exhibiting the greatest perturbation in chemical shift and/or line width.
Figure 10.
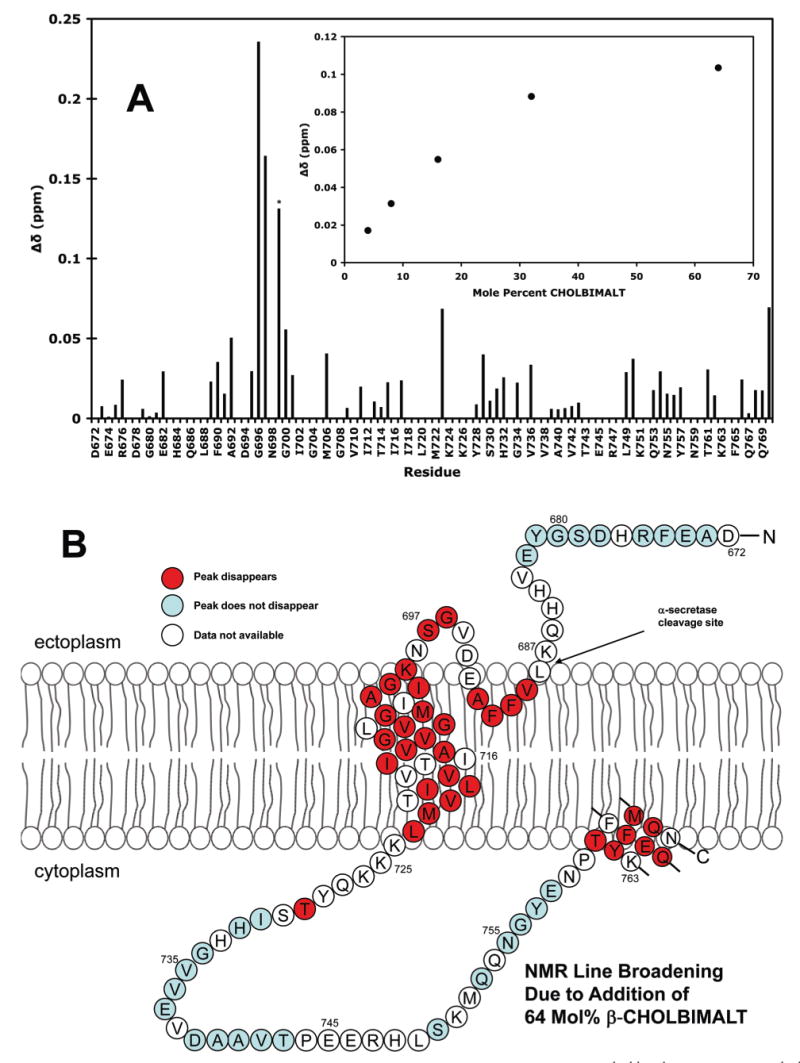
(A) Effect of 32 mol % β-CHOLBIMALT on the 1H,15N-TROSY chemical shifts of C99. Points lacking data represent unresolvable/undetectable peaks or peaks that disappeared during the course of the titration due to excessive broadening. The reported chemical shift changes represent a composite of the β-CHOLBIMALT-induced changes in the proton and nitrogen-15 shifts (see Methods). Inset: The average Δδ values for the three C99 peaks that undergo the greatest perturbation (S697, K699, G700) are plotted as a function of the mol % of β-CHOLBIMALT. The asterisk above K699 indicates that this peak split into multiple components at high β-CHOLBIMALT concentrations and that the value reported here is for the component that exhibited the greatest shift. (B) TROSY NMR peak disappearance (due to excessive line broadening) for C99 as a result of the presence of 64 mol % β-CHOLBIMALT. Sample and spectroscopic conditions are the same as described in the caption of Figure 9.
To assess whether the spectroscopic changes induced by the cholesterol analogue reflect changes in protein structure and/or local motions, the oligomeric state, dynamics, and paramagnetic probe accessibility of C99 were examined in samples that contained 16 mol % β-CHOLBIMALT. SDS–PAGE of C99 following cross-linking by glutaraldehyde demonstrated no difference in the amount of covalent dimer in the presence or absence of the cholesterol analogue, indicating β-CHOLBIMALT does not alter C99's propensity to homodimerize (data not shown). A full set of 900 MHz T1, T2, and 1H-15N steady-state NOE measurements indicated no major changes in local structural dynamics (Supporting Information Figure 2), although the membrane-associated domains exhibited uniformly elevated T1/T2 ratios that reflect slower overall tumbling of the C99/LMPG/CHOLBIMALT mixed micelles relative to C99/LMPG mixed micelles. This was confirmed by the observations that certain peaks were broadened beyond detection by the addition of β-CHOLBIMALT to 64 mol % and that these peaks were localized almost exclusively to the three micelle-associated helical segments (Figure 10B). Finally, a full set of 16-DSA and Gd-DTPA measurements revealed no major changes in the patterns of site accessibility to these paramagnetic probes (Supporting Information Figure 3). Altogether, these results indicate that interactions of C99 with cholesterol-like compounds do not result in any major changes in conformation or dynamics. However, C99 appears to bind β-CHOLBIMALT in a manner that is both saturable and appears to involve direct interactions with the loop connecting the extracellular helix to the transmembrane domain.
Discussion
The Aβ peptides that are derived from the N-terminus of C99 have been the subject of extensive structural characterization in solution, when bound to model membranes or as part of aggregates (25–33). Similarly, the cytosolic C-terminal domain of C99 has been examined in several careful structural studies in solution (38–40). A study of a polypeptide that corresponds to the TMD of C99 indicated that this peptide forms α-helical homodimers in SDS micelles (43). Also, recent CD, limited proteolysis, and solid-state NMR studies of a fragment containing the TMD and parts of the flanking juxtamembrane domains (residues 684–726) suggest that the secondary structure and dynamics of this polypeptide are sensitive to membrane fluidity (47). However, beyond a series of investigations that have suggested it forms homodimers (43–46), the structure of full-length C99 has not been examined. The results of this paper offer the first detailed insight into C99's conformation and motional dynamics and also set the stage for future studies to complete determination of its structure at high resolution. The results also establish that C99 binds cholesterol.
The studies of this paper were carried out using model membranes consisting of LMPG micelles. With its long (C14) saturated acyl chain, LMPG most likely forms large micelles with a diameter that approaches the hydrophobic span typical of lipid bilayers. LMPG is also a mild and nondenaturing detergent (54–56). For these reasons, we will tentatively assume, throughout the following discussion, that the structural conclusions reached in our studies of C99 in LMPG micelles can be extrapolated to C99 under bilayer conditions. We acknowledge, however, that only future structural studies will disclose the validity of this assumption.
The Transmembrane Domain of C99 Is a Classical α-Helix
The results showed that the transmembrane domain of C99 is encompassed by a 26-residue α-helix that starts at Asn698, entering the membrane at Gly700 (±1 residue) and ending after Leu723. This conclusion is consistent with the results of computational predictions (61), but is at variance with two studies that concluded the TMD of C99 spans only 12–18 residues (62, 63). Grziwa et al. argued that the TMD was only 12 residues long based on cysteine scanning mutagenesis of the TMD in conjunction with measurements of Cys site accessibility to thiol-reactive chemical probes (62). This approach likely underestimated the true length of the TMD because of helical “bobbing” along the bilayer normal that would allow water-soluble chemical probes to access and modify normally transmembrane sites when they become transiently exposed to the aqueous phase (see ref 64). In the second study, Tischer and Cordell concluded the TMD is 18 residues long (63) based on mutagenesis data showing that sites C-terminal to the sites of γ-secretase cleavage can be deleted or replaced with polar residues without disrupting proteolytic processing or interfering with membrane integration of the molecule as a whole. However, their “Asp walking” technique might not accurately predict the actual TMD length because the placement of polar residues near the boundary of the transmembrane region might be accommodated by “snorkeling” of the side chain to the membrane surface and/or by ion pairing of the Asp side chain with one of the three membrane-proximate WT lysine residues.
The transmembrane domain of C99 appears to be a single unbroken α-helix. Moreover, within the experimental uncertainty of the NMR relaxation measurements and as supported by both NOESY-based water–amide hydrogen-exchange measurements and paramagnetic probe accessibility measurements, the TMD of C99 appears to be conformationally well ordered. The sites of γ-secretase cleavage that lead to release of Aβ40 and Aβ42 (V711 and A713, respectively) are observed by paramagnetic probe measurements to be located in the most hydrophobic region of LMPG micelles, where their amide protons are maximally accessible to the hydrophobic probe 16-DSA (Figure 8). This indicates that these sites are not involved in the dimer interface (see below) and that these scissile sites are not recognized by γ-secretase on the basis of any unusual backbone conformational or dynamic features.
The γ-secretase complex includes the presenilin 1 (PS1) catalytic subunit and is a highly promiscuous protease that is capable of cleaving numerous type I membrane proteins (61). Extensive bioinformatic analyses of its substrates have failed to reveal any characteristics at the level of primary structure that would predispose a transmembrane (TM) protein for γ-secretase cleavage, except that known cleavage sites tend to be in parts of TM helices that are predicted to be subject to modest destabilization by surrounding residues (61). γ-Secretase appears to be capable of cleaving almost any type I transmembrane peptide provided that the substrate protein has undergone ectodomain shedding (61, 65). Mutagenesis studies have shown that a variety of APP mutations in proximity to the γ-secretase cleavage sites are tolerated (66, 67). Thus, it is not unexpected that the TMD of C99 would be structurally unremarkable. Both the local disruption of TM helices to expose the substrate scissile bond and the access of water to this site appear to be mediated by the γ-secretase complex (59, 61, 65, 68) rather than by any peculiar intrinsic properties of C99 that would facilitate water access to the scissile sites. For C99, the transmembrane helix surrounding the scissile sites is thought to be moderately destabilized by the presence of adjacent β-branched amino acids (61), thereby predisposing the helix to local unfolding within the γ-secretase active site to expose the scissile amide bonds to nucleophilic attack.
A number of FAD mutations within the APP TMD increase the Aβ42:Aβ40 production ratio (69). Several of these, such as Val715Ala, Ile716Val, Val717Ile, and Val717Leu, are very conservative and would not be predicted to significantly perturb the stability or structure of the transmembrane helix. This suggests that these mutations promote Aβ42 by exerting either very subtle conformational perturbations within the transmembrane domain or, more likely, that they alter the relative energetics of alternative binding modes by which the mutated substrate can interact with γ-secretase, favoring cleavage after Ala713 and release of Aβ42.
Homodimerization of the Transmembrane Domain of C99
C99 was observed to form dimers in LMPG micelles, which is consistent with results from other laboratories showing that C99 or its derived TMD is homodimeric under both membrane and model membrane conditions (43–46). At least three structural models have been proposed for homodimerization of C99, all involving its GXXXG motifs. GXXXG motifs are responsible for homodimerization of glycophorin A and many other membrane proteins (70, 71), sometimes singly and sometimes in tandem (72). C99 has three GXXXG motifs, one being G696SNKG700 in the juxtamembrane domain, with the others being located in the TMD (G700AIIG704, and G704LMVG708). In the model of Kielan-Campard et al., all three of these motifs are proposed to simultaneously participate in the helix–helix contacts that are responsible for homodimerization (44). Because our NMR data show that the helix leading into the TM segment starts at Asn698 and because Gly708 is observed to be fully exposed to the hydrophobic paramagnetic probe 16-DSA (see Figure 8), this model is not well supported by the results of this work.
In the dimerization model of Gorman et al., it is not the canonical GXXXG motifs that are thought to be critical but rather the G709XXXA713 motif that encompasses the sites of gamma γ-secretase cleavage that lead to Aβ40 and Aβ42 (43). The model does not well explain the data of the present work, where Figure 8 shows that the amide protons at both G709 and A713 are fully exposed to the hydrophobic paramagnetic probe 16-DSA. It should be noted that the Gorman et al. study was limited to the isolated TMD polypeptide of C99. This polypeptide appears to dimerize more avidly than full-length C99, running as a dimer on SDS–PAGE gels (43) unlike wild-type C99, which runs as a monomer unless covalently cross-linked (Figure 3). This indicates that the free energy in favor of dimer formation is reduced by the presence of the native extramembrane domains. Indeed, the possibility cannot be ruled out that location of the dimer interface is different for full-length C99 than for the isolated TMD polypeptide studied by Gorman et al.
In the third (Munter et al.) model of C99 homodimerization, G700XXXG704 is thought to form the critical helix–helix contacts responsible for homodimerization of C99 (46). This model is well supported by the observed paramagnetic topology mapping, since both Gly700 and Gly704 are fairly inaccessible to both 16-DSA and to the hydrophilic Gd-DTPA (Figure 8). The full accessibility of the central section of the TMD to 16-DSA suggests that the lower parts of each TM helix in the homodimer extend away from the G700XXXG704 dimerization interface in a scissor-like fashion. However, disfavoring the Munter model is the observation that mutations of Gly704 sometimes result in enhanced dimerization (44), which would indicate either that the Munter model is incorrect or that there are alternative modes of dimerization that become dominant when the primary G700XXXG704 motif is disrupted. This latter possibility does seem reasonable given the presence of the seemingly “unused” GXXXG and GXXXA motifs.
The Extramembrane Domain of C99 Includes a Surface-Associated Helix That May be Critical for Processing of C99
While the extreme N-terminus of C99 appears to be largely disordered, our data revealed the presence of a short α-helix that extends from residues Phe690 through Glu693 (see Figure 1). Moreover, it was observed that the consecutive Phe residues in this helix are buried in the membrane surface, with the preceding Val and following Ala residues also being partially buried. These results can be compared to corresponding results from studies of the amyloid-β polypeptides under micelle-associated conditions. Studies of the Aβ polypeptides bound to SDS micelles under neutral pH conditions concluded that these polypeptides include an extramembrane helix that starts at Tyr681 or Gln686 and then terminates at Val695 (26, 30), which is of greater length than observed in this study of C99. A second helix in the Aβ polypeptides starts at essentially the same position as for the TMD of C99 (Lys699) and terminates several residues from their C-termini (at Val711 and Ala713 for Aβ42 and Aβ40, respectively). The significant structural difference between LMPG-micellar C99 and the SDS-micellar Aβ polypeptides likely reflects different modes of membrane interactions for these molecules. The C-termini of the Aβ peptides are located at sites that are near the middle of C99's intact TMD. Therefore, the C-terminal helix of the Aβ peptides, although composed of apolar amino acids, is not nearly long enough to span a membrane or micelle. The Aβ polypeptides must therefore be accommodated by a mode of membrane interaction that allows the charged C-terminus to access the water-exposed micelle surface. This reasoning is supported by studies of Jarvet et al. (28) of Aβ40 in SDS micelles using NMR and paramagnetic probes. That the C-terminus of the Aβ polypeptides and the TMD of C99 have completely different modes of interaction with micelles appears to have profound consequences on the structure of the adjacent extramembrane domain. This probably explains the difference in length between the extramembrane helix in C99 and that in Aβ. Significant differences in the physicochemical properties of SDS and LMPG micelles constitute another likely contributor to the structural differences between C99 and Aβ (73). Unlike SDS, LMPG is a mild and nondenaturing detergent. Like native phospholipids and unlike SDS, LMPG includes a polar but uncharged spacer moiety (glycerol) between its charged headgroup and apolar tail. The electrostatic potential of the exposed sulfoanionic surface of SDS micelles is also probably very different than for LMPG micelles, where the anions associated with the phosphodiester moiety of LMPG will be much more buried by the glycerol moieties that flank it on both sides.
The surface helix of C99 at sites 690–693 may serve as an anchor to provide structural organization to parts of C99 that are critical for its processing by both α-secretase and γ-secretase. Immediately preceding this helix is the site of α-secretase cleavage, located between Lys687 and Leu688 (see Figure 1). Immediately following is a loop that connects the surface helix to the TMD. This connecting loop includes Ser697 and Lys699, residues known to be essential for γ-secretase cleavage at Val711 and Ala713 to release the Aβ peptides (74, 75). As described below, these loop residues also appear to constitute the focal point for cholesterol binding by C99. The insertion of helix-breaking residues at sites immediately following the α-secretase scissile site has been previously shown to confer APP with resistance to cleavage by this protease (76–79). Interestingly, replacement of the hydrophobic Val689, Phe690, and Ala692 with helix-compatible polar residues did not disrupt cleavage (79), suggesting that the extramembrane helix does not need to directly interact with the membrane surface in order for α-secretase cleavage to occur. Rather, the key additional requirement for scissile site recognition by α-secretase appears to be the number of residues from the transmembrane domain (78, 79). This suggests that during or immediately subsequent to association of APP with α-secretase, the extramembrane helix dissociates from the membrane surface in order to enter the active site, with the distance from the membrane surface serving as a “ruler” for enzymatic selection of the scissile site.
Several mutations associated with familial Alzheimer's disease (FAD) are located in the extracellular helix and just following it: A692G, E693G, E693Q, and D694N. These mutations have been studied most extensively in the context of the Aβ peptides, as some of these mutations are believed to enhance the tendency of Aβ to form pathogenic fibrils and aggregates (80–85). Of greater relevance to this paper is the study of Haass et al., who observed that the Flemish mutation caused impaired α-secretase cleavage of APP, an observation that reflects the above-noted requirement of α-secretase for substrates that contain an α-helix following the scissile site. This is expected to contribute to the etiology of Alzheimer's disease by tipping the balance between amyloidogenic β-secretase cleavage and benign α-secretase cleavage in favor of the former.
The C-Terminus of C99 Is Membrane Associated
The final ten residues of C99 were observed to form an α-helix that associates with moderate affinity with the surface of LMPG micelles. A helical wheel plot of residues 761–770 shows that this segment maps well as an amphipathic helix (Supporting Information Figure 4), which explains its preference for the micelle surface. While our studies were conducted in the presence of a C-terminal purification tag, the first eight residues of this tag are mostly polar and cannot be mapped as an amphipathic or apolar helix (Supporting Information Figure 4), an observation that strongly suggests the tag does not contribute to membrane association.
The association of the C-terminus of C99 with the membrane surface is of particular interest because this segment encompasses part of the Y757ENPTY motif that plays a central role as a Tyr-phosphorylation recognition element involved in the binding of APP and its derived fragments to several different adaptor proteins including Fe65, X11, and mDAB (86, 87). These interactions are believed to be closely linked to the endosomal trafficking of APP, to its putative transport of protein cargo in neurons, and to the role of the AICD in transcriptional regulation. We speculate that the interactions of various proteins with this motif may be modulated by the membrane association and dissociation of the C-terminus. Within intact C99, the C-terminus (sites 761–770) would be membrane associated, thus anchoring the YENPTY motif (sites 757–762) to the surface in a manner that might effectively sequester this site from recognition by some of its potential binding partners. Phosphorylation of either Tyr757 or Tyr762 (the latter especially) by regulatory tyrosine kinases could impose a strong electrostatic force capable of dissociating the C-terminus from the surface, thereby releasing this motif for facile access and binding by soluble interaction partners. AICD liberation by γ-secretase is also likely to trigger dissociation of the C-terminus from the membrane surface given that the avidity of this short amphipathic helix for the membrane is unlikely to be high enough to anchor AICD in the absence of the C99 TMD. Of additional note, other endosomal sorting/trafficking motifs in type I membrane protein have also been shown to be membrane surface-associated (88, 89), suggesting that modulation of protein–membrane interactions may be a common strategy for regulating these critical biological processes.
The site of APP phosphorylation that has been most extensively characterized (40) is Thr743, which is located in a largely disordered domain of C99 at a point almost equidistant from the end of the TMD and the beginning of the C-terminal amphipathic helix. Thr743 may therefore be rather prominently displayed at the most membrane-distal apex of the cytosolic membrane-to-membrane loop connecting these two helices. It has been shown that phosphorylated Thr743 is recognized by the prolyl isomerase, Pin1, which then catalyzes rapid interconversion between the cis and trans forms of Pro742, a phenomenon that may be linked to the manner by which Pin1 binding results in enhanced Aβ42 production (90).
Cholesterol Specifically Binds to C99 via the Loop Connecting the Extracellular Helix to the TMD
Titration of C99 by cholesterol and cholesterol-like compounds results in large chemical shift perturbations for residues G696, S697, and K699. For β-CHOLBIMALT, these changes are saturable, with half-maximal effect occurring at approximately 15 mol %. The changes in the NMR spectrum appear to result from specific and local cholesterol–C99 interactions rather than changes in the conformation or dynamics of C99, which appear to be minimal. Residues G696, S697, and K699 are located in the loop connecting the extracellular helix to the TMD, where it is also proximal to the putative interface for homodimerization.
These observations have important implications. First, C99 appears to have a specific affinity for cholesterol and related lipids. However, based on our results with β-CHOLBIMALT cholesterol's affinity for C99 appears to be only modest, such that cholesterol saturation of the protein may be approached only when the local cholesterol content in the membranes is >15 mol %. This affinity may be an important factor underlying C99's tendency to associate with cholesterol-rich lipid rafts. Under conditions where the cholesterol content in the membrane is low, C99 would generally not be complexed with cholesterol. However, under conditions where the membrane cholesterol content is sufficiently high such that it segregates to form cholesterol-rich microdomains, C99 would be expected to preferentially localize within such domains by virtue of its modest affinity for cholesterol. Because the amyloidogenic β- and γ-secretases are believed to reside in raft-like detergent-resistant membrane domains, C99's intrinsic affinity for cholesterol represents an inherently pro-amyloidogenic property.
It is widely believed that α- and β-secretases compete with one another for ectodomain shedding of APP. Accumulating evidence suggests the substrate partitioning of APP between these proteases is determined largely by intracellular trafficking of APP. Numerous studies have demonstrated that α-secretase processing of APP is generally localized to the bulk membrane phase of the cell surface (8), whereas β-secretase activity occurs primarily in cholesterol-rich raft-like domains of endosomal membranes. Therefore, the preferential cell surface partitioning of APP to cholesterol- and sphingomyelin-enriched lipid rafts (which can then internalize to endosomes enriched with β-secretase activity) is believed to be a decisive determinant of the competition between β-and α-secretase for initial proteolysis of APP (10, 91). Factors that promote raft association and internalization would tend to elevate β-secretase cleavage and contribute to the amyloid burden, while factors that inhibit internalization or promote cell surface localization would elevate α-secretase cleavage. The results of this study suggest that C99 (and most likely APP) can form a specific complex with cholesterol at physiologically relevant cholesterol levels, which suggests that the specific affinity of APP for cholesterol is likely to be one factor that promotes its association with raft-like membrane domains. This is consistent with previous results showing that covalent linkage of proteins to sterols is sufficient to induce raft localization (91). Providing further corroboration of the interaction between cholesterol and APP/C99 is the recent discovery that the ATP binding cassette transporter G1 (ABCG1), which redistributes cellular cholesterol to the plasma membrane, induces increased cell surface localization of APP (92). In addition to APP–cholesterol interactions, protein–protein interactions probably also contribute to APP's propensity to partition into raft-like lipid domains. For example, the flotillins are one family of proteins thought to facilitate clustering of both APP and cholesterol in raft-like membrane domains (93, 94). Recently, it was shown that the interaction between flotillin-2 and APP is cholesterol-dependent (94), suggestive of a specific complex between APP, cholesterol, and flotillin.
Does APP Function as a Cellular Cholesterol Sensor?
The AICD fragment liberated by cleavage near the C-terminus of APP was recently shown to suppress transcription of the LRP1 gene by translocating to the nucleus and physically binding to the LRP1 promoter (7). LRP1 is a major receptor for exogenous cholesterol (95). When the LPR1 gene expression is downregulated, the result is a reduction in cellular cholesterol levels (7). We propose that one function of APP and/or its C99 domain may be sensing sterol levels: when the local membrane concentration of cholesterol approaches levels at which membrane phase separation occurs, a significant fraction of cell surface APP will bind cholesterol and translocate to raft-like domains. These domains are then internalized to endosomes where APP is cleaved by BACE1, and the product (C99) is subsequently cleaved by γ-secretase to release two peptides, AICD and Aβ.3 The former downregulates LRP1 production at the transcriptional level, thereby decreasing intracellular cholesterol levels. The latter augments this effect by inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), the rate-limiting enzyme in cholesterol biosynthesis (96). Additionally, Aβ directly activates the catabolic sphingomyelinase, resulting in a reduction in sphingolipid levels (96). Grimm et al. demonstrated that the absence of presenilin in mouse embryonic fibroblast cells dramatically reduced Aβ, which, in turn, caused elevated cholesterol and sphingolipid levels, the latter two molecules being the primary lipid constituents of APP-containing rafts. By downregulating cholesterol and sphingolipid levels, Aβ may directly modulate its own production. In a sense, APP appears to be acting as a cholesterol sensor (Figure 11): after initial cleavage by BACE1 (a cholesterol-dependent phenomenon), the C-terminal stub (C99) is proteolyzed by γ-secretase. The C-terminal product of this reaction (AICD) downregulates the intake of exogenous cholesterol at the transcriptional level, whereas the N-terminal product (Aβ) downregulates cholesterol biosynthesis via direct or indirect inhibition of HMGR. In conjunction with previous findings, our data therefore support a novel function for APP as a regulator of neuronal cholesterol levels. The function(s) of APP is (are) incompletely understood. Our data shed light not only on its physiological role in cholesterol metabolism but also on the pathophysiological relation of cholesterol metabolism to Aβ production and Alzheimer's disease. In further support of this model is the observation that APP interacts at the cell surface with LRP1 and the two cointernalize (21, 97) and are subsequently degraded by BACE1 (98) and γ-secretase (99), suggesting that APP can further act to regulate cholesterol levels by promoting internalization of LRP1, thereby reducing its concentration at the plasma membrane.
Figure 11.
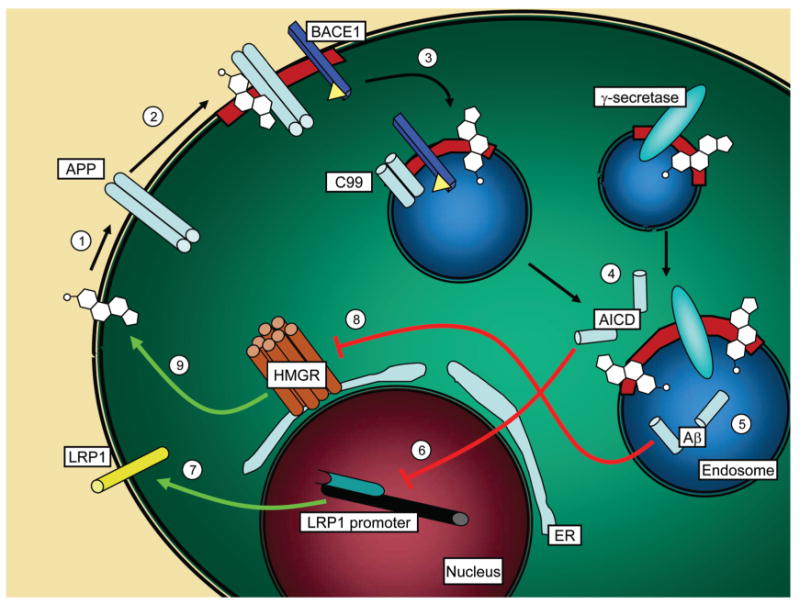
Schematic representation of the downstream effects of APP processing on cholesterol metabolism. APP is depicted as a dimer (light blue cylinders) residing at the plasma membrane. Initial binding to cholesterol (white polycyclic structure; 1), potentially in conjunction with flotillin-2 (not shown), induces translocation to lipid rafts (depicted as red membrane segments; 2) or nucleation of rafts around APP—cholesterol clusters (not shown). These rafts may contain BACE1 (blue rectangular prism), or APP may be presented to BACE1 at some later stage in the endocytic pathway (not shown). Internalization of the APP-containing lipid rafts delivers the protein to the more acidic endosomal system, conditions that activate BACE1 and promote release of the soluble APP ectodomain (3). γ-Secretase may have cointernalized with APP and BACE1 (not shown), or the C99-containing endosome may fuse with a γ-secretase-containing endosome (shown), following which γ-secretase (cyan oval) mediates cleavage of C99 to liberate AICD (4) and Aβ (5).3 The former forms a ternary complex with Fe65 and Tip60 (not shown), which subsequently translocate to the nucleus (maroon) where it physically binds to the promoter of the LRP1 gene (black cylinder of DNA), thereby suppressing its transcription (6) by RNA polymerase (teal structure). Reduced levels of LRP1 (yellow cylinder), a major neuronal apolipoprotein receptor, result in reduced intake of exogenous cholesterol (7). Simultaneously, Aβ inhibits HMG-CoA reductase (HMGR, brown cylinders), the rate-limiting enzyme in cholesterol biosynthesis, possibly via direct interaction (8). It is not known whether inhibition is mediated by direct interaction between HMGR and Aβ or whether Aβ effects inhibition by some other mechanism. HMG-CoA inhibition attenuates endogenous cholesterol production (9), reinforcing the transcriptional activity of AICD. Overall, both exogenous and endogenous sources of cholesterol are downregulated as a consequence of APP processing, which is initially stimulated by cholesterol—APP interaction.
Conclusions
The results of this paper provide the first detailed description of the structural properties of C99 and pave the way for future completion of its high-resolution structure in LMPG micelles using solution state NMR spectroscopy. While the transmembrane domain of C99 was found to be largely featureless, both the extracellular and intracellular domains contained structural elements that appear to be closely linked to APP function, proteolytic processing, and trafficking. Most importantly among these elements is a binding site for cholesterol, which is centered in an extramembrane segment of C99 that is also believed to be critical for γ-secretase cleavage at sites within the transmembrane domain. That C99 (and most likely full-length APP) specifically binds cholesterol, as opposed to having a less specific preference for raft-like membrane domains, suggests that cholesterol may be an important factor governing the trafficking of APP to rafts and may also play a direct role in the modulation of β- and/or γ-secretase cleavage. Moreover, when considered in the context of a wealth of recent data on APP trafficking, processing, and function, the observation of C99's cholesterol binding property suggests that one function of APP may be to serve as a cholesterol sensor/receptor that can downregulate cellular cholesterol uptake under conditions of high membrane cholesterol content.
Supplementary Material
Four supporting figures containing additional data and captions. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Anatrace, Inc. (Maumee, OH), for providing the CHOLBIMALT compounds used in this work. We also thank Dr. Stanley Howell for helpful discussion and assistance with data modeling and analysis.
Footnotes
This work was supported by NIH Grants R21 AG236581 and PO1 GM80513 and by Alzheimer's Association Grants IIRG-07-59379 and IIRG-05-14222. 900 MHz NMR data were collected at the NMR facility of the University of Georgia, which is supported in part by Grant 5P41GM066340-05.
NMR resonance assignments/chemical shifts for C99 have been deposited into the BioMagResBank (BMRB) as entry 15775.
Abbreviations: Aβ, amyloid-β; AICD, APP intracellular domain; APP, amyloid precursor protein; BACE1, β-site APP cleaving enzyme 1; β-CHOLBIMALT, β-cholesteryl-(maltosyl-β-(1,6)-maltoside); C99, 99-residue transmembrane C-terminal domain of APP; CD, circular dichroism; DRM, detergent-resistant membrane; DTT, dithiothreitol; FAD, familial Alzheimer's disease; Gd-DTPA, Gd(III)-diethylenetriaminepentaacetic acid; HMGR, 3-hydroxy-3-methylglutaryl-CoA reductase; LB, Luria broth; LMPG, lysomyristoylphosphatidylglycerol; LDLR, low-density lipoprotein (LDL) receptor; LRP1, LDL receptor-related protein 1; MTSL, 2,2,5,5-tetramethyl-3-pyrroline-3-methyl)-methanethiosulfonate; NMR, nuclear magnetic resonance spectroscopy; NOE, nuclear Overhauser effect; PAGE, polyacrylamide gel electrophoresis; 16-DSA, 16-doxylstearic acid; SDS, sodium dodecyl sulfate; TMD, transmembrane domain; TROSY, transverse relaxation-optimized NMR spectroscopy; WT, wild type.
While cholesterol sulfate and cholesterol hemisuccinate are also commercially available cholesterol derivatives with significantly more polar substituents than the hydroxyl moiety at the carbon-3 position, the experience of the authors has been that these compounds are much more difficult to solubilize either alone in aqueous solution or in detergent micelles. The CHOLBIMALT compounds, by contrast, are readily soluble in a variety of conditions and have the added advantage that they maintain the electroneutrality of cholesterol.
While not shown in Figure 11, the product of α-secretase cleavage, C83, would also be expected to bind cholesterol and to contribute to production of AICD (but not Aβ) after its internalization and cleavage by γ-secretase.
References
- 1.Cao X, Sudhof TC. A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 2.Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Protein aggregation in the brain—the molecular basis for Alzheimer's and Parkinson's Diseases. Mol Med. 2008;14:451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meredith SC. Protein denaturation and aggregation: Cellular responses to denatured and aggregated proteins. Ann NY Acad Sci. 2005;1066:181–221. doi: 10.1196/annals.1363.030. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 5.Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 2005;38:105–127. doi: 10.1007/0-387-23226-5_5. [DOI] [PubMed] [Google Scholar]
- 6.Fahrenholz F. Alpha-secretase as a therapeutic target. Curr Alzheimer Res. 2007;4:412–417. doi: 10.2174/156720507781788837. [DOI] [PubMed] [Google Scholar]
- 7.Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, Herz J, Muglia L, Bu G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56:66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM. Cleavage of Alzheimer's amyloid precursor protein by alpha-secretase occurs at the surface of neuronal cells. Biochemistry. 1999;38:9728–9734. doi: 10.1021/bi9906827. [DOI] [PubMed] [Google Scholar]
- 9.Chyung JH, Raper DM, Selkoe DJ. Gamma-secretase exists on the plasma membrane as an intact complex that accepts substrates and effects intramembrane cleavage. J Biol Chem. 2005;280:4383–4392. doi: 10.1074/jbc.M409272200. [DOI] [PubMed] [Google Scholar]
- 10.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riddell DR, Christie G, Hussain I, Dingwall C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol. 2001;11:1288–1293. doi: 10.1016/s0960-9822(01)00394-3. [DOI] [PubMed] [Google Scholar]
- 12.Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis. 2002;9:11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- 13.Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 1996;271:4436–4440. doi: 10.1074/jbc.271.8.4436. [DOI] [PubMed] [Google Scholar]
- 14.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buxbaum JD, Geoghagen NS, Friedhoff LT. Cholesterol depletion with physiological concentrations of a statin decreases the formation of the Alzheimer amyloid Abeta peptide. J Alzheimer's Dis. 2001;3:221–229. doi: 10.3233/jad-2001-3207. [DOI] [PubMed] [Google Scholar]
- 16.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 17.Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci USA. 2001;98:5815–5820. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah S, Yu G. sorLA: sorting out APP. Mol Interv. 2006:674–658. doi: 10.1124/mi.6.2.4. [DOI] [PubMed] [Google Scholar]
- 20.Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, Bales KR, Cappai R, Masters CL, Gliemann J, Mufson EJ, Hyman BT, Paul SM, Nykjaer A, Willnow TE. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakshmana MK, Chen E, Yoon IS, Kang DE. C-terminal 37 residues of LRP promote the amyloidogenic processing of APP independent of FE65. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00320.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cam JA, Zerbinatti CV, Knisely JM, Hecimovic S, Li Y, Bu G. The low density lipoprotein receptor-related protein 1B retains beta-amyloid precursor protein at the cell surface and reduces amyloid-beta peptide production. J Biol Chem. 2004;279:29639–29646. doi: 10.1074/jbc.M313893200. [DOI] [PubMed] [Google Scholar]
- 23.Fuentealba RA, Barria MI, Lee J, Cam J, Araya C, Escudero CA, Inestrosa NC, Bronfman FC, Bu G, Marzolo MP. ApoER2 expression increases Abeta production while decreasing Amyloid Precursor Protein (APP) endocytosis: Possible role in the partitioning of APP into lipid rafts and in the regulation of gamma-secretase activity. Mol Neurodegener. 2007;2:14. doi: 10.1186/1750-1326-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 25.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJ, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Solution NMR studies of the A beta(1–40) and A beta(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc. 2004;126:2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 26.Shao H, Jao S, Ma K, Zagorski MG. Solution structures of micelle-bound amyloid beta-(1–40) and beta-(1–42) peptides of Alzheimer's disease. J Mol Biol. 1999;285:755–773. doi: 10.1006/jmbi.1998.2348. [DOI] [PubMed] [Google Scholar]
- 27.Barrow CJ, Zagorski MG. Solution structures of beta peptide and its constituent fragments: relation to amyloid deposition. Science. 1991;253:179–182. doi: 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- 28.Jarvet J, Danielsson J, Damberg P, Oleszczuk M, Graslund A. Positioning of the Alzheimer Abeta(1–40) peptide in SDS micelles using NMR and paramagnetic probes. J Biomol NMR. 2007;39:63–72. doi: 10.1007/s10858-007-9176-4. [DOI] [PubMed] [Google Scholar]
- 29.Poulsen SA, Watson AA, Fairlie DP, Craik DJ. Solution structures in aqueous SDS micelles of two amyloid beta peptides of A beta(1–28) mutated at the alpha-secretase cleavage site (K16E, K16F) J Struct Biol. 2000;130:142–152. doi: 10.1006/jsbi.2000.4267. [DOI] [PubMed] [Google Scholar]
- 30.Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ. Solution structure of amyloid beta-peptide(1–40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry. 1998;37:11064–11077. doi: 10.1021/bi972979f. [DOI] [PubMed] [Google Scholar]
- 31.Tomaselli S, Esposito V, Vangone P, van Nuland NA, Bonvin AM, Guerrini R, Tancredi T, Temussi PA, Picone D. The alpha-to-beta conformational transition of Alzheimer's Abeta-(1–42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of beta conformation seeding. Chem Bio Chem. 2006;7:257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- 32.Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, Baumketner A, Shea JE, Urbanc B, Cruz L, Borreguero J, Stanley HE. Elucidating amyloid beta-protein folding and assembly: A multidisciplinary approach. Acc Chem Res. 2006;39:635–645. doi: 10.1021/ar050063s. [DOI] [PubMed] [Google Scholar]
- 33.Tycko R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Q Rev Biophys. 2006;39:1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 34.Dulubova I, Ho A, Huryeva I, Sudhof TC, Rizo J. Three-dimensional structure of an independently folded extracellular domain of human amyloid-beta precursor protein. Biochemistry. 2004;43:9583–9588. doi: 10.1021/bi049041o. [DOI] [PubMed] [Google Scholar]
- 35.Gralle M, Botelho MM, de Oliveira CL, Torriani I, Ferreira ST. Solution studies and structural model of the extracellular domain of the human amyloid precursor protein. Biophys J. 2002;83:3513–3524. doi: 10.1016/S0006-3495(02)75351-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heald SL, Tilton RF, Hammond LJ, Lee A, Bayney RM, Kamarck ME, Ramabhadran TV, Dreyer RN, Davis G, Unterbeck A. Sequential NMR resonance assignment and structure determination of the Kunitz-type inhibitor domain of the Alzheimer's beta-amyloid precursor protein. Biochemistry. 1991;30:10467–10478. doi: 10.1021/bi00107a015. [DOI] [PubMed] [Google Scholar]
- 37.Hynes TR, Randal M, Kennedy LA, Eigenbrot C, Kossiakoff AA. X-ray crystal structure of the protease inhibitor domain of Alzheimer's amyloid beta-protein precursor. Biochemistry. 1990;29:10018–10022. doi: 10.1021/bi00495a002. [DOI] [PubMed] [Google Scholar]
- 38.Kroenke CD, Ziemnicka-Kotula D, Xu J, Kotula L, Palmer AG., III Solution conformations of a peptide containing the cytoplasmic domain sequence of the beta amyloid precursor protein. Biochemistry. 1997;36:8145–8152. doi: 10.1021/bi9705669. [DOI] [PubMed] [Google Scholar]
- 39.Ramelot TA, Gentile LN, Nicholson LK. Transient structure of the amyloid precursor protein cytoplasmic tail indicates preordering of structure for binding to cytosolic factors. Biochemistry. 2000;39:2714–2725. doi: 10.1021/bi992580m. [DOI] [PubMed] [Google Scholar]
- 40.Ramelot TA, Nicholson LK. Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR. J Mol Biol. 2001;307:871–884. doi: 10.1006/jmbi.2001.4535. [DOI] [PubMed] [Google Scholar]
- 41.Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW. Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol. 1999;6:327–331. doi: 10.1038/7562. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 43.Gorman PM, Kim S, Guo M, Melnyk RA, McLaurin J, Fraser PE, Bowie JU, Chakrabartty A. Dimerization of the transmembrane domain of amyloid precursor proteins and familial Alzheimer's disease mutants. BMC Neurosci. 2008;9:17. doi: 10.1186/1471-2202-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kienlen-Campard P, Tasiaux B, Van HJ, Li M, Huysseune S, Sato T, Fei JZ, Aimoto S, Courtoy PJ, Smith SO, Constantinescu SN, Octave JN. Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GXXXG motifs. J Biol Chem. 2008;283:7733–7744. doi: 10.1074/jbc.M707142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Multhaup G. Amyloid precursor protein and BACE function as oligomers. Neurodegener Dis. 2006;3:270–274. doi: 10.1159/000095266. [DOI] [PubMed] [Google Scholar]
- 46.Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marenchino M, Williamson PT, Murri S, Zandomeneghi G, Wunderli-Allenspach H, Meier BH, Kramer SD. Dynamics and Cleavability at the {alpha}-Cleavage Site of APP(684–726) in Different Lipid Environments. Biophys J. 2008;95:1460–1473. doi: 10.1529/biophysj.108.129726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 50.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 51.Wishart DS, Sykes BD. Chemical-Shifts As A Tool for Structure Determination. Nucl Magn Reson. 1994;Part C 239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 52.Weigelt J. Single scan, sensitivity- and gradient-enhanced TROSY for multidimensional NMR experiments. J Am Chem Soc. 1998;120:10778–10779. [Google Scholar]
- 53.Chill JH, Louis JM, Delaglio F, Bax A. Local and global structure of the monomeric subunit of the potassium channel KcsA probed by NMR. Biochim Biophys Acta. 2007;1768:3260–3270. doi: 10.1016/j.bbamem.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang P, Liu Q, Scarborough GA. Lysophosphatidylglycerol: a novel effective detergent for solubilizing and purifying the cystic fibrosis transmembrane conductance regulator. Anal Biochem. 1998;259:89–97. doi: 10.1006/abio.1998.2633. [DOI] [PubMed] [Google Scholar]
- 55.Krueger-Koplin RD, Sorgen PL, Krueger-Koplin ST, Rivera-Torres IO, Cahill SM, Hicks DB, Grinius L, Krulwich TA, Girvin ME. An evaluation of detergents for NMR structural studies of membrane proteins. J Biomol NMR. 2004;28:43–57. doi: 10.1023/B:JNMR.0000012875.80898.8f. [DOI] [PubMed] [Google Scholar]
- 56.Tian C, Vanoye CG, Kang C, Welch RC, Kim HJ, George AL, Jr, Sanders CR. Preparation, functional characterization, and NMR studies of human KCNE1, a voltage-gated potassium channel accessory subunit associated with deafness and long QT syndrome. Biochemistry. 2007;46:11459–11472. doi: 10.1021/bi700705j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, van DA, Wang R, Selkoe DJ, Wolfe MS. Purification and characterization of the human gamma-secretase complex. Biochemistry. 2004;43:9774–9789. doi: 10.1021/bi0494976. [DOI] [PubMed] [Google Scholar]
- 58.Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, Ito A, Winblad B, Cowburn RF, Thyberg J, Ankarcrona M. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J Biol Chem. 2004;279:51654–51660. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- 59.Li YM, Xu M, Lai MT, Huang Q, Castro JL, Muzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi Y, Hayashi I, Tominari Y, Rikimaru K, Morohashi Y, Kan T, Natsugari H, Fukuyama T, Tomita T, Iwatsubo T. Sulindac sulfide is a noncompetitive gamma-secretase inhibitor that preferentially reduces Abeta 42 generation. J Biol Chem. 2003;278:18664–18670. doi: 10.1074/jbc.M301619200. [DOI] [PubMed] [Google Scholar]
- 61.Beel AJ, Sanders CR. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci. 2008;65:1311–1334. doi: 10.1007/s00018-008-7462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grziwa B, Grimm MO, Masters CL, Beyreuther K, Hartmann T, Lichtenthaler SF. The transmembrane domain of the amyloid precursor protein in microsomal membranes is on both sides shorter than predicted. J Biol Chem. 2003;278:6803–6808. doi: 10.1074/jbc.M210047200. [DOI] [PubMed] [Google Scholar]
- 63.Tischer E, Cordell B. Beta-amyloid precursor protein. Location of transmembrane domain and specificity of gamma-secretase cleavage. J Biol Chem. 1996;271:21914–21919. doi: 10.1074/jbc.271.36.21914. [DOI] [PubMed] [Google Scholar]
- 64.Czerski L, Sanders CR. Thiol modification of diacylglycerol kinase: dependence upon site membrane disposition and reagent hydrophobicity. FEBS Lett. 2000;472:225–229. doi: 10.1016/s0014-5793(00)01457-5. [DOI] [PubMed] [Google Scholar]
- 65.Wolfe MS. The gamma-secretase complex: membrane-embedded proteolytic ensemble. Biochemistry. 2006;45:7931–7939. doi: 10.1021/bi060799c. [DOI] [PubMed] [Google Scholar]
- 66.Lichtenthaler SF, Ida N, Multhaup G, Masters CL, Beyreuther K. Mutations in the transmembrane domain of APP altering gamma-secretase specificity. Biochemistry. 1997;36:15396–15403. doi: 10.1021/bi971071m. [DOI] [PubMed] [Google Scholar]
- 67.Lichtenthaler SF, Wang R, Grimm H, Uljon SN, Masters CL, Beyreuther K. Mechanism of the cleavage specificity of Alzheimer's disease gamma-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc Natl Acad Sci USA. 1999;96:3053–3058. doi: 10.1073/pnas.96.6.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lazarov VK, Fraering PC, Ye W, Wolfe MS, Selkoe DJ, Li H. Electron microscopic structure of purified, active gamma-secretase reveals an aqueous intramembrane chamber and two pores. Proc Natl Acad Sci USA. 2006;103:6889–6894. doi: 10.1073/pnas.0602321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 70.Curran AR, Engelman DM. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr Opin Struct Biol. 2003;13:412–417. doi: 10.1016/s0959-440x(03)00102-7. [DOI] [PubMed] [Google Scholar]
- 71.Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr Opin Struct Biol. 2004;14:465–479. doi: 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 72.Sulistijo ES, MacKenzie KR. Sequence dependence of BNIP3 transmembrane domain dimerization implicates side-chain hydrogen bonding and a tandem GxxxG motif in specific helix-helix interactions. J Mol Biol. 2006;364:974–990. doi: 10.1016/j.jmb.2006.09.065. [DOI] [PubMed] [Google Scholar]
- 73.Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem. 2006;44:Spec. No. S24–S40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- 74.Lichtenthaler SF, Beher D, Grimm HS, Wang R, Shearman MS, Masters CL, Beyreuther K. The intramembrane cleavage site of the amyloid precursor protein depends on the length of its transmembrane domain. Proc Natl Acad Sci USA. 2002;99:1365–1370. doi: 10.1073/pnas.032395699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ren Z, Schenk D, Basi GS, Shapiro IP. Amyloid beta-protein precursor juxtamembrane domain regulates specificity of gamma-secretase-dependent cleavages. J Biol Chem. 2007;282:35350–35360. doi: 10.1074/jbc.M702739200. [DOI] [PubMed] [Google Scholar]
- 76.Haass C, Hung AY, Selkoe DJ, Teplow DB. Mutations associated with a locus for familial Alzheimer's disease result in alternative processing of amyloid beta-protein precursor. J Biol Chem. 1994;269:17741–17748. [PubMed] [Google Scholar]
- 77.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maruyama K, Kametani F, Usami M, Yamao-Harigaya W, Tanaka K. “Secretase”, Alzheimer amyloid protein precursor secreting enzyme is not sequence-specific. Biochem Biophys Res Commun. 1991;179:1670–1676. doi: 10.1016/0006-291x(91)91767-7. [DOI] [PubMed] [Google Scholar]
- 79.Sisodia SS. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci USA. 1992;89:6075–6079. doi: 10.1073/pnas.89.13.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Watson DJ, Selkoe DJ, Teplow DB. Effects of the amyloid precursor protein Glu693→Gln “Dutch” mutation on the production and stability of amyloid beta-protein. Biochem J. 1999;340(Part 3):703–709. [PMC free article] [PubMed] [Google Scholar]
- 81.Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, Yamin G, Rigby AC, Teplow DB. Familial Alzheimer's disease mutations alter the stability of the amyloid beta-protein monomer folding nucleus. Proc Natl Acad Sci USA. 2007;104:16522–16527. doi: 10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L. The “Arctic” APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 83.Paivio A, Jarvet J, Graslund A, Lannfelt L, Westlind-Danielsson A. Unique physicochemical profile of beta-amyloid peptide variant Abeta1–40E22G protofibrils: conceivable neuropathogen in arctic mutant carriers. J Mol Biol. 2004;339:145–159. doi: 10.1016/j.jmb.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 84.Tsubuki S, Takaki Y, Saido TC. Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet. 2003;361:1957–1958. doi: 10.1016/s0140-6736(03)13555-6. [DOI] [PubMed] [Google Scholar]
- 85.Yamamoto N, Hasegawa K, Matsuzaki K, Naiki H, Yanagisawa K. Environment- and mutation-dependent aggregation behavior of Alzheimer amyloid beta-protein. J Neurochem. 2004;90:62–69. doi: 10.1111/j.1471-4159.2004.02459.x. [DOI] [PubMed] [Google Scholar]
- 86.King GD, Scott TR. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol. 2004;185:208–219. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 87.Russo T, Faraonio R, Minopoli G, De Candi P, De Renzis S, Zambrano N. Fe65 and the protein network centered around the cytosolic domain of the Alzheimer's beta-amyloid precursor protein. FEBS Lett. 1998;434:1–7. doi: 10.1016/s0014-5793(98)00941-7. [DOI] [PubMed] [Google Scholar]
- 88.Choowongkomon K, Carlin CR, Sonnichsen FD. A structural model for the membrane-bound form of the juxtamembrane domain of the epidermal growth factor receptor. J Biol Chem. 2005;280:24043–24052. doi: 10.1074/jbc.M502698200. [DOI] [PubMed] [Google Scholar]
- 89.Vinogradova O, Carlin C, Sonnichsen FD, Sanders CR. A membrane setting for the sorting motifs present in the adenovirus E3–13.7 protein which down-regulates the epidermal growth factor receptor. J Biol Chem. 1998;273:17343–17350. doi: 10.1074/jbc.273.28.17343. [DOI] [PubMed] [Google Scholar]
- 90.Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 91.Rietveld A, Neutz S, Simons K, Eaton S. Association of sterol- and glycosylphosphatidylinositol-linked proteins with Drosophila raft lipid microdomains. J Biol Chem. 1999;274:12049–12054. doi: 10.1074/jbc.274.17.12049. [DOI] [PubMed] [Google Scholar]
- 92.Tansley GH, Burgess BL, Bryan MT, Su Y, Hirsch-Reinshagen V, Pearce J, Chan JY, Wilkinson A, Evans J, Naus KE, McIsaac S, Bromley K, Song W, Yang HC, Wang N, DeMattos RB, Wellington CL. The cholesterol transporter ABCG1 modulates the subcellular distribution and proteolytic processing of beta-amyloid precursor protein. J Lipid Res. 2007;48:1022–1034. doi: 10.1194/jlr.M600542-JLR200. [DOI] [PubMed] [Google Scholar]
- 93.Chen TY, Liu PH, Ruan CT, Chiu L, Kung FL. The intracellular domain of amyloid precursor protein interacts with flotillin-1, a lipid raft protein. Biochem Biophys Res Commun. 2006;342:266–272. doi: 10.1016/j.bbrc.2006.01.156. [DOI] [PubMed] [Google Scholar]
- 94.Schneider A, Rajendran L, Honsho M, Gralle M, Donnert G, Wouters F, Hell SW, Simons M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J Neurosci. 2008;28:2874–2882. doi: 10.1523/JNEUROSCI.5345-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.May P, Woldt E, Matz RL, Boucher P. The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann Med. 2007;39:219–228. doi: 10.1080/07853890701214881. [DOI] [PubMed] [Google Scholar]
- 96.Grimm MO, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M, Tschape JA, De Strooper B, Muller U, Shen J, Hartmann T. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol. 2005;7:1118–1123. doi: 10.1038/ncb1313. [DOI] [PubMed] [Google Scholar]
- 97.Yoon IS, Chen E, Busse T, Repetto E, Lakshmana MK, Koo EH, Kang DE. Low-density lipoprotein receptor-related protein promotes amyloid precursor protein trafficking to lipid rafts in the endocytic pathway. FASEB J. 2007;21:2742–2752. doi: 10.1096/fj.07-8114com. [DOI] [PubMed] [Google Scholar]
- 98.von Arnim CA, von EB, Weber P, Wagner M, Schwanzar D, Spoelgen R, Strauss WL, Schneckenburger H. Impact of cholesterol level upon APP and BACE proximity and APP cleavage. Biochem Biophys Res Commun. 2008;370:207–212. doi: 10.1016/j.bbrc.2008.03.047. [DOI] [PubMed] [Google Scholar]
- 99.Lleo A, Waldron E, von Arnim CA, Herl L, Tangredi MM, Peltan ID, Strickland DK, Koo EH, Hyman BT, Pietrzik CU, Berezovska O. Low density lipoprotein receptor-related protein (LRP) interacts with presenilin 1 and is a competitive substrate of the amyloid precursor protein (APP) for gamma-secretase. J Biol Chem. 2005;280:27303–27309. doi: 10.1074/jbc.M413969200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Four supporting figures containing additional data and captions. This material is available free of charge via the Internet at http://pubs.acs.org.