

Abstract
Context
Prospective epidemiological studies consistently indicate that Parkinson’s disease (PD) risk declines with increasing serum urate.
Objective
To determine whether serum urate, a purine metabolite and potent antioxidant, predicts prognosis in PD.
Design, Setting, and Participants
Prospective study among 804 subjects with early PD enrolled in the PRECEPT study, a clinical trial of the neuroprotectant potential of CEP-1347, conducted between April 2002 and August 2005 (average follow-up time 21.4 months).
Main Outcome Measures
The primary study endpoint was progression to clinical disability sufficient to warrant dopaminergic therapy. Cox proportional hazards models were used to estimate the hazard ratio (HR) of reaching endpoint according to quintiles of baseline serum urate, adjusting for gender, age and other potential covariates. Change in striatal uptake of [123I]β-CIT, a marker for the presynaptic dopamine transporter, was assessed with linear regression for a subset of 399 subjects.
Results
The adjusted HR of reaching endpoint declined with increasing baseline concentrations of urate; subjects in the top quintile reached the endpoint at only half the rate of subjects in the bottom quintile (HR=0.51; 95% CI: 0.37 to 0.72; p=0.0002). This association was markedly stronger in men (HR=0.39; 95% CI: 0.26 to 0.60; p<0.0001) than in women (HR=0.77; 95% CI: 0.39 to 1.50; p=0.4). The percent loss in striatal [123I]β-CIT uptake also improved with increasing serum urate concentrations (overall p for trend=0.002; men, p=0.0008; women, p= 0.4).
Conclusion
These findings identify serum urate as the first molecular factor directly linked to the progression of typical PD and suggest that targeting urate or its determinants could be an effective disease modifying therapy in PD.
As a consequence of mutations in the urate oxidase gene early in primate evolution,1 urate in humans circulates at high concentrations near the limits of its solubility and constitutes the main end product of purine metabolism. Urate has an anti-oxidant efficacy comparable to that of ascorbate, and thus its high level serves as one of our major defenses against oxidative damage caused by reactive nitrogen and oxygen species.2 Because oxidative stress may contribute to the loss of dopaminergic neurons in the substantia nigra of individuals with Parkinson’s disease (PD)3 and to the pathophysiology of other neurodegenerative diseases,4 blood urate could be an important determinant of disease susceptibility and progression.
Supporting this notion, the results of prospective epidemiological studies consistently indicate that among healthy people the risk of PD declines with increasing uricemia.5,6,7 Whether uricemia also predicts a better prognosis in established PD had not, however, been investigated. To address this question efficiently we sought completed, rigorously conducted clinical studies of PD progression in which prospectively determined (baseline) levels of urate are available. We identified a large randomized clinical trial entitled, “Parkinson Research Examination of CEP-1347 Trial” (PRECEPT), which was originally designed to investigate a candidate neuroprotectant in PD using clinical and imaging assessments of neurodegeneration,8 as an ideal opportunity to evaluate the potential association between serum urate and subsequent rates of PD progression.
METHODS
Study design
We conducted a longitudinal cohort investigation among participants in the PRECEPT study, a two-year double-blind, randomized trial of oral CEP-1347, an antiapoptotic mixed lineage kinase inhibitor that had been found to be neuroprotective in animal models of PD.8 The PRECEPT study was designed to determine whether this drug could slow the progression of early PD. It was carried out by the Parkinson Study Group, and sponsored by Cephalon, Inc. and H. Lundbeck A/S. The participants (n=806) were enrolled between April 2002 and April 2004 at 65 sites across the United States and Canada. All participating sites obtained approval of the protocol by their institutional review boards, and all subjects gave written consent for study participation. The PRECEPT study was stopped early following a pre-specified interim analysis for futility.8
Study population
Subjects to be enrolled in the study had to have early PD (modified Hoehn and Yahr stage of ≤2.5 with two of the cardinal signs: resting tremor, bradykinesia or rigidity), not requiring the use of dopaminergic therapy, and age at diagnosis ≥30 years.8 Exclusion criteria included atypical parkinsonism, a diagnosis of PD ≥ 5 years duration, a tremor score ≥ 3, a Mini-Mental Status Exam score ≤ 26, a Beck Depression Inventory score ≥ 15, the use of symptomatic therapy within six months prior to randomization, or an expectation that dopaminergic therapy would be required within three months of study enrollment. Subjects were randomly assigned to receive placebo or CEP-1347 10 mg, 25 mg, or 50 mg twice daily.
Serum urate and covariates
Serum urate was measured at screening as well as the subsequent baseline visit (on average 4 weeks apart) as one component of the routine safety monitoring performed. The correlation between the screening and baseline serum urate was high (r=0.88; p<0.0001), and only baseline values, which were available for 99.8% of patients enrolled in the trial, have been used in the present analyses. Serum urate levels were determined using an enzymatic assay on non-fasting blood, performed at a central commercial clinical laboratory (Covance, Indianapolis, IN). Information on past medical history and regular use of medications was collected at the screening visit.
Clinical evaluation
Following the initial screening and baseline visits, subjects were seen one month after starting the study drug, and then every three months until 24 months had elapsed. At each visit the site investigator conducted a clinical assessment, and evaluated the subject for disability sufficient to require dopaminergic therapy, the primary endpoint for the study. A follow-up visit was performed one month after discontinuing the study medication. Of the 806 randomized patients, 95 (11.8%) were censored, 48 because of withdrawal and 47 because they requested dopaminergic treatment before this was considered required by the investigators. The average follow-up time was 21.4 months.8
Outcome
The primary endpoint was time to disability requiring dopaminergic therapy,9,8 determined by individual investigators masked to treatment assignment. Secondary endpoints included changes in the Unified PD Rating Scale (UPDRS) (sum of the motor, mentation, and activity of daily living subscales, termed “total” in this study) and β-CIT SPECT imaging of ligand binding to striatal dopamine transporter, a marker for nigrostriatal dopaminergic nerve terminals. Because the UPDRS is modified by the dopaminergic treatment instituted at endpoint, the annualized rate of change in UPDRS was determined based on change from baseline to endpoint for each subject, and was calculated as: ([total UPDRS at the last assessment before initiation of dopaminergic treatment – total UPDRS at baseline] / number of days between the two assessments) × (365 days/year).
Neuroimaging substudy
Single-photon emission computed-tomography (SPECT) of iodine-123-labeled 2-β-carboxymethoxy-3-β-(4-iodophenyl)tropane ([123I]β-CIT) uptake was used at baseline to measure striatal dopamine-transporter density among all subjects in the trial; imaging was carried out at the Institute for Neurodegenerative Disorders in New Haven, Connecticut with methods as described previously.10 All subjects were invited to repeat the SPECT at the end of the follow-up. The 399 subjects with repeated SPECT imaging completed as of May 2005 and with baseline serum urate were included in a subanalyses on the relation between baseline serum urate and percent change in the ratio of the specific striatal [123I]β-CIT uptake to the non-displaceable striatal [123I]β-CIT uptake between the two images. Mean interval between the two SPECT scans was 22 months.
Statistical analysis
Cox proportional hazards models were used to estimate the hazard ratios of reaching the endpoint according to quintiles of baseline serum urate, adjusting for gender and age (5-year groups). Initial analyses were conducted using quintiles based on the combined urate distribution in men and women (“common quintiles”). An important advantage of using common quintiles is that the hazard ratios in men and women estimate the effects of similar levels of serum urate. However, because of the expected higher level of urate in men, this categorization resulted in a markedly skewed distribution within gender, with most men in the top quintiles and most women in the bottom quintiles of serum urate, and thus in a loss of power of analyses within gender. These analyses were therefore complemented by estimating hazard ratios for gender-specific quintiles. In these analyses the advantage of a more balanced distribution of subjects across quintiles is in part offset by the lack of comparability of the hazard ratios; for example, in men the cut-offs for lowest and highest quintiles are< 4.9 and >7.0 mg/dL, versus <3.7 and >5.6 mg/dL in women. Tests for trend were conducted by including serum urate as a continuous variable in the proportional hazards models (Wald test). Potential confounding was assessed by adjusting the regression analyses for cigarette smoking (never, past, or current), body mass index (quintiles), serum cholesterol (continuous), and use of antihypertensive drugs or non-steroidal antiinflammatory drugs (use versus no-use). Because the adjusted results were not materially different from the unadjusted, and none of these covariates was significantly related to PD progression, only the age- and gender-adjusted results have been included in this report. Possible interactions between serum urate and age, gender, and treatment group were explored by including in the proportional hazards model the cross-product of serum urate as a continuous variable with the corresponding covariates (age: continuous in years; gender: 0,1; treatment: 0, 1, 2, 3, for placebo and each of the CEP-1347 doses). None of the interaction terms was significant, and only results not including these terms are reported. There was no significant deviation from the proportional hazards assumption, tested by adding the cross product of urate (continuous) with time of follow-up (0 for first year, 1 thereafter) to the Cox model.
The relation between serum urate and rate of change in UPDRS or percent change in striatal [123I]β-CIT uptake were assessed by linear regression. For each of these outcomes, we fitted regression models including age, gender (for analyses including men and women combined), and either common quintiles of serum urate or gender-specific quintiles, as outlined above. Because of the skewed distribution of UPDRS rates, analyses for this outcome were also conducted using Spearman correlation. All the p values presented are for 2-tailed tests with levels < 0.05 defined as significant.
RESULTS
Serum urate at baseline was available for 804 (517 men and 287 women) of the 806 subjects enrolled in the trial. Selected characteristics of these subjects are shown in Table 1. As expected, serum urate concentrations were positively correlated with male gender, body mass index, use of thiazide diuretics, and history of gout and hypertension (Table 1).
Table 1.
Characteristics of study participants according to quintiles of baseline serum urate (n=804).
| Quintile of baseline serum urate | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | All | |
| Serum urate (mg/dL) | <4.3 | 4.3–<5.1 | 5.1–<5.8 | 5.8–<6.7 | >=6.7 | |
| N. of patients | 177 | 157 | 147 | 169 | 154 | 804 |
| Female % | 74.6 | 44.6 | 25.2 | 15.4 | 14.3 | 35.7 |
| Age (median) | 57 | 60 | 59 | 61 | 60 | 59 |
| BMI | 25.0 | 26.2 | 27.8 | 28.7 | 29.9 | 27.5 |
| Current smokers % | 6.2 | 5.1 | 14.3 | 5.3 | 5.2 | 7.1 |
| Gout % | 0 | 0.6 | 0.7 | 1.2 | 5.2 | 1.5 |
| Hypertension (% use of antihypertensive drugs) | 19.8 | 26.8 | 36.1 | 37.3 | 57.1 | 35.0 |
| Thiazides % | 5.7 | 6.4 | 10.2 | 14.2 | 16.2 | 10.4 |
| NSAIDs use % | 27.7 | 29.3 | 29.9 | 20.7 | 30.5 | 27.5 |
| Endpoint: Yes % | 67.8 | 61.8 | 64.0 | 59.2 | 53.3 | 61.3 |
| UPDRS rate of change during follow-up (mean) | 16.9 | 17.0 | 14.8 | 14.1 | 14.3 | 14.9 |
| % change in striatal β-CIT uptake (mean) | − 10 | −9 | −10 | −8 | −4 | −8 |
Overall, 493 (61%) of participants reached the endpoint of disability sufficient to require dopaminergic therapy during follow-up. The hazard ratio of reaching the endpoint declined with increasing concentrations of serum urate; subjects in the top common quintile reached endpoint at approximately half the rate of subjects in the bottom quintile (HR = 0.51; 95% CI: 0.37 to 0.72; p< 0.0001) (Table 2). This association was markedly stronger in men than in women, although a test for interaction of urate with gender was not significant (Table 2). Results of analyses based on gender-specific quintiles were similar, and are summarized in Kaplan-Meier curves (Figure 1).
Table 2.
Hazard ratios (HR)† for reaching the endpoint according to common quintiles of baseline serum urate.
| Serum urate quintile | Median serum urate (mg/dL) | All (n=804) | Men (n=517) | Women (n=287) | |||||
|---|---|---|---|---|---|---|---|---|---|
| HR† (95% CI) | p value | n | HR‡ (95% CI) | p value | n | HR‡ (95% CI) | p value | ||
| 1 | 3.8 | 1.00 (Ref) | - | 45 | 1.00 (Ref) | - | 132 | 1.00 (Ref) | - |
| 2 | 4.8 | 0.80 (0.60–1.07) | 0.12 | 87 | 0.61 (0.40–0.94) | 0.03 | 70 | 0.93 (0.63–1.37 | 0.70 |
| 3 | 5.5 | 0.85 (0.63–1.15) | 0.29 | 110 | 0.66 (0.44–1.00) | 0.05 | 37 | 1.00 (0.61–1.64) | 0.99 |
| 4 | 6.3 | 0.65 (0.47–0.88) | 0.006 | 143 | 0.51 (0.34–0.76) | 0.001 | 26 | 0.76 (0.41–1.39) | 0.37 |
| 5 | 7.5 | 0.51 (0.37–0.72) | <0.0001 | 132 | 0.39 (0.26–0.60) | <0.0001 | 22 | 0.77 (0.39–1.50) | 0.44 |
|
| |||||||||
| p, for trend | 0.0002 | <0.0001 | 0.33 | ||||||
|
| |||||||||
| p, for genderurate interaction | 0.15 | ||||||||
Adjusted for age and gender;
adjusted for age only.
Figure 1.
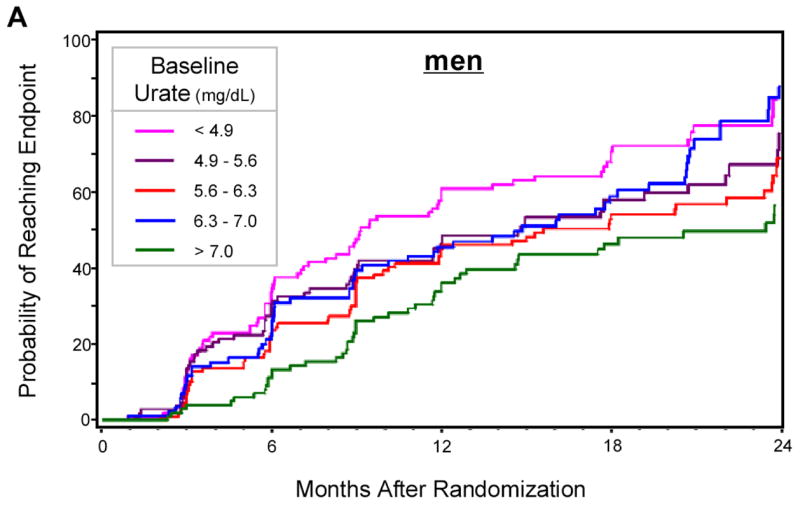
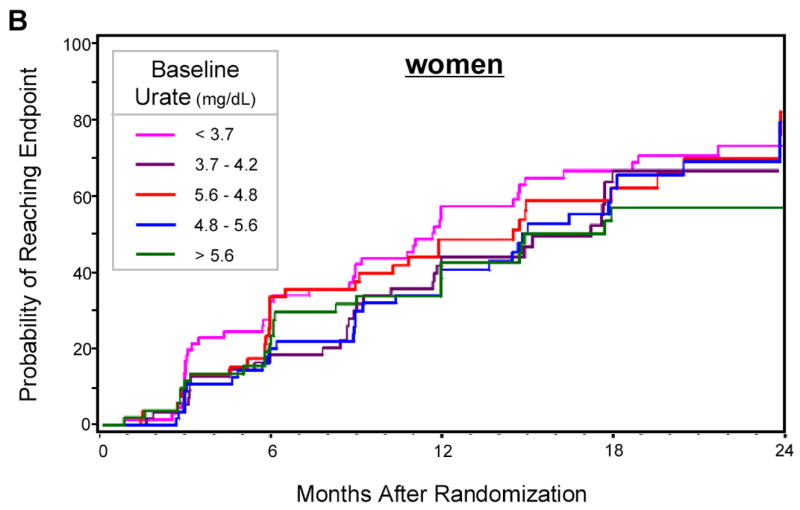
Kaplan-Meier estimates of the cumulative probability of reaching the end point by 24 months of follow-up, according to gender-specific quintiles of baseline serum urate. A. Men; B. Women. Log-rank tests: p=0.0011 in men, p=0.47 in women. At 24 months sample size was 46 in men and 21 in women.
The rate of change in UPDRS (points per year) in men and women combined was 16.9 among patients in the lowest quintile of baseline urate, and 14.3 among those in the highest quintile (p for trend = 0.09). Among men, there was a modest but significant inverse association between baseline serum urate and rate of UPDRS change (Spearman correlation coefficient = −0.10; p=0.02). A significantly lower rate of change in UPDRS was observed among patients in the highest as compared with those in the lowest gender-specific quintile of serum urate (adjusted difference = 7.0; p = 0.02). In contrast, no significant association was found in women (Spearman r = −0.03, p = 0.5).
The percent change in striatal [123I]β-CIT uptake also declined with increasing serum urate concentrations (p for trend = 0.002), although the trend was largely driven by a lower percent change among subjects in the top quintile of serum urate, with little or no differences between quintiles 1 through 4 (Figure 2). Because there were only 4 women in the top quintile of serum urate when the cut-offs for quintiles were generated from men and women combined, stratified analyses were only conducted using gender-specific quintiles. As in the endpoint analyses, a significant association was only seen in men (Figure 2).
Figure 2.

Age-adjusted percent change in striatal [123I]β-CIT uptake according to overall and gender-specific quintiles of baseline serum urate. Median serum urate by quintiles (1 to 5) mg/dL: All: 3.8, 4.8, 5.5, 6.3, 7.5; Men: 4.4, 5.3, 6.0, 6.6, 7.8; Women: 3.1, 4.0, 4.5, 5.2, 6.6. * p < 0.05; ** p < 0.001 compared to corresponding quintile 1. P for linear trend: all, p= 0.002; men, p=0.0008; women, p=0.4.
COMMENT
In this large prospective investigation among subjects in the early stages of PD enrolled in a randomized clinical trial, we found that the rate of progression to the primary clinical endpoint declined with increasing levels of baseline serum urate. There was a clear dose-response relationship with a 35% reduction in rate of progression among patients in the fourth quintile of serum urate and a 49% reduction among those in the highest quintile, as compared with those in the lowest quintile. These associations were highly significant, and corroborated by the finding that patients with higher urate also had a lower percent loss of striatal [123I]β-CIT uptake during the follow-up.
Strengths of this study include the longitudinal design, the measurement of serum urate at baseline and before starting any anti-parkinsonian treatment, the large number of participants, and the rigorous clinical assessment of all patients. We specifically examined the relation between serum urate and PD progression because of the strong a priori evidence that individuals with high levels of serum urate have a markedly reduced risk of developing PD.5–7 The convergence between the results of previous epidemiological studies and those of the present investigation is striking. Combined, these results support the continuity of the neurodegenerative process before and after the onset of the first motor symptoms that lead to the diagnosis of PD, and imply that either higher serum urate itself is neuroprotective, or it serves as an indirect marker of protection of the dopaminergic neurons that are lost in PD.
The inverse association between uricemia and PD progression could be explained if both were affected by a common factor, or, in epidemiological lexicon, a confounder. In subjects without PD, the strongest correlates of serum urate are male gender, obesity, and arterial hypertension.11 Further, use of thiazide diuretics is known to increase urate levels.12 These correlations were also found among participants in our study, suggesting that the main determinants of serum urate are the same in individuals with or without PD. However, the relation between serum urate and PD progression in our study was independent from these factors. Also, adjustment for cigarette smoking and use of non-steroidal anti-inflammatory drugs, which have been related to PD risk, did not appreciably change the results. Genetic factors could also affect both serum urate and PD progression, and thus act as confounders. Heritability of serum urate is estimated to range from 25% to 70%,13 and several genetic mutations that affect uricemia have been identified.14–16 Known mutations with marked effects on uricemia, however, are rare and seem unlikely to fully explain the strong inverse associations between uricemia and both PD risk and PD progression. Finally, dietary factors should also be considered. High dairy consumption has been associated with an increased risk of PD17–19 and with decreased serum urate,20 but the latter association is weak and unlikely to account for much variation in urate levels. On the other hand, high alcohol consumption increases serum urate,21 but in longitudinal studies alcohol consumption was not consistently related to PD risk.22 Purine 20 and fructose 23 intakes also increase serum urate, but these effects are modest and there is no evidence that these nutrients would affect PD risk or progression independently from their effects on uricemia. Overall, it seems therefore unlikely that the inverse relation between uricemia and PD progression is due to confounding by known factors. As in all observational studies, however, a role for unknown factors cannot be excluded.
Although several clinical features of PD (e.g., prominent asymmetry, rest tremor predominance, absence of early cognitive or gait dysfunction) have been identified previously as predictors of a slower rate of clinical progression24, 25 these are complex behavioral characteristics of the disease and are thus likely to result from, rather than influence pathogenic mechanisms. By contrast, as an antioxidant with peroxynitrite scavenging and metal chelating29 properties, urate is well positioned to serve as a neuroprotectant against the underlying neurodegeneration of PD. Considerable evidence from genetic as well as idiopathic forms of PD has implicated oxidative and nitrative stress as central pathogentic mechanisms.26,27 Urate at physiological concentrations is as effective an antioxidant as ascorbate.2 It also stabilizes ascorbate,28 possibly by forming complexes with iron ions,29 and scavenges nitrogen radicals.30, 31 Further, administration of urate reduced the exacerbation of the oxidative stress and mitochondrial dysfunction in human dopaminergic cells exposed to the pesticide rotenone or to iron ions.32
Alternatively, the predictive association between urate and PD progression could reflect a neuroprotective effect of a urate precursor, rather than urate itself. For example, adenosine and its deaminated metabolite inosine (which is in turn deribosylated and oxidized to urate) both modulate neuronal death on their own. Adenosine can have either neuroprotective or neurotoxic effects on dopaminergic neurons via adenosine A1 and A2A receptors, respectively.33,34 Inosine has also shown potential as a neuroprotectant in models of stroke and multiple sclerosis.35,36 Whether urate, its metabolic precursors or other determinants modulate neurodegeneration in PD, their potential is supported by lower levels of urate in cerebrospinal fluid37 and post-mortem substantia nigra38 of patients with PD.
Whereas the concentration-dependent inverse relationship was robust and highly significant statistically in men, it appeared as a weak non-significant trend amongst women. This difference between men and women (also noted for the association between urate and [123I]β-CIT uptake neuroimaging) could result in part from a biological effect of gender on urate mechanisms in PD.39 Alternatively, it may reflect the substantially lower average urate concentrations in women, who comprise only 16% of the subjects in the two uppermost quintiles in which the substantially slower rates of disease progression were observed.
That urate and its metabolic pathway are particularly amenable to existing pharmacological and dietary manipulations enhances the potential therapeutic significance of the present findings. A purine-rich diet can elevate serum urate, and the purine supplement inosine, used as a potential therapy for multiple sclerosis in a phase II randomized clinical trial, markedly and chronically raised urate concentrations without inducing gout or other adverse effects.40,41 It is also well known that thiazide diuretics even at low doses elevate urate by reducing its renal clearance.12 Individuals with higher serum urate, however, have an increased risk of hypertension, coronary heart disease, and stroke.5,42 Although these associations may in part be confounded by obesity and other risk factors,43,44 a long-term neuroprotective effect of urate or its precursors would have to be weighed against potential adverse cardiovascular effects.
It should be noted that measurement of urate on its own in newly diagnosed PD patients as an indicator of an individual patient’s future rate of progression is likely to be of modest clinical utility.45 On the other hand, urate testing may aid the rational design of neuroprotective trials in PD, particularly those targeting mechanisms (antioxidant or purinergic) that are potentially shared with urate. For example, coenzyme Q10, creatine and rasagiline – potential neuroprotectants targeting oxidative stress pathways in planned neuroprotection trials – might be most effective in PD patients whose endogenous antioxidant pool (including urate) is lowest at baseline, and thus controlling for an interaction with or stratifying by urate levels at baseline may improve the power of such trials.
Of note, the present discovery of a urate link to PD progression was achieved through additional analyses of a rigorously conducted clinical trial whose database was made available to test unforeseen hypotheses upon conclusion of the primary investigation.8 The findings thus reflect a broader opportunity to retrospectively explore a growing repository of high quality data from neuroprotection trials for PD, Alzheimer’s disease and other progressive degenerative disorders.
Acknowledgments
Funding/Support: Supported by NIH grants NS048517 and ES010804, and the Beeson Scholars Program of the American Federation for Aging Research.
We thank Emily Gorbold for her assistance in coordinating this collaborative project, Leslie Unger for technical assistance in preparing the manuscript, and John Ondrasik for conducting a secondary review of statistical programs and reported results.
APPENDIX
The following members of the Parkinson Study Group were investigators in PRECEPT (Parkinson Research Examination of CEP-1347 Trial) and authored this report
Colorado Neurological Institute, Englewood, CO: Rajeev Kumar, MD; London Health Sciences Center, London, Canada: Mandar Jog, MD, Cheryl Horn, RN; Rush-Presbyterian-St. Luke’s Medical Center, Chicago, IL: Kathleen Shannon, MD; University of Colorado Health Sciences Center, Denver, CO: Maureen Leehey, MD, Teresa Derian, RN; Ottawa Hospital Civic Site, Ottawa, Ontario, Canada: David Grimes, MD, Melodie Mortensen, BSCN; University of Minnesota/Minnesota VA Medical Center, Minneapolis, MN: Paul Tuite, MD; University of California Irvine, Irvine, CA: Neal Hermanowicz, MD, Shari Niswonger, RN; University of Rochester, Rochester, NY: Roger Kurlan, MD, Irenita Gardiner, RN, CCRC; Toronto Western Hospital, University Health Network, Toronto, Ontario, Canada: Janis Miyasaki, MD, FRCPC, Lisa Johnston, RN, BSCN, CNN; The Parkinson’s Institute, Sunnyvale, CA: James Tetrud, MD; NeuroHealth Parkinson’s Disease Movement Disorders Center, Warwick, RI: Joseph Friedman, MD, Hubert Fernandez, MD; University of Iowa, Iowa City, IA: Robert Rodnitzky, MD, Judith Dobson, RN, CCRC; Mayo Clinic Arizona, Scottsdale, AZ: Virgilio Evidente, MD, Marlene Lind, RN; Oregon Health & Science University, Portland, OR: Pamela Andrews, BS; Chum-Hotel Dieu/McGill Center for Studies in Aging, Montreal, Quebec, Canada: Michel Panisset, MD; Washington University School of Medicine, St. Louis, MO: Brad Racette, MD, Patricia Deppen, RN; Baylor College of Medicine, Houston, TX: Joseph Jankovic, MD, Christine Hunter, RN, CCRC; Albany Medical College, Albany, NY: Eric Molho, MD, Stewart Factor, MD; Indiana University School of Medicine, Indianapolis, IN: Joanne Wojcieszek, MD, JoAnn Belden, RN, LPN; University of California Davis, Sacramento, CA: Lisa Wilson, MS, CCRP, Patricia Campbell, RN; Duke University Medical Center, Durham, NC: Burton Scott, MD; Cleveland Clinic, Cleveland, OH: Thyagarajan Subramanian, MD, Ruth Kolb, CCRP; University of Pennsylvania, Philadelphia, PA: Andrew Siderowf, MD, Heather Maccarone, RN, BSN; University of South Florida, Tampa, FL: Robert Hauser, MD, Diana Delaney, BHS, Joanne Nemeth, RN; Johns Hopkins University, Baltimore, MD: Joseph Savitt, MD, PhD, Melissa Gerstenhaber, RNC, MSN; University of Cincinnati/Cincinnati Children’s Hospital, Cincinnati, OH: Alok Sahay, MD, Maureen Gartner, RN; Mayo Clinic Jacksonville, Jacksonville, FL: Ryan Uitti, MD, Margaret Turk, RN; University of Sherbrooke, Sherbrooke, Quebec, Canada: Jean Rivest, MD, Daniel Soucy, RN; University of Virginia, Charlottesville, VA: Frederick Wooten, MD, Elke Rost-Ruffner, RN, BSN; Massachusetts General Hospital, Boston, MA: Michael Schwarzschild, MD, PhD, Marsha Tennis, RN; Medical College of Georgia, Augusta, GA: Kapil Sethi, MD, Lisa Hatch, RN, BSN; University of Tennessee-Memphis, Memphis, TN: Ronald Pfeiffer, MD, Brenda Pfeiffer, RN, BSN; North Shore-LIJ Health System, Manhasset, NY: Andrew Feigin, MD, Jean Ayan, RN, Barbara Shannon, RN; Northwestern University, Chicago, IL: Tanya Simuni, MD, Karen Williams, BA, Annette Kaczmarek, CCRC, Michele Wolff, BA; Medical University of Ohio, Toledo, OH: Lawrence Elmer, MD, PhD, Kathy Davis, RN; University of Connecticut, Glastonbury, CT: Antonelle deMarcaida, MD, Sheila Thurlow, RN; Hotel-Dieu Hospital-CHUM, Montreal, Quebec, Canada: Sylvain Chouinard, MD, Hubert Poiffaut, RN; Barrow Neurological Institute, Phoenix, AZ: Holly Shill, MD, Mark Stacy, MD, Lynn Marlor, BSN, MSHS; The Parkinson’s & Movement Disorder Institute, Fountain Valley, CA: Daniel Truong, MD; LSU Health Science Center Shreveport, Shreveport, LA: Richard Zweig, MD, Rhonda Feldt, RN; Columbia University Medical Center, New York, NY: Cheryl Waters, MD, Angel Figueroa, BBA; University of Kansas Medical Center, Kansas City, KS: Rajesh Pahwa, MD, Amy Parsons, RN, BSN; University of Southern California, Los Angeles, CA: Jennifer Hui, MD, Allan Wu, MD; University of Alberta, Edmonton, AB, Canada: Richard Camicioli, MD, Pamela King, BScN, RN; University of Chicago, Chicago, IL: Arif Dalvi, MD, Un Jung Kang, MD; University of Maryland School of Medicine, Baltimore, MD: Stephen Reich, MD, Lisa Shulman, MD, Kelly Dustin, RN: UMDNJ Robert Wood Johnson Medical School, New Brunswick, NJ: Margery Mark, MD; Saskatoon Dist Health Board Royal University Hospital, Saskatoon SK, Canada: Ali Rajput, MD; Boston University, Boston, MA: Peter Novak, MD; University of California San Diego, La Jolla, CA: David Song, MD; Creighton University, Omaha, NE: Carolyn Peterson, RN; Medical College of Wisconsin, Milwaukee, WI: Karen Blindauer, MD, Jeanine Petit, ANP; Scott & White Hospital/Texas A&M University, Temple, TX: Bala Manyam, MD, Danielle McNeil-Keller, LMSW; Clinical Neuroscience Center, Southfield, MI: Peter LeWitt, MD; University of Calgary, Calgary, AB, Canada: Ranjit Ranawaya, MD, Oksana Suchowersky, MD, Carol Pantella, RN; Brigham & Women’s Hospital, Boston, MA: Lewis Sudarsky, MD; Beth Israel Deaconess Medical Center, Boston, MA: Daniel Tarsy, MD; Long Island Jewish Medical Center, New Hyde Park, NY: Mark Forrest Gordon, MD; Beth Israel Medical Center, New York, NY: Alessandro DiRocco, MD; Stanford University Medical Center, Stanford, CA: Amy Andrzejewski, BS; UMDNJ School of Osteopathic Medicine, Stratford, NJ: Gerald Podskalny, DO; Cleveland Clinic Florida-Weston, Weston, FL: Nestor Galvez-Jimenez, MD; University of Arkansas for Medical Sciences, Little Rock, AR: Sami Harik, MD, Samer Tabbal, MD, Jana Patterson, RN.
Footnotes
The final, edited, formatted, and published article can be found online: http://archneur.ama-assn.org/cgi/content/full/2008.65.6.nct70003v1
Author contributions: Dr. Alberto Ascherio had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design, drafting of manuscript, supervision: Schwarzschild and Ascherio.
Data analysis: Schwarzschild, Ascherio, Watts, Oakes.
Obtaining funding: Schwarzschild and Ascherio.
Acquisition and interpretation of data, and critical revision of manuscript: Schwarzschild, Schwid, Marek, Watts, Lang, Oakes, Shoulson and Ascherio
Financial disclosures: None.
Role of the Sponsors: The PRECEPT study was supported by Cephalon, Inc. and Lundbeck A/S. The analyses of the fully accessible PRECEPT database were carried out independently by the Parkinson Study Group Biostatistics Center in Rochester, NY.
References
- 1.Oda M, Satta Y, Takenaka O, Takahata N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol. 2002 May;19(5):640–653. doi: 10.1093/oxfordjournals.molbev.a004123. [DOI] [PubMed] [Google Scholar]
- 2.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A. 1981;78(11):6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53 Suppl 3:S26–36. S36–28. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005 Oct;58(4):495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 5.Davis JW, Grandinetti A, Waslien CI, Ross GW, White LR, Morens DM. Observations on serum uric acid and the risk of idiopathic Parkinson’s disease. Am J Epidemiol. 1996;144:480–484. doi: 10.1093/oxfordjournals.aje.a008954. [DOI] [PubMed] [Google Scholar]
- 6.de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum uric acid levels and the risk of Parkinson disease. Ann Neurol. 2005 Oct 20;58(5):797–800. doi: 10.1002/ana.20663. [DOI] [PubMed] [Google Scholar]
- 7.Weisskopf MG, O’Reilly E, Chen H, Schwarzschild MA, Ascherio A. Plasma urate and risk of Parkinson’s disease. Am J Epidemiol. 2007 doi: 10.1093/aje/kwm127. [In Press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Parkinson Study Group PRECEPT Investigators. The mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson’s disease. Neurology. 2007;69:1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- 9.Parkinson Study Group. DATATOP: a multicenter controlled clinical trial in early Parkinson’s disease. Arch Neurol. 1989 Oct;46(10):1052–1060. doi: 10.1001/archneur.1989.00520460028009. [DOI] [PubMed] [Google Scholar]
- 10.Marek K, Innis R, van Dyck C, et al. [123I]beta-CIT SPECT imaging assessment of the rate of Parkinson’s disease progression. Neurology. 2001 Dec 11;57(11):2089–2094. doi: 10.1212/wnl.57.11.2089. [DOI] [PubMed] [Google Scholar]
- 11.Roubenoff R, Klag MJ, Mead LA, Liang KY, Seidler AJ, Hochberg MC. Incidence and risk factors for gout in white men. JAMA. 1991 Dec 4;266(21):3004–3007. [PubMed] [Google Scholar]
- 12.Reyes AJ. Cardiovascular drugs and serum uric acid. Cardiovasc Drugs Ther. 2003 Sep–Nov;17(5–6):397–414. doi: 10.1023/b:card.0000015855.02485.e3. [DOI] [PubMed] [Google Scholar]
- 13.Yang Q, Guo CY, Cupples LA, Levy D, Wilson PW, Fox CS. Genome-wide search for genes affecting serum uric acid levels: the Framingham Heart Study. Metabolism. 2005 Nov;54(11):1435–1441. doi: 10.1016/j.metabol.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002 May 23;417(6887):447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 15.Graessler J, Graessler A, Unger S, et al. Association of the human urate transporter 1 with reduced renal uric acid excretion and hyperuricemia in a German Caucasian population. Arthritis Rheum. 2006 Jan;54(1):292–300. doi: 10.1002/art.21499. [DOI] [PubMed] [Google Scholar]
- 16.Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004 Mar 16;555(Pt 3):589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Zhang SM, Hernan MA, Willett WC, Ascherio A. Diet and Parkinson’s disease: A potential role of dairy products in men. Ann Neurol. 2002 Dec;52(6):793–801. doi: 10.1002/ana.10381. [DOI] [PubMed] [Google Scholar]
- 18.Park M, Ross GW, Petrovitch H, et al. Consumption of milk and calcium in midlife and the future risk of Parkinson disease. Neurology. 2005 Mar 22;64(6):1047–1051. doi: 10.1212/01.WNL.0000154532.98495.BF. [DOI] [PubMed] [Google Scholar]
- 19.Chen H, O’Reilly E, McCullough ML, et al. Consumption of dairy products and risk of Parkinson’s disease. Am J Epidemiol. 2007;165:998–1006. doi: 10.1093/aje/kwk089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi HK, Liu S, Curhan G. Intake of purine-rich foods, protein, and dairy products and relationship to serum levels of uric acid: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2005 Jan;52(1):283–289. doi: 10.1002/art.20761. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto T, Moriwaki Y, Takahashi S. Effect of ethanol on metabolism of purine bases (hypoxanthine, xanthine, and uric acid) Clin Chim Acta. 2005 Jun;356(1–2):35–57. doi: 10.1016/j.cccn.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 22.Hernán MA, Chen H, Schwarzschild MA, Ascherio A. Alcohol consumption and the incidence of Parkinson’s disease. Ann Neurol. 2003;54(2):170–175. doi: 10.1002/ana.10611. [DOI] [PubMed] [Google Scholar]
- 23.Mayes PA. Intermediary metabolism of fructose. Am J Clin Nutr. 1993 Nov;58(5 Suppl):754S–765S. doi: 10.1093/ajcn/58.5.754S. [DOI] [PubMed] [Google Scholar]
- 24.Marras C, Rochon P, Lang AE. Predicting motor decline and disability in Parkinson disease: a systematic review. Arch Neurol. 2002 Nov;59(11):1724–1728. doi: 10.1001/archneur.59.11.1724. [DOI] [PubMed] [Google Scholar]
- 25.Gasparoli E, Delibori D, Polesello G, et al. Clinical predictors in Parkinson’s disease. Neurol Sci. 2002 Sep;23 Suppl 2:S77–78. doi: 10.1007/s100720200078. [DOI] [PubMed] [Google Scholar]
- 26.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 27.Jenner P, Olanow CW. The pathogenesis of cell death in Parkinson’s disease. Neurology. 2006 May 23;66(10 Suppl 4):S24–36. doi: 10.1212/wnl.66.10_suppl_4.s24. [DOI] [PubMed] [Google Scholar]
- 28.Stocker R, Frei B. Endogenous antioxidant defences in human blood plasma. In: Sies H, editor. Oxidative Stress: Oxidants and Antioxidants. San Diego: Academic Press; 1991. pp. 213–243. [Google Scholar]
- 29.Davies KJ, Sevanian A, Muakkassah-Kelly SF, Hochstein P. Uric acid-iron ion complexes. A new aspect of the antioxidant functions of uric acid. Biochem J. 1986 May 1;235(3):747–754. doi: 10.1042/bj2350747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Squadrito GL, Cueto R, Splenser AE, et al. Reaction of uric acid with peroxynitrite and implications for the mechanism of neuroprotection by uric acid. Arch Biochem Biophys. 2000 Apr 15;376(2):333–337. doi: 10.1006/abbi.2000.1721. [DOI] [PubMed] [Google Scholar]
- 31.Whiteman M, Ketsawatsakul U, Halliwell B. A reassessment of the peroxynitrite scavenging activity of uric acid. Ann N Y Acad Sci. 2002 May;962:242–259. doi: 10.1111/j.1749-6632.2002.tb04072.x. [DOI] [PubMed] [Google Scholar]
- 32.Duan W, Ladenheim B, Cutler RG, Kruman II, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J Neurochem. 2002;80(1):101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- 33.Delle Donne KT, Sonsalla PK. Protection against methamphetamine-induced neurotoxicity to neostriatal dopaminergic neurons by adenosine receptor activation. J Pharmacol Exp Ther. 1994 Dec;271(3):1320–1326. [PubMed] [Google Scholar]
- 34.Xu K, Bastia E, Schwarzschild M. Therapeutic potential of adenosine A(2A) receptor antagonists in Parkinson’s disease. Pharmacol Ther. 2005 Mar;105(3):267–310. doi: 10.1016/j.pharmthera.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Scott GS, Spitsin SV, Kean RB, Mikheeva T, Koprowski H, Hooper DC. Therapeutic intervention in experimental allergic encephalomyelitis by administration of uric acid precursors. Proc Natl Acad Sci U S A. 2002 Dec 10;99(25):16303–16308. doi: 10.1073/pnas.212645999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen H, Chen GJ, Harvey BK, Bickford PC, Wang Y. Inosine reduces ischemic brain injury in rats. Stroke. 2005 Mar;36(3):654–659. doi: 10.1161/01.STR.0000155747.15679.04. [DOI] [PubMed] [Google Scholar]
- 37.Tohgi H, Abe T, Takahashi S, Kikuchi T. The urate and xanthine concentrations in the cerebrospinal fluid in patients with vascular dementia of the Binswanger type, Alzheimer type dementia, and Parkinson’s disease. J Neural Transm Park Dis Dement Sect. 1993;6(2):119–126. doi: 10.1007/BF02261005. [DOI] [PubMed] [Google Scholar]
- 38.Church WH, Ward VL. Uric acid is reduced in the substantia nigra in Parkinson’s disease: effect on dopamine oxidation. Brain Res Bull. 1994;33(4):419–425. doi: 10.1016/0361-9230(94)90285-2. [DOI] [PubMed] [Google Scholar]
- 39.Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr Drug Targets CNS Neurol Disord. 2004 Aug;3(4):297–313. doi: 10.2174/1568007043337193. [DOI] [PubMed] [Google Scholar]
- 40.Spitsin S, Hooper DC, Leist T, Streletz LJ, Mikheeva T, Koprowskil H. Inactivation of peroxynitrite in multiple sclerosis patients after oral administration of inosine may suggest possible approaches to therapy of the disease. Mult Scler. 2001 Oct;7(5):313–319. doi: 10.1177/135245850100700507. [DOI] [PubMed] [Google Scholar]
- 41.Koprowski H, Spitsin SV, Hooper DC. Prospects for the treatment of multiple sclerosis by raising serum levels of uric acid, a scavenger of peroxynitrite. Ann Neurol. 2001 Jan;49(1):139. doi: 10.1002/1531-8249(200101)49:1<139::aid-ana28>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 42.Bos MJ, Koudstaal PJ, Hofman A, Witteman JC, Breteler MM. Uric Acid Is a Risk Factor for Myocardial Infarction and Stroke. The Rotterdam Study. Stroke. 2006 Jun;37(6):1503–1507. doi: 10.1161/01.STR.0000221716.55088.d4. [DOI] [PubMed] [Google Scholar]
- 43.Wheeler JG, Juzwishin KD, Eiriksdottir G, Gudnason V, Danesh J. Serum uric acid and coronary heart disease in 9,458 incident cases and 155,084 controls: prospective study and meta-analysis. PLoS Med. 2005 Mar;2(3):e76. doi: 10.1371/journal.pmed.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forman JP, Choi H, Curhan GC. Plasma uric acid level and risk for incident hypertension among men. J Am Soc Nephrol. 2007 Jan;18(1):287–292. doi: 10.1681/ASN.2006080865. [DOI] [PubMed] [Google Scholar]
- 45.Ware JH. The limitations of risk factors as prognostic tools. N Engl J Med. 2006 Dec 21;355(25):2615–2617. doi: 10.1056/NEJMp068249. [DOI] [PubMed] [Google Scholar]