

Abstract
Mycoplasma arthritidis induces an acute to chronic arthritis in rodents. Arthritis induced in mice histologically resembles human rheumatoid arthritis and can be associated with lethal toxicity following systemic injection. The M. arthritidis mitogen (MAM) superantigen has long been implicated as having a role in pathogenesis, but its significance with respect to toxicity and arthritogenicity in mycoplasma-induced disease is unclear. To study the pathogenic significance of MAM, M. arthritidis mutants that overproduced or failed to produce MAM were developed. MAM overproduction and knockout mutants were more and less mitogenic, respectively, than the wild-type strain. The degree of mitogenic activity correlated with lethal toxicity in DBA/2J mice. In contrast, histopathological studies detected no correlation between MAM production and the severity of arthritis induced in DBA/2J and CBA/J mice.
Mycoplasma arthritidis is a naturally occurring arthritogen of rats (12). Experimental arthritis can be induced in mice by intravenous injection (4) and in rabbits by intra-articular injection (48). Disease in rats and mice is associated with persistence of viable organisms in the joints, but disease in rabbits persists even in the absence of viable organisms (12). Arthritis induced in mice is chronic and relapsing and histologically resembles human rheumatoid arthritis (10, 22). M. arthritidis secretes a superantigen (SAg), M. arthritidis mitogen (MAM), which is a potent activator of murine and human T cells (14, 23). MAM is in many aspects a typical SAg. It does not bind to the antigen groove of the major histocompatibility complex (MHC) molecule as do conventional antigens, and T-cell recognition of MAM is not MHC restricted (11). The presence of a functional H-2E molecule is required to present MAM to T cells (14) with presentation mediated by Eα-containing molecules (13). T-cell responses to MAM stimulation are clonally expressed (16). Responsiveness is associated with expression of particular subsets of Vβ chains present on the T-cell receptor.
MAM has unique features that differentiate it from other bacterial SAgs. Sequence analysis of MAM revealed that it is phylogenetically distinct (17). MAM, but not other SAgs, contains the consensus legume lectin motif that is important for T-cell activation by concanavalin A. Unlike most other SAgs, which have a similar three-dimensional structure consisting of a small β-barrel domain and a large domain with a β-grasp motif, MAM adopts a novel fold composed of two completely α-helical domains (33). It interacts with the third complementarity-determining region (CDR3) of the T-cell receptor (31), a feature of nominal peptide antigens rather than SAgs. It is much more mitogenic for murine cells than are staphylococcal SAgs (19). Both Toll-like receptor 2 and 4 (TLR2 and TLR4) on macrophages are engaged by MAM (34). At least one of these TLRs must be present for macrophage activation by MAM. It was also suggested that TLR4 signaling downregulates the MAM/TLR2 inflammatory response. The interaction between MAM and TLRs of macrophages and the subsequent release of cytokines appear to determine the susceptibility of mice to M. arthritidis-induced disease. The cytokine profile induced by MAM in C3H/HeJ mice, a strain bearing a missense mutation in TLR4, is different from that in C3H/SnJ mice, a strain bearing an intact TLR4 (36). As a result, these strains of mice display different susceptibilities to disease induced by M. arthritidis. MAM is the first SAg reported to have DNase activity (25). These unique characteristics demonstrate the novelty of MAM and warrant further research on its role in the development of SAg-mediated diseases.
The role of MAM in the pathogenesis of rodent arthritis is unclear. Lymphocytes from some strains of mice that are resistant to M. arthritidis-induced arthritis respond to MAM (14, 21, 35). Arthritis in rats can be induced by direct intra-articular injection of MAM, but the arthritis induced does not reproduce the disease phenotype induced by infection with live organisms (5). Significant toxicity but not necessarily significant arthritis has been associated with MAM (18, 20, 36). In this study, we developed mutants that failed to produce MAM or overproduced MAM with the goal of elucidating the role of MAM in disease induced by M. arthritidis.
MATERIALS AND METHODS
Strains of bacteria and medium.
Mutants of M. arthritidis strain 158 (8, 43) were developed in this study (Table 1) by using the minitransposon Tn4001TF1 as an insertional mutagen and as a vector. Strain TnCtrl was developed as a control that has the empty minitransposon vector inserted at an intergenic position in the mycoplasma genome. M. arthritidis was cultured in EA (agar) or EB (broth) at 37°C as described previously (44, 46). For antibiotic selection in M. arthritidis, medium was supplemented with 5 μg of tetracycline/ml. Escherichia coli One Shot TOP10 (Invitrogen, Carlsbad, CA) grown in Luria-Bertani medium was used as the host strain to construct plasmids, with tetracycline selection at 10 μg/ml. DNA from Mycoplasma pulmonis strain CTp12 was used as a control for some PCR experiments (40).
TABLE 1.
M. arthritidis strains used in this study
| Strain | Parent | Transposon | Transposon positiona | Disrupted gene product | Truncation of gene productb |
|---|---|---|---|---|---|
| 158 | NAc | NA | NA | NA | NA |
| TnCtrl | 158 | Tn4001TF1 | 37911 | NA | NA |
| KO1 | 158 | Tn4001TF1 | 38870 | MAM | 101/238 |
| KO2 | 158 | Tn4001TF1 | 38784 | MAM | 72/238 |
| Over1 | 158 | Tn4001TF1-mia-mam2 | 398742 | NA | NA |
| Over2 | 158 | Tn4001TF1-mia-mam1 | 95959 | NA | NA |
| Over3 | 158 | Tn4001TF1-mia-mam1 | 412672 | MspD | 2353/2592 |
The nucleotide positions correspond to the completed M. arthritidis genome sequence (GenBank accession number CP001047).
Number of amino acids in the truncated product/number of amino acids in the full-length product.
NA, not applicable.
Development of mam knockout mutants.
Oligonucleotides used for plasmid construction and PCRs are listed in Table 2. To minimize mutations caused by PCR amplification, Expand High FidelityPlus PCR system (Roche Applied Science, Indianapolis, IN) was used for all plasmid construction. Plasmid pMT85 carrying mini-Tn4001 was provided courtesy of R. Herrmann (53). To construct a mini-Tn4001 carrying a marker that would function in M. arthritidis, a 2.3-kb region of transposon Tn916 carrying the tetM gene was amplified by PCR using primer pair TetM-F-Bam and TetM-R-Bam. The product was cloned into the BamHI site of the mini-Tn4001 portion of pMT85 to generate Tn4001TF1 in plasmid pTF85 (Fig. 1A).
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide | Sequencea |
|---|---|
| TetM-F-Bam | GGTTGTGGATCCTTGTGGGTACTTTTAGGGC |
| TetM-R-Bam | CTTATTGGATCCGAAACCATATTTATATAACAAC |
| iPCR-F | CTGATTCTGTGGATAACCGTATTACCGC |
| iPCR-R | CAGTATTTGGTATCTGCGCTCTGCTGAAGCCAGTTA |
| iPCR-S | TTTGAGTGAGCTGATACCGCTCGC |
| mia-PF-Not | CGGCGGCCGCAACCTTTGGAGATTGCTTC |
| mia-PR-Eco | CATTATTTTATTGTGAATTCCTTTTC |
| mam-F-Eco | CACAAATTTAAAAATCATAAGGAATTCAAAAATGAAAAC |
| mam-R-Not | CGGCGGCCGCCCCCAACCTTTTAGATTG |
| mia-RT-F | AAATTTATTAAAACAGGTAACTCCG |
| mam-RT-F | GCTGTGAAAATGAAATTCTTCAC |
| mia-mam-RT-R | GCTAAAGTGACGGTTGCG |
| Tn-F | TTTAGCAAATCATATATAAAAGCGCG |
| Tn-R | TTTTAAATAGTTTCTTTGTTTACCCGC |
| Over1-F | ACTACCGAAGGCGAAACCGC |
| Over1-R | ACATCTTCGCAAGAAGCCGC |
Underlined nucleotides are the nucleotides in restriction sites used for cloning of PCR products.
FIG. 1.

(A) Schematic of plasmid pTF85 illustrating transposon Tn4001TF1 with the direction of transcription of the genes (open bars) indicated by arrows. tetM, tetracycline resistance marker; gm-r, gentamicin resistance marker; tnp, transposase gene; ori, origin of plasmid replication; B, BamHI restriction site; N, NotI site into which the mia-mam gene was inserted for overexpression studies; S, rightmost Sau3A1 site in Tn4001TF1 (other Sau3A1 sites not shown). The orientations of primers iPCR-F, iPCR-R, and iPCR-S used for inverse PCR to map the genomic location of the transposon are indicated. (B) Agarose gel of direct PCR products obtained from amplification of genomic DNA isolated from the mam knockout mutants KO1 and KO2 and using the mam-F-Eco and mam-R-Not primers that flank mam. Genomic DNA from wild-type M. arthritidis 158 and M. pulmonis strain CTp12 was used as positive and negative control, respectively. PCRs amplifying the mia promoter with primers mia-PF-Not and mia-PR-Eco were performed to serve as quality controls of DNA template preparation. The direct PCR products of the mam gene and the mia promoter are 805 bp and 369 bp, respectively.
To obtain mutants that fail to produce MAM, a transposon library was constructed using minitransposon Tn4001TF1. Mycoplasma strain 158 was transformed using the polyethylene glycol-mediated method as described previously (45). Plasmid pTF85 does not replicate in mycoplasmas, and transformants are obtained only when the minitransposon transposes into the mycoplasma chromosome. Individual transformant colonies were picked and grown at 37°C in 1 ml broth containing tetracycline. Each culture was frozen at −80°C in broth supplemented with 15% glycerol.
The genomic location of the minitransposon was determined for each transformant by PCR amplification of the junction between the transposon and the adjacent genomic DNA. The sequence of the PCR product was compared to the complete genome sequence for identification of the transposon insertion site. The inverse PCR conditions and primers were similar to those used to map the transposon location in transformants of M. pulmonis (41) except that genomic DNA was digested with Sau3AI instead of NlaIII and inverse PCR amplifications were performed with primers iPCR-F and iPCR-R. The inverse PCR product was purified from an agarose gel, and the nucleotide sequence was determined using primer iPCR-S. Two mutants, KO1 and KO2, were identified that had the minitransposon inserted within the mam gene (Table 1).
Development of mam overexpression mutants.
To construct mutants that overproduce MAM, the mam gene was inserted downstream of a strong promoter derived from the M. arthritidis mia gene (Fig. 2A). The mia promoter was thought to have strong activity because MIA is one of the most abundant proteins produced by M. arthritidis strain 158 (43). The mia promoter including its ribosomal binding site was amplified by PCR by using the primer pair mia-PF-Not and mia-PR-Eco and inserted into plasmid pCR2.1-Topo (Invitrogen) to generate plasmid Topo-mia. The mam coding region including its signal peptide coding sequence was amplified from strain 158 genomic DNA using primer pair mam-F-Eco and mam-R-Not and inserted into a separate pCR2.1-Topo vector, resulting in plasmid Topo-mam. The fragment containing the mia promoter was excised from Topo-mia by digestion with NotI and inserted into the NotI site of pBluescript to generate plasmid pBS-mia. The fragment containing the mam coding region was excised from Topo-mam by digestion with EcoRI and inserted into EcoRI site of pBS-mia. The resulting plasmid, pBS-mia-mam, has the mam coding region immediately downstream of the mia promoter. The mia-mam construct was excised from pBS-mia-mam by digestion with NotI and inserted into the NotI site of the Tn4001TF1 portion of pTF85. Two constructs were obtained, Tn4001TF1-mia-mam1 has the mia-mam gene in the forward orientation and Tn4001TF1-mia-mam2 has mia-mam in the reverse orientation in the transposon. The resulting plasmids, pTF85-mia-mam1 and pTF85-mia-mam2, were confirmed to be absent of any mutations by DNA sequence analysis and transformed into wild-type M. arthritidis 158 by the polyethylene glycol method.
FIG. 2.

(A) Schematic of the mam (top) and mia-mam (bottom) genes showing the locations of primers used for RT-PCR. mam-F, primer mam-RT-F; R, primer mia-mam-RT-R; mia-F, mia-RT-F; E, EcoRI restriction site used for combining the mia promoter with the mam coding region. (B) Agarose gels of RT-PCR and regular PCR products obtained from amplification of the mam (91-bp product) and mia-mam (117-bp product) genes using the three primers mia-mam-RT-R, mam-RT-F, and mia-RT-F mixed together. (Top) The template was genomic DNA from wild-type strain 158 (WT) or strain Over1. (Bottom) The template was Over1 genomic DNA (Over1 PCR), Over1 RNA (Over1 RT), Over2 RNA (Over2 RT), or Over3 RNA (Over3 RT).
The genomic location of the minitransposon carrying the mia-mam gene was determined to identify transformants that might have the minitransposon inserted into an innocuous site that would not adversely affect the virulence of the mycoplasma. The protocol for identifying the nucleotide insertion site of Tn4001TF1-mia-mam1 or Tn4001TF1-mia-mam2 in transformants of strain 158 was the same as described above for mapping the location of Tn4001TF1. Three independent overexpression mutants, Over1, Over2, and Over3, each with the transposon inserted at what appeared to be an innocuous site were chosen for further study.
RT-PCR.
Bacteria were grown to stationary phase, harvested by centrifugation, and washed once in 1× phosphate-buffered saline (PBS). Genomic DNA was isolated by using the EasyDNA kit (Invitrogen) according to the manufacturer's instruction. Total RNA was isolated using RNAqueous-4PCR kit (Applied Biosystems/Ambion, Austin, TX) according to the manufacturer's instructions. Turbo DNase (Ambion) was used to remove DNA contamination present in the samples. One-step reverse transcriptase PCR (RT-PCR) was performed using the MessageSensor RT kit (Ambion) and SuperTaq DNA polymerase (Ambion) according to the manufacturer's instructions. A 117-bp fragment covering a portion of the sequence downstream of the transcription start site of the mia promoter and mam coding region of the exogenous mia-mam transcripts was amplified using primers mia-RT-F and mia-mam-RT-R. A 91-bp fragment covering portions of natural mam promoter and mam coding region of the endogenous mam transcripts was amplified with primers mam-RT-F and mia-mam-RT-R (Fig. 2B). Transcripts of both mia-mam and mam were amplified in the same reaction. Reaction mixtures without RT or RNA template were used as negative controls. RNA samples were reverse transcribed for 15 min at 42°C. cDNA samples were predenatured for 5 min at 95°C, followed by 35 cycles, with 1 cycle consisting of denaturation at 95°C for 15 s, annealing at 45°C for 40 s, and extension at 72°C for 60 s. A final extension step was performed at 72°C for 10 min. PCR products were electrophoresed in 10% polyacrylamide gels and visualized by ethidium bromide staining. Experiments were repeated three times to ensure the same trend was reproduced.
Mitogen assays.
Cultures of M. arthritidis were grown in sterile microcentrifuge tubes to stationary phase, and the number of CFU was determined for each culture. M. arthritidis culture supernatant filtrates (MAS-F) was obtained by centrifuging the fresh culture at 12,000 × g for 15 min to remove cells, followed by filtration of the supernatant through a 0.2-μm sterile filter (VWR International, West Chester, PA). The filtrates were dialyzed overnight in Slide-A-Lyzer 7,000-molecular-weight-cutoff dialysis cassettes (Pierce, Rockford, IL) against 1× PBS at pH 7.2 to remove toxic metabolites that might interfere with the assay. Filtrates were stored at −80°C. Prior to the performance of mitogen assays, filtrates were adjusted to normalize the filtrates to the culture that had the lowest CFU concentration. For some assays, the normalized filtrates were diluted 10 times with 1× PBS (10× dilutions).
Splenocyte suspensions were prepared from two DBA/2J or CBA/J mice by pressing spleens through a 70-μm Nitex fabric (Sefar Filtration, Depew, NY) into Hank's buffered salt solution (HBSS) (Invitrogen/Gibco). Red blood cells were lysed in 10 ml Gey's solution (6). After red blood cell lysis, splenocytes were washed and counted using a Z2 Coulter Counter (Beckman Coulter, Fullerton, CA). Cells were harvested and suspended at a concentration of 5 × 106 cells/ml in RPMI 1640 (Invitrogen/Gibco) containing 10% fetal bovine serum (HyClone, Logan, UT) plus 0.05 mM 2-mercaptoethanol and 2 mM l-glutamine (Invitrogen) (RPMI-2ME).
The ability of MAS-F to stimulate T cells was assessed by the dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA). One hundred microliters of splenocyte suspension was dispensed into each well of a 96-well flat-bottom cell culture plate (Corning, Inc., Corning, NY). Five microliters of diluted MAS-F was added to each test well. As a negative control, 5 μl of dialyzed mycoplasma culture medium was added to wells in place of MAS-F to measure the background fluorescence. Positive controls contained 5 μl of 10 μg/ml phorbol-12-myristyl-13-acetate (PMA) (Sigma) and 5 μl of 20 μg/ml A23187 ionophore (Sigma) instead of MAS-F. The total volume of each well was adjusted to 200 μl with RPMI 1640 containing 10% fetal bovine serum plus 0.05 mM 2-mercaptoethanol and 2 mM l-glutamine. All test samples and controls were analyzed in triplicate. After incubation at 37°C in a humidified 5% CO2 atmosphere for 24 h, cell proliferation was assessed using the DELFIA cell proliferation kit (PerkinElmer) according to the manufacturer's instructions. Briefly, cells were labeled with 5-bromo-2-deoxyuridine (BrdU) by adding 20 μl of 100 μM BrdU labeling solution to each well and reincubated for an additional 8 h. The cells were fixed for 30 min at room temperature and stained with 100 μl of 0.5 μg/ml europium-labeled anti-BrdU. After the cells were incubated at room temperature for 2 h, they were washed four times and fluorescence was induced by adding 200 μl DELFIA inducer to each well. After 15 min of incubation, europium fluorescence was measured in a Wallac 1420 multilabel counter (PerkinElmer) in its time-resolved fluorometry (DELFIA) europium mode. Undiluted filtrates were used for the mitogenic comparison between wild-type M. arthritidis, the TnCtrl strain, and the mam knockout mutants KO1 and KO2. Because of the high level of SAg activity produced by the Over1, Over2, and Over3 overexpression mutants, it was necessary to assay 10× diluted filtrates to compare wild-type M. arthritidis, TnCtrl, and the mam overexpression mutants. Each experiment was repeated twice to ensure that the same trend was obtained.
Animal experimentation.
In preparation for infection of animals, cells were harvested by centrifugation, washed once in fresh EB medium, suspended in EB with 15% glycerol, and stored at −70°C. Before inoculation, an aliquot was thawed, and the number of CFU was determined.
The authors' institutional review boards approved all animal studies. Female CBA/J (Hk Eα+, 6 to 8 weeks old) and DBA/2J mice (Hd Eα+, 6 to 8 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME). CBA/J or DBA/2J mice were infected with 4 × 108 CFU or 1 × 109 CFU, respectively, by tail vein injection. For CBA/J mice, animals were infected in groups of five. Arthritis was assessed by histological analysis of the joints (wrist and ankle) tissue at 2 weeks postinoculation. The virulence of wild-type M. arthritidis strain 158, the TnCtrl strain, the two independent mam knockout mutants KO1 and KO2, and the three independent mam overexpression mutants Over1, Over2, and Over3 were assessed in CBA/J mice. Negative-control mice were injected with sterile EB. Similar experiments were performed in DBA/2J mice infected in groups of eight, but with a more comprehensive assessment of the induced disease. Visible arthritis in wrists, ankles, and metatarsal and metacarpal joints was scored as 0 to 3, and arthritis in digits were scored as 0 to 1.5 based on the severity of inflammation. Deaths in each group were recorded and compared by statistical analysis. The body weight of each mouse was measured and recorded. Other manifestations of the disease, such as dermal necrosis and paralysis of the legs, were also observed and recorded. Arthritis in DBA/2J mice was also assessed by histological analysis of the wrist and ankle joint tissue at 2 weeks postinoculation. For these experiments, the virulence levels of the transposon control, the mam knockout mutant KO1, and the overexpression mutant Over1 were assessed. Again, one group of mice was injected with sterile EB as a control.
Histopathological evaluation of arthritis.
Wrist and ankle joints were excised from mice sacrificed 2 weeks postinoculation. The joints were fixed in 70% alcoholic formalin, demineralized, sectioned at a thickness of 5 μm, and stained with hematoxylin-eosin. The severity of arthritis was assessed by a pathologist (T.R.S.) who was not provided with the treatment information of the different experimental groups. For each joint, the following lesion characteristics were scored: joint cavity exudate, synovial and capsular inflammation, synovial and capsular fibrosis, articular cartilage erosion, intra-articular bone destruction/remodeling, ankylosis, extra-articular osteomyelitis, periostitis and periosteal bone proliferation, tendon sheath exudate, tendon sheath inflammation, and tendon sheath fibrosis. These lesion characteristics were scored 0, 1, 2, or 3 for absent/normal, mild, moderate, or severe, respectively. Distribution (single or multiple joints and/or tendon sheaths) of the above changes was also scored 0, 1, 2, 3, or 4 for none or approximately one-fourth, one-half, or three-fourths or all of the joint structures affected, respectively. The total score for the joint was calculated as the product of the extent score and the sum of the component scores, with scores for cartilage destruction, osteomyelitis, and periostitis weighted by multiplying by 2, since bone lesions indicate severe disease. The total score for a mouse was the sum of the individual joint scores.
Growth in vitro and colonization of mouse joints.
The protocol for measuring the growth of mutants in vitro is adapted from that described elsewhere (27). Briefly, fresh mycoplasma cultures were diluted 1,000 times in EB in a sealed sterile tube and incubated at 37°C. Cultures were sampled every 2 h and continued until stationary phase was reached. At each time point, 50-μl samples were removed for CFU analysis. Growth curves were plotted as the log CFU versus time to calculate the doubling times of wild-type and mutant strains.
The level of colonization or load of M. arthritidis in the joints was also studied. DBA/2J mice were infected with 1 × 109 CFU in groups of three. Mice were sacrificed at 3 weeks. Ankle and wrist joints were collected under sterile conditions with the fur removed. Joints from each mouse were minced in 1 ml EB and sonicated in a sonifier (model 250/450; Branson, Danbury, CT) at a power level of 5 and duty cycle of 50% for 45 s. The suspension was subjected to CFU analysis. Some of the mycoplasmas recovered from the joints were analyzed as described below to assess the stability of the transposon insertion in the genome.
Stability of transposon integration.
To study the stability of the mini-Tn4001 transposon in the mycoplasma genome, mycoplasmas recovered from animal joints were grown in EB medium without antibiotic selection, and genomic DNA was isolated. Inverse PCR (procedure as described above) combined with DNA sequencing was first performed on the recovered TnCtrl isolates to confirm the presence of a single copy of the transposon in the genome. Regular PCR with primers mam-F-Eco and iPCR-R was used to confirm the presence of the transposon in the mam coding region of the KO1 mutant. Regular PCRs using primers flanking transposon insertion sites were then performed to exclude the presence of any revertants in these isolates that would arise from loss of the transposon. Primer pair mam-F-Eco and mam-R-Not and primer pair Tn-F and Tn-R were used to assay for loss of the transposon from its resident location in the KO1 mutant and the TnCtrl strain, respectively. Genomic DNA from wild-type strain 158 was used throughout as a control.
Protein profiles.
Cultures (10 ml) of M. arthritidis were grown until stationary phase was reached. Cells were harvested, washed twice in 1× PBS, and suspended in Western blot lysis buffer (52). The samples were boiled at 95°C for 10 min, and the protein concentration was quantified using a bicinchoninic acid protein assay kit (Pierce). Thirty micrograms of protein from each strain was resolved on a 4 to 15% gradient sodium dodecyl sulfate-polyacrylamide gel (ready gel; Bio-Rad). The separated proteins were transferred to a nitrocellulose membrane (Whatman). The major antigens of M. arthritidis were detected on the immunoblots using previously described serum and methods (43).
Nuclease assays.
DNase assays were performed as described elsewhere (25). Briefly, genomic DNA was isolated from E. coli One Shot TOP10 by using the EasyDNA kit. Three micrograms of E. coli genomic DNA was incubated at 37°C for 30 min with EB or diluted MAS-F from M. arthritidis strain TnCtrl, KO1, or Over1. Serial 10-fold dilutions of MAS-F of strain KO1 were used to digest the genomic DNA from E. coli. The MAS-F of strains TnCtrl, KO1, and Over1 was diluted with the dilution factor at which no DNase activity was detected in the MAS-F of strain KO1. As a control, a sample of genomic DNA was incubated with 7 units of DNase I (Qiagen, Hilden, Germany). To each reaction mixture, 5 μl EB medium or diluted filtrates and 8 μl RDD buffer (Qiagen) were added. The total volume for each reaction mixture was adjusted to 35 μl with water. DNA degradation was analyzed by agarose gel electrophoresis, followed by staining with ethidium bromide.
Statistical analysis.
All statistical analyses were performed with SigmaStat version 3.1 (Systat Software, San Jose, CA). Mitogen assay data were normally distributed and had equal variance and were analyzed by one-way analysis of variance. Pairwise comparison was performed using Tukey and Student-Newman-Keuls tests. Only the results that were confirmed significant by two tests were reported as significant. The Kruskal-Wallis one-way analysis of variance test was used to analyze the histopathological results. Survival data were compared by using the log rank test and the Gehan-Breslow test, and only the results that were confirmed significant by both tests were reported significant. Pairwise comparison of survival rates between groups was performed using the Holm-Sidak test. The t test was used for the comparison of the loads of mycoplasmas in the joints of infected animals. For all tests, a P of <0.05 was used as the criterion of significance.
RESULTS
MAM knockout mutants.
From a library of M. arthritidis strain 158 mutants created by using the minitransposon Tn4001TF1 as an insertional mutagen, two independent mutants with disruptions in the mam gene were isolated (Fig. 1). The full-length MAM protein has 238 amino acids. In the knockout mutants KO1 and KO2, the length of the truncated mam gene product would be only 42% (101 amino acids) or 30% (72 amino acids) of the full-length MAM protein (Table 1). Truncated MAM proteins from both mutants lack amino acids 71 to 95, a domain critical for lymphocyte activation (17).
MAM overproduction mutants.
To overproduce MAM, the mam coding region was inserted downstream of the strong promoter isolated from the M. arthritidis mia gene. Transformants containing the mia-mam gene cloned into Tn4001TF1 were screened to identify those that had the minitransposon inserted into the mycoplasma genome at a site that was likely to be innocuous (Fig. 2A and Table 2). Strain Over1 has the transposon integrated in an intergenic region downstream of ORF459, 460 bp upstream of the next gene, ORF462, which codes for a hypothetical lipoprotein. Strain Over2 has the transposon integrated within an intergenic region downstream of ORF109, 159 bp upstream of ORF110, which codes for a predicted ABC transporter ATP-binding protein. Based on its location, it is unlikely that the transposon in either mutant would alter the expression of neighboring genes. Strain Over3 has the transposon inserted into the 3′ end of the mspD gene, coding for a large surface protein of 2,592 amino acids (28). Strain Over3 would produce a MspD protein 91% the length of the wild-type protein. The Msp proteins are a 12-member family of predicted phase-variable surface proteins that may have redundant functions, and it is not known which of the msp genes is expressed in the parent strain. We view it unlikely that the Over3 strain would have altered virulence due to disruption of the 3′ terminus of mspD.
To compare the expression of exogenous mia-mam and endogenous mam, RT-PCR experiments were designed to amplify mia-mam and endogenous mam in the same reaction. Conventional PCR with DNA template demonstrated that the primers chosen amplified mia-mam and the native mam genes with comparable efficiency. About 10 times more mia-mam transcripts were detected than endogenous mam in the overexpression mutants (Fig. 2B). Controls demonstrated that no RT-PCR product was detected in reaction mixtures in which RT or RNA template was omitted (data not shown).
Mitogenic activity.
Splenocytes from both CBA/J and DBA/2J mice were used to study the mitogenic activity of MAS-F. Using undiluted filtrates, the overexpression mutants stimulated the splenocytes to such high levels that the cells proliferated and died during the 24-hour incubation period before the incorporation of BrdU was examined as a measure of T-cell stimulation. This suggested a level of mitogenic activity higher than that of controls stimulated with PMA and the calcium ionophore A23187, which lacked significant cell death. Using undiluted filtrates, wild-type strain 158 stimulated splenocytes from CBA/J and DBA/2J mice as predicted. The mitogenic activity of the TnCtrl strain was indistinguishable from that of strain 158, the parent. In contrast, the KO1 and KO2 mutants lacked mitogenic activity, stimulating splenocytes to about the same degree as controls that were incubated with mycoplasma growth medium (EB) only (Fig. 3A and C). Accordingly, there is no evidence of a T-cell mitogen secreted by M. arthritidis other than MAM. Using 10× diluted filtrates (Fig. 3B and D), significant increased mitogenic activity was detected in each of the mam overexpression mutants compared to wild-type M. arthritidis and TnCtrl. No difference in mitogenic activity was detected between the three mam overexpression mutants. These results indicate that the MAM protein produced from mia-mam acts as a mitogen and stimulates the production of splenocytes from CBA/J and DBA/2J mice.
FIG. 3.

Mitogenic activity of mam knockout and overexpression mutants on splenocytes of CBA/J (A and B) and DBA/2J (C and D) mice. T-cell proliferation was represented as fluorescence counts. Data have had background (splenocytes stimulated with EB) subtracted. Experiments were repeated twice to ensure that the same trend was obtained. The data represent the means of three replicates. The standard deviations are indicated by the error bars. Undiluted filtrates were used for the mitogenic comparison between the wild-type strain (WT), transposon control (TnCtrl), and mam knockout mutants (panels A and C). Tenfold-diluted filtrates were used for the mitogenic comparison between the wild-type strain, strain TnCtrl, and mam overexpression mutants. Values that are statistically significant are indicated by an asterisk.
MAM is not required to cause arthritis in CBA/J mice.
The ability of MAM knockout and overexpression strains to induce arthritis in CBA/J mice was compared to that of the wild-type and TnCtrl strains. Mice were sacrificed 2 weeks postinjection, and the wrists and ankle joints were collected for histopathological studies. Lesions induced by all strains of M. arthritidis were primarily synovitis and tenosynovitis with profound synovial proliferation and accumulation of polymorphonuclear leukocytes and mononuclear cells in synovia and tendon sheaths (Fig. 4A). Modest neutrophil and fibrin exudates in the articular lumina were also observed in some cases. Cartilage damage and osteomyelitis, which occur in arthritis induced in rats by M. arthritidis (22), were not observed. No arthritis (median arthritis score of 0) was induced by EB medium, consistent with previous studies (42). The median arthritis scores induced by the different strains of mycoplasma were compared. Mice infected with the parent 158 strain, mam knockout mutants, or overexpression mutants all had mild to moderate arthritis. The median arthritis scores for mice infected with the wild-type strain 158, TnCtrl, KO1, KO2, Over1, Over2, and Over3 strains were 1.1, 2.4, 3.1, 1.2, 2.9, 2.9, and 1.9, respectively (Fig. 4B). No significant differences in arthritis score between any of the groups infected with mycoplasma were detected by statistical analysis, indicating that MAM is not associated with arthritogenicity in CBA/J mice.
FIG. 4.

Arthritis induced in CBA/J mice. (A) Representative histopathology of arthritis induced in CBA/J mice. (a) 4× amplification of healthy metacarpal joints from a control mouse injected with EB medium; (b) 20× amplification of synovium of a control mouse injected with EB, showing healthy synovial cells; (c) 4× amplification of metacarpal joints of a mouse inoculated with a mutant, showing moderate synovitis (s) and tendonitis (t) with exudates (e) of neutrophils and fibrin in the joint space; (d) 20× amplification of synovium of a mouse inoculated with a mutant, showing moderate synovitis with cell proliferation (cp) and exudate of neutrophils and fibrin (e) overlying synovial cells. (B) Histological scores of CBA/J mice infected with M. arthritidis. The box encompasses the 25th through the 75th percentiles of the range of data (interquartile range [IQR]). The solid line in the box shows the median. The whisker shows up to 1.5 times the IQR. Data points outside the whisker are indicated by x's. WT, wild type.
MAM is associated with mortality but not with arthritis in DBA/2J mice.
A more comprehensive study of the association between MAM and disease was performed in DBA/2J mice, which have a defect in the C5 component of the complement system that may contribute to the high susceptibility of these animals to infection. For these experiments, the severity of disease induced by strains KO1, Over1, and TnCtrl and sterile EB medium was determined by assessment of clinical arthritis, histopathological examination of the joints, body weight, and signs relating to toxicity including time of death. Throughout the 2-week course of infection, all groups of mice infected with mycoplasma exhibited similar dynamic arthritic scores (Fig. 5A). All groups of infected animals had similar numbers of inflamed joints (Fig. 5B). The experimental protocol was to collect wrist and ankle joints at 2 weeks postinoculation for histopathological analysis, but all eight mice infected with strain Over1 and four of eight mice infected with strain TnCtrl died prior to the date of sacrifice. Histological analysis was limited to the surviving animals, all eight mice injected with EB medium, all eight mice infected with KO1, and four of eight mice infected with TnCtrl. No arthritis was induced by EB medium. All of the infected mice had mild to moderate arthritis. Lesions did not differ from those in CBA/J mice. The median arthritis score of animals infected with TnCtrl or KO1 was 8.6 or 9.7, respectively, a difference which is not statistically significant (Fig. 5C).
FIG. 5.
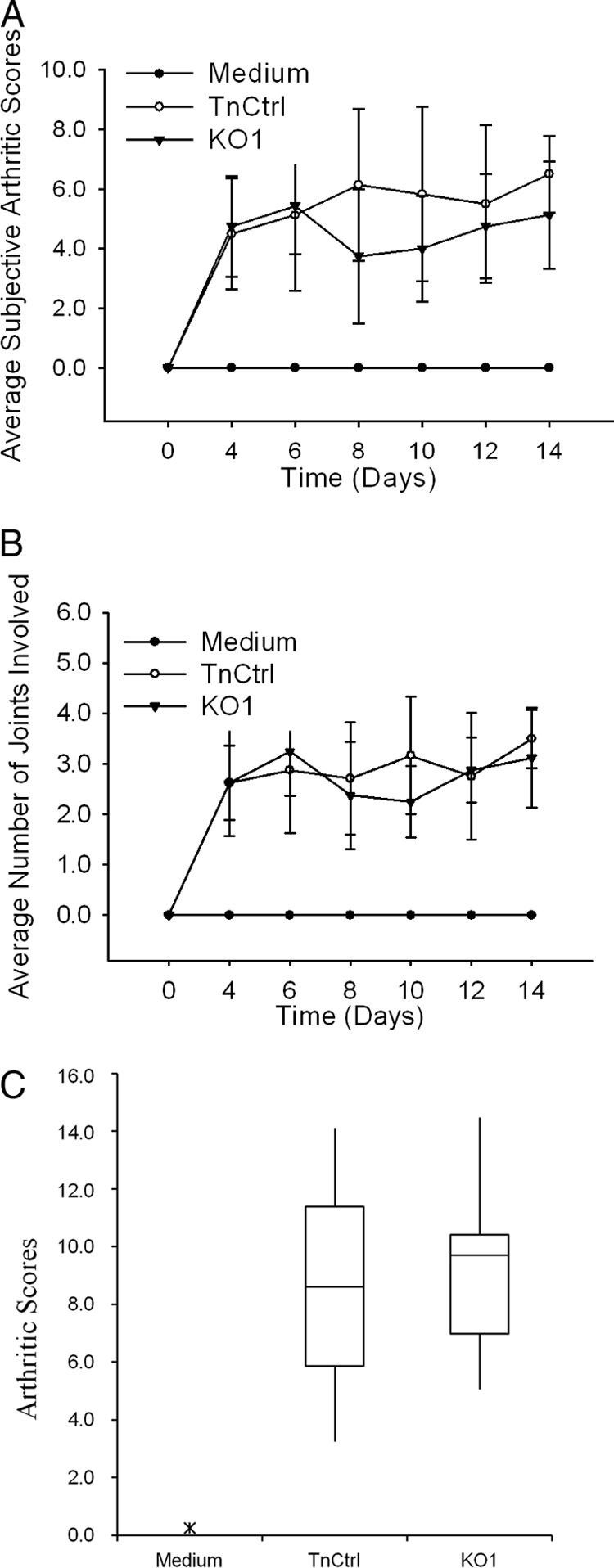
Arthritis induced in DBA/2J mice. (A) Average subjective scores for visible arthritis. Visible arthritis developed in wrists, ankles, and metatarsal and metacarpal joints was scored as 0 to 3, and visible arthritis in the digits were scored as 0 to 1.5 based on the severity of inflammation. (B) Average number of joints involved. Standard deviations are indicated by error bars. (C) Histological scores of arthritis in joints (wrist and ankle) tissue collected at 2 weeks postinoculation. The box encompasses the 25th through the 75th percentiles of the interquartile range (IQR). The solid line in the box shows the median. The whisker shows up to 1.5 times the IQR. Data points outside the whisker are indicated by x's.
Though there were no significant differences in arthritis detected between the mam mutants and strain TnCtrl, significantly different mortality was observed (Fig. 6A). This finding was first noted in a small pilot study in which we found that mice infected with strain Over1 were more prone to die than mice infected with strain KO1 (data not shown). In follow-up experiments, six of eight mice infected with Over1 died at day 3 and the remaining two mice died by day 6. Half of the mice infected with the TnCtrl strain died, all at day 7 or later. No mice infected with KO1 died. A comparison of the survival rates between the three groups yielded a statistically significant result. Consistent with the mortality data, significant greater weight loss occurred in mice infected with TnCtrl than with KO1. The weight losses in these two groups of mice were comparable for the first 6 days, but the weight loss for mice infected with strain KO1 subsequently leveled off, while weight loss for mice infected with strain TnCtrl continued (Fig. 6B). Additionally, more severe ruffling of fur, lethargy, and seizures were observed in mice infected with TnCtrl than in mice infected with KO1. Although similar dermal necrosis developed under the neck skin in the first week, the dermal necrosis that developed in mice infected with KO1 was shorter in duration and began to recede starting at day 7. For mice infected with TnCtrl, the severity of dermal necrosis was constant until the end of experiment. Three mice infected with TnCtrl exhibited paralysis in both hind legs 1 day before their death, which was never observed in mice infected with KO1 or in CBA/J mice. No disease was induced by EB medium under any conditions.
FIG. 6.

Toxic effect of M. arthritidis on DBA/2J mice. (A) Survival curve plotted as survival rate versus time (in days) over a 2-week period. Groups of mice infected with EB medium and strain KO1 were combined and are shown as the MAM (−) group. The difference between groups was statistically significant. (B) Weight change as a function of time over a 2-week period. Standard deviations are indicated by error bars. Mice infected with strain Over1 are not presented due to early death.
Transposon integrations are stable in vivo.
The loss of the transposon vector would lead to reversion of the mycoplasma's phenotype that might interfere with the interpretation of in vivo results. It was anticipated that the vector would be maintained in the genome because of its being a minitransposon, but the stability of Tn4001TF1 during infection had not been previously examined. A PCR-based assay was performed to confirm the presence and location of the transposon, excluding the possibility of transposon loss or jumping, in mam mutants. Seven isolates from each of three DBA/2J mice that had been infected with strain KO1 or TnCtrl were isolated from the joints of the animals at 21 days postinoculation. For each of these strains of mycoplasma, all 21 isolates had the transposon located at the same genomic site as did the inoculum. Using primers that bound to mycoplasma DNA that flanked the location of the transposon, no PCR product was obtained that would be indicative of even a minor subpopulation of template lacking the transposon. All evidence suggests that Tn4001TF1 is stably integrated into the mycoplasma genome.
MAM does not affect growth of mycoplasmas or colonization of mouse joints.
It has been reported that mice bearing different MHC molecules have different abilities to clear M. arthritidis, resulting in different susceptibilities to M. arthritidis-induced toxicity and death (20). It has also been reported that Escherichia coli producing the MAM protein has reduced growth (25). Because the possibility exists that MAM might alter growth or affect clearance, experiments were performed to study whether the load of mycoplasmas in the joints was altered in the mutants. The mam knockout mutants and the three mam overexpression mutants had the same growth parameters as the wild-type M. arthritidis 158 strain and strain TnCtrl did, with a doubling time of about 2 h (Fig. 7A). Colonization of M. arthritidis in vivo was assessed by measuring the CFU recovered from the joints of infected mice. For this purpose, DBA/2J mice were infected with 1 × 109 CFU. Joints were collected 3 weeks postinoculation, and the number of CFU was determined. A similar number of mycoplasmas were recovered from the mice infected with the KO1 mutant and TnCtrl strain, suggesting that the mam mutant survives in the joints as efficiently as does TnCtrl (Fig. 7B).
FIG. 7.

Effects of MAM on the growth of M. arthritidis in vitro and mycoplasma load in vivo. (A) Forty-hour growth curves of M. arthritidis strains. WT, wild type. (B) CFU recovered from the ankles and wrist joints at 3 weeks postinoculation from DBA/2J mice infected with 1 × 109 M. arthritidis. The log of the total CFU recovered from each mouse is presented. Standard deviations are indicated by error bars.
The antigenic profile of phase-variable genes and DNase activity of mycoplasma filtrates is not altered in mam mutants.
Several potential phase- or length-variable genes have been identified in the M. arthritidis genome (28). Some of these genes encode surface proteins that might conceivably affect virulence. For example, the MIA protein has been implicated as being associated with virulence of M. arthritidis (43). To examine the antigenic profile of wild-type and mutant strains, Western blots of M. arthritidis proteins were reacted with sera from mice that have been infected with M. arthritidis strain 158L3-1. The protein profiles, including the MIA protein profile, were essentially identical among all mutants and wild-type strains (data not shown).
MAM reportedly has DNase activity (25). Being a nuclease provides MAM with another possible mechanism to affect fitness in the host. Because several M. arthritidis genes are predicted to code for nucleases, the absence of MAM may not substantially reduce the total DNase activity of mycoplasma filtrates. To examine whether the DNase activity of filtrates correlated with MAM, genomic DNA from E. coli was digested with serial 10-fold dilutions of MAS-F from strain KO1. DNase activity was detected in 104-diluted MAS-F. When diluted 105, no DNase activity was detected in MAS-F prepared from strain TnCtrl, KO1, or Over1. We conclude that MAM is not the major nuclease present in preparations of MAS-F.
DISCUSSION
MAM has been studied for many years, but its definitive role in murine disease has remained unclear. Especially unclear is the arthritogenicity of MAM. All strains of M. arthritidis produce MAM, even strains that fail to induce significant arthritis (10, 46, 51). Some studies have indicated that arthritis developed only in strains of mice that had macrophages with a functional H-2E molecule to present MAM to T cells (14). However, mouse strains lacking H-2E can develop acute arthritis, and the incidence of arthritis in H-2E-negative strains at times can be higher than in H-2E-positive strains (21). One difficulty in sorting out the literature is that for some strains of mice, the lymphocytes might be more responsive to MAM than was realized at the time the experiments were performed (19). Similarly, there is no clear association between lymphocyte response to MAM and arthritis in rats. Athymic nude rats can develop polyarthritis in response to M. arthritidis infection (3). Although collagen-induced arthritis in mice can be triggered or exacerbated by systemic administration of MAM (15), arthritis in rats could be induced only with purified MAM by direct injection into the joints (5). The disease induced was transient and characterized by hypertrophy and hyperplasia in the subsynovium. In contrast, rats infected with live M. arthritidis organisms develop a purulent, sustained disease phenotype characterized by infiltration of the synovium and subsynovium with polymorphonuclear neutrophilic leukocytes. Unlike the unclear relationship between arthritis and MAM, a strong association between toxicity and MAM has been found. Toxic shock syndrome (20) and dermal necrosis (18) associated with M. arthritidis infection are dependent on MAM, since they occur only in mouse strains that have the appropriate MHC haplotype to respond to MAM. The association between toxicity and MAM was revisited recently in a study in which C3H/HeJ mice, which lack functional TLR4, were found to be more susceptible to toxic death induced by infection of live M. arthritidis organisms than C3H/HeSnJ mice, which have an intact TLR4 (36). The different susceptibilities to toxic death were attributed to different cytokine profiles induced by MAM, but no clear prediction as to the susceptibility to arthritis could be made because many of the C3H/HeJ mice died.
To study the role of MAM in disease induced by M. arthritidis, we developed stable mutants that failed to produce or overproduced MAM and demonstrated that MAM is the major mitogen secreted by M. arthritidis. The absence of MAM led to significantly reduced mitogenic activity, while excessive MAM production led to significantly enhanced mitogenic activity. We demonstrated that MAM was not associated with arthritis but was associated with toxic syndrome and mortality. All the mycoplasma isolates recovered from animals infected with mutants of M. arthritidis harbored a single copy of the same transposon in the same genomic location as in the inoculum. No revertants, emerging mutants, or contamination of other mutants was detected. The altered virulence in these mutants was associated with the level of mitogenic activity. Mutants and wild-type controls had no apparent differences in growth and had essentially the same antigenic profile.
Although the mitogenic activity of MAM is associated with lethal toxicity, the mechanism is unclear. Recent studies suggest that the cytokines elicited by M. arthritidis play a key role. The elicitation of type 1 cytokines and tumor necrosis factor alpha is associated with host susceptibility to M. arthritidis-induced disease, while a predominant induction of type 2 cytokines is associated with host resistance (34-36). Although one report (35) demonstrated that C3H/HeJ, a mouse strain mainly secreting type 1 cytokines in response to MAM, is more susceptible to M. arthritidis-induced arthritis than BALB/c, a mouse strain mainly producing a type 2 cytokine response to MAM, the difference in arthritis severity may not be due to the cytokines that are produced. Different responses other than cytokines may be elicited by MAM in mouse strains that have different genetic backgrounds. Also, all the cytokine changes discovered were systemic. It is unclear whether local cytokine expression, especially the cytokines in the joints, differs between strains of mice. Mu et al. compared infections in C3H/HeJ and C3H/SnJ mice, which differ only at the lpsd gene, encoding TLR4 (36). These mice exhibited different susceptibilities to toxicity and death induced by MAM as a result of the different cytokine profiles elicited. C3H/HeJ mice have a spontaneous mutation in the lpsd gene that results in the dominant secretion of type 1 cytokines, making these mice more prone to toxicity and death. The different cytokine profiles can be attributed to the different status of Toll-like receptors (34). Though TLR4 and TLR2 are both engaged by MAM, TLR4 appears to be an antagonist of TLR2 activity, since the absence of TLR4 leads to overactivation of TLR2 and subsequent release of type 1 cytokines. The systemic effects of MAM seen in this current study are reminiscent of those in these previous studies. DBA/2J mice infected with the mam overexpression mutant had significantly increased mortality, while mice infected with the mam knockout mutant had no mortality and reduced symptoms of toxicity, such as fatigue, ruffled fur, dermal necrosis, and weight loss. Further studies are needed to examine the cytokine profiles elicited during infection with the mam overexpression and knockout mutants to investigate the cytokine responses that are associated with lethal toxicity.
Although MAM may conceivably contribute in minor respects to arthritis induced by M. arthritidis, it is clear that MAM is not a major arthritogenic factor for CBA/J and DBA/2J mice because MAM is not required to cause arthritis in either strain. Mice infected with mam knockout mutants developed moderate arthritis indistinguishable from that of mice infected with control strains. The virulence of M. arthritidis is probably a multifactorial process (51). Several potential virulence factors have been identified in M. arthritidis. We no longer believe that bacteriophage MAV1 is associated with virulence (7, 8). MAA1 and MAA2 are two adhesion molecules that have been identified in M. arthritidis and that are involved in attachment to host lung cells in vitro (47, 49, 50). Rats that were actively immunized with MAA1 or MAA2 protein or passively immunized with antibodies against these proteins were partially protected from M. arthritidis-induced arthritis (51). However, all M. arthritidis strains have the ability to adhere to rat cells in vitro regardless of their virulence (51), and whether the MAA proteins have a role in mouse arthritis is unknown. Another candidate virulence factor is the MIA protein, which was the only noted antigenic difference between a spontaneous mutant (strain 158-1) of M. arthritidis 158 that has a drastic reduction in arthritogenicity (43). Further studies are needed to clarify the role of MIA in pathogenesis.
Our studies demonstrate the advantage of using Tn4001TF1 rather than previously described derivatives of Tn4001 as a mycoplasma mutagen. Tn4001 transposes actively in mycoplasmas. Revertants can arise at a high frequency and complicate phenotypic analysis. Tn4001TF1 lacks the transposase gene, which is located elsewhere on pTF85, eliminating many problems associated with instability of Tn4001 (37). Indeed, our results suggest that Tn4001TF1 is stably integrated in the M. arthritidis genome even during growth in animals for a period of at least 3 weeks. Analysis of the transposon libraries suggests that Tn4001TF1 inserts into the mycoplasma genome at essentially random sites (30). Unlike libraries constructed using Tn4001, no examples were encountered in which more than one copy of Tn4001TF1 was inserted in the genome of a single transformant.
We also demonstrated that Tn4001TF1 is a useful vector for mediating expression of cloned genes in mycoplasmas. Previous research has demonstrated that MIA is an immunodominant protein of M. arthritidis (43). By RT-PCR, we demonstrated that the strength of mia promoter is roughly 10 times that of the natural mam promoter. A stretch of 8 A nucleotides is found in the putative −35 and −10 regions of the mia promoter. We were initially concerned that phase variation due to slipped-strand mispairing within this poly(A) region might affect the spacing between the −10 and −35 sites, resulting in phase variation of promoter activity. RT-PCR experiments and mitogen assays performed on different stocks of the overexpression mutants that had been passaged in vitro did not detect any loss of mia-mam expression. Animal experiments demonstrated that overproduction of MAM leads to enhanced virulence, suggesting that mam is overexpressed in vivo from the mia-mam promoter. Therefore, the frequency of phase variation in mia promoter activity, if any, would be low.
Whether MAM-like SAgs are involved in human disease is unclear. M. arthritidis antigens have been detected in human patients (9). Antibody against MAM has also been detected in sera from patients with rheumatic diseases (39). The major T-cell subset identified in the synovium of several human rheumatoid arthritis patients expressed the exact Vβ that has been found to be responsive to MAM (1, 32). It has also been demonstrated that MAM can stimulate human T-cell proliferation (2), promote human B-cell differentiation (24), and upregulate human NK cell activity (26). Rheumatoid factors can be produced by human B cells in response to MAM stimulation (29). Therefore, MAM-like SAgs have the potential to perturb human immunity and contribute to disease. As demonstrated in our study, an acute severe toxic syndrome is associated with MAM. This is a reminiscence of acute diseases of human induced by other SAgs (38). Therefore, MAM-like SAgs, when encountered in vivo, may cause similar clinical syndromes or exacerbate the disease by further disturbing the immune system. Studies on MAM and other virulence factors of M. arthritidis will provide clues as to the role of SAgs in inflammatory diseases in humans and give insight into the pathogenesis of human rheumatic diseases, which may lead to novel therapeutic targets for these diseases.
Acknowledgments
This work was supported by NIH grant AR44252.
Editor: J. B. Bliska
Footnotes
Published ahead of print on 8 September 2008.
REFERENCES
- 1.Baccala, R., L. R. Smith, M. Vestberg, P. A. Peterson, B. C. Cole, and A. N. Theofilopoulos. 1992. Mycoplasma arthritidis mitogen. V beta engaged in mice, rats, and humans, and requirement of HLA-DR alpha for presentation. Arthritis Rheum. 35434-442. [DOI] [PubMed] [Google Scholar]
- 2.Bhardwaj, N., A. S. Hodtsev, A. Nisanian, S. Kabak, S. M. Friedman, B. C. Cole, and D. N. Posnett. 1994. Human T-cell responses to Mycoplasma arthritidis-derived superantigen. Infect. Immun. 62135-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Binder, A., H. J. Hedrich, K. Wonigeit, and H. Kirchhoff. 1993. The Mycoplasma arthritidis infection in congenitally athymic nude rats. J. Exp. Anim. Sci. 35177-185. [PubMed] [Google Scholar]
- 4.Cahill, J. F., B. C. Cole, B. B. Wiley, and J. R. Ward. 1971. Role of biological mimicry in the pathogenesis of rat arthritis induced by Mycoplasma arthritidis. Infect. Immun. 324-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cannon, G. W., B. C. Cole, J. R. Ward, J. L. Smith, and E. J. Eichwald. 1988. Arthritogenic effects of Mycoplasma arthritidis T cell mitogen in rats. J. Rheumatol. 15735-741. [PubMed] [Google Scholar]
- 6.Carsetti, R. 2004. Characterization of B-cell maturation in the peripheral immune system. Methods Mol. Biol. 27125-35. [DOI] [PubMed] [Google Scholar]
- 7.Clapper, B., A.-H. T. Tu, A. Elgavish, and K. Dybvig. 2004. The vir gene of bacteriophage MAV1 confers resistance to phage infection on Mycoplasma arthritidis. J. Bacteriol. 1865715-5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clapper, B., A.-H. T. Tu, W. L. Simmons, and K. Dybvig. 2004. Bacteriophage MAV1 is not associated with virulence of Mycoplasma arthritidis. Infect. Immun. 727322-7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark, H. W., M. R. Coker-Vann, J. S. Bailey, and T. M. Brown. 1988. Detection of mycoplasmal antigens in immune complexes from rheumatoid arthritis synovial fluids. Ann. Allergy 60394-398. [PubMed] [Google Scholar]
- 10.Cole, B. C. 1991. The immunobiology of Mycoplasma arthritidis and its superantigen MAM. Curr. Top. Microbiol. Immunol. 174107-119. [DOI] [PubMed] [Google Scholar]
- 11.Cole, B. C., and C. L. Atkin. 1991. The Mycoplasma arthritidis T-cell mitogen, MAM: a model superantigen. Immunol. Today 12271-276. [DOI] [PubMed] [Google Scholar]
- 12.Cole, B. C., and G. H. Cassell. 1979. Mycoplasma infections as models of chronic joint inflammation. Arthritis Rheum. 221375-1381. [DOI] [PubMed] [Google Scholar]
- 13.Cole, B. C., C. S. David, D. H. Lynch, and D. R. Kartchner. 1990. The use of transfected fibroblasts and transgenic mice establishes that stimulation of T cells by the Mycoplasma arthritidis mitogen is mediated by E alpha. J. Immunol. 144420-424. [PubMed] [Google Scholar]
- 14.Cole, B. C., R. A. Daynes, and J. R. Ward. 1981. Stimulation of mouse lymphocytes by a mitogen derived from Mycoplasma arthritidis. I. Transformation is associated with an H-2-linked gene that maps to the I-E/I-C subregion. J. Immunol. 1271931-1936. [PubMed] [Google Scholar]
- 15.Cole, B. C., and M. M. Griffiths. 1993. Triggering and exacerbation of autoimmune arthritis by the Mycoplasma arthritidis superantigen MAM. Arthritis Rheum. 36994-1002. [DOI] [PubMed] [Google Scholar]
- 16.Cole, B. C., D. R. Kartchner, and D. J. Wells. 1989. Stimulation of mouse lymphocytes by a mitogen derived from Mycoplasma arthritidis. VII. Responsiveness is associated with expression of a product(s) of the V beta 8 gene family present on the T cell receptor alpha/beta for antigen. J. Immunol. 1424131-4137. [PubMed] [Google Scholar]
- 17.Cole, B. C., K. L. Knudston, A. Oliphant, A. D. Sawitzke, A. Pole, M. Manohar, L. S. Benson, E. Ahmed, and C. L. Atkin. 1996. The sequence of the Mycoplasma arthritidis superantigen, MAM: identification of functional domains and comparison with microbial superantigens and plant lectin mitogens. J. Exp. Med. 1831105-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole, B. C., M. W. Piepkorn, and E. C. Wright. 1985. Influence of genes of the major histocompatibility complex on ulcerative dermal necrosis induced in mice by Mycoplasma arthritidis. J. Investig. Dermatol. 85357-361. [DOI] [PubMed] [Google Scholar]
- 19.Cole, B. C., A. D. Sawitzke, E. A. Ahmed, C. L. Atkins, and C. S. David. 1997. Allelic polymorphisms at the H-2A and HLA-DQ loci influence the response of murine lymphocytes to the Mycoplasma superantigen MAM. Infect. Immun. 654190-4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole, B. C., R. N. Thorpe, L. A. Hassell, L. R. Washburn, and J. R. Ward. 1983. Toxicity but not arthritogenicity of Mycoplasma arthritidis for mice associates with the haplotype expressed at the major histocompatibility complex. Infect. Immun. 411010-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cole, B. C., J. R. Ward, and L. Golightly-Rowland. 1973. Factors influencing the susceptibility of mice to Mycoplasma arthritidis. Infect. Immun. 7218-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cole, B. C., J. R. Ward, K. S. Jones, and J. F. Cahill. 1971. Chronic proliferative arthritis of mice induced by Mycoplasma arthritidis. I. Induction of disease and histopathological characteristics. Infect. Immun. 4344-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole, B. C., L. R. Washburn, G. J. Sullivan, and J. R. Ward. 1982. Specificity of a mycoplasma mitogen for lymphocytes from human and various animal hosts. Infect. Immun. 36662-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crow, M. K., G. Zagon, Z. Chu, B. Ravina, J. R. Tumang, B. C. Cole, and S. M. Friedman. 1992. Human B cell differentiation induced by microbial superantigens: unselected peripheral blood lymphocytes secrete polyclonal immunoglobulin in response to Mycoplasma arthritidis mitogen. Autoimmunity 1423-32. [DOI] [PubMed] [Google Scholar]
- 25.Diedershagen, M., S. Overbeck, S. Arlt, B. Plumakers, M. Lintges, and L. Rink. 2007. Mycoplasma arthritidis-derived superantigen (MAM) displays DNase activity. FEMS Immunol. Med. Microbiol. 49266-271. [DOI] [PubMed] [Google Scholar]
- 26.D'Orazio, J. A., B. C. Cole, and J. Stein-Streilein. 1996. Mycoplasma arthritidis mitogen up-regulates human NK cell activity. Infect. Immun. 64441-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dybvig, K., J. W. Simecka, H. L. Watson, and G. H. Cassell. 1989. High-frequency variation in Mycoplasma pulmonis colony size. J. Bacteriol. 1715165-5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dybvig, K., C. Zuhua, P. Lao, D. S. Jordan, C. T. French, A.-H. T. Tu, and A. E. Loraine. 2008. Genome of Mycoplasma arthritidis. Infect. Immun. 764000-4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emery, P., G. S. Panayi, K. I. Welsh, and B. C. Cole. 1985. Rheumatoid factors and HLA-DR4 in RA. J. Rheumatol. 12217-222. [PubMed] [Google Scholar]
- 30.French, C. T., P. Lao, A. E. Loraine, B. T. Matthews, H. Yu, and K. Dybvig. 2008. Large-scale transposon mutagenesis of Mycoplasma pulmonis. Mol. Microbiol. 6967-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodtsev, A. S., Y. Choi, E. Spanopoulou, and D. N. Posnett. 1998. Mycoplasma superantigen is a CDR3-dependent ligand for the T cell antigen receptor. J. Exp. Med. 187319-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howell, M. D., J. P. Diveley, K. A. Lundeen, A. Esty, S. T. Winters, D. J. Carlo, and S. W. Brostoff. 1991. Limited T-cell receptor beta-chain heterogeneity among interleukin 2 receptor-positive synovial T cells suggests a role for superantigen in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 8810921-10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, H., Y. Zhao, Y. Guo, S. J. Vanvranken, Z. Li, L. Eisele, and W. Mourad. 2007. Mutagenesis, biochemical, and biophysical characterization of Mycoplasma arthritidis-derived mitogen. Mol. Immunol. 44763-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mu, H. H., N. D. Pennock, J. Humphreys, C. J. Kirschning, and B. C. Cole. 2005. Engagement of Toll-like receptors by mycoplasmal superantigen: downregulation of TLR2 by MAM/TLR4 interaction. Cell. Microbiol. 7789-797. [DOI] [PubMed] [Google Scholar]
- 35.Mu, H. H., A. D. Sawitzke, and B. C. Cole. 2000. Modulation of cytokine profiles by the mycoplasma superantigen Mycoplasma arthritidis mitogen parallels susceptibility to arthritis induced by M. arthritidis. Infect. Immun. 681142-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mu, H. H., A. D. Sawitzke, and B. C. Cole. 2001. Presence of Lpsd mutation influences cytokine regulation in vivo by the Mycoplasma arthritidis mitogen superantigen and lethal toxicity in mice infected with M. arthritidis. Infect. Immun. 693837-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pour-El, I., C. Adams, and F. C. Minion. 2002. Construction of mini-Tn4001tet and its use in Mycoplasma gallisepticum. Plasmid 47129-137. [DOI] [PubMed] [Google Scholar]
- 38.Proft, T., and J. D. Fraser. 2003. Bacterial superantigens. Clin. Exp. Immunol. 133299-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawitzke, A., D. Joyner, K. Knudtson, H. H. Mu, and B. Cole. 2000. Anti-MAM antibodies in rheumatic disease: evidence for a MAM-like superantigen in rheumatoid arthritis? J. Rheumatol. 27358-364. [PubMed] [Google Scholar]
- 40.Simmons, W. L., and K. Dybvig. 2003. The Vsa proteins modulate susceptibility of Mycoplasma pulmonis to complement killing, hemadsorption, and adherence to polystyrene. Infect. Immun. 715733-5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teachman, A. M., C. T. French, H. Yu, W. L. Simmons, and K. Dybvig. 2002. Gene transfer in Mycoplasma pulmonis. J. Bacteriol. 184947-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tu, A.-H., J. R. Lindsey, T. R. Schoeb, A. Elgavish, H. Yu, and K. Dybvig. 2002. Role of bacteriophage MAV1 as a mycoplasmal virulence factor for the development of arthritis in mice and rats. J. Infect. Dis. 186432-435. [DOI] [PubMed] [Google Scholar]
- 43.Tu, A.-H. T., B. Clapper, T. R. Schoeb, A. Elgavish, J. Zhang, L. Liu, H. Yu, and K. Dybvig. 2005. Association of a major protein antigen of Mycoplasma arthritidis with virulence. Infect. Immun. 73245-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voelker, L. L., and K. Dybvig. 1998. Characterization of the lysogenic bacteriophage MAV1 from Mycoplasma arthritidis. J. Bacteriol. 1805928-5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voelker, L. L., and K. Dybvig. 1996. Gene transfer in Mycoplasma arthritidis: transformation, conjugal transfer of Tn916, and evidence for a restriction system recognizing AGCT. J. Bacteriol. 1786078-6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voelker, L. L., K. E. Weaver, L. J. Ehle, and L. R. Washburn. 1995. Association of lysogenic bacteriophage MAV1 with virulence of Mycoplasma arthritidis. Infect. Immun. 634016-4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Washburn, L. R., D. W. Bird, and K. Dybvig. 2003. Restoration of cytoadherence to an adherence-deficient mutant of Mycoplasma arthritidis by genetic complementation. Infect. Immun. 71671-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Washburn, L. R., B. C. Cole, M. I. Gelman, and J. R. Ward. 1980. Chronic arthritis of rabbits induced by mycoplasmas. I. Clinical, microbiologic, and histologic features. Arthritis Rheum. 23825-836. [DOI] [PubMed] [Google Scholar]
- 49.Washburn, L. R., S. Hirsch, and L. L. Voelker. 1993. Mechanisms of attachment of Mycoplasma arthritidis to host cells in vitro. Infect. Immun. 612670-2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Washburn, L. R., E. J. Miller, and K. E. Weaver. 2000. Molecular characterization of Mycoplasma arthritidis membrane lipoprotein MAA1. Infect. Immun. 68437-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Washburn, L. R., and E. J. Weaver. 1997. Protection of rats against Mycoplasma arthritidis-induced arthritis by active and passive immunizations with two surface proteins. Clin. Diagn. Lab. Immunol. 4321-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu, J., Z. Liu, T. L. Clemens, and J. L. Messina. 2006. Insulin reverses growth hormone-induced homologous desensitization. J. Biol. Chem. 28121594-21606. [DOI] [PubMed] [Google Scholar]
- 53.Zimmerman, C.-U., and R. Herrmann. 2005. Synthesis of a small, cysteine-rich, 29 amino acids long peptide in Mycoplasma pneumoniae. FEMS Microbiol. Lett. 253315-321. [DOI] [PubMed] [Google Scholar]