

Abstract
Rtt109, also known as KAT11, is a recently characterized fungal-specific histone acetyltransferase (HAT) that modifies histone H3 lysine 56 (H3K56) to promote genome stability. Rtt109 does not show sequence conservation with other known HATs and depends on association with either of two histone chaperones, Asf1 or Vps75, for HAT activity. Here we report the X-ray crystal structure of an Rtt109–acetyl coenzyme A complex and carry out structure-based mutagenesis, combined with in vitro biochemical studies of the Rtt109–Vps75 complex and studies of Rtt109 function in vivo. The Rtt109 structure reveals noteworthy homology to the metazoan p300/CBP HAT domain but exhibits functional divergence, including atypical catalytic properties and mode of cofactor regulation. The structure reveals a buried autoacetylated lysine residue that we show is also acetylated in the Rtt109 protein purified from yeast cells. Implications for understanding histone substrate and chaperone binding by Rtt109 are discussed.
HAT enzymes form a superfamily of proteins that transfer an acetyl group from the acetyl coenzyme A (acetyl-CoA) cofactor to the ε-amino group of histone or sometimes non-histone proteins to promote gene activation1. HATs also contain a structurally conserved core region that plays an analogous role in acetyl-CoA cofactor binding. Paradoxically, despite the similar chemistry carried out by these enzymes, and their structurally related core regions, they fall into subfamilies with very limited to no sequence homology. They also have structurally divergent core flanking regions and use divergent mechanisms for catalysis2–6. Three well-studied HAT families include Gcn5/PCAF, MYST and p300/CBP. Many of the HATs from these three subfamilies are catalytically active on their own but require associated protein subunits to target acetylation to nucleosomal histone substrates7.
Rtt109 (also known as KAT11) was first identified as a budding yeast regulator of Ty1 transposition that also plays an important role in the DNA damage response to genotoxic agents8,9. More recently, genomic and proteomic screens as well as a more directed effort led to the observation that Rtt109 promotes genome stability and resistance to a variety of DNA-damaging agents through the direct acetylation of H3K56 during S phase10–12. Notably, Rtt109 has no detectable sequence homology to other known HATs and, also unlike other HATs, was subsequently shown to carry out catalysis only in the presence of either the Asf1 or the Vps75 histone chaperone13–15.
To obtain molecular insights into the basis for HAT activity by Rtt109, we determined the X-ray crystal structure of Saccharomyces cerevisiae Rtt109 in complex with the acetyl-CoA cofactor and carried out structure-based mutagenesis together with in vitro biochemical studies of the Rtt109–Vps75 complex and studies of Rtt109 function in vivo. We found that the fungal-specific Rtt109 has substantial structural homology to metazoan p300/CBP. However, Rtt109 exhibits catalytic properties that are uncharacteristic of other HAT proteins, and the Rtt109 structure reveals a buried acetylated lysine residue that we show occurs through autoacetylation and that is present in the Rtt109 protein purified from yeast cells. We discuss implications for understanding histone substrate and chaperone binding by Rtt109.
RESULTS
Overall structure of the Rtt109–acetyl-CoA complex
We initially prepared full-length recombinant budding yeast Rtt109 in bacteria but were unable to prepare suitable crystals for X-ray structure determination. Based on the observation that residues 125–188 were poorly conserved among Rtt109 homologs from other fungal species and absent in fission yeast (Supplementary Fig. 1 online), we prepared a recombinant budding yeast deletion construct of Rtt109 (Δ130–179) that readily formed crystals in complex with acetyl-CoA that diffracted to 3.1 Å resolution. The structure was determined using a two-wavelength MAD experiment from a mercury-soaked crystal and refined to residual Rfree/Rwork values of 25.8%/19.4% with excellent geometry, except for one poorly ordered loop region (described below). Though Asf1 and Vps75 are necessary for Rtt109 catalytic activity, we showed using fluorescence-based assays with acetonyl-CoA6 and affinity-labeling studies with a sulfoxy-CoA analog16 that Rtt109 binding to acetyl-CoA is similar to Rtt109 in complex with Vps75 (Supplementary Fig. 2 online). Comparison of the coenzyme A analog affinities for Rtt109 and the Rtt10–Vps75 complex reveals less than a three-fold difference, which suggests that the histone chaperone is unlikely to significantly alter enzyme-CoA interactions. Thus, our structure probably captures a mechanistically relevant form of Rtt109, although we cannot rule out some conformational change occurring in the active complex.
The overall structure of the Rtt109 HAT domain consists of a central β-sheet comprising all eight β-strands of the molecule surrounded by nine α-helices and several loops (Fig. 1a). Helices α1–α4 line the bottom of the molecule, whereas helices α5–α7 plus the η1 310 helix form a helical bundle along one end of the molecule, and helices α8 and α9 form a helical bundle at the opposite end of the molecule. The acetyl-CoA cofactor is positioned between the β4-α2 segment on one side and the β5-α4 segment on the opposite side, with the 21-residue L1 loop connecting β5 to α4 also sitting on the side of the cofactor that is pointing away from the protein (Fig. 1a). A shallow groove is formed near the center and top face of the β-sheet (β2-β4) and flanked by the helical bundles at opposite ends of the molecule and the acetyl-CoA and L1 loop on the back. The current structure extends to residue 404 following the β8-strand, with residues 405–436 not visible in the electron density map and presumably disordered. The position of the α3-helix and β5-strand would place the 130–179 segment that is deleted in the current structure on the bottom surface of the molecule.
Figure 1.
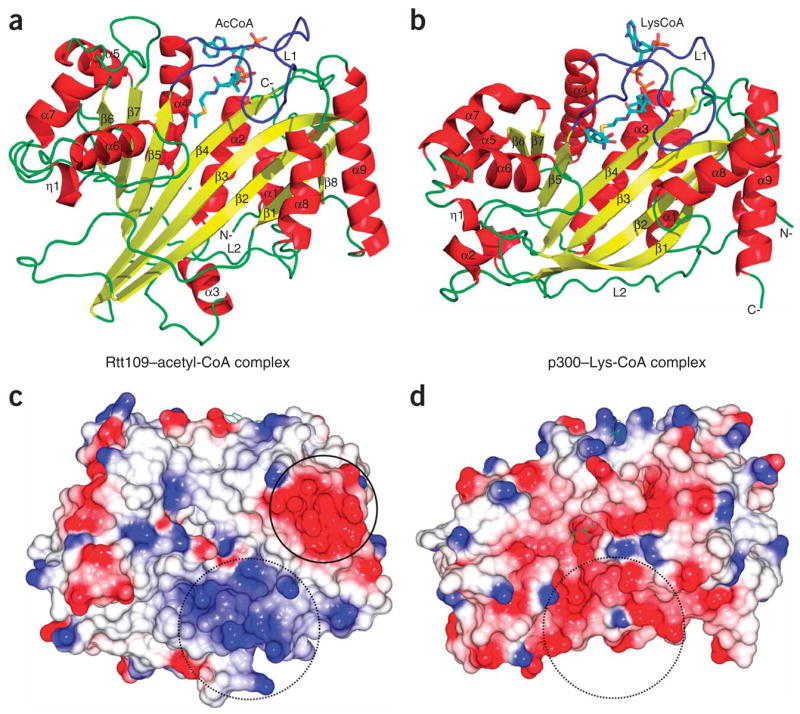
Overall structure of the Rtt109–acetyl-CoA complex and its relationship to p300. (a) Cartoon of the Rtt109 structure with bound acetyl-CoA. Secondary structural elements are numbered with the acetyl-CoA cofactor shown in stick representation in CPK coloring (with carbon atoms in aqua), with “N-” and “C-” denoting the respective polypeptide termini. (b) Cartoon of the p300 structure with bound Lys-CoA inhibitor. (c) Electrostatic surface of the Rtt109 HAT domain with blue, red and white representing electropositive, electronegative and neutral areas, respectively. (d) Electrostatic surface of the p300 HAT domain shown on the same scale as in c. Open circles in c and d highlight the divergent surface-charge features proximal to the lysine binding pockets. Closed circle highlights a conserved acidic patch on Rtt109 homologs.
Comparison to other HATs
A comparison of the Rtt109 HAT domain with other structurally characterized HATs including the Gcn5/PCAF, MYST and p300/CBP families shows a shared structurally conserved central region encompassing β2-β4 and α2 of Rtt109. Outside of this shared core region, Rtt109 shows divergence from the Gcn5/PCAF and MYST HATs but notable homology to the p300/CBP HAT domain with an overall r.m.s. deviation of 3.5 Å for 236 out of 354 Cα atoms (Fig. 1a,b and Supplementary Fig. 3a online). This is particularly surprising given the fact that Rtt109 homologs do not exist in metazoans that contain p300/CBP homologs, and fungal species do not contain p300/CBP homologs. Both HAT domains have a very similar configuration of secondary structural elements, including a structurally overlapping central αβ core region and similar helical bundles at opposite ends of the molecule, although these flanking regions do not overlay as well as the central core region. In addition, the unusually long L1 and L2 loops that sit over the acetyl-CoA cofactor to make important acetyl-CoA interactions (Figs. 1 and 2a,b) and track under the molecule connecting the two helical bundle regions, respectively, are conserved in both proteins. Although some of the secondary structural elements between the Rtt109 and p300 HAT domains are aligned at the primary structure level (for example, β5-α4-β6), other aligned secondary structural elements are shifted in the primary sequence (for example, β2-β3-β4) (Supplementary Fig. 1). Together, these findings suggest an evolutionary link between the HAT domains of Rtt109 from fungus and p300/CBP from metazoans, resulting in similar structural folds despite divergence in amino acid sequence.
Figure 2.

Detailed view of the Rtt109–acetyl-CoA structure. (a) Surface conservation of the Rtt109 HAT domain among the first four sequences in Supplementary Figure 1. The color-coding indicates the degree of conservation as shown in the bar key. Left: the box highlights a patch of high surface conservation possibly involved in histone substrate binding; the circle highlights another conserved patch of surface conservation on the α8-α9 segment. Right: the box highlights the highly conserved acetyl-CoA binding site. (b) The acetyl-CoA binding site. Acetyl-CoA is shown in stick model with CPK coloring and with a simulated annealing omit map (omitting acetyl-CoA) contoured at 2.5σ. Hydrogen bonds are shown as yellow dashed lines. (c) The putative histone H3K56 substrate binding site. Residues proximal to the active site are shown as stick models in CPK coloring in a cartoon of the active site, with potential catalytic residues labeled in red. The Lys-CoA inhibitor from the p300–Lys-CoA structure (magenta) is overlaid for reference to indicate the putative H3K56 entry site.
Unique aspects of the Rtt109 structure
Despite the similar overall architecture between the HAT domains of Rtt109 and p300/CBP, there are several notable differences that suggest functional divergence. In particular, an electrostatic surface proximal to the active site cavity of Rtt109 that must accommodate the lysine-bearing protein substrate is relatively apolar with a nearby basic patch (Fig. 1c). The apolar nature of this region is more similar to the corresponding substrate binding surfaces of Gcn5/PCAF and MYST proteins but different from the largely acidic binding region of p300/CBP (Fig. 1d). This is consistent with the divergent substrate preferences for these two HATs, whereby Rtt109 appears to be specific for H3K56 while p300/CBP is more promiscuous for several lysine sites within histone and non-histone proteins6,13. The residues that define the Rtt109 active site are highly conserved within the Rtt109 family (Fig. 2a) but differ from the residues that form the active site of p300/CBP (Supplementary Fig. 1a). In particular, potential catalytic residues that are proximal to the acetyl group of the acetyl-CoA cofactor include Ser86, Tyr199, Trp222 and Asp288 (Fig. 2c). In addition, Phe84, Phe192, Tyr199, Pro289 and the aliphatic regions of Lys87 and Arg194 form a hydrophobic tunnel that likely accommodates the aliphatic region of the target lysine (Fig. 2c). Although Pro84, Tyr199 and Trp222 show conservation in p300, the rest of the residues differ. Furthermore, two key catalytic residues in p300 (Trp1436 and Tyr1467) are not conserved in Rtt109 (Phe192 and Trp222) (Supplementary Fig. 1a). In addition, inhibition studies reveal that the potent p300/CBP inhibitor Lys-CoA (ref. 17), and a Lys56 peptide-CoA conjugate, are very poor inhibitors of Rtt109 (Supplementary Fig. 3a,b). Taken together, this is consistent with the fact that Rtt109 and p300/CBP target divergent lysine substrates and likely use different catalytic mechanisms.
Mutagenesis and structure-function correlations
A handful of debilitating mutations of Rtt109 conserved residues have been described in the literature and include D89A, D89N and a D287A D288A double mutation10,12. The Rtt109–acetyl-CoA structure reveals that Asp89 of the β4-strand is unlikely to play a direct catalytic role as it is too far from the scissile bond of acetyl-CoA. Instead, Asp89 forms a salt bridge with Lys210 and two hydrogen bonds with the backbone nitrogens of Tyr199 and Leu200 of the L1 loop that forms multiple interactions with acetyl-CoA (Fig. 2b). Consistent with the importance of this interaction, Lys210 is strictly conserved among the Rtt109 homologs (Supplementary Fig. 1). In addition, a similar salt bridge is seen between Asp1445 of the β4-strand and His1451 of the L1 loop of the p300 HAT, and Asp1445 is also mutationally sensitive for p300 activity6. Aspartic acid residues Asp287 and Asp288 are located in the loop that connects helix η1 to α6, and although these residues also appear to be too far away to directly participate in catalysis, they may participate in binding the lysine-bearing histone substrate. The loop connecting α7 to β7 is poorly ordered between residues 319 and 325 in our structure, as inferred from the relatively weak electron density and high atomic B-factors in this area of the structure (Supplementary Fig. 4 online). Notably, a triple mutation of a highly conserved motif in this loop, R318A Q319A E320A, does not compromise Rtt109 HAT activity13; this observation will be discussed in more detail below.
To further probe catalysis by Rtt109 and the molecular role of active site residues that potentially play catalytic roles, we first prepared a wild-type active Rtt109–Vps75 recombinant complex as well as complexes harboring several active site mutations in Rtt109. Whereas the wild-type Rtt109–Vps75 complex showed standard Michaelis-Menten kinetics with constant Xenopus laevis histone H3 and variable acetyl-CoA, an unusual sigmoidal concentration dependence was observed with varied H3 levels (Fig. 3a). The data was best fit by including a Hill coefficient of 3, which suggests a high degree of cooperativity of the histone substrate in the acetylation reaction. We observed related, albeit less pronounced, cooperativity using a yeast H3/H4 tetramer substrate, which showed comparable kcat and KM parameters (Supplementary Fig. 5a online). We interpret this kinetic behavior in a model where more than one histone H3 molecule is involved in the reaction complex, with additional histone H3 molecules mimicking the role of histone H4 in enhancing H3 acetylation by the Rtt109–Vps75 complex14—a cooperativity in acetylation that has not been reported with other HATs. The lack of inhibition of the Rtt109–Vps75 HAT by peptide-CoA conjugate bisubstrate analogs, which block other HATs, is consistent with its complex nature of protein substrate recognition (Supplementary Fig. 3a,b).
Figure 3.

Structure-guided mutagenesis of the active site. (a) Kinetics for 30 nM of Rtt109–Vps75 complex with saturating levels of acetyl-CoA (25 μM) and full-length histone H3 at specified concentrations. The averages of two duplicate experiments are shown for each data point. The solid line is fit to the Hill equation, and the dashed line is fit to the classical Michaelis-Menten equation (see Methods). (b) Kinetics for 30 nM of Rtt109 W222F–Vps75 mutant complex with 21 μM of acetyl-CoA and full-length histone H3 at specified concentrations. (c) Kinetics for 30 nM of Rtt109 D287A–Vps75 mutant complex with 21 μM of acetyl-CoA and full-length histone H3 at specified concentrations. (d) The table summarizes the kinetic parameters measured in Figure 3a–c and Supplementary Figure 5. N.D., not determined due to experimental considerations.
We next measured the steady state parameters of mutants in several active site residues that could potentially play catalytic roles. We examined the single-site mutants D89N, Y199S, W222F, D287A and Y68F (a residue that could potentially move to the active site upon Vps75 binding) (Figs. 2c and 3). We found that the D89N, Y199S and W222F single point mutants showed reduced catalytic efficiency compared with wild-type Rtt109–Vps75. The W222F mutant behaved most similarly to the D89N mutant, which had been previously reported to have a debilitating effect on catalysis10 (Fig. 3b and Supplementary Fig. 5d). W222F and D89N displayed apparent KM effects that resulted in a ~25-fold rate decrease at 8 μM H3 (Fig. 3d). The Y199S mutant had an intermediate effect on catalysis, with a rate decrease of about ten-fold, whereas the D287A mutant had a more mild three-fold decrease in rate at 8 μM H3 (Fig. 3 and Supplementary Fig. 5c). These results indicate that residues Trp222 and Asp89 in particular, and also Tyr199 and Asp287, have important roles in catalysis and/or substrate binding and validate the structural interactions observed.
To assess the functional relevance of these residues in vivo, Flag-tagged wild-type and mutant forms of Rtt109 were expressed from ARS CEN plasmids containing the natural RTT109 promoter in S. cerevisiae cells carrying a deletion of the RTT109 gene (Fig. 4). The Rtt109 mutants Y68F, Y199S, D287A, D288A and W222F were expressed at levels similar to those of wild-type Rtt109-Flag3, but the D89N mutant was expressed at a lower level (Fig. 4b). The Rtt109 D287A and D288A mutants had nearly normal levels of H3K56 acetylation and were not sensitive to genotoxic agents at the doses that we tested (Fig. 4a). Thus, individually, residues Asp287 and Asp288 are not essential for Rtt109 function in vivo, unlike the D287A D288A double mutant that was previously shown to cripple Rtt109 function in vivo10. In contrast, two of the mutants (D89N and W222F) had barely detectable levels of histone H3K56 acetylation based on immunoblotting, and the Y199S mutation also showed a strong decrease in acetylation (Fig. 4b). Consistent with this, Rtt109 D89N and W222F mutant cells were as sensitive as rtt109 null mutants to low doses of genotoxic agents (the alkylating agent methyl methane sulfonate (MMS) and hydroxyurea) that damage DNA during replication (Fig. 4a). The Rtt109 Y199S mutants showed moderate sensitivity to the same agents, in accordance with the presence of residual H3K56 acetylation (Fig. 4a,b). In keeping with its wild-type in vitro enzymatic activity, the Y68F mutant showed no defect in H3K56 acetylation or genotoxic agent sensitivity in vivo. In general, the in vivo data correlate well with the in vitro kinetic studies, with two caveats. First, the Rtt109 D287A mutant showed moderately compromised H3K56 acetylation properties in vitro (Fig. 3) but not in vivo (Fig. 4b), and this mutant had no detectable genotoxic agent sensitivity in vivo (Fig. 4a), which suggests that a more modest loss in H3K56 acetylation in vitro can be tolerated in vivo. Second, in addition to its lower catalytic activity in vitro, the Rtt109 D89N mutant shows reduced protein levels in vivo, which suggests that residue Asp89 also has a role in Rtt109 protein stability in cells.
Figure 4.

H3K56 acetylation and sensitivity to genotoxic agents in S. cerevisiae strains expressing wild-type and mutant forms of Rtt109. (a) Six-fold serial dilutions of the indicated yeast strains were plated onto YPD or YPD containing the indicated concentrations of MMS or hydroxyurea (HU). (b) Immunoblots of whole-cell lysates derived from the indicated yeast strains were probed to detect Rtt109-Flag3 and histone H3K56 acetylation. The Ponceau S staining and the immunoblot with an anti-H4 antibody were used as loading controls.
Autoacetylation of Rtt109
In vitro, budding yeast10,14 and fission yeast18 Rtt109, like p300 (ref. 19), can be autoacetylated. A sequence alignment of over 20 nonredundant Rtt109 homologs revealed three highly conserved lysine residues: Lys87, Lys210 and Lys290 (Supplementary Fig. 1). This suggests that these residues are potential autoacetylation sites. Analysis of the experimental electron density map does not reveal additional electron density that might correspond to an acetyl group at the tips of Lys87 or Lys210. However, Lys290, the most buried of the three lysine residues, does show additional density at its tip that could be modeled with an acetyl group that refines well (Fig. 5a). We used mass spectrometry analysis to confirm that Rtt109 Lys290 is indeed acetylated and that this modification arose from autoacetylation. Specifically, we carried out LC-MS/MS of trypsinized recombinant Rtt109 (Δ130–179), used for the current structure determination, and a recombinant Rtt109 D89N mutant in complex with Vps75. Analysis of this data shows that the recovery of an acetylated Lys290 peptide over a corresponding non-acetylated peptide is about three orders of magnitude greater for the Δ130–179 protein (Supplementary Fig. 6a online), whereas this ratio is roughly reversed for the Rtt109 D89N mutant (Supplementary Fig. 6b). This acetyl group makes hydrogen bonds to the mutationally sensitive Asp288, thus accommodating the negatively charged side chain that otherwise would not be well positioned within a highly conserved hydrophobic pocket defined by residues Phe270, Pro283, Phe285, Phe84, Phe192, Val329 and Trp312 (Fig. 5a). This interaction helps establish the conformation of Asp287, a solvent-exposed residue that is sensitive to mutation (Fig. 3). Given that the current position of the Lys290 acetyl group is only about 4.5 Å away from the putative Lys56 binding pocket, we speculate that this autoacetylation may occur intramolecularly.
Figure 5.
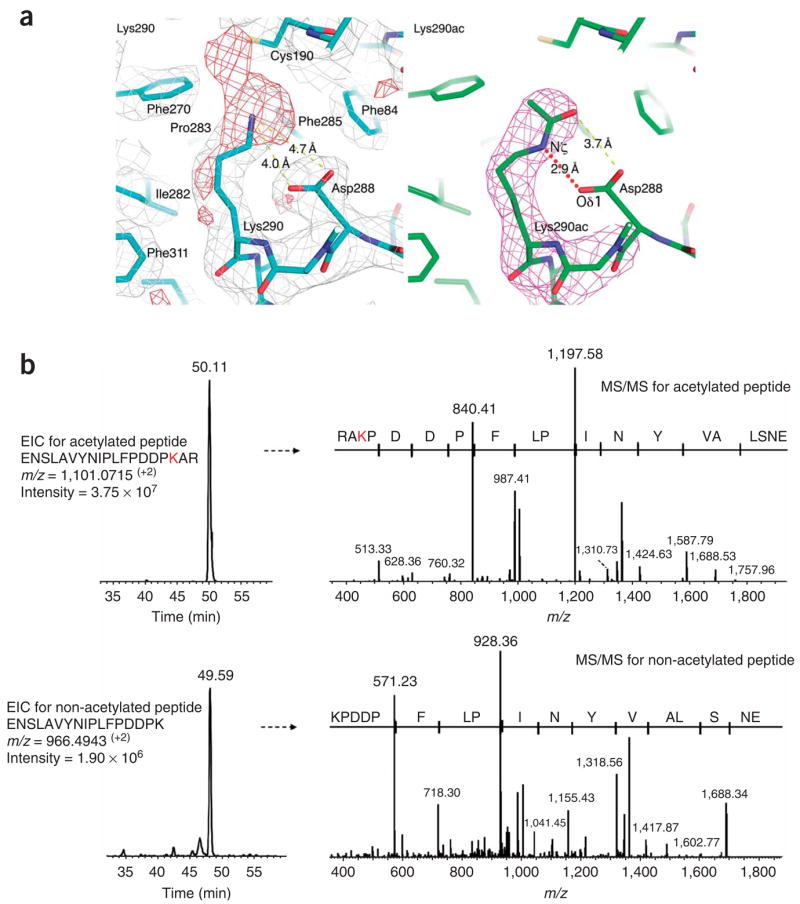
Rtt109 autoacetylation at Lys290. (a) Close-up view of acetyllysine residue 290 of Rtt109. Left: 2Fo − Fc (silver, 1σ) and Fo − Fc (red, 2.5σ) difference maps are shown after refinement of a Lys290 side chain. Right: simulated annealing omit (magenta, 2.5σ) maps are shown after refinement of an acetylated Lys290 side chain. Selected interatomic distances are highlighted in yellow dotted lines. Note the formation of a hydrogen bond mediated by the acetylated lysine side chain, between Lys290 Nζ and Asp288 Oδ1, as shown in red dotted line. (b) Extracted ion chromatogram (EIC) from LC-MS/MS analysis of the Rtt109 tryptic peptides containing acetylated Lys290 (upper left) and its corresponding MS/MS spectrum (upper right), confirming the location of the modified Lys290 residue. Lower panels show the unmodified Lys290 tryptic peptide from the same Rtt109 protein sample. Comparison of the relative intensities of the modified and nonmodified Lys290 residue further confirmed the prevalent in vivo acetylation of this residue.
To address whether Rtt109 is acetylated in vivo, we affinity purified TAP-tagged Rtt109 from asynchronous S. cerevisiae cells. After SDS-PAGE, the Rtt109 band was trypsinized and subsequently analyzed by LC-MS/MS (Fig. 5b). Database search results indicated that tryptic peptides comprising the Lys290 residue were present as both acetylated and non-acetylated forms, with the acetylated form being about an order of magnitude more abundant. The MS/MS spectrum of the acetylated tryptic peptide showed a characteristic y4 fragment ion, which is consistent with acetylated Lys290 (Fig. 5b). These data demonstrate that Lys290 is indeed acetylated in vivo in yeast cells. To determine the role of Rtt109 Lys290 acetylation in vivo, we generated an Rtt109 K290R mutant strain and tested it for H3K56 acetylation and sensitivity to genotoxic agents (Fig. 4). We found that this mutant is wild type for both H3K56 acetylation and sensitivity to genotoxic stress, which suggests that Rtt109 Lys290 acetylation is not essential for H3K56 acetylation in vivo. Alternatively, acetylated Lys290, as seen in the structure in the absence of Vps75 or Asf1 histone chaperone, may represent an autoinhibitory state of the enzymes that is inactive for H3K56 acetylation. Thus, a K290R mutation would maintain Rtt109 in a conformation that is compatible for H3K56 acetylation in vivo.
DISCUSSION
Although the structure of the Rtt109–acetyl-CoA complex does not provide direct insight into Vps75 or histone H3 interaction, it does suggest how aspects of these interactions might occur. Specifically, a mapping of surface conservation within the Rtt109 family onto the structure reveals that in addition to surface conservation in the catalytic and acetyl-CoA binding sites, another separate patch of surface conservation maps to the α8- and α9-helices (Figs. 1a and 2a and Supplementary Fig. 1). This surface patch overlaps with a highly electronegative patch on the Rtt109 HAT domain (Fig. 1c and Supplementary Fig. 7a online). This observation suggests that this region of Rtt109 might be used for interaction with an electropositive surface of an Rtt109 interacting protein such as Vps75 or histone H3. Correlating with a possible Vps75 interaction through this region of Rtt109, an alignment of Vps75 homologs revealed a highly conserved basic patch of residues that are not present within other histone chaperones (Supplementary Fig. 7b). We hypothesize that this segment of Vps75, with predicted helical secondary structure, would interact with the α8-α9 region of Rtt109 via helix-helix charge interactions. Notably, both motifs (from Rtt109 and Vps75) are either missing or not conserved in fission yeast and a number of other fungi (Supplementary Fig. 7b), which is consistent with previous reports that an Rtt109–Vps75 interaction is not essential for H3K56 acetylation in vivo and that fission yeast Rtt109 is functional for Lys56 acetylation in vivo18.
In light of (i) the fact that the 130–179 loop of Rtt109, deleted in our current structure, is not present in fission yeast and some other fungi, and (ii) the argument above that Rtt109 may not form an interaction with Vps75 in fission yeast, we addressed the role of this loop for Rtt109 association with budding yeast Vps75. To this end, we used pull-down studies to compare the ability of full-length Rtt109 and the (Δ130–179) deletion construct as glutathione S-transferase (GST) fusion proteins to associate with full-length Vps75 protein. We find that while full-length GST-Rtt109 interacts strongly with Vps75, the GST-Rtt109 (Δ130–179) deletion construct interacts very weakly under the same conditions (Supplementary Fig. 7c), which demonstrates that the 130–179 loop of Rtt109 from budding yeast is used for Vps75 interaction. This data, taken together with the lack of conservation of the α8-α9 region of Rtt109 between fission and budding yeast noted above, suggests that Rtt109 uses some unique features for interaction with Vps75 in budding yeast that are not conserved in fission yeast.
To explore what role Vps75 might play in H3K56 acetylation by the Rtt109–Vps75 complex, we titrated an excess amount of free Vps75 along with yeast H3/H4 tetramers into an Rtt109–Vps75 complex. We found that H3K56 acetylation was substantially inhibited (~ten-fold) compared to the same assay without added Vps75 (Supplementary Figs. 7d and 5a). We propose that this observation results from a competition between the Rtt109–Vps75 complex and free Vps75 for the H3/H4 tetramer, with slow exchange of the free and Rtt109-bound Vps75 due to a strong interaction between Rtt109 and Vps75 (data not shown). Based on this result, we propose that one role that Vps75 plays during H3K56 acetylation is to bind H3/H4 substrate for presentation to Rtt109 and acetylation.
The loop connecting α7 to β7 is highly conserved within the Rtt109 homologs, but surprisingly a R318A Q319A E320A mutation in this loop was found not to compromise catalytic activity13. This loop is only about 9 Å away from the putative H3K56 substrate binding site, which suggests that it may play some role in histone H3 substrate binding. Residues 319–325 of this loop are poorly ordered in our structure (Supplementary Fig. 4), which suggests that the loop is highly mobile. Notably, removal of this loop in the structure reveals a highly conserved hydrophobic core (Phe192, Pro245, Trp312 and Val329) that was formally occupied by Phe321 of the loop. Given that the histone H3K56 substrate of Rtt109 contains a phenylalanine (in budding yeast) or tyrosine (in most other species) two residues N-terminal to the Lys56 substrate, it is possible that an extended H3 peptide could bind to Rtt109 such that Tyr/Phe54 and Lys56 of H3 can occupy the position of Phe321 and the lysine binding tunnel of Rtt109, respectively. A docking exercise shows that a single side chain rotamer rotation (of Arg194) could accommodate such a binding mode (Supplementary Fig. 8 online). Based on this observation, we hypothesize that the Rtt109 region proximal to the α7-β7 loop participates in histone H3 substrate binding specificity, and that the α7-β7 loop could serve as a lid to mask the hydrophobic pocket from solvent exposure when the enzyme is not bound to histone H3 substrate.
Our structural studies suggest that yeast Rtt109 is the evolutionary precursor of metazoan p300/CBP. However, the structural similarity is not matched by an overlap in key catalytic residues. In addition, although it is tempting to suggest that p300/CBP might be a functional paralog of Rtt109, there is no strong evidence for H3K56 acetylation in human cells20, and we have found that H3K56 is not a good substrate for p300/CBP in vitro (data not shown). Taken together, this suggests that fungal Rtt109 and metazoan p300/CBP are not functional homologs, at least for the only H3K56 substrate that has been identified for Rtt109 so far. Nonetheless, this distinction between fold and mechanism underscores the point that the conserved fold or templating function of the HAT domain supercedes in importance the precise catalytic strategy, presumably because of the chemical simplicity of the reaction of a thioester with an amine. It should be noted that p300/CBP uses a series of protein-protein interaction domains within one long intact polypeptide for localization of the substrate. In contrast, this substrate-targeting function in Rtt109 is likely mediated by its intermolecular binding partners Asf1 and Vps75, thus exploiting modularity in chromatin regulation.
METHODS
Protein preparation
We subcloned DNA encoding full-length Rtt109 from yeast genomic DNA into an N-terminal GST fusion plasmid, which we further modified to encode the Rtt109 (Δ130–179) deletion construct by PCR mutagenesis using the Kunkel method21. We overexpressed full-length Rtt109 and its (Δ130–179) deletion mutant in Escherichia coli BL21(DE3) Gold cells as GST fusion proteins at 20 °C overnight upon induction with 0.8 mM IPTG at OD600 = 0.8. For purifying the Rtt109(Δ130–179) deletion mutant for crystallization, we harvested the cells and lysed them by sonication in phosphate-buffered saline (PBS)–β-mercaptoethanol (β-ME) buffer (150 mM NaCl, 8.3 mM Na2HPO4, 1.85 mM NaH2PO4, pH 7.4, 5 mM β-ME) supplemented with protease inhibitors. After centrifugation to isolate the supernatant, we purified the protein to homogeneity using a combination of GST affinity, on-resin cleavage with TEV protease, and further purification with MonoS cation exchange (in 20 mM HEPES pH 7.0 buffer with a NaCl gradient from 50 mM to 1,000 mM and 5 mM β-ME) and Superdex 200 gel filtration chromatography (in 20 mM HEPES pH 7.0, 150 mM NaCl and 5 mM β-ME). We concentrated the protein to about 15 mg ml−1 in the gel filtration buffer and flash froze it in liquid nitrogen until further use.
We subcloned DNA encoding full-length Vps75 from yeast genomic DNA with a single intron removed by standard mutagenesis. We then subcloned the DNA encoding the intronless Vps75 protein downstream of DNA encoding the N-terminal GST fusion protein expression plasmid pRSF-GST-Vps75. GST-cleaved Vps75 full-length protein was prepared as described above for the Rtt109 (Δ130–179) deletion mutant used for crystallization. For preparation of the Rtt109–Vps75 complex, we co-transformed the pRSF-GST-Vps75 plasmid described above with a pCDF-6xHis-Rtt109 plasmid into BL21(DE3) Gold cells. Protein co-expression was induced at 37 °C for 3 h upon induction with 0.8 mM IPTG. We harvested the cells and lysed them by sonication in PBS–β-ME supplemented with protease inhibitors. After centrifugation, we recovered the supernatant harboring the tightly associated complex, and the protein complex was purified to homogeneity using a combination of GST affinity followed by on-resin TEV cleavage, MonoQ anion exchange (using 20 mM HEPES pH 7.5 buffer and a NaCl gradient from 50 mM to 1,000 mM and 5 mM β-ME) and Superdex S200 gel filtration chromatography (using 20 mM HEPES pH 7.5, 150 mM NaCl and 5 mM β-ME). We then concentrated the protein complex to about 10 mg ml−1 in the gel filtration buffer and flash froze it until further use.
Crystallization and structure determination of Rtt109(Δ130–179)
We obtained small cubic-looking crystals of Rtt109(Δ130–179) using hanging-drop vapor-diffusion at room temperature (20 °C) from a reservoir solution containing 12.5% (v/v) polyethylene glycol (PEG)4000 and 100 mM HEPES pH 7.5 buffer. We prepared a cryoprotected heavy atom derivative of the crystals by soaking the crystals with harvest buffer containing saturated mercury acetate and supplemented with increasing amounts of glycerol cryoprotecting solution (0%, 10% and 20% (v/v)). We soaked protein crystals for about 5 min each during the step-wise soaking (from low to high glycerol concentration) and flash froze the resultant crystals in liquid nitrogen.
We collected crystallographic data from a mercury acetate-derivatized crystal at beamline X6A at the National Synchrotron Light Source (NSLS, Brookhaven National Laboratories). We collected two-wavelength anomalous (MAD)22 diffraction datasets, and the data were processed and scaled with the HKL2000 suite23. We identified two mercury atoms per protein molecule to obtain initial phases using the programs SOLVE/RESOLVE24. We built the protein model with Coot25 using the REFMAC library26 and refined with CNS27 with iterative model building, simulated annealing and positional and B-factor refinement strategies. Inspection of Fo − Fc difference Fourier maps at the later stage of refinement facilitated the unambiguous placement of the acetyl-CoA, which was modeled using the HI-CUp server28. After the complete model was checked for errors with composite simulated annealing omit maps, we further refined the structure to a final resolution of 3.1 Å with the addition of 28 waters (Table 1). We evaluated the final model with PROCHECK29, which revealed good stereochemical parameters and Ramachandran plot statistics for most residues, except several disordered residues primarily in the loop connecting α7 to β7, which we have discussed in the main text.
Table 1.
Data collection, phasing and refinement statistics
| Rtt109 (Δ130–179)–acetyl-CoA complex | ||
|---|---|---|
| Data collection | ||
| Space group | F432 | |
| Cell dimensions a, b, c (Å) | 233.91, 233.91, 233.91 | |
| Peak | Inflection | |
|
|
||
| Wavelength (Å) | 1.0072 | 1.0090 |
| Resolution (Å)a | 50–3.1 (3.21–3.1) | 50–3.3 (3.42–3.3) |
| Rsym | 0.123 (0.414) | 0.125 (0.394) |
| I/sI | 51.1(14.5) | 53.6 (16.5) |
| Completeness (%) | 100 (100) | 100 (100) |
| Redundancy | 45.5(44.9) | 46.8 (46.9) |
| Refinement | ||
| Resolution (Å) | 15–3.1 (3.21–3.1) | |
| No. reflections | 18,820 | 15,549 |
| Rwork/Rfree | 0.194/0.258 | |
| No. atoms | ||
| Protein | 2,893 | |
| AcCoA/Hg | 51/2 | |
| Water | 28 | |
| B-factors | ||
| Protein | 37.7 | |
| Ligand/ion | 64.1/37.8 | |
| Water | 24.6 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.0081 | |
| Bond angles (1) | 1.48 | |
Values in parentheses are for the highest resolution shell.
HAT enzyme assay
We adapted the radioactive HAT assay from previously published methods30,31. We determined the kinetic parameters for the Rtt109–Vps75 complex and mutants using this gel-based assay that measures the incorporation of a 14C-labeled acetyl group into the X. laevis histone H3 substrate. Enzyme activity for both the wild type and mutant Rtt109 complexes was linear with respect to time and enzyme concentration. We performed all assays in duplicate, and these generally agreed within 20%. We fitted the data to either the Michaelis-Menten equation (v = Vmax × [S]/KM + [S]) or the Hill equation (v = (Vmax × [S]n)/(Kn + [S]n)), where n is the Hill coefficient, by using a nonlinear least-squares analysis (computed with GraFit, version 5.0). For inhibition studies, we used previously synthesized Lys-CoA and the H3K56-CoA-bisubstrate conjugate that we newly synthesized on solid phase (H3 residues 49–63) (Supplementary Fig. 3b) by previously published methods17.
DNA damage sensitivity assays
Yeast strains and plasmids are described in the Supplementary Methods online. We grew yeast strains overnight at 30 °C in minimal medium lacking leucine for plasmid selection. We diluted cultures to equivalent cell densities, and five-fold serial dilutions were plated onto rich medium (YPD) containing MMS (0.002%, 0.004% or 0.008%) or hydroxyurea (20, 40 or 80 mM) and incubated for 3 d at 30 °C.
Immunoblotting
We grew yeast cells to exponential phase in YPD medium at 30 °C and prepared whole-cell lysates from 107 cells using an alkaline extraction method32. We separated proteins using SDS polyacrylamide gels, and the proteins were transferred to nitrocellulose membranes. We used an immunoblot with anti-H4 antibody and Ponceau S staining as loading controls. We probed blots with antibodies against the Flag epitope (anti-Flag M2, Sigma), Lys56 acetylated H3 (ref. 33) or H4 (anti-H4 AV94).
Histone preparation, pull-down studies, mass spectrometry analysis and figure rendering can be found in the Supplementary Methods.
Accession codes
Protein Data Bank: Atomic coordinates of the Rtt109 (Δ130–179)–acetyl-CoA complex and structure factors are deposited under accession code 3D35.
Supplementary Material
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
Acknowledgments
We thank M. Allaire for assistance with crystallographic data collection and K. Speicher, T. Beer and D. Speicher in the Wistar Institute Proteomics Core Facility for performing the in-gel digestion, LC-MS/MS, database searching and analysis on the recombinant Rtt109 proteins. This work was supported by US National Institutes of Health grants to R.M. and P.A.C. Part of this research was conducted on beamline X6A at the National Synchrotron Light Source at Brookhaven National Laboratory, which is supported by the US Department of Energy under contract No.DE-AC02-98CH10886. Beamline X6A is funded by the US National Institutes of Health, National Institute of General Medical Sciences under agreement Y1 GM-0080-03. Research in the A.V. and P.T. laboratories is funded by the Canadian Institutes for Health Research (CIHR) operating funds. The Institute for Research in Immunology and Cancer receives infrastructure funds from Fonds de Recherche en Sante du Quebec and a multi-resources facility grant from CIHR.
Footnotes
AUTHOR CONTRIBUTIONS
Y.T., M.A.H., H.W., K.M., W.R. and M.G. designed and carried out experiments reported in the manuscript, and prepared manuscript figures and text; E.J. carried out preliminary studies that led to experiments reported in the manuscript; P.T., A.V., P.A.C. and R.M. designed and supervised experiments and prepared manuscript text. All authors read and approved the submitted manuscript.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Marmorstein R. Structure and function of histone acetyltransferases. Cell Mol Life Sci. 2001;58:693–703. doi: 10.1007/PL00000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan Y, Harper S, Speicher DW, Marmorstein R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat Struct Biol. 2002;9:862–869. doi: 10.1038/nsb849. [DOI] [PubMed] [Google Scholar]
- 3.Trievel RC, et al. Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proc Natl Acad Sci USA. 1999;96:8931–8936. doi: 10.1073/pnas.96.16.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berndsen CE, Albaugh BN, Tan S, Denu JM. Catalytic mechanism of a MYST family histone acetyltransferase. Biochemistry. 2007;46:623–629. doi: 10.1021/bi602513x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanner KG, Langer MR, Kim Y, Denu JM. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J Biol Chem. 2000;275:22048–22055. doi: 10.1074/jbc.M002893200. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–850. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- 7.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 8.Bennett CB, et al. Genes required for ionizing radiation resistance in yeast. Nat Genet. 2001;29:426–434. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- 9.Chang M, Bellaoui M, Boone C, Brown GW. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci USA. 2002;99:16934–16939. doi: 10.1073/pnas.262669299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han J, et al. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–655. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- 11.Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315:649–652. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider J, Bajwa P, Johnson FC, Bhaumik SR, Shilatifard A. Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J Biol Chem. 2006;281:37270–37274. doi: 10.1074/jbc.C600265200. [DOI] [PubMed] [Google Scholar]
- 13.Tsubota T, et al. Histone H3–K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 2007;25:703–712. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han J, Zhou H, Li Z, Xu RM, Zhang Z. Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J Biol Chem. 2007;282:28587–28596. doi: 10.1074/jbc.M702496200. [DOI] [PubMed] [Google Scholar]
- 15.Han J, Zhou H, Li Z, Xu RM, Zhang Z. The Rtt109-Vps75 histone acetyl-transferase complex acetylates non-nucleosomal histone H3. J Biol Chem. 2007;282:14158–14164. doi: 10.1074/jbc.M700611200. [DOI] [PubMed] [Google Scholar]
- 16.Hwang Y, et al. A selective chemical probe for coenzyme A-requiring enzymes. Angew Chem Int Ed. 2007;46:7621–7624. doi: 10.1002/anie.200702485. [DOI] [PubMed] [Google Scholar]
- 17.Lau OD, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 18.Xhemalce B, et al. Regulation of histone H3 lysine 56 acetylation in Schizosaccharomyces pombe. J Biol Chem. 2007;282:15040–15047. doi: 10.1074/jbc.M701197200. [DOI] [PubMed] [Google Scholar]
- 19.Thompson PR, et al. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol. 2004;11:308–315. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- 20.Garcia BA, et al. Organismal differences in post-translational modifications in histones H3 and H4. J Biol Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 21.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendrickson WA, Ogata CM. Phase determination from multiwavelength anomalous diffraction measurements. Methods Enzymol. 1997;276:494–523. doi: 10.1016/S0076-6879(97)76074-9. [DOI] [PubMed] [Google Scholar]
- 23.Dauter Z, Dauter M, Rajashankar KR. Novel approach to phasing proteins: derivatization by short cryo-soaking with halides. Acta Crystallogr D Biol Crystallogr. 2000;56:232–237. doi: 10.1107/s0907444999016352. [DOI] [PubMed] [Google Scholar]
- 24.Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Vagin AA, et al. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 27.Brunger AT, et al. Crystallography & NMR system: a new software suite for macro-molecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 28.Kleywegt GJ. Crystallographic refinement of ligand complexes. Acta Crystallogr D Biol Crystallogr. 2007;63:94–100. doi: 10.1107/S0907444906022657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 30.Lau OD, et al. p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Kinetic analysis of the catalytic mechanism. J Biol Chem. 275:21953–21959. doi: 10.1074/jbc.M003219200. 2000. [DOI] [PubMed] [Google Scholar]
- 31.Thompson PR, Kurooka H, Nakatani Y, Cole PA. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J Biol Chem. 2001;276:33721–33729. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- 32.Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 33.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.