

Abstract
The DNA glycosylase hNEIL1 initiates the base excision repair (BER) of a diverse array of lesions, including ring-opened purines and saturated pyrimidines. Of these, the hydantoin lesions, guanidinohydantoin (Gh) and the two diastereomers of spiroiminodihydantoin (Sp1 and Sp2) have garnered much recent attention due to their unusual structures, high mutagenic potential and detection in cells. In order to provide insight into the role of repair, the excision efficiency by hNEIL1 of these hydantoin lesions relative to other known substrates was determined. Most notably, quantitative examination of the substrate specificity with hNEIL1 revealed that the hydantoin lesions are excised much more efficiently (> 100-fold faster) than the reported standard substrates thymine glycol (Tg) and 5-hydroxycytosine (5-OHC). Importantly, the glycosylase and β,δ-lyase reactions are tightly coupled such that the rate of the lyase activity does not influence the observed substrate specificity. The activity of hNEIL1 is also influenced by the base pair partner of the lesion, with both Gh and Sp removal being more efficient when paired with T, G or C than when paired with A. Notably, the most efficient removal is observed with the Gh or Sp paired in the unlikely physiological context with T; indeed, this may be a consequence of the unstable nature of base pairs with T. However, the facile removal via BER in promutagenic base pairs that are reasonably formed after replication (such as Gh:G) may be a factor that modulates the mutagenic profile of these lesions. In addition, hNEIL1 excises Sp1 faster than Sp2 indicating the enzyme can discriminate between the two diastereomers. This is the first time that a BER glycosylase has been shown to be able to preferentially excise one diastereomer of Sp. This may be a consequence of the architecture of the active site of hNEIL1 and the structural uniqueness of the Sp lesion. These results indicate that the hydantoin lesions are the best substrates identified thus far for hNEIL1, and suggest that repair of these lesions may be a critical function of the hNEIL1 enzyme in vivo.
Cells experiencing oxidative stress have an overabundance of reactive oxygen species (ROS) such as superoxide, hydrogen peroxide, and hydroxyl radicals (1, 2). ROS are present in cells as byproducts of endogenous metabolic reactions or as a result of external sources such as ionizing radiation. The reactions mediated by ROS can lead to various types of DNA damage including strand breaks, DNA-protein cross-links, abasic sites, and base lesions, which are potentially detrimental to cells (3–6). Oxidative DNA damage is mitigated by a variety of DNA repair pathways (7–9). The importance of repairing DNA damage has been highlighted by the correlation between defects in DNA repair pathways and cancer (7, 10–12).
Although a variety of guanine oxidation products have been identified (13), most studies have focused on the oxidized lesion, 7,8-dihydro-8-oxo-guanine (OG)1,2 (7, 14). Two oxidized guanine lesions that have recently been attracting attention are the hydantoin lesions, spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh) (Figure 1) due to their interesting structures and high mutagenic potential (7, 13). These lesions can be formed from G or OG under a variety of conditions with a plethora of oxidants, such as singlet oxygen, peroxynitrite, and high-valent metal compounds (13, 15–19). Their facile formation is highly suggestive of the presence of such lesions in cells. Moreover, Hailer et al. detected Sp by ESI-MS in E. coli cells deficient in the base excision repair glycosylase Nei after treatment with chromate (20). In vitro, these lesions can be produced conveniently by oxidation of OG with the one-electron oxidant sodium hexachloroiridate (IV) (21–24). This has allowed detailed examination of the unique structural properties of these lesions, their mutagenic effects in vivo, and their processing by DNA polymerases and repair enzymes in vitro (7, 13, 21). Single nucleotide insertion and primer extension experiments using Escherichia coli (E. coli) Klenow fragment exonuclease I (KF exo−) DNA polymerase indicate that dAMP and dGMP are inserted opposite these oxidized lesions (25, 26). Moreover, in E. coli based mutagenesis assays with single-stranded lesion-containing viral DNA, Gh and Sp are potently mutagenic causing both G→ T and G→ C transversion mutations (27, 28).
Figure 1. Structures of various lesions used in this study.
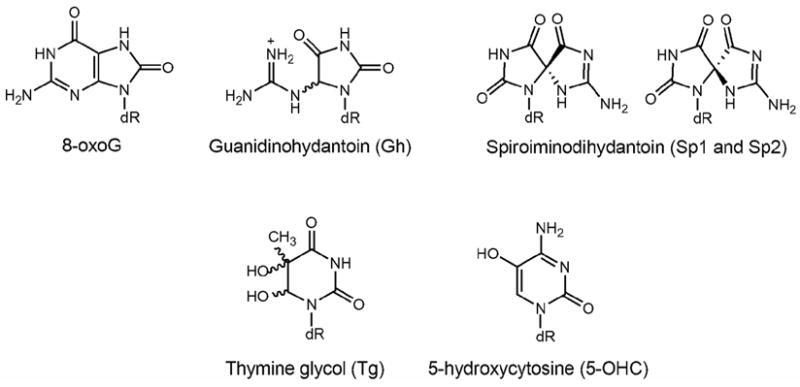
DNA duplexes containing the lesion base OG, the hydantoin products, guanidinohydantoin (Gh) and the two diastereomers of spiroiminodihydantoin (Sp) derived from oxidation of OG, thymine glycol (Tg) and 5-hydroxycytosine (5-OHC) were used as substrates to evaluate the relative processing of these lesions by hNEIL1.
Oxidative damage to individual DNA bases may be repaired via the base excision repair (BER) pathway that is initiated by damage-specific glycosylases that excise damaged DNA bases (7). Other enzymes in the BER pathway remove the remaining sugar fragment and incorporate an undamaged nucleotide at the site of the original damaged base. In E. coli, the prevention of mutations associated with OG formed within duplex DNA requires two base excision repair (BER) glycosylases, MutM and MutY (7, 29, 30). MutM, also known as Fpg, removes OG from OG:C mispairs in the DNA duplex while MutY removes A from an OG:A mismatch to prevent propagation of this inappropriate base pair and provide an opportunity to recreate a proper substrate for Fpg/MutM. Two well-characterized mammalian repair enzymes, OGG1 and the endonuclease III homolog 1 (NTH1), are known to be responsible for repairing oxidatively damaged purines and pyrimidines, respectively (8). Recently, a new set of BER enzymes has been identified that are structural homologues of the Fpg/Endonuclease VIII (Nei) family. These mammalian homologs are designated as the Nei-like or ‘NEIL’ family of enzymes (NEIL1,2,3) (31–34).
The human NEIL1 (hNEIL1) enzyme is the best characterized of the hNEIL enzymes and is structurally similar to Nei and Fpg (35). A distinct feature of hNEIL1 is the presence of a “zincless finger DNA binding motif that effectively mimics the zinc finger motif found in Fpg and Nei (35). Like Fpg and Nei, hNEIL1 is a bifunctional glycosylase/lyase that utilizes the N-terminal proline residue to catalyze β- and δ-elimination reactions leading to strand scission (31, 32, 34). Damaged bases that have been shown to be removed by hNEIL1 include thymine glycol (Tg), 5-hydroxycytosine (5-OHC), dihydrothymine (DHT), 5-hydroxyuracil (5-OHU), the ring-opened formamidopyrimidines (FapyG and FapyA) and oxanine (31, 34, 36, 37) Notably, the activity of NEIL enzymes towards OG-containing duplexes has been a source of debate (31, 32, 34, 38).
The hydantoin lesions, Gh and Sp have been shown to be substrates for the E. coli enzymes Fpg (24, 39) and Nei (40), and the murine enzymes NEIL1 and NEIL2 (38). In order to elucidate the processing of the hydantoin lesions relative to OG, Tg and 5-OHC, we have performed detailed kinetic analysis of duplexes containing these embedded lesions with hNEIL1. These studies reveal that hNEIL1 catalyzes the removal of the hydantoin lesions opposite all four DNA bases, while there is minimal OG removal activity. Interestingly, quantitative examination of the substrate specificity with hNEIL1 reveals that the hydantoin lesions are excised very efficiently, considerably more so (< 100-fold) than the well-established substrate Tg (Figure 1). Indeed, our data suggests that the hydantoin lesions are the best substrates identified for the enzyme so far. In addition, the opposite base influences the removal of Gh, Sp1 and Sp2 by hNEIL1. The efficient activity of hNEIL of the hydantoin lesions opposite G suggests that similarly robust activity in vivo may potentially enhance, rather than reduce mutagenesis mediated by these lesions. We also observed that hNEIL1 can differentiate between the two diastereoisomers of spiroiminodihydantoin and catalyzes the removal of Sp1 at a much faster rate than Sp2 in all base pair contexts. This work provides insight into the role of hNEIL1 in the recognition of the Gh and Sp lesions and suggests that these lesions are likely important in vivo substrates for these enzymes.
Materials and methods
General materials and instrumentation
Rosetta (DE3) pLysS cells strains (Novagen) were used for the purification of hNEIL1 using a pET30a plasmid containing the hNEIL1 gene (31). 7,8-Dihydro-8-oxo-2′-deoxyguanosine phosphoramidite was purchased from Glen Research. DNA oligonucleotides were synthesized at the core facility at the University of Utah Medical School. These samples were purified via HPLC on a Beckman Gold Nouveau system with a Protein-Pak DEAE 8HR column. The oligonucleotide containing 5-hydroxy cytosine (5-OHC) or thymine glycol (Tg) was purchased from Midland Reagents. Radiolabeling was done using [γ-32P]-ATP purchased from ICN with T4 polynucleotide kinase obtained from New England BioLabs. Labeled oligonucleotides were purified using ProbeQuant G-50 spin columns from Amersham Pharmacia according to the manufacturer’s protocol. A Milli-Q PF system was used to purify distilled, deionized water that was used to make all the buffers. All buffers were passed through a 0.45 μm filter before use. Storage phosphor autoradiography was performed on either a Typhoon 9400 phosphorimager or a Molecular Dynamics Storm 840 phosphorimager system. Data analysis was performed using ImageQuaNT software (version 5.2) and the rate constants were determined using GraFit 5.0 software. All other chemicals used for these experiments were purchased from Fisher Scientific, VWR, or Sigma.
Substrate DNA preparation
Gh- and Sp- containing oligonucleotides were synthesized from the OG-containing oligonucleotide as previously described (41). The identity of lesion-containing oligomers was confirmed by ESI-MS analysis. The two diastereomers of Sp were separated and purified by HPLC using a Dionex DNAPac PA-100 column. Separation of the Sp diastereomers was achieved with buffer systems consisting of 35% solvent A and 65% B at the beginning and gradually changed to 100% solvent B. (Solvent A was 10% acetonitrile and 90% H2O, while solvent B was 10% acetonitrile and 90% 1.5 M ammonium acetate (pH 7). The 30-nucleotide (nt) sequence that was used is listed below: d(5′–TGTTCATCATGGGTCXTCGGTATATCCCAT–3′) in which X = OG, Gh, Sp1, Sp2, 5-OHC or Tg; and the complementary strand d(3′–ACAAGTAGTACCCAGYAGCCATATAGGGTA–5′) in which Y = C, A, T or G. For all experiments, 2.5 pmoles of the X-containing strand was radiolabeled on the 5′ end using [γ-32P]-ATP by T4 kinase at 37 °C. Excess [γ-32P]-ATP was removed using a Pharmacia Microspin G-50 spin column, according to the manufacturer’s protocol. Additional nonradioactive X-containing DNA was added to the labeled strand to allow 5 % labeled DNA, which was then annealed to the complement (added at 20 % excess) by heating to 90 °C for 5 min and allowing to cool overnight in annealing buffer (20 mM Tris-HCl (pH 7.6), 10 mM EDTA, and 150 mM NaCl).
Purification of hNEIL1
Purification of recombinant C-terminally His-tagged hNEIL1 was as reported previously (31, 42) with the slight modification that the cells were grown overnight at 30 º C instead of 16 °C. The purified protein was dialyzed as reported and stored in liquid nitrogen. Total concentration of the protein was determined by the Bradford assay using BSA as the standard. Active site titrations (43, 44) were performed using a Sp1:G duplex and all concentrations are listed as active concentrations.
Glycosylase Assays
Single-turnover experiments, where [Enz] > [DNA], were performed using the 30 base pair duplexes to evaluate the glycosylase activity of the enzymes. In each case, the total reaction volume was 60 μL with a final duplex DNA concentration of 20 nM. The duplex was incubated with 200 nM active hNEIL1 in an assay buffer (20 mM Tris-HCl, pH 7.6, 10 mM EDTA, 0.1 mg/mL BSA, and 150 mM NaCl) at 37 °C. For measurement of lyase activity, aliquots were removed at various times (15 s to 60 min) and quenched by the addition of 5 μL of formamide denaturing dye (80% formamide, 0.025% xylene cyanol, 0.025% bromophenol blue in TBE buffer). In order to measure the glycosylase activity of the enzyme, aliquots were removed at various times and the reactions were quenched by the addition of 5 μL of 0.5 N NaOH, heated to 90 °C for two minutes and then placed on dry ice. Control experiments without enzyme also established that all the lesions were cleaved minimally by the buffer conditions and NaOH quenching (< 5%). The extent of background cleavage in the control reactions (no enzyme) was subtracted during quantification of the storage phosphor autoradiograms.
For experiments under single-turnover conditions in which the glycosylase reaction was too fast to measure manually, a Rapid Quench Flow instrument (RQF-3) from Kintek was used. The reaction buffer and conditions and overall analysis are similar to the manual experiments. However, addition of a nonspecific duplex (25 nM) to the hNEIL1 (400 nM) while in the injection loop was found to be necessary to stabilize the enzyme. The enzyme was mixed with the DNA duplex (20 nM final), and samples were removed at various time points between 0.1 s and 5 min, quenched with 0.5 M NaOH, heated to 90 °C for two minutes and placed on dry ice. The aliquots were treated with 5μL of formamide denaturing dye (80 % formamide, 0.025 % xylene cyanol, 0.025 % bromophenol blue in TBE buffer) and were then run on a 15 % denaturing polyacrylamide gel in 1X TBE at 1600 V for 2 hours. The separation of the 15-nucleotide DNA fragment arising from the product and the 30-nucleotide fragment originating from the substrate was visualized using autoradiography by exposure to a storage phosphor screen overnight.
Multiple-turnover experiments were performed using the same buffer conditions as the single-turnover assays, except the active enzyme concentration was lower than the concentration of the substrate. Active-site concentration was determined using 20 nM Sp1:G-containing duplex, incubated with 2–12 nM total protein in an assay buffer as in the case of the single-turnover assays. Aliquots were removed at various times (15 s to 40 min) and quenched by the addition of 5 μL of formamide denaturing dye (80 % formamide, 0.025 % xylene cyanol, 0.025 % bromophenol blue in TBE buffer). As a control, the multiple-turnover experiments were also quenched with a NaOH/heat treatment to measure the glycosylase activity. The samples were then run on a 15 % denaturing polyacrylamide gel in 1X TBE at 1600 V for 2 hours. Stability assay experiments were performed similarly to the active site determination with the modification of pre-incubating the enzyme at 37 °C in the glycosylase buffer for times ranging from 0 to 40 min, prior to addition of labeled substrate. Based on the amplitude of the burst phase, those conditions were chosen for the active site concentration determination. In addition, the concentration of the non-specific duplex was varied from 0 to 100 nM in order to determine the optimal concentration of non-specific duplex to stabilize hNEIL1.
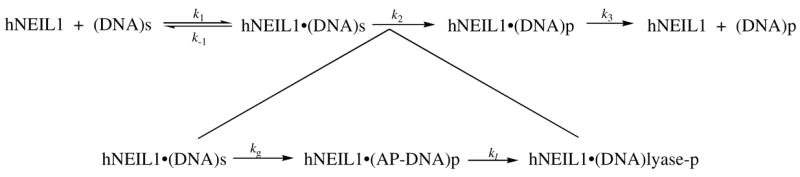
Fitting of the data was performed with GraFit 5.0 software with the appropriate equations in a manner analogous to that reported previously by our laboratory with Fpg and MutY (24, 44). Data from multiple –turnover experiments were fitted with equation 1, to determine the amplitude of the burst (A0), kobs (rate constant for the burst) and kss (rate of the linear phase). Under conditions where [E] > Kd (44), A0 = [active hNEIL1].
| (1) |
Data from single-turnover experiments were used to determine the observed rate of product formation (kobs) from fitting of the data using eq. 2.
| (2) |
Under conditions of single-turnover where [E] > Kd, kobs = k2 (Scheme 1). In all cases, the data are reported as the average of at least four separate experiments, and the error is reported as the standard deviation of the sample set.
Results
Glycosylase Activity of hNEIL1
The glycosylase activity of hNEIL1 was analyzed using a 30-bp duplex containing OG, 5-OHC, Tg, Gh, Sp1 or Sp2. The general method involved end-labeling the lesion-containing strand with γ-32P-ATP, and determining the extent of strand scission at the lesion site as a function of time. To ensure observance of strand scission at all abasic sites produced by the glycosylase reaction, the initial test reactions were quenched with NaOH and heat, rather than relying on the lyase activity of hNEIL to provide strand scission. With the OG duplex, hNEIL1 mediated minimal strand cleavage (< 5 % after 60 minutes) irrespective of the base opposite the lesion (Figure 2A). Even with the duplex containing the reported “good” substrate, 5-OHC base paired with G (31, 32), only 13 % of the substrate was converted to product in the same time frame (Figure 2B). With the Tg-containing duplexes, the reaction was observed to go to completion within the 60 minute incubation time period (Figure 3A). However, the most striking feature of the initial substrate evaluation was the extremely efficient removal of the hydantoin lesions (Gh, Sp1 and Sp2) by hNEIL1, with complete conversion to the product being observed by the first acquired time point of 30 sec (Figure 3B). Since the initial evaluation of a variety of substrates indicated that Sp1:G was an excellent substrate, this base pair duplex was used for detailed characterization of the features of the base excision reaction mediated by hNEIL1 and optimization of the assay conditions.
Figure 2. Figure 2A and B: Storage phosphor autoradiogram of hNEIL1 with a 30-bp OG:G and 5-OHC:G duplexes over a 60 min. time course.

Representative autoradiograms of product formation as a function of time, under single-turnover conditions with hNEIL1 on a OG:G (A) and 5-OHC:G (B) 30- bp duplexes (C) at 37 °C. Quantification of the storage phosphor autoradiogram reveals that the OG:G substrate is minimally cleaved by hNEIL1 (< 5%), while the 5-OHC:G is cleaved ~ 13%.
Figure 3. Figure 3A and B: Storage phosphor autoradiogram of hNEIL1 with a 30-bp Tg:G and Sp1:G duplexes over a 60 min. time course.
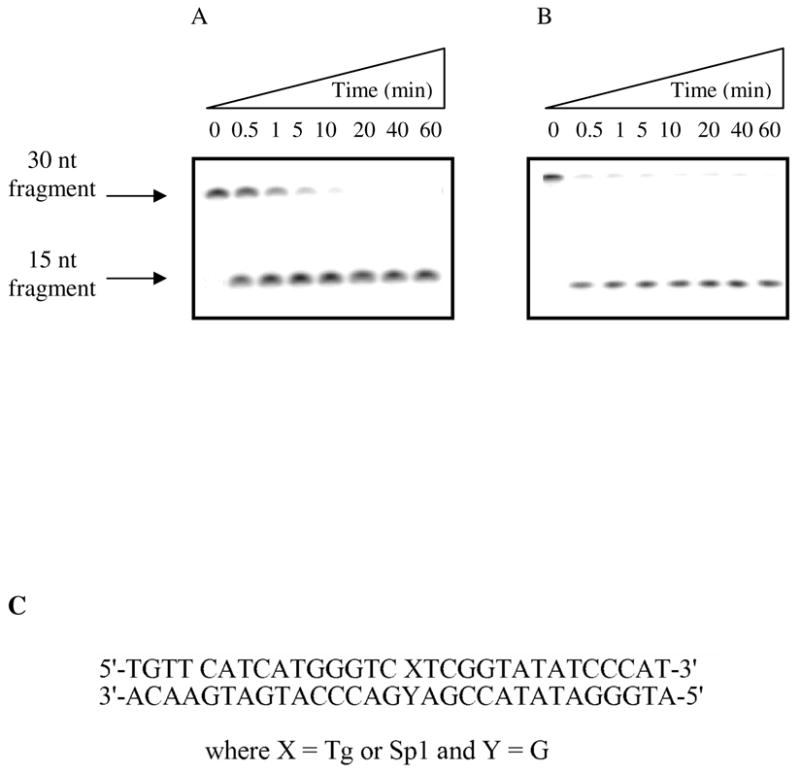
Representative autoradiograms of product formation as a function of time, under single-turnover conditions with hNEIL1 on a Tg:G (A) and Sp1:G (B) 30 bp duplexes (C) at 37 °C. Quantification of the storage phosphor autoradiogram reveals that both the Tg:G and the Sp1:G substrates are completely cleaved by hNEIL1. However, the rate of excision with Sp1:G substrate is complete within the first time point acquired (30 sec).
Analysis of hNEIL1 activity under multiple-turnover conditions
Under multiple-turnover conditions, in which the enzyme concentration is less than the substrate, the reaction of Sp1:G with hNEIL1 was characterized by an exponential burst of product formation, followed by a slower steady-state phase (Figure 4A). This biphasic kinetic behavior of hNEIL1 is similar to that previously observed with hOGG1 (45) and the adenine glycosylase, MutY (44). Since hNEIL1 catalyzes an associated β and δ-lyase activity, it is important to determine if these steps may be influencing the observed reaction profile. The combined glycosylase/lyase activity of the enzyme was evaluated by quenching the reactions with a formamide dye while the glycosylase activity alone was analyzed by quenching with a NaOH/heat treatment. The nature of the quench did not seem to alter the biphasic profile or alter the rate of the overall reaction establishing that the glycosylase and lyase steps of the enzyme are tightly coupled (Figure 4A).
Figure 4. Characterization of the Activity and Stability of hNEIL1.
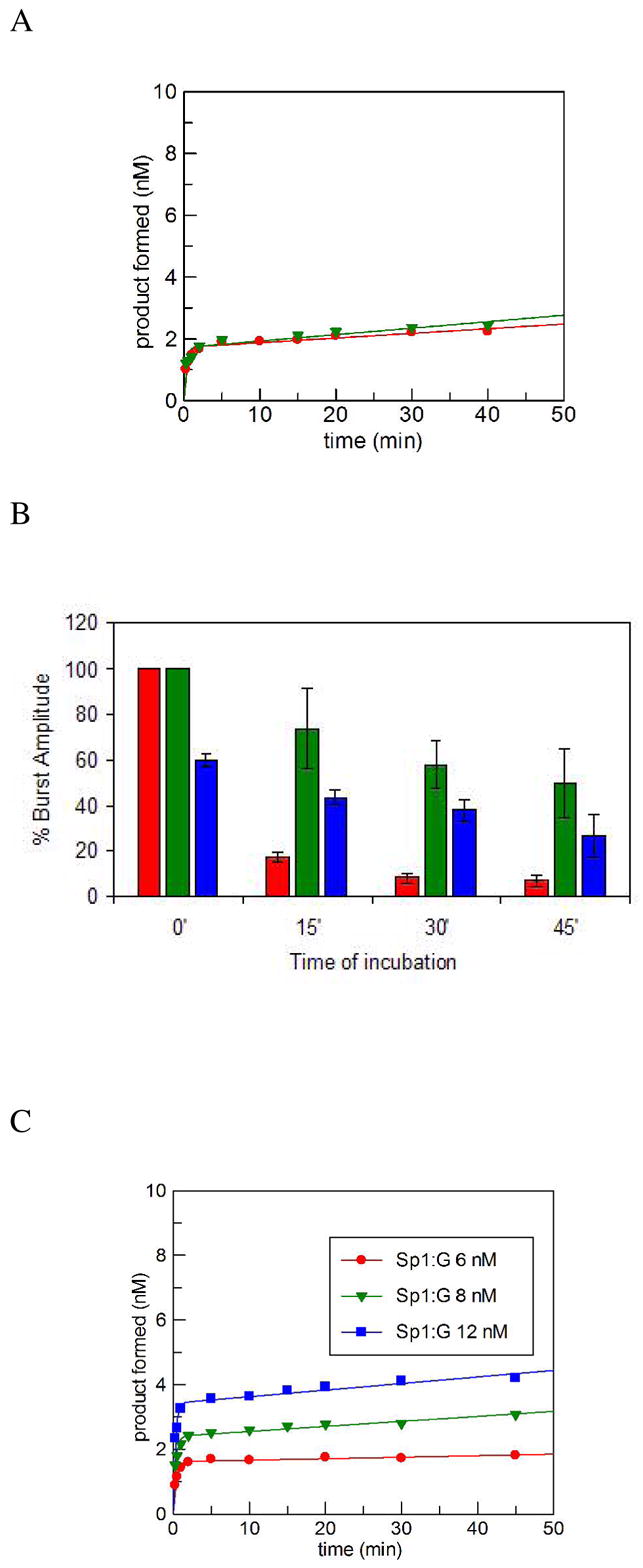
(A) Representative plots of product formation as a function of time, under multiple-turnover conditions with hNEIL1 on a Sp1:G containing DNA duplex at 37 °C quenched with either a formamide dye (red circles) or a NaOH/heat treatment (green triangles). The graph illustrates a biphasic behavior for hNEIL1 with an initial burst of product formation followed by a slower steady-state phase. The formamide dye quench determines the glycosylase/lyase activity while the glycosylase activity alone is analyzed by NaOH/heat quench.
(B) Bars show the burst amplitude of hNEIL1 after incubation with 0 nM (red), 25 nM (green), and 50 nM nonspecific DNA (blue)
(C) Active site titration of hNEIL1 with 20 nM duplex, and 6 (red circles), 8 (green triangles), 12 nM (blue squares) enzyme (total)
In cases where there is a “burst” of product formed due to a slow step following chemistry, the amplitude of the burst phase may be used to determine the amount of active enzyme (43). The amplitude of the burst phase increased proportionally to the amount of hNEIL1 enzyme, indicating that the burst is related to the amount of active enzyme. The burst amplitude observed with the Sp1:G duplex was also used to determine the stability of hNEIL1 during the time course of these experiments. Based on this analysis, hNEIL1 loses activity over the time course of the experiment of 60 minutes. The steady loss of enzyme activity may contribute to incomplete conversion of substrate to product observed in other studies with NEIL enzymes (38). Various concentrations of non-specific DNA were added during the enzyme incubation period to potentially provide stability to hNEIL1. Aliquots were removed at regular intervals and the amplitude of the burst was determined. Interestingly, the presence of 25 nM non-specific DNA duplex stabilized the enzyme to the greatest extent (Figure 4B). However, a further increase in the concentration of the non-specific duplex led to a decrease in burst amplitude, presumably due to competition between the specific and non-specific DNA duplex. Importantly, the burst amplitude did not diminish with pre-incubation in the reaction conditions until after several minutes (~ 5 min). Importantly, the burst phase is complete within the first minute of the reaction, and therefore reports accurately on the concentration of active sites in hNEIL1. The active-site titration performed under conditions determined for optimal enzyme stability with the Sp1:G substrate (Figure 4C) indicated that the active hNEIL1 enzyme concentration was 28 % relative to the total protein concentration determined by Bradford assays (46) in the preparation of the enzyme used herein.
Determination of rate constants for the glycosylase activity of hNEIL1
Since hNEIL1 displays biphasic behavior similar to the OG glycosylase hOGG1 and MutY, we have analyzed the glycosylase activity using a similar minimal kinetic scheme (Scheme 1) (45). The data from experiments performed under single-turnover conditions, where the enzyme concentration is in excess to the substrate, were fitted with the appropriate rate equations to isolate the rate constant k2. The rate of product formation mediated by hNEIL1 is not influenced by base-quenching indicating that k2 = kg. Rate constants (k2) from single-turnover experiments for the base removal reaction of hNEIL1 with duplexes containing site-specific lesions in various-base pairing contexts are listed in Table 1. In the excision of OG by hNEIL1 from the OG:C-, OG:G- and OG:A-containing 30 bp duplex, the reaction does not go to completion, and therefore only an upper limit on the rate constant ( e.g. k2 < 0.02 min−1 with OG:C) could be obtained. The incomplete reaction and very slow rate establish the poor activity of hNEIL1 for removal of OG. Interestingly, the reaction of hNEIL1 for removal of Tg from within the same duplex context proceeds to completion with a respectable rate (Table 1). The rate constants for Tg removal by hNEIL1 are influenced by the opposite base, with activity for Tg removal from Tg:G base pairs (k2 = 2.6 ± 0.2 min−1) that is 2-fold greater than from Tg:A base pairs (k2 =1.3 ± 0.1 min−1).
Table 1.
Rate constants Determined at 37 °C Under Single Turnover Conditions for hNEIL1 with oxidized lesions
| k2(min−1) | ||||
|---|---|---|---|---|
| Base opposite | G | A | C | T |
|
| ||||
| Lesion | ||||
| OG | < 0.01 | < 0.01 | < 0.02 | N.D |
| Tg | 2.6 ± 0.2 | 1.3 ± 0.1 | N.D | N.D |
| Gh | 189 ± 18 | 98 ± 10 | 104 ± 14 | 356 ± 36 |
| Sp1 | 356 ± 13 | 82 ± 7 | 177 ± 11 | > 500 |
| Sp2 | 152 ± 11 | 63 ± 8 | 139 ± 8 | 224 ± 27 |
N.D. = Not determined. Duplex and conditions of the assays are outlined in the methods section.
The most striking values in the series of rate constants measured listed in Table 1 are those for the reaction of hNEIL1 with the hydantoin lesions (Gh, Sp1 and Sp2). Indeed, these rate constants reveal that these lesions are removed at least a 100-fold faster than Tg in the same base pair contexts. Moreover, the hydantoin lesions are excised very efficiently opposite all four bases. Measurable differences in rates of excision were observed depending on the identity of the base opposite the lesion. The quantitative data reveals that in all cases, the lesion was removed the fastest when paired with T and least efficiently when paired with A. The Gh:T substrate (356 ± 36 min−1) is processed at least three fold faster than the Gh:A substrate (98 ± 10 min−1). Also, hNEIL1 catalyzed removal of the biologically relevant Gh:G base pair at a rate constant of 189 ± 18 min−1, a value that is nearly two-fold higher than that observed for lesion removal from Gh:A. Overall, the rates of excision of the Gh lesion followed the pattern T > G > C ~ A. Similar to the Gh lesion, the Sp lesions are excised with a clear dependence on the identity of the opposite base. The Sp1:T lesion is processed at such a high rate (> 500 min−1 ) that it is only possible to estimate a lower limit. Also, hNEIL1 excises Sp1:G (356 ± 13 min−1) fourfold and Sp1:C (177 ± 11 min−1) at least two-fold more efficiently than it does Sp1:A (82 ± 7 min−1). The effect of opposite base on removal of the Sp2 lesions is similar, with rate constants for hNEIL1 mediated removal from Sp2:G (152 ± 11 min−1) and Sp2:C (139 ± 8 min−1) that are two-fold larger than from Sp2:A (63 ± 8 min−1). Like the Gh lesions, both of the Sp lesions are removed most efficiently when when paired with T, with the preference of the base opposite the lesion of T > G > C > A.
Interestingly, hNEIL1 has a preference for one Sp isomer over the other. As is evident from Table 1, in all base pair contexts, the Sp1 diastereomer is removed more efficiently than the Sp2 diastereomer. For example, excision of Sp1 opposite G by hNEIL1 is 356 ± 13 min−1, a value that is at least two-fold higher than the excision of Sp2 opposite G (152 ± 11 min−1). Also, the rate of base removal with Sp1:A (82 ± 7 min−1) is higher than Sp2:A (63 ± 8 min−1). Indeed, this is the first time that significant differences in relative removal of the two diastereomers of Sp by a glycosylase have been revealed.
Discussion
Excision of oxidized bases by BER glycosylases provides a mechanism to prevent mutagenesis and toxicity resulting from oxidative DNA damage. This work represents the first quantitative measurements under single-turnover conditions of the glycosylase activity of the newly discovered mammalian glycosylase, hNEIL1 with the hydantoin lesions, Gh and the two diastereomers of Sp (Sp1 and Sp2). To elucidate the processing of these hydantoin lesions relative to other known substrates such as OG, Tg and 5-OHC, we have performed detailed kinetic analysis of the processing of hNEIL1 with 30 bp duplexes containing these embedded lesions in various base-pairing contexts with hNEIL1.
In contrast to some literature reports (33, 47), quantitative analysis of the activity of hNEIL1 with an OG-containing duplex reveals that irrespective of the base opposite OG, the enzyme does not catalyze the removal of the lesion effectively (Table 1) (Figure 2A). Recent simulation studies of hNEIL1 by Broyde and co-workers with a modeled DNA substrate in the crystal structure of hNEIL1 are completely consistent with this data (48). The simulations indicated that the planar structure of OG does not permit the base to be fully accommodated within the recognition pocket of hNEIL1, and this may account for its inefficient removal. The study also indicated that the inserted portion of OG would have few hydrogen-bonding interactions, and moreover that the six-membered ring portion of OG would be solvent exposed. While OG is a poor substrate, Tg is effectively removed by hNEIL1. Indeed, the molecular simulations data suggested that the hNEIL1 lesion-specific pocket would be able to form favorable interactions with the endocyclic imide portion of the pyrimidine ring of Tg (48).
The quantitative determinations of the rates of lesion removal reveal the extremely robust activity for removal of the hydantoin lesions by hNEIL1. Rate constants indicate that both Gh and the two diastereomers of Sp (Sp1 and Sp2) are excised efficiently by hNEIL1, with values that are two orders of magnitude greater than Tg. Though derived from guanine, both Gh and Sp effectively mimic “T” and possess an endocyclic imide functionality. Moreover, Gh and Sp have the imide functionality localized in a smaller ring that may be more effectively docked within the hNEIL1 active site. Indeed, the modeling and dynamics studies with hNEIL1 by Broyde and coworkers (48) suggested that the nonplanar structure of Sp allows the A-ring of Sp to be docked within the enzyme, while the B ring may make other contacts (Figure 5). Previous analysis of the activity of mNEIL1 with high molecular weight DNA containing multiple lesions indicated that FapyG and FapyA lesions are more efficiently removed than Tg (49). The Fapy lesions also possess a smaller ring than OG with a more flexible amide linkage that may be more effectively docked within the hNEIL1 active site like Sp and Gh. Additional quantitative kinetic studies with hNEIL1 with FapyG and FapyA embedded in the same duplex context as Gh and Sp may reveal the relative activity of these two types of lesions. Recent quantitative kinetic studies of the activity of Fpg with FapyG- and FapyA-containing substrates indicated many similarities to the activity with the hydantoin lesions (50).
Figure 5.
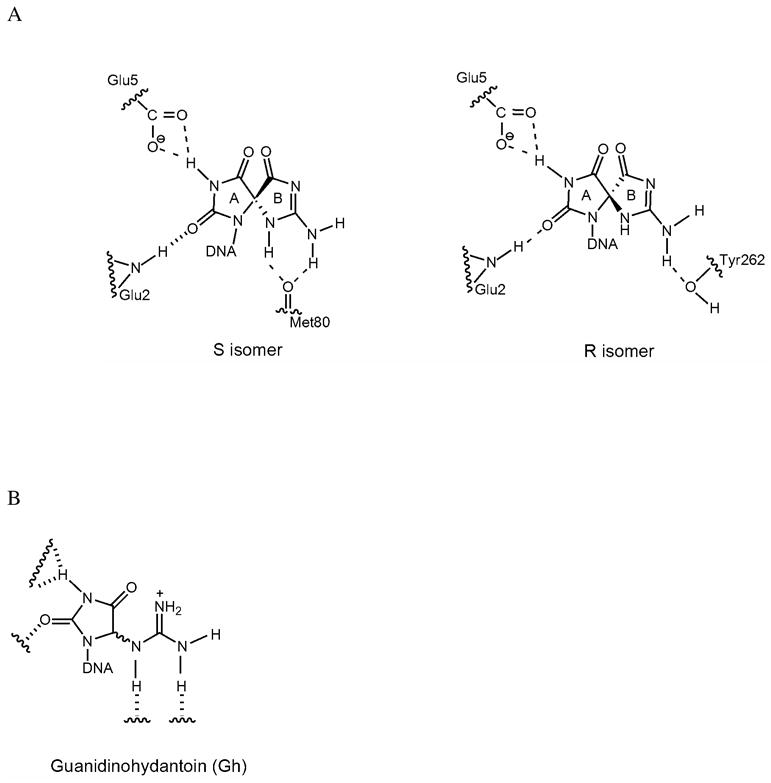
Schematic based on simulation studies (48) and X-ray of hNEIL1(35) illustrating potential interaction of the two diastereomers of Sp within the active site pocket of hNEIL1 (A). Based on the simulation studies with Sp, we also propose the manner in which Gh may be accommodated in the hNEIL active site (B).
An important feature of the activity of hNEIL1 with the hydantoin lesions, is the superior activity toward one of the two diastereomers of Sp; this is the first study to reveal significant differential processing of the two diastereomers of Sp. In fact, the removal of Sp1 from both Sp1:G and Sp1:T base pairs is at least two-fold faster than the corresponding rates for Sp2 removal. This suggests that the enzyme can differentiate between the two diastereomers, leading to different rates of removal. The previous dynamics and modeling studies (48) had predicted that the distinct structural properties of the two diastereomers of Sp would result in differential processing by hNEIL1. Specifically, these studies showed that one stereoisomer (S) would have more favorable electrostatic interactions within the hNEIL enzyme than the other isomer (R). The superior recognition of the S isomer may translate to more efficient removal than the R isomer. From our data and the modeling studies, it is tempting to suggest that the R diastereomer is Sp2, while S is the Sp1 diastereomer. (Figure 5) However, we note that naming of Sp1 and Sp2 is based on elution order of Sp-containing oligonucleotides on an ion exchange column which may not correlate to the same Sp1 and Sp2 assignments of nucleosides on a Hypercarb (graphite) HPLC column (51, 52). Further work is necessary to confirm the R and S designations of the monomer and oligomers. Of note, Gh also exists as a mixture of diastereomers, but these interconvert on a time scale that is too rapid to allow isolation and independent study (53).
Thermal denaturation experiments have found that the thermodynamic stability of Sp-containing oligonucleotides is significantly reduced (ΔΔG37 ~ 6 kcal/mol) compared to OG or G counterparts (25, 26, 54). Molecular dynamics simulations also indicated that the Sp nucleotide would be destabilizing to the DNA duplex and that the R isomer of Sp would be less destabilizing than the S isomer within a DNA duplex (55). Local destabilization of the duplex may aid in recognition and extrusion of the damaged base lesion by DNA glycosylases and therefore may be an important feature leading to the more facile removal of some lesions by hNEIL1. Indeed, we have previously observed that base pair stability is an important feature in the recognition and removal process with other BER glycosylases (39, 50, 56, 57). Facile disruption of the base pair coupled with more suitable contacts within the base pocket by hNEIL1 may lead to the superior activity toward Sp over Gh, and Sp1 over Sp2.
In all cases, the single-turnover rate constants (Table 1) indicate that the base opposite the lesion influences the efficiency of the lesion removal; in fact, the lesion (Gh, Sp1 and Sp2) is always excised more efficiently when paired opposite T, than opposite G, C and A. Gh:T is removed nearly four-fold faster than Gh:A and two-fold faster than Gh:G. The Sp diastereomers also exhibit similar trends with the lesions removed the most efficiently when paired with T than with G or A. Previous work by Hafalla et al. showed similar trends in the influence of the base opposite in hNEIL1 cleavage of Tg (58). Though there is a modulation depending on the opposite base, the lesions are removed efficiently in all contexts and the extent of discrimination is not as great as other glycosylases. For example, hOGG1 removes OG approximately 3000-fold more efficiently opposite C than A, showing a large degree of specificity for the opposite base(45, 50). Structural studies of hOGG1 have also revealed that recognition of “C” is a major component in the recognition and “finding” of OG (59). With hNEIL1, the opposite base pair dependence may be more indirect and due to the stability of the bp and susceptibility to deformation by the enzyme. In fact both Gh and the Sp diastereomers can mimic “T” and thus Ghsyn or Spsyn can base pair with G in the same fashion as a Tanti:Ganti wobble pair or with A as a normal Tanti:Aanti base pair (39, 41). Less stable base pairing of the hydantoin lesions with T, relative to G and A may translate into more ready disruption of the Gh/Sp:T bp and more facile recognition and excision by hNEIL1 (26). Though the Gh/Sp: T containing duplexes are the best substrates, they are unlikely to be present in a cellular context since the hydantoins direct for incorporation of G or A (25, 26), or are formed initially from G in normal G:C bps.
The mutational spectra associated with oxidative damage to guanine results from the large number of lesions formed as well as the possibility of multiple base insertions opposite a specific lesion by DNA polymerases (13, 60). In general, the predominant types of mutations observed are G → T and G → C transversion mutations. The observation of G → T transversions is usually thought to arise from misinsertion of A opposite OG (60). Other lesions, such as FapyG, Gh and Sp, are also likely to contribute to this type of mutation (13, 28, 61, 62). Fewer lesions can lead to G → C transversion mutations and the hydantoin lesions, in particular Gh, are reasonable candidates for these mutations. Capture of Gh and Sp lesions by repair enzymes prior to replication would be expected to prevent mutagenesis. However, the high miscoding properties of Gh and Sp lesions may be worsened by attempts to repair these lesions in the inappropriate base pairing context. In fact, these lesions are removed efficiently when opposite the non-mutagenic partner C and the mutagenic partner G. This is particularly interesting in light of the in vivo mutagenesis studies since formation of Gh:G and Sp:G base pairs would be intermediates leading to G → C transversion mutations. Removal of the lesion from such base pairing contexts would therefore enhance mutagenesis mediated by these lesions. Thus, repair may have an interesting role in altering the mutagenic profile of a given lesion. This modulation may also be a factor influencing the mutagenic outcome of the two diasteromers of Sp; in fact, the in vivo mutagenesis studies provided a mixture of both G → T and G → C transversion mutations, the ratio being dependent on the diastereomer involved (27, 28).
We also observe that under multiple turnover conditions with hNEIL1, an exponential burst phase of product formation is observed, followed by a slower linear phase. This biphasic behavior has been observed previously with many other glycosylases (63). In the reaction with human OG glycosylase hOGG1, this type of behavior is only observed when monitoring the reaction where the enzyme provides strand scission due to the slow β–lyase reaction(50, 64, 65). This is in contrast to hNEIL1, where the glycosylase and β,δ-lyase functions are tightly coupled. In both enzymes, it is the product of the β– or β,δ-lyase reaction that remains tightly bound to glycosylase rather than the intact abasic site-duplex. Generally the abasic site or its cleaved product is more toxic than the lesion itself; so many glycosylases remain associated with the product duplex. Interestingly, hNEIL1 is capable of excision of 3′ end proximal lesions (66) and has been shown to remove 5-OHU and OG within single-stranded DNA and bubble structures containing DNA (67). These studies indicate catalytic behavior for hNEIL1 distinct from other human glycosylases, such as hOGG1 and NTH1.
The catalytic turnover of many glycosylases can be stimulated by APE1, the major AP endonuclease in humans (63, 64, 68, 69). Indeed, APE1 plays an essential role in NTH1 and hOGG1-initiated BER (8). In contrast, NEIL1-initiated BER does not require APE1, and instead relies on polynucleotide kinase to remove the 3′-phosphate left after the β.δ lyase reaction of NEIL1 (70). The glycosylase activity of NEIL1 is also stimulated by a variety of proteins, including the 9-1-1 complex (71–73) and Werner Syndrome protein (74). In addition, hNEIL1 also stimulates the activity of OGG1 on substrate DNA containing OG (75). These various results indicate that hNEIL interacts with a variety of different proteins, and that excision of oxidized bases may be correlated with other processes in the cell. These results also imply that hNEIL1 does not merely serve as a backup for hOGG1 or NTH1, but has a unique and significant function in vivo.
Previous studies with hOGG1 have shown that this human OG glycosylase does not catalyze the removal of the hydantoin lesions (45). Thus, the potent activity of hNEIL for the hydantoin substrates prompts the question: are these lesions the relevant substrates for these enzymes in vivo? Indeed, although hydantoin lesions may not be present at as high levels as OG, their strong miscoding ability, and potentially increased mutagenicity due to inappropriate repair may make them much more mutagenic and toxic than OG in cells. Of note, neil1 knockout mice (neil1−/−) exhibit symptoms consistent with metabolic malfunction similar to metabolic syndrome in humans (76, 77). This is in contrast to ogg1 (−/−) knockout mice that were shown to have only a minimal phenotype though the accumulation of OG lesions was observed (78, 79). This suggests that ineffective repair of oxidized bases may have some unusual consequences. Could the very strong phenotype due to the absence of hNEIL1 be related to its importance in removing highly oxidized and mutagenic lesions, like Gh and Sp?? This is a provocative question that will require innovative chemical and biological approaches to answer.
Supporting Information Available
Figure S1 shows the HPLC trace of separation of Sp diastereomers that are separated on a Dionex DNAPac PA-100 column as described in the methods section. This material is available free of charge via the Internet at http://pubs.acs.org
Scheme 1.
Acknowledgments
We gratefully acknowledge Dr. Viswanath Bandaru and Prof. Susan Wallace for providing the hNEIL1 expression plasmid and for helpful suggestions. We also thank Sucharita Kundu for help with the purification of the hNEIL1 and Dr. James Muller for assistance with mass spectrometric characterization of oligonucleotides.
Footnotes
This work was supported by a grant from the National Cancer Institute of the National Institutes of Health (CA90689).
Abbreviations: BER, base excision repair; bp, base pair; EDTA, ethylenediaminetetraacetic acid; Fpg/MutM; bacterial formamidopyrimidine glycosylase; Gh, guanidinohydantoin; hNEIL1, human Nei-like glycosylase 1; Nei; endonuclease VIII; NTH1, endonuclease III homologue 1; OG, 7,8-dihydro-8-oxoguanine; 5-OHC, 5-hydroxycytosine; PAGE, polyacrylamide gel electrophoresis; ROS, reactive oxygen species, Sp, spiroiminodihydantoin; Tg, thymine glycol.
For the sake of simplicity, we are using the one or two letter abbreviation to refer to both the base and the nucleotide. This should be evident by the context with which the abbreviation is used.
References
- 1.Ames BN, Gold LS. Endogenous mutagens and the causes of aging and cancer. Mutat Res. 1991;250:121. doi: 10.1016/0027-5107(91)90157-j. [DOI] [PubMed] [Google Scholar]
- 2.Klaunig JE, Kamendulis LM. The Role of Oxidative Stress in Carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–67. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 3.Henle ES, Linn S. Formation, prevention and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem. 1997;272:19095–19098. doi: 10.1074/jbc.272.31.19095. [DOI] [PubMed] [Google Scholar]
- 4.Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutation Research. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Kamiya H. Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: approaches using synthetic oligonucleotides and nucleotides. Nuc Acids Res. 2003;31:517–531. doi: 10.1093/nar/gkg137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beckman KB, Ames BN. Oxidative Decay of DNA. J Biol Chem. 1997;272:19633–19636. doi: 10.1074/jbc.272.32.19633. [DOI] [PubMed] [Google Scholar]
- 7.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes DE, Lindahl T. Repair and Genetic Consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 9.Lindahl T, Wood RD. Quality Control by DNA Repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 10.Hoeijmakers JHJ. Genome Maitenance mechanisms for preventing cancers. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 11.Sweasy JB, Lang T, Dimaio D. Is Base Excision Repair a Tumor Suppressor Mechanism? Cell Cycle. 2006;5:250–259. doi: 10.4161/cc.5.3.2414. [DOI] [PubMed] [Google Scholar]
- 12.Cooper PK, Fuss JO. DNA Repair: Dynamic Defenders against Cancer and Aging. PLOS Biology. 2006;4:899–903. doi: 10.1371/journal.pbio.0040203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neeley WL, Essigmann JM. Mechanisms of Formation, Genotoxicity, and Mutation of Guanine Oxidation Products. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 14.Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL, Sauvaigo S. Hydroxyl Radicals and DNA Base Damage. Mutation Res. 1999;424:9–21. doi: 10.1016/s0027-5107(99)00004-4. [DOI] [PubMed] [Google Scholar]
- 15.Ravanat JL, Saint-Pierre C, Di Mascio P, Martinez GR, Medeiros MHG, Cadet J. Damage of isolated DNA mediated by singlet oxygen. Helv Chim Acta. 2001;84:3702–3709. [Google Scholar]
- 16.Misiaszek R, Crean C, Joffe A, Geacintov NE, Shafirovich V. Oxidative DNA damage associated with combination of guanine and superoxide radicals and repair mechanisms via radical trapping. J Biol Chem. 2004;279:32106–32115. doi: 10.1074/jbc.M313904200. [DOI] [PubMed] [Google Scholar]
- 17.Luo W, Muller JG, Burrows CJ. The pH-Dependent Role of Superoxide in Riboflavin-Catalyzed Photooxidation of 8-Oxo-7,8-dihydroguanosine. Org Lett. 2001;3:2801–2804. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 18.Sugden KD, Campco CK, Martin BD. Direct oxidation of guanine and 7,8-dihydro-8-oxoguanosine in DNA by high valent chromium complex: A possible mechanism for chromate genotoxicity. Chem Res Toxicol. 2001;14:1315–1322. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- 19.Slade PG, Hailer MK, Martin BD, Sugden KD. Guanine-specific oxidation of double stranded DNA by Cr(VI) and ascorbic acid forms spiroiminodihydantoin and 8-oxo-2′deoxyguanosine. Chem Res Toxicol. 2005;18:1140–1149. doi: 10.1021/tx050033y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hailer MK, Slade PG, Martin BD, Sugden KD. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem Res Toxicol. 2005;18:1378–1383. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burrows CJ, Muller JG, Kornyushyna O, Luo W, Duarte V, Leipold MD, David SS. Structure and potential mutagenicity of new hydantoin products from guanosine and 8-oxo-7,8-dihydroguanosine oxidation by transition metals. Environ Health Perspect. 2002;110:713–717. doi: 10.1289/ehp.02110s5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of Spiroiminodihydantoin as a Product of One-Electron Oxidation of 8-Oxo-7,8-dihydroguanosine. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 23.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of Hydantoin Products from One-Electron Oxidation of 8-Oxo-7,8-dihydroguanosine in a Nucleoside Model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 24.Leipold MD, Muller JG, Burrows CJ, David SS. Removal of Hydantoin Products of 8-Oxoguanine Oxidation by the Escherichia coli DNA Repair Enzyme, Fpg. Biochemistry. 2000;39:14984–14992. doi: 10.1021/bi0017982. [DOI] [PubMed] [Google Scholar]
- 25.Kornyushyna O, Burrows CJ. Effect of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin on proofreading by Escherichia coli DNA polymerase I (Klenow Fragment) in different sequence contexts. Biochemistry. 2003;42:13008–13018. doi: 10.1021/bi0350755. [DOI] [PubMed] [Google Scholar]
- 26.Kornyushyna O, Berges AM, Muller JG, Burrows CJ. In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow fragment) Biochemistry. 2002;41:15304–15314. doi: 10.1021/bi0264925. [DOI] [PubMed] [Google Scholar]
- 27.Delaney S, Neeley WL, Delaney JC, Essigmann JM. The Substrate Specificity of MutY for Hyperoxidized Guanine Lesions in Vivo. Biochemistry. 2007;46:1448–1455. doi: 10.1021/bi061174h. [DOI] [PubMed] [Google Scholar]
- 28.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 29.David SS, Williams SD. Chemistry of Glycosylases and Endonucleases Involved In Base-Excision Repair. Chem Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 30.Michaels ML, Tchou J, Grollman AP, Miller JH. A Repair System for 8-oxo-7,8-dihydrodeoxyguanosine. Biochemistry. 1992;31:10964–10968. doi: 10.1021/bi00160a004. [DOI] [PubMed] [Google Scholar]
- 31.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair. 2002;1:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 32.Morland I, Rolseth V, Luna L, Rognes T, Bjoras M, Seeberg E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002;30:4926–4936. doi: 10.1093/nar/gkf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapanti SK, Mitra S, Izumi T. Identification of a Novel Human DNA Glycosylase for Repair of Cytosine-derived Lesions. J Biol Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 34.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and Characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci USA. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci USA. 2004;101:10284–10289. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong L, Meira LB, Hazra TK, Samson LD, Cao W. Oxanine DNA glycosylase activities in mammalian systems. DNA Repair. 2008;7:128–134. doi: 10.1016/j.dnarep.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakano T, Katafuchi A, Shimizu R, Terato H, Suzuki T, Tauchi H, Makino K, Skorvaga M, Van Houten B, Ide H. Repair activity of base and nucleotide excision repair enzymes for guanine lesions induced by nitrosative stress. Nucleic Acids Res. 2005;33:2181–2191. doi: 10.1093/nar/gki513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hailer KM, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the base excision repair glycosylases NEIL1 and NEIL2. DNA Repair. 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Krishnamurthy N, Muller JG, Burrows CJ, David SS. Unusual Structural Features of Hydantoin Lesions Translate into Efficient Recognition by Escherichia coli Fpg. Biochemistry. 2007;46:9355–9365. doi: 10.1021/bi602459v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hazra TK, Muller JG, Manuel RC, Burrows CJ, Lloyd RS, Mitra S. Repair of hydantoins, one electron oxidation product of 8-oxoguanine, by DNA glycosylases of Escherichia coli. Nucleic Acids Res. 2001;29:1967–1974. doi: 10.1093/nar/29.9.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X, Muller JG, Halasyam M, David SS, Burrows CJ. In vitro ligation of oligodeoxynucleotides containing C8-oxidized purine lesions using bacteriophage T4 DNA ligase. Biochemistry. 2007;46:3734–3744. doi: 10.1021/bi062214k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bandaru V, Blaisdell JO, Wallace S. Oxidative DNA glycosylases:Recipes from cloning to characterization. Methods Enzymol. 2006;408:15–31. doi: 10.1016/S0076-6879(06)08002-5. [DOI] [PubMed] [Google Scholar]
- 43.Fersht A. Enzyme Structure and Mechanism. W. H. Freeman; New York: 1985. [Google Scholar]
- 44.Porello SL, Leyes AE, David SS. Single-turnover and Pre-Steady-State Kinetics of the Reaction of the Adenine Glycosylase MutY with Mismatch-Containing DNA substrates. Biochemistry. 1998;37:14756–14764. doi: 10.1021/bi981594+. [DOI] [PubMed] [Google Scholar]
- 45.Leipold MD, Workman H, Muller JG, Burrows CJ, David SS. Recognition and Removal of Oxidized Guanines in Duplex DNA by the Base Excision Repair Enzymes hOGG1, yOGG1 and yOGG2. Biochemistry. 2003;42:11373–11381. doi: 10.1021/bi034951b. [DOI] [PubMed] [Google Scholar]
- 46.Bradford MM. Anal Chemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 47.Wiederhold L, Leppard JB, Kedar P, Busheri FK, Nia AR, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Molecular Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Jia L, Shafirovich V, Geacintov NE, Broyde S. Lesion specificity in the base excision repair enzyme hNeil1: Modeling and dynamics studies. Biochemistry. 2007;46:5305–14. doi: 10.1021/bi062269m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaruga P, Birincioglu M, Rosequist TA, Dizdaroglu M. Mouse NEIL1 Protein is Specific for Excision of 2,6-Diamino-4-hydroxy-5-formamidopyrimidine and 4,6-Diamino-5-formamidopyrimidine from Oxidatively Damaged DNA. Biochemistry. 2004;43:15909–15914. doi: 10.1021/bi048162l. [DOI] [PubMed] [Google Scholar]
- 50.Krishnamurthy N, Haraguchi K, Greenberg MM, David SS. Efficient Removal of Formamidopyrimidines by 8-Oxoguanine Glycosylases. Biochemistry. 2008;47:1043–1050. doi: 10.1021/bi701919u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Durandin A, Jia L, Crean C, Kolbanovskiy A, Ding S, Shafirovich V, Broyde S, Geacintov N. Assignment of the Absolute Configurations of the Enatiomeric Spiroiminodihydantoin Nucleobases by Experimental and Computational Optical Rotatory Dispersion Methods. Chem Res Toxicol. 2006;19:908–913. doi: 10.1021/tx060078e. [DOI] [PubMed] [Google Scholar]
- 52.Karwowski B, Dupeyrat F, Bardet M, Ravanat JL, Krajewski P, Cadet J. Nuclear Magnetic Resonance Studies of the 4R and 4S Diastereomers of Spiroiminodihydantoin 2′-Deoxyribonucleosides: Absolute Configuration and Conformational Features. Chem Res Toxicol. 2006;19:1357–1365. doi: 10.1021/tx060088f. [DOI] [PubMed] [Google Scholar]
- 53.Ye Y, Muller JG, Luo W, Mayne CL, Shallop AJ, Jones RA, Burrows CJ. Formation of 13C-15N- and 18O-labeled Guanidinohydantoin from Guanosine Oxidation with Singlet Oxygen. Implications for Structure and Mechanism. J Am Chem Soc. 2003;125:13926–13927. doi: 10.1021/ja0378660. [DOI] [PubMed] [Google Scholar]
- 54.Chinyengetere F, Jamieson ER. Impact of the Oxidized Guanine Lesion Spiroiminodihydantoin on the Conformation and Thermodynamic Stability of a 15-mer DNA Duplex. Biochemistry. 2008;47:2584–2581. doi: 10.1021/bi701502t. [DOI] [PubMed] [Google Scholar]
- 55.Jia L, Shafirovich V, Shapiro R, Geacintov NE, Broyde S. Structural and Thermodynamic Features of Spiroiminodihydantoin Damaged DNA Duplexes. Biochemistry. 2005;44:13342–13353. doi: 10.1021/bi050790v. [DOI] [PubMed] [Google Scholar]
- 56.Francis AW, Helquist SA, Kool ET, David SS. Probing the Requirements for Recognition and Catalysis in Fpg and MutY with Nonpolar Adenine Isosteres. J Am Chem Soc. 2003;125:16235–16242. doi: 10.1021/ja0374426. [DOI] [PubMed] [Google Scholar]
- 57.Chepanoske CL, Langelier CR, Chmiel NH, David SS. Recognition of the nonpolar base 4-methylindole in DNA by the DNA repair adenine glycosylase MutY. Org Lett. 2000;2:1341–1344. doi: 10.1021/ol005831o. [DOI] [PubMed] [Google Scholar]
- 58.Hafalla MTO, Altamirano A, Basu AK, Chan MK, JEA O, A CJ, Boorstein RJ, Cunningham RP, Teebor GW. Repair of thymine glycol by hNth1 and hNeil1 is modulated by base pairing and cis-trans epimerization. DNA. 2006;5:444–454. doi: 10.1016/j.dnarep.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 60.Wang D, Kreutzer DA, Essigmann JM. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutation Research. 1998;400:99–115. doi: 10.1016/s0027-5107(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 61.Kalam MA, Haraguchi K, Chandani S, Loechler EL, Maasaki M, Greenberg MM, Basu AK. Genetic effects of oxidative DNA damages:comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 2006;34:2305–2315. doi: 10.1093/nar/gkl099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patro JN, Wiederholt CJ, Jiang YL, Delaney JC, Essigmann JM, Greenberg MM. Studies on the Replication of the Ring Opened Formamidopyrimidine, Fapy•dG in E. coli. Biochemistry. 2007;46:10202–10212. doi: 10.1021/bi700628c. [DOI] [PubMed] [Google Scholar]
- 63.Hang B, Singer B. Protein-Protein Interactions Involving DNA Glycosylases. Chem Res Toxic. 2003;16:1181–1195. doi: 10.1021/tx030020p. [DOI] [PubMed] [Google Scholar]
- 64.Vidal AE, Hickson ID, Boiteux S, Radicella JP. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase step. Nucleic Acids Res. 2001;29:1285–1292. doi: 10.1093/nar/29.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sidorenko VS, Nevinsky GA, Zharkov DO. Mechanisms of interactin between human 8-oxoguanine-DNA glycosylase and Ap endonuclease. DNA Repair. 2007;6:317–328. doi: 10.1016/j.dnarep.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 66.Parsons JL, Zharkov DO, Dianov GL. NEIL1 excises 3′ end proximal oxidative DNA lesions resistant to cleavage by NTH1 and OGG1. Nucleic Acids Res. 2005;33:4849–4856. doi: 10.1093/nar/gki816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duo H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 68.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps of base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pope MA, David SS. DNA damage recognition and repair by the murine MutY homologue. DNA Repair. 2005;4:91–102. doi: 10.1016/j.dnarep.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 70.Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: A new perspective. DNA Repair. 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toueille M, El-Andaloussi N, Frouin I, Freire R, Funk D, Shevelev I, Friedrich-Heineken E, Villani G, Hottiger MO, Hubscher U. The human Rad9/Rad1/Hus1 damage sensor clamps interacts with DNA polymerase b and increases its DNA substrate utilisaztion efficiency: implications for DNA repair. Nucleic Acids Res. 2004;32:3316–3324. doi: 10.1093/nar/gkh652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Freidrich-Heineken E, Toueille M, Tannler B, Burki C, Ferrari E, Hottiger MO, Hubscher U. The Two DNA Clamps Rad9/Rad1/Hus1 Complex and Proliferating Cell Nuclear Antigen Differentially Regulate Flap Endonuclease I Activity. J Mol Biology. 2005;353:980–989. doi: 10.1016/j.jmb.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 73.Shi G, Chang DY, Cheng CC, Guan X, Veclovas V, Lu AL. Physical and Functional Interactions between MutY Glycosylase homologue (MYH) and Checkpoint proteins Rad9-Rad1-Hus1. Biochem J. 2006;400:53–62. doi: 10.1042/BJ20060774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Das A, Boldogh I, Lee JW, Harrigan JA, Hegde ML, Piotrowski J, De Souza Pinto N, Ramos W, Greenberg MM, Hazra TK, Mitra S, Borh VA. The Human Werner Syndrome Protein Stimulates Repair of Oxidative DNA Base Damage by the DNA Glycosylase NEIL1. J Biol Chem. 2007;282:26591–26602. doi: 10.1074/jbc.M703343200. [DOI] [PubMed] [Google Scholar]
- 75.Mokkapanti SK, Wiederholdt L, Hazra TK, Mitra S. Stimulation of DNA Glycosylase Activity of OGG1 by NEIL1: Functional Collaboration between two Human DNA Glycosylases. Biochemistry. 2004;43:11596–11604. doi: 10.1021/bi049097i. [DOI] [PubMed] [Google Scholar]
- 76.Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK, Lloyd RS. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. PNAS. 2006;103:1864–1869. doi: 10.1073/pnas.0507444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roy LM, Jaruga P, Wood TG, McCullough AK, Dizdaroglu M, Lloyd RS. Human polymorphic variants of the NEIL1 DNA glycosylase. J Biol Chem. 2007 doi: 10.1074/jbc.M610626200. [DOI] [PubMed] [Google Scholar]
- 78.Klunglund A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe G, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Minowa O, Arai T, Hirano M, Monden Y, Nakai S, Fakuda M, Itoh M, Takano H, Hippou Y, Aburatani H, Masumura K, Nohmi T, Nishimura S, Noda T. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. PNAS. 2000;97:4156–4161. doi: 10.1073/pnas.050404497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 shows the HPLC trace of separation of Sp diastereomers that are separated on a Dionex DNAPac PA-100 column as described in the methods section. This material is available free of charge via the Internet at http://pubs.acs.org