

Abstract
The development of novel human vaccines would be greatly facilitated by the development of in vivo models that permit preclinical analysis of human immune responses. Here, we show that nonobese diabetic severe combined immunodeficiency (NOD/SCID) β2 microglobulin−/− mice, engrafted with human CD34+ hematopoietic progenitors and further reconstituted with T cells, can mount specific immune responses against influenza virus vaccines. Live attenuated trivalent influenza virus vaccine induces expansion of CD8+ T cells specific to influenza matrix protein (FluM1) and nonstructural protein 1 in blood, spleen, and lungs. On ex vivo exposure to influenza antigens, antigen-specific CD8+ T cells produce IFN-γ and express cell-surface CD107a. FluM1-specific CD8+ T cells can be also expanded in mice vaccinated with inactivated trivalent influenza virus vaccine. Expansion of antigen-specific CD8+ T cells is dependent on reconstitution of the human myeloid compartment. Thus, this humanized mouse model permits preclinical testing of vaccines designed to induce cellular immunity, including those against influenza virus. Furthermore, this work sets the stage for systematic analysis of the in vivo functions of human DCs. This, in turn, will allow a new approach to the rational design and preclinical testing of vaccines that cannot be tested in human volunteers.
Introduction
Vaccination represents one of the major successes of medicine as it has spared countless people from polio, tetanus, and other acute infections.1 At present, no less than 26 infectious diseases can be prevented through vaccination.2 With a notable exception of smallpox3–5 and yellow fever6 vaccines, which generate cellular immunity, classic preventive vaccines are designed to generate neutralizing antibodies. Yet, some viruses that cause considerable morbidity and mortality in humans escape the immune control elicited by these vaccines.7,8 For example, no universally effective vaccines have yet been developed for respiratory syncytial virus (RSV), hepatitis C virus, and human immunodeficiency virus (HIV).8,9 Among possible causes for the escape from neutralizing antibodies are genetic diversity and mutational evolution of these viruses. Therefore, novel strategies for protective vaccination against these viruses need to take into consideration other immune effectors (ie, CD8+ T cells).10 Indeed, CD8+ T cell–mediated protective responses might prove beneficial by, for example, elimination of infected cells, which will limit viral replication and, consequently, disease development.11 CD8+ T cells also appear as important players in therapeutic vaccination in conditions, such as chronic infections and cancer.
An essential component of vaccination are dendritic cells (DCs), antigen-presenting cells (APCs) of skin and mucosal surfaces that capture vaccine antigens and present them to lymphocytes.12 DCs constitute a system of professional APCs, which initiate, maintain, and regulate adaptive immune responses.13 Recent studies corroborate a concept of distinct DC subsets generating quantitatively and qualitatively distinct types of adaptive immunity.14,15 This is fundamental for the rational design of new, improved, vaccines. However, studies of human DC biology are mostly confined to in vitro systems and are hampered by the lack of in vivo models. These shortcomings cannot be fully addressed by murine studies because mice and humans differ in several aspects of DC biology, an example of this being the pattern of toll-like receptor (TLR) 9 expression, which is present on all DCs in the mouse but only on plasmacytoid DCs (pDCs) in the human.16 Thus, mice would not accurately predict how certain TLR ligands would impact vaccine immunogenicity in humans. These differences in DC biology might also explain considerable discrepancies between mouse and human in the outcomes of DNA vaccination.17 Thus, although highly effective in mouse models, DNA vaccines are clearly less immunogenic in humans.18
To test human vaccines in vivo, we embarked on the construction of mice with human immune system following the pioneering studies of the late 1980s.19–21 To this end, NOD/SCID β2m−/− immunodeficient mice are transplanted with human CD34+ hematopoietic progenitor cells (HPCs).22 Such mice develop all human DC subsets and B cells.22 pDCs and myeloid DCs populate the bone marrow and spleen, Langerhans cells (LCs) are found in the epidermis, and interstitial DCs (intDCs) in peripheral tissues.22 In this model, T cells are adoptively transferred, thereby permitting the analysis of T-cell subsets.
To analyze the in vivo function of human APCs in humanized mice, we used influenza virus vaccines. Influenza virus is the cause of an acute rather than chronic infection. Approximately 36 000 people die each year because of influenza virus infection.23 Optimizing a vaccine against influenza virus represents therefore a public health priority. Thus far, studies in mouse have not allowed for an improved vaccine. Neutralizing antibodies are traditionally regarded as the most important outcome of vaccination against influenza.24,25 However, although antibodies specific to hemagglutinin can neutralize the virus, hemagglutinin undergoes rapid mutations, leading to virus escape and necessitating the design and manufacture of a new vaccine for each season.26 It is plausible that vaccination strategies that enhance the spectrum of effector cells (ie, recruitment of influenza antigen-specific CD8+ T cells) could improve the current influenza vaccines. This concept is corroborated by the demonstration of the protective role of CD8+ T cells in the course of natural influenza virus infection.26 Thus, the generation of effector T cells able to eliminate infected epithelial cells could represent an important component of protective vaccination against influenza virus.
We show herein that humanized mice can mount antigen-specific recall CD8+ T-cell immunity on vaccination with seasonal trivalent influenza vaccines (ie, live attenuated influenza virus [LAIV] vaccine and inactivated influenza virus [TIV] vaccine).
Methods
Antibodies and reagents
Antibodies to human CD3 (SK7), CD4 (SK3), CD8 (SK1), CD11c (S-HCL-3), CD19 (HIB19), CD20 (2H7), CD28 (L293), CD34 (8G12), CD45 (HI30), CD45RA (HI100), CD49d (L25), CD56 (B159), CD107a (H4A3), CD123 (9F5), HLA-DR (L243), IFN-γ (25723.11), IL-2 (5344.111), Linage cocktail 1, and TNF-α (6401.1111) were obtained from BD Biosciences (San Jose, CA). Anti–human CD3 (UCHT1; Beckman Coulter, Fullerton, CA) and CD27 (CLB-27/1; Invitrogen, Carlsbad, CA) were also used. Human HLA-ABC (W6/32) antibody was from Dako Denmark (Glostrup, Denmark).
Human FLT3 ligand IgG, Fc fusion protein was expressed and purified as described.27
Peptides were HLA-A*0201–restricted (Bio-Synthesis, Lewisville, TX) with purity more than 95%: influenza A virus M1 58-66 (GILGFVFTL), influenza A virus NP 383-391 (SRYWAIRTR), influenza A virus NS1 122-130 (AIMDKNIIL), and HIV gag 77-85 (SLYNTVATL). The HLA-A*0201 tetramers loaded with influenza A virus M1 58-66, NS1 122-130, and HIV-1 gag 77-85 were purchased from Beckman Coulter.
Trivalent LAIV vaccine FluMist (2006-2007 season; MedImmune, Gaithersburg, MD), TIV vaccine Fluzone (2006-2007 season; Sanofi Pasteur, Swiftwater, PA), Alum-precipitated tetanus toxoid (TT; Sanofi Pasteur) were all obtained from the hospital pharmacy.
Humanized mice
NOD/SCID β2m−/− mice (The Jackson Laboratory, Bar Harbor, ME) at the age of 4 to 5 weeks were sublethally irradiated (12 cGy per gram of body weight) using a 137Cs gamma irradiator (MDS Nordion, Ottawa, ON). CD34+ HPCs (3 × 106) from granulocyte colony-stimulating factor mobilized peripheral blood apheresis of healthy volunteers (Table S1, available on the Blood website; see the Supplemental Materials link at the top of the online article) were given in 200 μL of phosphate-buffered saline (PBS) into the tail-vein. Mice were used between 4 and 8 weeks after HPC transplantation. All protocols were reviewed and approved by the institutional review board and institutional animal care and use committee at Baylor Research Institute.
T cells were isolated from peripheral blood mononuclear cells (PBMCs) autologous to CD34+ HPCs using total human T-cell isolation kit (StemCell Technologies, Vancouver, BC) following the manufacturer's protocol by using antibody cocktail: CD14, CD16, CD19, CD20, CD36, CD56, CD123, and glycophorin A and following magnetic bead separation. Isolated total T cells had purity more than 95%. In the experiment with sorted B and T cells, PBMCs were sorted as CD11c−CD14−CD16−CD56−CD123−CD141− cells by FACSAria (BD Biosciences) with purity more than 99%. T cells were given by intraperitoneal injection.
Vaccination
Humanized mice were treated with 5 doses of 10 μg human FLT3 Ligand subcutaneously for a period of 10 days. After T-cell transfer intraperitoneally, humanized mice were vaccinated with TT (one-tenth of human dose) or influenza virus vaccine (one-fifth of human dose) via intraperitoneal and intravenous injections or intranasal inoculation. Mice were bled at different time points before and after vaccination to monitor the immune response and harvested according to individual experimental design.
Analysis
Mice were killed and blood was collected with heparin. After flushing out blood, the lungs were harvested for single cell suspension. The lungs were digested with 2 mg/mL of collagenase D (Roche Diagnostics, Indianapolis, IN) for 30 minutes at 37°C. Single cell suspension was made with 2 frosted slides, and the debris was removed by filtering through a 70-μm cell strainer (BD Biosciences). Spleen was digested for 10 minutes using the same method. Single cell suspensions were further purified for ex vivo analysis.
Phenotype analysis
Cells were first treated with purified antibody against murine CD16/32 (2.4G2, BD Biosciences; to block nonspecific FcR interactions) and then stained on ice with fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorophyll protein (PerCP), and allophycocyanin (APC) conjugated specific antibodies, or appropriate isotype controls. After washing twice with PBS, cells were fixed with FACSlysis solution (BD Biosciences) or 1% paraformaldehyde and analyzed for up to 6 parameters on a FACSCalibur (BD Biosciences) using CellQuest software (BD Biosciences) or FlowJo software (TreeStar, Ashland, OR).
Tetramer analysis
Heparinized blood or single cell suspension from different tissues was resuspended in PBS/2% fetal calf serum. Each sample was stained with FITC, PE, PerCP, or APC conjugated antibody to CD8 and CD3 for surface marker and PE or APC conjugated FluM1-HLA-A*0201 tetramer for FluM1-specific CD8+ T cells and HIVgag-HLA-A*0201 tetramer for the negative control. Each sample was stained at room temperature for 30 minutes and washed twice with PBS. Afterward, the sample was resuspended with FACSlysis buffer and incubated at room temperature for 10 minutes to lyse red blood cells. Finally, FACSlysis buffer was washed away and the sample was resuspended with 1% paraformaldehyde. The sample was analyzed for the frequency of FluM1-tetramer positive cells in the CD8+ T-cell population.
Intracellular cytokine staining
Intracellular cytokine and degranulation of CD8+ T cells were examined using a modified protocol as described.28 Briefly, total cell suspension from vaccinated mice was stimulated with 2.5 μg/mL of specific peptide in the presence of anti-CD28 and anti-CD49d antibodies. Monesin and FITC-conjugated anti-CD107a was included during the culture period. After T-cell stimulation, cells were surface-labeled with surface antibodies and then intracellularly labeled with antibodies specific for IFN-γ, IL-2, and TNF-α using BD Biosciences' Cytofix/Cytoperm and Perm/Wash reagents. Samples were analyzed up to 6 parameters on a FACSCalibur and up to 12 parameters on a FACSAria (BD Biosciences).
Results
Live attenuated trivalent influenza vaccine expands FluM1-specific CD8+ T cells in blood and tissues
Sublethally irradiated NOD/SCID β2m−/− immunodeficient mice were transplanted with 3 × 106 CD34+ HPCs from HLA-A*0201+ healthy donors, and at 4 to 8 weeks after transplantation were injected intraperitoneally with 20 × 106 autologous T cells. These humanized mice were vaccinated once (one-fifth of the human dose) with LAIV vaccine composed of H1N1 (A/New Caledonia/20/99), H3N2 (A/Wisconsin/67/2005), and Influenza B (B/Malaysia/2506/2004) viruses (2006-2007 season) at equal hemagglutinin (HA) ratio. To establish vaccination parameters and determine the magnitude and the breath of influenza-specific CD8+ T-cell immunity, mice were vaccinated intraperitoneally and intravenously (Figure 1A). Induction of influenza-specific CD8+ T-cell response was assessed by staining blood and tissues with HLA-A*0201 tetramer loaded with the matrix protein 1, FluM158-66 (GILGFVFTL) peptide, as this epitope is common to both H1N1 and H3N2 viruses. Vaccinated mice demonstrated, at day 12 after vaccination (Figure 1B), circulating human CD8+ T cells binding FluM1-tetramer with high intensity (3/3 mice: 0.22%, 0.37%, and 0.5% CD8+ T cells). These results were obtained in 4 independent cohorts of humanized mice reconstituted with cells from 4 different HLA-A*0201+ healthy volunteers (Figure 1C; Table S2). Altogether, 9 of 14 mice showed expansion of FluM1-specific CD8+ T cells in the blood (mean = 0.26% ± SEM = 0.07%, n = 14 mice, P = .005; Figure 1C; Table S2; positive response was defined as the percentage of FluM1 tetramer+CD8+ T cells in the group vaccinated with LAIV 2-fold higher than the mean + 2SEM in the group of control mice vaccinated with PBS or TT). Control mice vaccinated with the TT vaccine did not show expansion of FluM1-specific CD8+ T cells (mean = 0.03% ± SEM = 0.007%, n = 7; Figure 1C; Table S2). Thus, the expansion of FluM1-specific CD8+ T cells in LAIV-vaccinated mice is driven by the presentation of influenza antigen indicating specificity.
Figure 1.

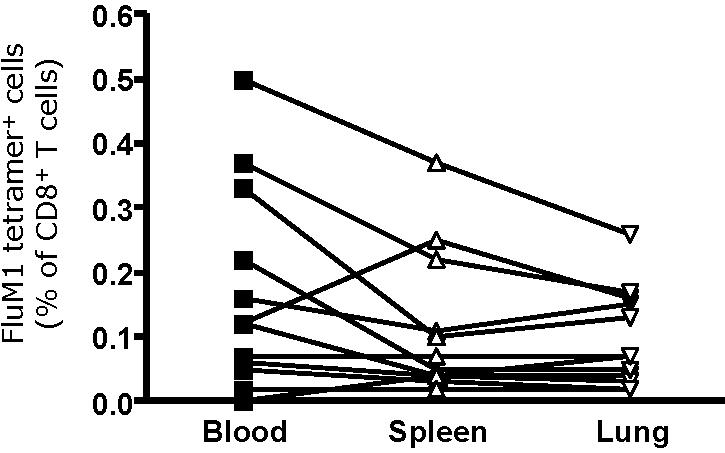
The expansion FluM1-specific CD8+ T cells in LAIV-vaccinated humanized mice. (A) LAIV vaccine was given intraperitoneally/intravenously; TT vaccine was used as the control. (B) At day 12 after vaccination, the frequency of CD8+ T cells binding FluM1-tetramer and HIVgag-tetramer in the blood was analyzed by flow cytometry. Analysis gates are set for high intensity tetramer staining. Representative data from an experiment with 3 mice vaccinated with LAIV or TT. (C) The frequency of FluM1-specific CD8+ T cells in the blood at day 12 after vaccination from 4 experiments with 4 different donors. Two-tailed nonparametric Mann-Whitney test. (D) The frequency of CD8+ T cells binding FluM1-tetramer and HIVgag-tetramer in the spleen and lungs at day 14 after vaccination. (E) The frequency of FluM1-tetramer+ CD8+ T cells in the blood correlates with their frequency in the spleen (Spearman correlation, P < .001) and (F) in the lung (Spearman correlation, P < .001).
FluM1-specific CD8+ T cells could also be detected in the spleen and in the lung of LAIV-vaccinated mice (mean percentage of CD8+ T cells ± SEM = 0.26% ± 0.09%, 0.17% ± 0.06%, and 0.16% ± 0.06% in the blood, spleen, and lung, respectively; Figures 1D, S1). The percentages of FluM1-specifc CD8+ T cells in the lung and in the spleen correlated with the expansion of CD8+ T cells in the blood (Spearman r = 0.84 and r = 0.79; P < .001 and P < .001; Figure 1E and 1F, respectively). FluM1-specific CD8+ T cells were not detected in tissues of TT-vaccinated mice (Figure 1D), further demonstrating the specificity of the response to vaccine antigens.
Thus, vaccination of humanized mice with live attenuated influenza virus results in expansion of FluM1-specific human CD8+ T cells.
Inactivated trivalent influenza vaccine expands FluM1-specific CD8+ T cells
The expansion of human CD8+ T cells after vaccination with LAIV could result from either a direct presentation of viral antigens by infected human APCs or indirect presentation of viral peptides by human APCs (ie, DCs) that captured infected cells or infected cell products (eg, pMHC complexes). Thus, to determine whether human APCs in humanized mice are capable of indirect antigen presentation, we vaccinated mice with trivalent inactivated vaccine (TIV; 2006-2007 season) and analyzed the induction of influenza-specific CD8+ T cells. TIV is composed of 3 viruses that are formalin-fixed, purified, and chemically disrupted to generate a “split virus” that is not infective. As a result of this process, the vaccine is enriched in HA and neuraminidase (NA) antigens; contains M1 and nuclear protein (NP) structural proteins; but lacks nonstructural NS1 protein. At day 12 after vaccination, circulating FluM1-specific CD8+ T cells could be detected in 4 of 4 mice: 0.88%, 0.49%, 1.2%, and 0.44% (Figure 2A). Such results were reproduced in 5 independent cohorts of humanized mice generated using cells from 4 different HLA-A*0201 healthy volunteers (Figure 2B; Table S3). Altogether, 15 of 17 mice vaccinated with TIV showed expansion of FluM1-specific CD8+ T cells (mean = 0.50% ± SEM = 0.12, n = 17; Figure 2B; Table S3). FluM1-specific CD8+ T cells binding tetramers with high intensity could also be detected in the spleen and lungs (Figure 2C). These results suggest that antigens of the inactivated vaccine are captured by human APCs and processed for indirect presentation in vivo to antigen-specific CD8+ T cells.
Figure 2.

The expansion of FluM1-specific CD8+ T cells in TIV-vaccinated humanized mice. Experiment as in Figure 1, except that mice received TIV vaccine. (A) Specific CD8+ T cells binding FluM1-tetramer and HIVgag-tetramer by flow cytometry. Representative mice from an experiment with 4 mice vaccinated with TIV. (B) The frequency of FluM1-specific CD8+ T cells in the blood at day 12 after vaccination from 5 experiments with 4 different donors. Two-tailed nonparametric Mann-Whitney test. (C) CD8+ T cells in the spleen and lungs at day 14 after vaccination.
To further analyze the role of human APCs in the expansion of influenza-specific CD8+ T cells in vaccinated mice, TIV vaccine was administered to NOD/SCID β2m−/− mice that were reconstituted with T and B cells but did not have human DCs because they were not transplanted with human CD34+ HPCs (see mice construction and experimental scheme in Figure 3A,B). Both cohorts were reconstituted with the same number of T cells (20 × 106 per mouse). As shown in Figure 3C, these partially reconstituted mice did not expand FluM1-specific CD8+ T cells in response to TIV vaccine, whereas the 3 of 3 control humanized mice did (Figure 3D). FluM1-specific CD8+ T cells could not be detected at day 27 (Figure 3E), indicating a complete absence of response rather than a delayed response. The lack of FluM1-specifc CD8+ T-cell response in the blood of partially reconstituted mice was not related to the absence of CD8+ T cells, which could be detected at all time points and increased with time (Figure 3F). Furthermore, even after a boost vaccination at day 27, spleens of NOD/SCID β2m−/− mice that were transferred with T and B cells did not harbor FluM1-specific CD8+ T cells (Figure S2). The kinetics analysis in fully reconstituted humanized mice showed a peak response in blood at day 12 followed by a gradual disappearance of influenza antigen-specific CD8+ T cells by day 20. The disappearance of FluM1-specific CD8+ T cells in fully reconstituted mice was not related to the disappearance of human CD8+ T cells (Figure 3F), suggesting contraction of the antigen-specific CD8+ T cell pool. Thus, human APCs other than B cells, most probably DCs, are able to present vaccine antigens in vivo to CD8+ T cells, thereby allowing their expansion.
Figure 3.

Cross-presentation of FluM1 in humanized mice. Experimental protocols: (A) NOD/SCID β2m−/− mice were reconstituted with 20 × 106 sorted T cells and with 10 × 106 sorted B cells (purity > 99%) and vaccinated with TIV intraperitoneally/intravenously. (B) NOD/SCID β2m−/− mice were transplanted with CD34+ HPCs and reconstituted with 20 × 106 autologous sorted total T cells (purity > 99%) before vaccination. TIV was given intraperitoneally/intravenously. (C,D) The frequency of FluM1-specific CD8+ T cells in NOD/SCID β2m−/− mice (C) or (D) in humanized mice at day 12 after vaccination. (E) The frequency of FluM1-specific CD8+ T cells (mean ± SEM, n = 3 for each cohort at each time point) at different time points after vaccination. (F) The numbers of total human CD8+ T cells (mean ± SEM, n = 3 for each cohort at each time point) measured in the same volume of blood at different time points after vaccination.
Trivalent influenza vaccines expand CD8+ T cells with broad specificities
To determine the breadth of elicited responses, human CD8+ T cells, isolated from spleens 14 days after vaccination, were exposed for 8 hours to either the HIV gag77-85 peptide (SLYNTVATL) or 3 HLA-A*0201–binding influenza antigen-derived peptides (Flu-M158-66, nonstructural protein 1 [NS1122-130 AIMDKNIIL], and nucleoprotein [NP383-391 SRYWAIRTR]) in the presence of anti-CD28 and anti-CD49d. CD8+ T-cell activation was assessed by measuring intracellular IFN-γ with flow cytometry (Figure 4A). This analysis showed responses to FluM1 (0.8% CD8+ T cells in the spleen, Figure 4A), thus confirming the tetramer binding data (Figure 1). NS1-specific CD8+ T-cell expansion could also be detected (0.3% CD8+ T cells in the spleen; Figure 4A,B). No expansion of NP-specific CD8+ T cells could be detected, possibly because of the low affinity binding of this peptide for HLA-A*0201 (not shown). Less than 0.1% CD8+ T cells expressed IFN-γ in response to HIV gag peptide, further demonstrating the specificity of influenza responses.
Figure 4.

The breadth and effector phenotype of elicited influenza-specific CD8+ T cells. Vaccination as indicated in figure panels. Single-cell suspensions from tissues harvested at day 14 after vaccination were stimulated for 8 hours with indicated peptides and antibodies against CD28 and CD49d. The frequency of IFN-γ–secreting CD8+ T cells by flow cytometry. (A-D) Spleen analysis. (A) Representative experiment. (B) IFN-γ–secreting CD8+ T cells (mean ± SEM, n = 3) after stimulation with indicated peptides. (C) Analysis after vaccination with TIV or TT. (D) The frequency of IFN-γ–secreting CD8+ T cells (mean, n = 4) specific to indicated peptides. (E) Blood: specific CD8+ T cells binding FluM1 and NS1 tetramer in humanized mice vaccinated with LAIV or TIV. (F) Blood and lungs of TIV or TT-vaccinated mice. IFN-γ and CD107a expression by CD8+ T cells in response to HIVgag or FluM1 peptides.
Thus, mice vaccinated with LAIV vaccine generate recall CD8+ T-cell immunity to at least 2 of the vaccine antigens (Figure 4A). The presence of responses to NS1 peptide derived from nonstructural protein further suggests in vivo infection of cells by the LAIV vaccine. Vaccination of mice with TIV also resulted in the expansion of FluM1-specific CD8+ T cells secreting IFN-γ (0.75% CD8+ T cells, Figure 4C). In contrast to mice vaccinated with LAIV, mice vaccinated with TIV did not show the expansion of NS1-specific CD8+ T cells (Figure 4C), consistent with the lack of NS1 protein expression on vaccination with killed influenza virus.29 These findings were reproduced in 2 independent cohorts of mice reconstituted with cells from 2 different healthy volunteers (Figure 4D). The lack of NS1-specifc CD8+ T-cell differentiation was further confirmed by the lack of CD8+ T cells binding NS1 peptide-loaded tetramer in the blood of mice vaccinated with TIV (Figure 4E).
Trivalent influenza vaccines expand CD8+ T cells with different effector phenotypes
To further establish differentiation of antigen-specific CD8+ T cells into effector cells, we analyzed cytokine expression together with surface expression of CD107a, a surrogate marker of CTL function.30 Blood cells of TIV-vaccinated mice showed 2 subsets of antigen-specific CD8+ T cells, ie, double IFN-γ+ CD107a+ and single IFN-γneg CD107a+ cells (1.57% and 1.77% of total CD8+ T cells, respectively, in the representative experiment, Figure 4F). A similar pattern of CD8+ T-cell differentiation was found in the lung from the same mice (0.17% of IFN-γ+ CD107a+ CD8+ T cells, Figure 4F). Only very few FluM1-specific CD8+ T cells expressed IL-2 and/or TNF-α (data not shown). Thus, the predominant phenotype of CD8+ T cells expanded by vaccination is that of highly differentiated effector cells. A similar phenotype of FluM1-specific CD8+ T cells was found in LAIV-vaccinated mice that were reconstituted with cells from the same healthy donor (not shown). Thus, humanized mice vaccinated with trivalent influenza vaccines generate FluM1-specific effector CD8+ T cells.
Intranasal vaccination with LAIV leads to expansion of FluM1-specific CD8+ T cells in blood and tissues
Because humans are administered LAIV intranasally, we tested this route of vaccination in humanized mice. In this experiment, 3 of 4 mice vaccinated with LAIV intranasally demonstrated expansion of FluM1-specific CD8+ T cells in the blood: 0.12%, 0.23%, and 0.29% of CD8+ T cells at day 12 after vaccination (Figure 5A). Analysis of draining lymph nodes (pooled cervical and mediastinal lymph nodes) revealed the presence of FluM1-specific CD8+ T cells (Figure 5B). CD8+ T cells with FluM1 specificity could also be detected in control lymph nodes (pooled axillary and inguinal lymph nodes; Figure 5B), further indicating the ability of human antigen-specific CD8+ T cells to circulate through lymphatic and peripheral tissues in vaccinated mice. Accordingly, FluM1-specific CD8+ T cells could also be detected in the blood, spleen, and lungs of the same mice (Figure 5B from representative mouse; Figure S3). Thus, mucosal delivery of vaccine antigens permits the expansion of influenza-specific CD8+ T cells in humanized mice.
Figure 5.

Intranasal vaccination with LAIV permits expansion of FluM1-specific CD8+ T cells. (A) Blood of mice vaccinated with LAIV intraperitoneally/intravenously (i.v./i.p.) as in Figure 1 or intranasally (i.n.). Frequency of CD8+ T cells binding FluM1 tetramer at day 12 after vaccination. (B) Flu-M1 specific CD8+ T cells in draining and control lymph node (LN) suspension pooled from 3 mice; blood, spleen, and lung from representative mouse.
Discussion
Herein we analyzed the breadth, magnitude, and quality of CD8+ T-cell responses in humanized mice vaccinated with seasonal influenza vaccines. Our humanized mouse is based on NOD/SCID β2m−/− mice, which, when transplanted with human CD34+ HPCs, develop in vivo all human DC subsets and B cells.22 In this model, T cells are adoptively transferred, thereby permitting us to exploit the influenza-specific memory T-cell compartment to measure the in vivo function of human APCs in humanized mice.
Here we show that fully reconstituted humanized mice vaccinated with LAIV vaccine expand CD8+ T cells specific to at least 2 influenza virus antigens, matrix protein 1 and nonstructural protein 1. The expansion of CD8+ T cells with at least 2 specificities demonstrates the capacity of human APCs to process and present multiple vaccine antigens in vivo in humanized mice. Because nonstructural protein is expressed only on cell infection, the expansion of NS1-specific CD8+ T cells indicates active infection in vivo on LAIV vaccination. Importantly, immune responses are generated via the natural route of vaccination with LAIV (ie, via mucosal intranasal delivery). It remains to be established whether the expansion of human CD8+ T cells after vaccination with LAIV result from direct presentation of viral antigens by lung-resident human APCs that become infected and/or from indirect presentation of viral peptides by human APCs (ie, DCs) that captured infected murine lung epithelial cells.
Humanized mice vaccinated with killed influenza virus (TIV) vaccine show expansion of FluM1-specific CD8+ T cells. Because this vaccine is composed of killed viruses, generation of specific responses demonstrates the capacity of human APCs of indirect presentation of vaccine antigens. In line with this, NOD/SCID β2m−/− mice reconstituted with T and B cells but lacking myeloid cells cannot support expansion of influenza-specific CD8+ T cells on TIV vaccination. Thus, in this scenario, B cells are not able to present vaccine antigens in vivo to CD8+ T cells, suggesting that myeloid cells, most probably DCs, play this role. These results are consistent with murine studies in mice depleted of CD11c+ DCs, which fail to cross-present exogenous antigens and therefore cannot prime antigen-specific CD8+ T cells.31
The exact nature of immune responses generated by LAIV and TIV vaccines as well as mechanisms of protection remains to be understood, and our current model might help in doing so. Classically, TIV vaccine is considered more efficient in generation of humoral rather than CD8+ T-cell responses.32 For example, in the analysis of vaccinated children 5 to 9 years of age, the mean percentages of IFN-γ–secreting influenza antigen-specific T cells (both CD4+ and CD8+) increased significantly after LAIV, but not after TIV immunization.33 However, the phenotypic changes of influenza-specific CD8+ T cells on vaccination appear to differ depending on the age of vaccine as well as on the type of vaccine.34 This might be related to the standard intramuscular route of vaccination with TIV. Herein, both vaccines delivered intravenously and intraperitoneally generate comparable magnitudes of FluM1-specific CD8+ T-cell responses.
Protective vaccination against influenza continues to be a public health issue. New approaches to vaccination are being developed, including the use of reverse genetics for rapid production of virus preparation containing the HA and NA of circulating strains,32 attenuated virus with altered NS1 gene,35 adjuvants,36,37 and strategies aimed to target DC subsets.12 Current models for influenza vaccine testing rely on ferrets, and more recently guinea pigs, for clinical protection.38 There is, however, a need for models to test the immune efficacy of these novel vaccination strategies. This need may now be at least partially alleviated by the use of humanized mice as we demonstrate herein. This in turn will allow a new approach to the rational design and preclinical testing of much-needed influenza vaccines as well as other vaccines that cannot be tested in human volunteers.
Supplementary Material
Acknowledgments
The authors thank volunteers for participation in our studies; Dr Joseph Fay and the clinical group as well as Lynette Walters and the personnel at Cell and Tissue Procurement core at Baylor Institute for Immunology Research; Albert Barnes and the personnel at Bioscience Center at BRI; Flow cytometry and Luminex Core at Baylor Institute for Immunology Research; Anne-Laure Flamar, Dr Xiao-Hua Li, Sandra Zurawski, and Dr Gouchen He for FLT3 Ligand; Dr SangKon Oh and Dr Hideki Ueno for discussion; Cindy Samuelsen, Dr Carson Harrod, and Nicolas Taquet for administrative and facility support; and Dr Michael Ramsay and Dr William Duncan for continuous support.
This work was supported by BHCS Foundation, DANA Foundation, and National Institutes of Health (U19 AI057234, U19 AI062623, AIO56001, and CA78846). J.B. holds the Caruth Chair for Transplant Immunology Research. A.K.P. holds the Ramsay Chair for Cancer Immunology Research.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.I.Y. designed and performed research, collected, analyzed, and interpreted data, performed statistical analysis, and wrote the manuscript; M.G. performed research and collected data and analyzed data; F.M. designed and performed research and collected and analyzed data; G.Z., O.R., and A.G.-S. contributed vital reagents, designed research, and wrote the manuscript; J.B. designed research and wrote the manuscript; and A.K.P. designed and performed research, analyzed and interpreted data, performed statistical analysis, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: A. Karolina Palucka, Baylor Institute for Immunology Research, 3434 Live Oak Street, Dallas, TX 75204; e-mail: karolinp@baylorhealth.edu.
References
- 1.Germain RN. An innately interesting decade of research in immunology. Nat Med. 2004;10:1307–1320. doi: 10.1038/nm1159. [DOI] [PubMed] [Google Scholar]
- 2.Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3:630–641. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 3.Fauci AS. Smallpox vaccination policy: the need for dialogue. N Engl J Med. 2002;346:1319–1320. doi: 10.1056/NEJM200204253461711. [DOI] [PubMed] [Google Scholar]
- 4.Amanna IJ, Slifka MK, Crotty S. Immunity and immunological memory following smallpox vaccination. Immunol Rev. 2006;211:320–337. doi: 10.1111/j.0105-2896.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- 5.Precopio ML, Betts MR, Parrino J, et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J Exp Med. 2007;204:1405–1416. doi: 10.1084/jem.20062363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Querec T, Bennouna S, Alkan S, et al. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203:413–424. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMichael AJ. HIV vaccines. Annu Rev Immunol. 2006;24:227–255. doi: 10.1146/annurev.immunol.24.021605.090605. [DOI] [PubMed] [Google Scholar]
- 8.Letvin NL. Correlates of immune protection and the development of a human immunodeficiency virus vaccine. Immunity. 2007;27:366–369. doi: 10.1016/j.immuni.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Pantaleo G, Koup RA. Correlates of immune protection in HIV-1 infection: what we know, what we don't know, what we should know. Nat Med. 2004;10:806–810. doi: 10.1038/nm0804-806. [DOI] [PubMed] [Google Scholar]
- 10.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 11.Johnston MI, Fauci AS. An HIV vaccine: evolving concepts. N Engl J Med. 2007;356:2073–2081. doi: 10.1056/NEJMra066267. [DOI] [PubMed] [Google Scholar]
- 12.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 13.Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell. 2001;106:263–266. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 14.Pulendran B, Smith JL, Caspary G, et al. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci U S A. 1999;96:1036–1041. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudziak D, Kamphorst AO, Heidkamp GF, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 16.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 17.Kutzler MA, Weiner DB. Developing DNA vaccines that call to dendritic cells. J Clin Invest. 2004;114:1241–1244. doi: 10.1172/JCI23467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanke T. On DNA vaccines and prolonged expression of immunogens. Eur J Immunol. 2006;36:806–809. doi: 10.1002/eji.200635986. [DOI] [PubMed] [Google Scholar]
- 19.Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. 1988;335:256–259. doi: 10.1038/335256a0. [DOI] [PubMed] [Google Scholar]
- 20.McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science. 1988;241:1632–1639. doi: 10.1126/science.241.4873.1632. [DOI] [PubMed] [Google Scholar]
- 21.Kamel-Reid S, Dick JE. Engraftment of immune-deficient mice with human hematopoietic stem cells. Science. 1988;242:1706–1709. doi: 10.1126/science.2904703. [DOI] [PubMed] [Google Scholar]
- 22.Palucka AK, Gatlin J, Blanck JP, et al. Human dendritic cell subsets in NOD/SCID mice engrafted with CD34+ hematopoietic progenitors. Blood. 2003;102:3302–3310. doi: 10.1182/blood-2003-02-0384. [DOI] [PubMed] [Google Scholar]
- 23.Thompson WW, Shay DK, Weintraub E, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 24.Subbarao K, Murphy BR, Fauci AS. Development of effective vaccines against pandemic influenza. Immunity. 2006;24:5–9. doi: 10.1016/j.immuni.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed R, Oldstone MB, Palese P. Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat Immunol. 2007;8:1188–1193. doi: 10.1038/ni1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doherty PC, Turner SJ, Webby RG, Thomas PG. Influenza and the challenge for immunology. Nat Immunol. 2006;7:449–455. doi: 10.1038/ni1343. [DOI] [PubMed] [Google Scholar]
- 27.Hannum C, Culpepper J, Campbell D, et al. Ligand for FLT3/FLK2 receptor tyrosine kinase regulates growth of haematopoietic stem cells and is encoded by variant RNAs. Nature. 1994;368:643–648. doi: 10.1038/368643a0. [DOI] [PubMed] [Google Scholar]
- 28.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 29.Birch-Machin I, Rowan A, Pick J, Mumford J, Binns M. Expression of the nonstructural protein NS1 of equine influenza A virus: detection of anti-NS1 antibody in post infection equine sera. J Virol Methods. 1997;65:255–263. doi: 10.1016/s0166-0934(97)02189-7. [DOI] [PubMed] [Google Scholar]
- 30.Rubio V, Stuge TB, Singh N, et al. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med. 2003;9:1377–1382. doi: 10.1038/nm942. [DOI] [PubMed] [Google Scholar]
- 31.Jung S, Unutmaz D, Wong P, et al. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palese P, Garcia-Sastre A. Influenza vaccines: present and future. J Clin Invest. 2002;110:9–13. doi: 10.1172/JCI15999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He XS, Holmes TH, Zhang C, et al. Cellular immune responses in children and adults receiving inactivated or live attenuated influenza vaccines. J Virol. 2006;80:11756–11766. doi: 10.1128/JVI.01460-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He XS, Holmes TH, Mahmood K, et al. Phenotypic changes in influenza-specific CD8+ T cells after immunization of children and adults with influenza vaccines. J Infect Dis. 2008;197:803–811. doi: 10.1086/528804. [DOI] [PubMed] [Google Scholar]
- 35.Ferko B, Stasakova J, Romanova J, et al. Immunogenicity and protection efficacy of replication-deficient influenza A viruses with altered NS1 genes. J Virol. 2004;78:13037–13045. doi: 10.1128/JVI.78.23.13037-13045.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyle J, Eastman D, Millar C, et al. The utility of ISCOMATRIX adjuvant for dose reduction of antigen for vaccines requiring antibody responses. Vaccine. 2007;25:2541–2544. doi: 10.1016/j.vaccine.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 37.Baras B, Stittelaar KJ, Simon JH, et al. Cross-protection against lethal H5N1 challenge in ferrets with an adjuvanted pandemic influenza vaccine. PLoS ONE. 2008;3:e1401. doi: 10.1371/journal.pone.0001401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowen AC, Palese P. Influenza virus transmission: basic science and implications for the use of antiviral drugs during a pandemic. Infect Disord Drug Targets. 2007;7:318–328. doi: 10.2174/187152607783018736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}