

Abstract
DNA mismatch repair (MMR) proteins are ubiquitous players in a diverse array of important cellular functions. In its role in post-replication repair, MMR safeguards the genome correcting base mispairs arising as a result of replication errors. Loss of MMR results in greatly increased rates of spontaneous mutation in organisms ranging from bacteria to humans. Mutations in MMR genes cause hereditary nonpolyposis colorectal cancer, and loss of MMR is associated with a significant fraction of sporadic cancers. Given its prominence in mutation avoidance and its ability to target a range of DNA lesions, MMR has been under investigation in studies of ageing mechanisms. This review summarizes what is known about the molecular details of the MMR pathway and the role of MMR proteins in cancer susceptibility and ageing.
Keywords: Mismatch repair, HNPCC, Ageing, Genome instability, MutS, MutL
1. Introduction
Ensuring the fidelity of DNA replication is central to preserving genomic integrity, and DNA mismatch repair (MMR) is critical for maintaining the fidelity of replication. We review what is known concerning the molecular mechanism of MMR, its role in DNA damage signalling, and its relationship to cancer and ageing. The recent literature is emphasized; for more comprehensive discussions, please see Harfe and Jinks-Robertson (2000), Jiricny (2006), Kunkel and Erie (2005), Modrich (2006), Schofield and Hsieh (2003).
Replication is an extraordinarily faithful process (Iyer et al., 2006); mutations occur at a frequency of roughly 1 in 109 to 1010 base pairs per cell division. Nucleotide selection at the base incorporation step and the proofreading function of DNA polymerases collectively result in an error rate of approximately 10-7 per bp per genome. The MMR pathway, a DNA repair pathway conserved from bacteria to humans, targets base substitution mismatches and insertion-deletion mismatches (IDLs) that arise as a result of replication errors that escape the proofreading function of DNA polymerases. In doing so, MMR contributes an additional 50–1000-fold to the overall fidelity of replication. Thus, inactivation of MMR confers a strong mutator phenotype in which the rate of spontaneous mutation is greatly elevated. A hallmark of many MMR-deficient cells is instability at microsatellite regions consisting of mono- and di-nucleotide repeats. Strand slippage during replication through microsatellite regions gives rise to IDLs that are normally repaired by MMR; hence, microsatellite instability (MSI) is widely used as a diagnostic marker for loss of MMR activity in tumour cells (Umar et al., 2004).
1.1. Mutation avoidance and post-replication repair
In Escherichia coli, the methyl-directed MMR system has been extensively studied, and the entire pathway has been reconstituted in vitro from purified proteins in the Modrich laboratory (Table 1, reviewed in Kunkel and Erie, 2005; Schofield and Hsieh, 2003). MMR in prokaryotes is initiated when mismatches are recognized by a highly conserved MMR protein, MutS. MutS and a second conserved protein, MutL, act in concert to license the excision repair pathway by activating endonucleolytic cleavage by a third MMR protein, MutH. MutH directs its nicking activity to the unmethylated strand at transiently hemimethylated CATC sites shortly after replication. This methyl-directed nicking by MutH ensures that MMR in E. coli is directed to the newly synthesized DNA strand containing the error. In vitro studies helped to establish that MMR is bidirectional with respect to the excision step. In other words, MMR can utilize a nick on either side of the mismatch. With the help of MutL, this nick in the nascent strand acts as a point of entry for helicase II that unwinds the nascent strand, a process that is facilitated by single-strand binding protein (SSB) (Matson and Robertson, 2006; Robertson et al., 2006b). This exposes the strand to digestion by one of four single-strand exonucleases having either 3′-5′ or 5′-3′ polarity: ExoI, ExoVII, ExoX, or RecJ (Burdett et al., 2001). The resulting DNA gap is repaired in a reaction involving pol III and ligase thereby restoring the duplex to its intact parental genotype.
Table 1.
Identity and functions of E. coli and eukaryotic proteins involved in MMR of replication errors
| E. coli protein | Function | Homologues | Function |
|---|---|---|---|
| MutS | Binds mismatches | MSH2-MSH6 (MutSα) | Repairs single base-base and 1 –2 base IDL mismatches |
| MSH2-MSH3 (MutSβ) | Repair of some single base IDLs and IDLs ≥2 bases
Partially redundant with Msh2-Msh6 |
||
| MutL | Matchmaker that coordinates multiple steps in MMR | MLH1-PMS2 (yPMS1) (MutLα)
MLH1-MLH2 (hPMS1) (MutLβ) MLH1-MLH3 (MutLγ) |
Matchmaker for coordinating events from mismatch binding by MutS homologues to DNA repair synthesis Endonuclease (PMS2)
Function of human heterodimer unknown Suppresses some IDL mutagenesis in yeast Suppresses some IDL mutagenesis Participates in meiosis |
| MutH | Nicks nascent unmethylated strand at hemimethylated CATC sites | None | |
| γ-δ complex | Loads β-clamp onto DNA | RFC complex | Loads PCNA, modulates excision polarity |
| β-Clamp | Interacts with MutS and may recruit it to mismatches and/or the replication fork | PCNA | Interacts with MutS and MutL homologues |
| Enhances processivity of DNA pol III | Recruits MMR proteins to mismatches Increases MM binding specificity of Msh2–Msh6 Participates in excision and probably in signalling Participates in DNA repair synthesis Participates in DNA re-synthesis | ||
| Helicase II | Loaded onto DNA at nick by MutS and MutL | None | |
| Unwinds DNA to allow excision of ssDNA | |||
| ExoI, ExoX | Perform 3′ –5′ excision of ssDNA | EXOI(Rth1) | Excision of dsDNA |
| RecJ | Perform 5′ –3′ excision of ssDNA (also 3′-5′ excision by ExoVII) | 3′ exo of polδ | Excision of ssDNA |
| ExoVII | 3′ exo of polε | Synergistic mutator with Exo1 mutant | |
| DNA pol III | Accurate re-synthesis of DNA | DNA polδ | Accurate repair synthesis |
| SSB | Participates in excision and DNA synthesis | RPA | Participates in excision and in DNA synthesis |
| DNA ligase | Seals nicks after completion of DNA synthesis | DNA ligase | Seals nicks after completion of DNA synthesis |
Adapted from Kunkel and Erie (2005) with permission.
MMR in eukaryotes follows the broad outline described above for the E. coli methyl-directed MMR pathway (see Fig. 1 and discussion below), and reconstitutions in the Li and Modrich laboratories of MMR reactions from purified proteins possess many of the key features associated with MMR in vivo (Constantin et al., 2005; Dzantiev et al., 2004; Zhang et al., 2005). These studies were predicated on a large body of earlier work that identified individual components from active fractions of cell extracts and characterized partial reactions (reviewed in Jiricny, 2006). Zhang et al. (2005) have demonstrated MMR of a G-T mismatch in 5′-directed repair reactions containing MutSα, MutLα, RPA, EXO1, PCNA, RFC, HMGB1, DNA polymerase δ, and DNA ligase I (Yuan et al., 2004). Substitution of MutSβ for MutSα allowed repair of a 3 nt IDL. MutLα was not required for 5′-directed repair, but did regulate EXO1-catalysed excision. 3′-Directed repair was not supported in this system. Constantin et al. (2005) reconstituted a nick-directed, bidirectional reaction involving seven components: MutSα, MutLα, RPA, EXO1, PCNA, RFC, and DNA polymerase δ. Again, MutLα was not required for 5′-directed repair, but was essential for 3′-directed repair. EXO1 was required for both 3′- and 5′-directed repair.
Fig. 1.
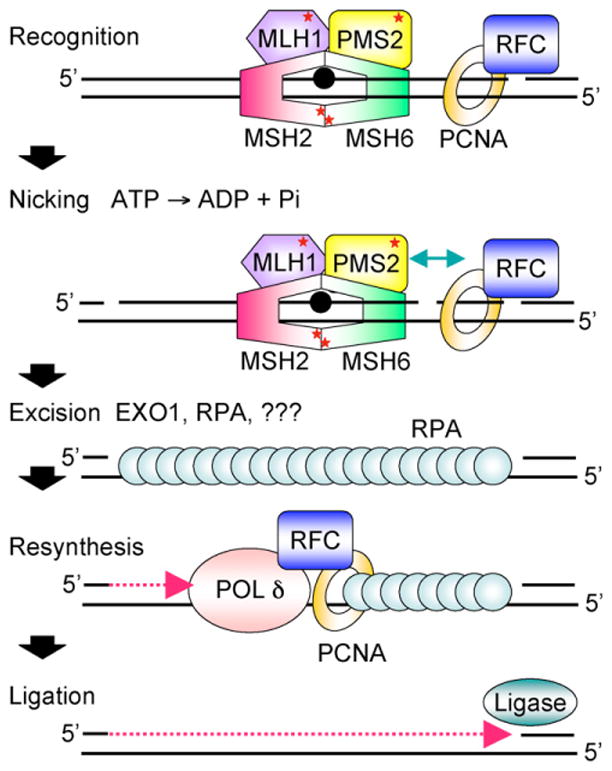
Cartoon scheme for 3′-directed eukaryotic MMR. Recognition of a mismatch by MutSα (MSH2-MSH6) or MutSβ (MSH2-MSH3, not shown) and MutLα (MLH1-PMS2) results in the formation of a ternary complex whose protein-protein and protein-DNA interactions are modulated by ATP/ADP cofactors bound by MutSα and MutLα (indicated by red *). PCNA may play an important role in the recruitment of MMR proteins to the vicinity of the replication fork via a PIP motif on MSH6 and MSH3. Nicking by the endonuclease function of PMS2 stimulated by ATP, PCNA, and RFC and relevant protein-protein interactions (indicated by green arrow) may establish strand discrimination targeting repair to the newly synthesized strand. MMR is bidirectional and can be 5′-directed as well; this is not shown. HMGB1, a nonhistone chromatin protein that bends DNA also facilitates MMR in vitro at or before the excision step (not shown). Excision by EXO1 and possibly other as yet unidentified exonucleases leads to the formation of an RPA-coated single-strand gap. Resynthesis by replicative polδ and ligation restore the integrity of the duplex. See text for details.
Some notable differences between MMR in E. coli and MMR in eukaryotes are readily apparent (reviewed in Modrich, 2006). First, whereas bacterial MutS and MutL proteins function as homodimeric proteins, their eukaryotic counterparts are invariably heterodimers composed of two related, but distinct protein subunits. In fact, eukaryotic cells possess several MutS and MutL homologues, and the choice of subunit partner dictates substrate specificity and cellular function (see Table 1; Kunkel and Erie, 2005). MSH2-MSH6, or MutSα, targets base-base mispairs and +1 IDLs; MSH2-MSH3, or MutSβ, targets primarily IDLs though recent genetic and biochemical data support a role for yMsh3 in the repair of certain base-base mispairs in vivo (Harrington and Kolodner, 2007). Second, although the E. coli methyl-directed MMR system has been completely defined, a minimal human system has only been recently reconstituted with purified proteins (see below), and many aspects of the pathway remain unclear. Third, while E. coli and closely related Gram-negative bacteria can take advantage of dam methylation to direct strand-specific repair, such signals are not available to other prokaryotic or eukaryotic cells.
A number of key issues concerning the molecular mechanism of MMR remain unresolved. Chief among them are: (i) How are MMR proteins recruited to newly replicated DNA most likely in the context of the replication machinery, and how does MutS recognize mismatches? (ii) What is the nature of the MutS (MutSα)-MutL (MutLα)-heteroduplex DNA complex that licenses MMR? (iii) How does MMR couple the mismatch recognition step with a strand-specific excision step? The last is critical as the gapped DNA intermediate formed during MMR, if not repaired, renders the cell vulnerable to replication arrest and the formation of lethal double-strand breaks (DSBs).
1.1.1. Mismatch recognition
E. coli MutS recognizes seven of eight possible base-base mismatches, C:C mismatches being refractory to this repair pathway, and IDLs of up to four bases. Co-crystal structures of MutS proteins containing short, C-terminal truncations from E. coli and T. aquaticus bound to a variety of mispairs and IDLs reveal a homodimeric protein clamped around the DNA at the mismatch (see Fig. 2; Junop et al., 2001; Lamers et al, 2000; Natrajan et al, 2003; Obmolova et al., 2000). The homodimers form a θ where the mismatched DNA resides in the lower channel while the upper channel is unoccupied. Interestingly, in the bacterial MutS proteins, binding of a mismatch induces asymmetry in the two protein subunits such that only one makes direct contact with the mismatch. Recent biochemical and genetic studies reveal that the dimer is the biologically functional unit of E. coli MutS although the protein exists in vitro as a dimer-tetramer equilibrium (Mendillo et al., 2007). In the crystal structures, the DNA is sharply kinked about 60° towards a narrowed major groove at the mismatch. A conserved Phe-X-Glu motif at the N-terminus of bacterial MutS, originally identified in cross-linking studies, and eukaryotic MSH6 constitutes the mismatch binding motif with the Phe residue involved in an aromatic ring stack with the mismatched base displaced 2–3 Å into a widened minor groove (Dufner et al., 2000; Malkov et al., 1997). Mutation of the Phe abrogates mismatch binding in vitro and confers loss of MMR in vivo (Bowers et al., 1999; Das Gupta and Kolodner, 2000; Drotschmann et al., 2001; Yamamoto et al., 2000). The carboxyl group of Glu is hydrogen-bonded to a mismatched base. Mutation of this residue results in more complicated phenotypes, but the Glu residue appears to contribute specificity to mismatch interactions and is involved in nucleotide cofactor-mediated allosteric regulation of MutS proteins (Drotschmann et al., 2001; Lebbink et al., 2006; Schofield et al., 2001 a).
Fig. 2.
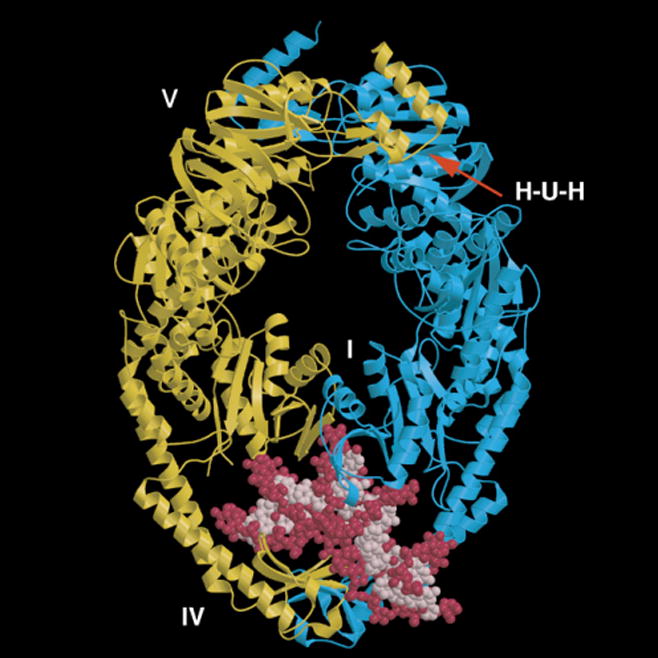
Structural model for T. aquaticus MutS bound to a mismatched DNA. The two protein monomers containing a deletion of the C-terminal 43 amino acids are shown in yellow and blue. The mismatched DNA containing a single unpaired T is shown in pink and red. Domains I and IV constitute the mismatch binding site. Two composite nucleotide binding sites reside in domain V. The H-U-H helix-u-turn-helix motif is essential for subunit dimerization.
Crystal structures of human MutSα bound to several mismatched DNAs have recently been determined (see Fig. 3; Warren et al., 2006, 2007). The structure utilized full-length MSH2 and a truncated MSH6 missing the N-terminal 340 amino acids. The resulting MSH6 truncation is more or less collinear with the bacterial proteins as the N-terminal region that is absent is specific to MSH6. Thus, like the bacterial structures, the MSH2 and truncated MSH6 subunits of the hMutSα-mismatch co-complex each retain the five-domain configuration and, together, form a θ-shaped clamp on the DNA. There is asymmetric utilization of a Phe-X-Glu motif in MSH6 but not MSH2 at the mismatch binding site, DNA kinking at the mismatch, and, at the opposite end of MutSα heterodimer, two composite ABC transporter ATPase sites that, like the bacterial proteins, are connected to the distant mismatch binding site via long α-helical levers. Interestingly, in the hMutSα structure, the upper channel that could, in principle, accommodate a second DNA helix in bacterial MutS, is largely occluded by disordered loops in the case of hMutSα.
Fig. 3.
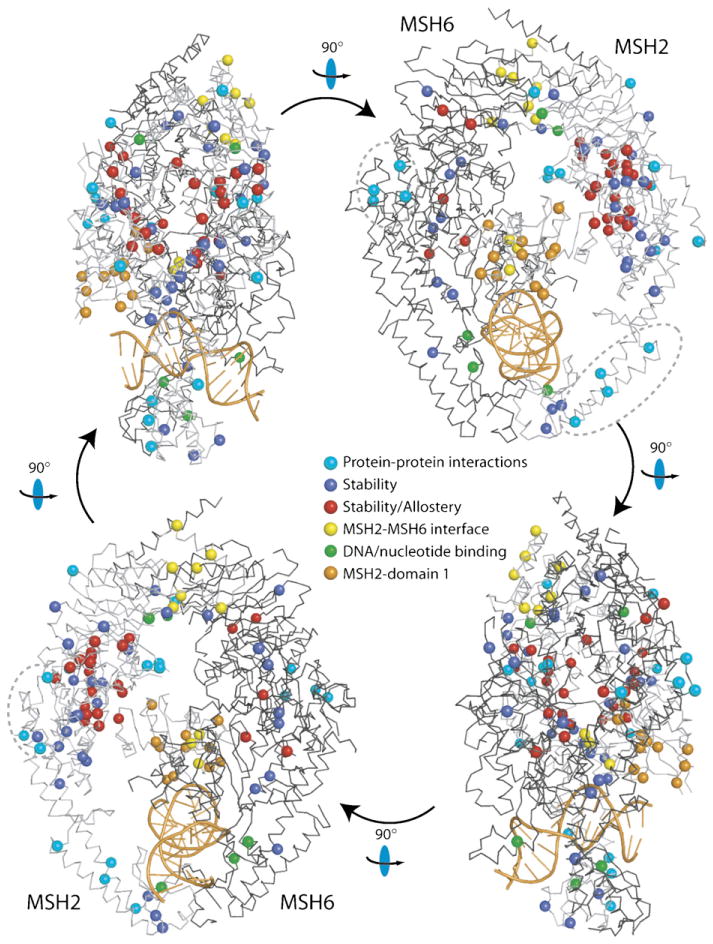
Structural model for human MutSα with HNPCC mutations. Four views of MutSα related by 90° rotations as indicated, with positions of HNPCC missense mutations indicated by spheres. Hypothetical functional classification of mutations is indicated by sphere colour (see legend). MSH2 and MSH6 are shown as light and dark grey Cα chain traces, respectively, and the DNA is coloured orange. Three clusters of surface mutations, which may correspond to sites of protein-protein interactions are indicated with dashed ovals. Reproduced with permission (Warren et al., 2007).
The N-terminal domains of MSH3 and MSH6 proteins present intriguing possibilities with regard to function. PCNA-interacting protein (PIP) boxes are located here (see below). In yMsh6 and human MSH6, the N-terminal region possesses DNA binding activity, a PWWP domain characteristic of proteins that associate with chromatin and a conserved motif at the extreme C-terminus of the domain required for both MMR and DNA damage signalling (Clark et al., 2007). Clearly, these N-terminal domains of MutSα and MutSβ function in multiple contexts.
MutS proteins have ATP binding domains that are essential for MMR (Haber et al., 1988), and are members of the ABC transporter ATPase superfamily (Gorbalenya and Koonin, 1990). Each dimeric MutS protein has two composite nucleotide binding sites each of which are comprised of six conserved motifs include Walker A and Walker B motifs from one protein subunit and an ABC signature motif contributed by the opposing subunit. These ATP binding sites are located at the opposite end of the molecule from the mismatch binding site. The composite nature of the ATP binding sites explains why loss of subunit dimerization results in the simultaneous loss of ATPase activity (Biswas et al., 2001). The importance of these nucleotide binding sites for MMR is reflected in the fact that mutations in these sites frequently turn up as HNPCC alleles and confer strong mutator phenotypes in E. coli and Saccharomyces cerevisiae. In addition, the two nucleotide binding sites are asymmetric with binding constants in the 1 –20 μM range. A number of biochemical studies has established that the two ATP binding sites are nonidentical with MSH6 (and presumably its bacterial counterpart) having a high affinity for ATP and MSH2 having a higher affinity for ADP (Antony and Hingorani, 2003, 2004; Antony et al., 2006; Bjornson and Modrich, 2003; Martik et al., 2004). Seemingly identical mutations in the ATP binding sites of either MSH2 or MSH6 can have different effects on MMR in vivo and DNA binding and ATP hydrolysis in vitro supporting the notion that nucleotide binding/hydrolysis plays a central role in regulating the activities of MutS proteins (Jacobs-Palmer and Hingorani, 2007). For example, dominant mutations in or near the ATP binding site of yMsh2 and yMsh6 exert their effects by either occluding the mismatch binding site thereby inhibiting other mismatch repair pathways from functioning or alter the interaction with MLH1-PMS1 such that MLH1-PMS1 function is compromised (Hess et al, 2006). The nucleotide binding domain and the mismatch binding domain are connected by long α-helices that act as levers in which small movements in the nucleotide binding domain triggered by nucleotide occupancy/hydrolysis are amplified by the levers resulting in larger movements in the mismatch binding domain (Junop et al., 2001; Lamers et al., 2004).
The importance of the nucleotide binding sites of MutS proteins for function is mirrored by the presence of HNPCC alleles in key conserved residues in the nucleotide binding domain of hMSH2 (see Fig. 3). Similarly, cadmium, an ubiquitous environmental carcinogen, targets the ATP binding site of MutS and inhibits MMR in vivo in yeast and in human cell extracts (Jin et al., 2003). Subsequent work has established that cadmium metal inhibits the ATPase activity of both MutSα and MutSβ and disrupts the interaction of MutSα with mismatched DNA (Banerjee and Flores-Rozas, 2005; Clark and Kunkel, 2004). In addition, cadmium alters the MMR-dependent response to DNA alkylating agents suppressing cell cycle arrest in treated cells (Lutzen et al., 2004).
The C-terminus of MutS proteins contains a helix-u-turn-helix motif (see Fig. 2) that is essential for subunit dimerization, mismatch binding, ATP hydrolysis, and in vivo MMR (Biswas et al., 2001). The extreme C-terminus was truncated in the bacterial proteins used in structural studies, and the corresponding E. coli mutSΔ800 mutant in single copy behaves like a mutS null strain for mutation avoidance, anti-recombination and sensitivity to cytotoxic agents in a dam background (Calmann et al., 2005a,b). Full-length MutS proteins exist in a dimer-tetramer equilibrium whereas the Δ800 truncation proteins are largely dimers raising the question of the functional relevance of the tetrameric form and the C-terminal “tetramerization domain”. Biochemical and genetics studies of C-terminal mutants of E. coli MutS, while not unequivocal suggested that the tetrameric form might not be essential for MMR (Manelyte et al., 2006). A confounding factor in the analyses was the fact that C-terminal truncations could not only abolish tetramerization but could also destablize dimers. Structural, biochemical, and genetic characterization of two C-terminal mutants of E. coli MutS, D835R, and R840E, each of which disrupts tetramerization but not dimer formation, revealed that MMR requires stable dimers, but not tetramers of MutS (Mendillo et al., 2007).
Our understanding of mismatch recognition by MutS proteins is incomplete. Since discrimination between mismatched DNA and perfectly paired DNA is not large, most commonly no more than two orders of magnitude (reviewed in Jiricny, 2006; Kunkel and Erie, 2005; Schofield and Hsieh, 2003), the issue of finding a mismatch is not trivial. Total internal reflection fluorescence microscopy of yMSH2-MSH6 reveals that the protein travels along undamaged DNA duplexes via ID diffusion (Gorman et al., 2007). Tethering of MMR proteins to the replication machinery via interactions with the β clamp or PCNA replication processivity factors in E. coli and eukaryotes, respectively, may also help mediate an efficient search for errors in newly replicated DNA (reviewed in Kunkel and Erie, 2005). Despite co-crystal structures of both bacterial and human MutS proteins bound to a variety of mismatched DNAs, we still do not understand how MutS proteins discriminate between mismatches and perfectly paired DNAs. To date, all the solved structures bear a great resemblance to each other with very similar interactions between the Phe-X-Glu motif of only one subunit and the mismatch, DNA kinking, and nonspecific contacts between the protein and sugar-phosphate backbone of flanking DNA regions. This may reflect a universal recognition mechanism, but may also be a consequence of the requirements for crystallization that preclude obtaining high-resolution structures of more dynamic conformations. Recent evidence suggests that the preference for base-base mismatches as opposed to IDLs resides exclusively at the mismatch binding domain of yMsh3 and yMsh6 as a chimeric protein bearing the Msh3 mispair-binding domain grafted in place of the corresponding Msh6 structure targets IDLs like Msh3, but otherwise, behaves like Msh6 (Shell et al., 2007a).
Any mechanism for mismatch recognition by MutS proteins must take into account the inverse correlation between the efficiency of repair for any given mismatch and its propensity to introduce distortion in the DNA helix (G:T mismatches are well repaired and induce relatively little distortion whereas C:C mismatches are poorly or not at all repaired and are the most destabilizing mismatch). Atomic force microscopy studies of MutS-mismatched DNA complexes unexpectedly revealed the existence of a population of “unbent” DNA complexes apparent only when MutS is specifically bound at a mismatch (Wang et al., 2003). These findings and others suggest that simple kinking of the mismatched DNA by MutS is insufficient to confer specificity on the MMR reaction and that the recognition process is likely to involve multiple conformational changes in both protein and mismatched DNA (see discussion in Kunkel and Erie, 2005). Whatever the precise mechanism, alterations in base stacking interactions at DNA lesions are likely to play an important role (Yang, 2006).
Finally, the efficiency of MMR in vivo is not uniform across the genome with some loci being well repaired while others are rather poorly repaired in S. cerevisiae (Hawk et al., 2005). This could reflect local sequence context effects on mismatch recognition as has been described for MutS proteins (see, e.g. Marsischky and Kolodner, 1999) as well as longer range chromatin structure effects on mismatch recognition or other steps of MMR.
1.1.2. MutL—”reach out and touch someone”
The function of MutL has long been enigmatic. It is a weak ATPase and binds nonspecifically to DNA. It has been deemed a “matchmaker” as it interacts with and, in some cases stimulates the activity of, a large number of proteins. In E. coli, this includes MutS, MutH, UvrD helicase, and the β clamp (reviewed in Kunkel and Erie, 2005; Modrich, 2006). Recently, Cannavo et al. (2007) have reported the results of a proteomics approach to defining the interactome of hMLH1, hPMS1, and hPMS2 MutL homologues. In addition to identifying the “usual suspects” such as the MutS and MutL homologues, EXO1, and PCNA, the study points to potential new players in MMR, and implicates MutL homologues in a diverse range of processes including intracellular transport, cell signalling, and cell morphology.
MutLα, MLH1-PMS1 in budding yeast and MLH1-PMS2 in mammals, plays the major role in post-replication repair. Other MutL homologues, MutLβ (MLH1/PMS1) and MutLγ (MLH1/MLH3), are surmised to participate in the repair of some base-base mispairs and IDLs and may be partially redundant with MutLα (see (Cannavo et al., 2005).
Crystal structures of large fragments of MutL and their eukaryotic counterparts reveal a very dynamic molecule composed of two protein subunits. MutL homologues belong to a superfamily of ATPases typified by HSP90, type II DNA topoisomerases, and histidine kinases known as the GHKL superfamily. The structure of a 40 kDa N-terminal ATPase domain of E. coli MutL reveals that binding of AMPPNP, but not ADP, induces ordering of peptide loops and dimerization leading to the formation of two complete ATPase active sites characteristic of the GHKL superfamily of ATPases (reviewed in Kunkel and Erie, 2005; Modrich, 2006). Opening and closing of the N-terminal ATPase domain involving a movable “lid” is regulated by nucleotide binding and hydrolysis with the ATP-bound form being closed and hydrolysis resulting in opening. In contrast to E. coli MutL, the N-terminal fragment of hPMS2 remains a monomer in the crystal structure, even in the presence of ATP, and the monomeric fragment is capable of ATP hydrolysis (Guarne et al., 2001). A crystal structure of a C-terminal dimerization domain of E. coli MutL reveals a V-shaped dimer that plays critical roles in the activation of UvrD by MutL and in DNA binding by MutL (Guarne et al., 2004). The DNA binding activity of MutL has been shown to be essential for methyl-directed MMR in E. coli (Robertson et al., 2006a). The N-terminal ATPase domain and the C-terminal dimerization domain are connected to each other via a flexible peptide linker with the elbow of the dimerization domain at one end and the lid of the ATPase domain at the other.
Like MutS proteins, the MutL proteins possess two asymmetric nucleotide binding sites that regulate many if not all aspects of MutL function and interaction. MLH1 has a high affinity ATP binding site whereas PMS1/PMS2 has a lower affinity site (Hall et al., 2002). Recent atomic force microscopy (AFM) studies of full-length yeast and human MutLα reveal four different conformations: a Y-shaped extended conformation, an asymmetrical one-armed conformation, a semi-condensed state with two equal masses, and a condensed, globular conformation (Sacho et al., 2008). These different conformations reflect large-scale movements of the N-terminal ATPase domains with respect to the C-terminal dimerization domains, and relative occupancies of these four states are modulated by nucleotide cofactor binding. The picture that emerges from a very large body of evidence is that of MutL as a dynamic ATP-activated clamp with a central channel that can capture DNA. Thus, in a very qualitative sense, MutL mirrors the complexities presented by MutS. The task at hand is to understand how nucleotide cofactor-induced conformational changes dictate function for these dynamic MMR proteins particularly in the context of large, multi-protein-DNA complexes.
1.1.3. A ternary complex: MutS-MutL-mismatch
A large number of studies have been directed at understanding how nucleotide binding and hydrolysis modulate the activity of MutS proteins including its interaction with DNA and with MutL (see Kunkel and Erie, 2005). The formation of a ternary complex involving MutS, MutL, and a mismatched DNA has been examined using a variety of techniques, but our understanding of what this complex looks like, how it is modulated by nucleotide occupancy, in fact, how it functions in MMR is woefully primitive despite atomic resolution structures of MutS and MutL proteins and their homologues.
Several confounding factors have made studies of a ternary complex difficult. A complex containing MutS and MutL has a total of four distinct, but interdependent ATP binding sites, and MutS by itself can assume, in principle, nine different nucleotide occupancy states (Sixma, 2001)! Both MutS and MutL bind nonspecifically to DNA, and MutS has an appreciable affinity for DNA ends (Acharya et al., 2003; Plotz et al., 2006, 2002; Wang et al., 2003). While an intact ATPase site in MutLα is not required for its interaction with MutS(α) (Acharya et al., 2003; Selmane et al., 2003), the requirement for ATP binding or hydrolysis by MutS(α) in forming a ternary complex with MutL(α) has been extensively studied, but with no definitive results yet (see Iyer et al., 2006). In addition, DNasel footprinting studies reveal that whereas MutS protects approximately one turn of the DNA helix to either side of the mismatch consistent with the crystal structures, the MutS-MutL interaction in the presence of ATP yields a very large DNasel footprint indicative of multiple proteins bound to the DNA (Grilley et al., 1989; Selmane et al., 2003). Formation of a ternary complex involving either the E. coli or human MMR proteins also requires DNA heteroduplexes in excess of 60 bp reflecting multiple binding events in vitro (Blackwell et al., 2001; Plotz et al., 2002; Schofield et al., 2001b). Whether this is an artefact of in vitro conditions or reflects multiple loadings of MMR proteins, either multiple loading of diffusing clamps or cooperative binding of a protein filament, is not definitively established. Characterization of the MutS-MutL interaction remains an important goal.
Any proposed model must explain how information is propagated from the mismatch site to the excision start site located some distance away, and this has led to two general classes of models in which MutS and MutL and their homologues communicate with downstream players in cis or in trans (see recent discussions in Kolodner et al., 2007; Kunkel and Erie, 2005; Modrich, 2006). In a trans activation model, communication occurs between MutS-MutL complexes at a mismatch and proteins that operate downstream like MutH by protein-protein interactions accompanied by DNA bending. This proposal was based in part on the ability to activate MutH for cleavage “in trans”, i.e. when the mismatch signal and the dGATC hemimethylated site resided on two different DNA duplexes or were separated by a physical barrier such as a four-way DNA junction (Junop et al., 2001; Schofield et al., 2001 b). In addition, E. coli MutS and MutL were observed to form a stable complex at a mismatch in the presence of ATP (Selmane et al., 2003). In these studies, the activation of MutH was measurable, but modest, and nonspecific DNA binding was a potentially confounding issue. Stronger support came from the observation that in vitro MMR reactions using HeLa cell extracts were not substantially inhibited by the presence of a physical barrier such as a DNA hairpin between the mismatch and the initiating nick (Wang and Hays, 2003, 2004).
In a cis activation model, MutS-MutL complexes utilize the DNA helix and travel as a series of sliding clamps or possibly polymerize to form a protein filament until they encounter a signal, i.e. a strand break possibly in the context of other auxiliary proteins such as PCNA and/or RFC in eukaryotic cells at which time they activate the excision step and/or the endonuclease activity of MutLα (see Iyer et al., 2006; Kolodner et al., 2007).
In vitro experiments using mismatched DNA duplexes with blocked ends have firmly established that in the presence of ATP, MutS and its homologues behave as sliding clamps (reviewed in Iyer et al., 2006). While movement of the clamp was originally proposed to occur via an ATP-hydrolysis driven model or a diffusion-driven model, subsequent work supports the latter. ATP-induced dissociation of yMsh2-Msh6 from a mismatch occurs preferentially from DNA ends though direct dissociation from the DNA is also observed (Mazur et al., 2006; Mendillo et al., 2005). Finally, although MutS and MutL (and their eukaryotic homologues) have been observed to form quasi-stable complexes at sites of mismatches in vitro (Blackwell et al., 2001; Räschle et al., 2002; Schofield et al., 2001 b), yeast and human complexes have also been observed to move along the DNA contour in the presence of ATP in surface plasmon resonance experiments (Blackwell et al., 2001; Mendillo et al., 2005).
Recently, Pluciennik and Modrich (2007) have demonstrated that a block to movement along the DNA in the form of a tight-binding EcoRI variant significantly reduced MMR by E. coli MutS and MutL consistent with a “cis” model. However, physical barriers separating a mispair from a 5′ strand break did not seem to inhibit MMR in human cell extracts (Wang and Hays, 2004). These differences may be attributable to multiple pathways or possibly more than one distinct “activation” step (see discussion in Kolodner et al., 2007; Wang and Hays, 2007). A definitive experiment is hard to come by in this system, but the weigh of the evidence thus far argues for some type of cis model in which information is communicated along the DNA helix. The possibility of protein-protein interactions through space has not been ruled out, however. Unravelling the mechanism MMR utilizes for signalling over long distances remains a big challenge.
1.1.4. Excision and gap filling
The excision step of MMR in eukaryotes has been studied in vitro using purified proteins or nuclear extracts (for a comprehensive discussion, see Kunkel and Erie, 2005). Essentially, we know that excision is mismatch-provoked, initiates at a preexisting nick or gap, is capable of bidirectionality, generally proceeds along the shortest path to the mismatch, and terminates about 150 nucleotides beyond the mismatch. In a 5′-directed reconstituted MMR reaction, MutSα activates 5′-3′ excision by EXO1 conferring high processivity on the exonuclease (Genschel and Modrich, 2003). A model for the termination of excision involves the inhibition of EXO1 activity by MutSα and MutLα once the mismatch has been removed and displacement from DNA of EXO1 by RPA. Consistent with these observations, MutSα and MutLα can inhibit EXO1 activity in the absence of a mismatch, but not in its presence (Nielsen et al., 2004).
A long-standing conundrum in the field is how strand specificity of excision repair is assured in eukaryotes and prokaryotes other than E. coli and closely related Gram-negative bacteria since there is no comparable MutH homologues and DNA methylation system to target excision to the nascent strand. A second puzzle is the absence of a requirement for a 3′-5′ exonuclease in reconstituted 3′-nick-directed MMR reactions and a surprising requirement for EXO1, a 5′-3′ exonuclease, in the 3′-directed reaction (Genschel et al., 2002; Dzantiev et al., 2004). How is the excision step being carried out in this case? Based on an examination of repair products in Xenopus egg extracts, Varlet et al. (1996) proposed the existence of a mismatch-activated, strand-specific endonuclease operating in MMR. Recently, a cryptic endonuclease activity in the eukaryotic PMS2 subunit of MutLα has been identified (Kadyrov et al., 2006,2007). A conserved metal-binding motif in PMS2 (or yPMS1) DQHA(X)2-E(X)4E constitutes part of the active site for the endonuclease activity in both PMS2 and MLH3. Not only is the endonuclease function of MutLα required for MMR, it is also required for other functions of MutLα including suppression of homologous recombination and DNA damage signalling (Deschenes et al., 2007; Erdeniz et al., 2007). The requirement for only a single 5′-3′ exonuclease in MMR can now be reconciled with the discovery of the endonuclease activity of MutLα although it is still possible that other exonucleases including those with a 3′-5′ polarity also participate.
A notable feature of the PMS2/PMS1 endonuclease activity is its potentiation in the presence of both RFC and PCNA to function as a strand-specific nicking enzyme in which the break is directed to the strand containing a pre-existing nick. In this way, interactions between MutLα and PCNA loaded on DNA may direct the excision step of MMR to newly synthesized strands containing single strand breaks in the case of leading strands and Okazaki fragments in the case of lagging strands. This provides an explanation for how strand discrimination is achieved in cells lacking a MutH-like system though the exact mechanism remains to be worked out.
Exo1−/− mice have a slightly increased incidence of lymphoma at advanced age and modestly reduced survival which stands in sharp contrast to the corresponding phenotypes of Msh2−/− mice (Wei et al., 2003). EXO1-deficient cells are deficient for MMR activity in vitro and exhibit elevated MSI at some markers. This suggests that there may be redundant exonuclease functions for MMR in eukaryotes as is the case for E. coli. Genetic studies in yeast have implicated exonucleases with a 3′-5′ polarity including DNA polymerase δ and/or DNA polymerase ε (Tran et al., 1999a,b); and the 3′-5′ exonuclease activity of hMRE1 1 may also play a role (Vo et al., 2005). Alternatively, in some cases, the combined activities of MutLα and EXO1 may be sufficient to confer bidirectional MMR in cells lacking the MutH-directed pathway. Kadyrov et al. (2006) has shown that nicking by MutLα precedes excision of the nascent strand by EXO1.
EXO1 may also have multiple roles in MMR as genetic evidence supports a structural or chaperone role for EXO1 in the formation and/or stabilization of MMR protein complexes in addition to its catalytic role as an exonuclease (Amin et al., 2001; Nielsen et al., 2004; Tishkoff et al., 1997; Tran et al., 2007). Recent studies of EXO1 function in telomerase dysfunctional mTerc−/− mice reveal that the exonuclease domain of EXO1 has important roles in inducing DNA damage signalling, cell cycle arrest, and apoptosis in telomere-dysfunctional mice presumably due to the formation of ssDNA (Schaetzlein et al., 2007). EXO1 contributes to the formation of chromosomal fusions leading eventually to chromosome instability and increased cancer risk (see additional discussion below).
A critical player in MMR is the ubiquitous replication processivity factor, PCNA (Tsurimoto, 1999). PCNA, with the help of a clamp loader, RFC, loads onto the 3′-end of Okazaki fragments or the 3′-end of the leading strand. Its three identical protein subunits form a tripartite clamp with two nonequivalent faces, one of which can interact with a bewildering array of protein partners. PCNA not only associates with replicative polymerases, but also functions in processing Okazaki fragments as well as participates in several DNA repair pathways. While a role for PCNA in the gap filling step of MMR catalysed by polδ was expected, early studies revealed a surprising role for PCNA in MMR prior to gap synthesis (Umar et al., 1996). Conserved PIP boxes at the N-termini of both MSH3 and MSH6 (Qxxhxxaa, where x is any residue, h is hydrophobic and a is aromatic) mediate the interaction of MutSα and MutSβ with PCNA; mutation of the PIP site diminishes MMR in vivo (Bowers et al., 2001; Clark et al., 2000; Flores-Rozas et al., 2000; Gu et al., 1998; Johnson et al., 1996; Kleczkowska et al., 2001). The interaction between MMR proteins and clamp proteins is also observed in prokaryotes. Colocalization studies in Bacillus subtilis place MutS and MutL at replication foci (Smith et al., 2001), and E. coli MutS and MutL interact with the β clamp (Lopez de Saro and O′Donnell, 2001; Lopez de Saro et al., 2006).
PCNA by virtue of its multiple interactions with proteins involved in MMR and replication, may act to coordinate activities at sites of newly replicated DNA. Small-angle-X-ray scattering studies of the N-terminal region of yMsh6 by itself or bound to PCNA revealed that it is an unstructured “tether” to PCNA (Shell et al., 2007b). This flexible tether may aid MutSα in its interaction with PCNA by providing some breathing space for what is likely to be a very crowded environment given the number of different partners that interact with PCNA. Aside from its role in DNA resynthesis, biochemical studies suggest that PCNA bound to newly replicated DNA may serve to deliver Msh2-Msh6 to mismatches thereby increasing the efficiency of the mismatch search (Lau and Kolodner, 2003). Interestingly, studies in yeast have demonstrated preferential MMR on the lagging strand compared to the leading strand (Pavlov et al., 2003). One interpretation is that the higher density of PCNA rings on lagging strands facilitates recruitment of MMR proteins. A weak interaction between yeast and human MutLα and PCNA has been reported and mutation of the putative PIP motif in MLH1 confers a mutator phenotype (Lee and Alani, 2006; Dzantiev et al., 2004; Umar et al., 1996). PCNA also interacts with EXO1 and has important functions in the excision step of MMR (Dzantiev et al., 2004; Nielsen et al., 2004). Understanding how the various proteins involved in MMR gain orderly access to newly replicated DNA remains an important goal.
An area that has not received extensive attention is the nuclear import of MMR proteins. Knudsen et al. (2007) have identified a nonpartitite nuclear localization signal (NLS) in hEXO1 418KRPR421 that localizes to a region also required for the interaction of EXO1 with hMLH1. Nuclear import of EXO1 most likely involves the formation of functional complexes with other MMR proteins and utilizes the importin αs 1, 3, and 7. Perhaps not surprisingly, nuclear import of hMLH1 and hPMS2 requires dimerization to form MutLα mediated by the C-terminus of each protein (Wu et al, 2003b). Mutations in putative NLS motifs in hMLH1, K471, and R472, and hPMS2, K577, and R578, resulted in impaired nuclear import though no functional studies of these mutant proteins were carried out (Brieger et al., 2005).
1.2. MMR and DNA damage signalling
The MutS proteins not only recognize mismatches arising from replication errors but also recognize mismatches involving damaged or modified bases that result from exposure of cells to certain DNA damaging agents (reviewed in Jiricny, 2006). These include O6methylguanine (O 6meG) resulting from modification by SN1 alkylators, 8-oxoguanine, halogenated pyrimidines such as 5-fluorouracil (FdU), adducts from environmental carcinogens like benzo[c]phenanthrene dihydrodiol epoxide (Wu et al., 2003a), UV photoproducts (Wang et al., 2006), and cisplatin adducts. In several cases, the loss of MMR activity renders cells less sensitive to cell killing by DNA damaging agents reflecting the role(s) of the MMR system in DNA damage signalling that triggers cell cycle checkpoints, arrest, and apoptosis.
Tolerance to the cytotoxic effects of SN1 alkylators such as N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) was first observed in MMR-deficient strains of E. coli and subsequently shown to occur in MMR-deficient animal cell lines (reviewed in Karran, 2001). Alkylation damage signalling and the induction of apoptosis require the MMR proteins MutSα and MutLα and result in the activation of a large number of downstream targets including p53, Chk1, Chk2, SMC1, and CDC25A. During replication, O 6-meG templates incorporation of C or more commonly, T. These might serve as targets for MMR. In vitro studies reveal O 6-meG mispairs are recognized by MutSα and activate its ATPase activity (Berardini et al., 2000; Duckett et al., 1996).
The cell cycle arrest induced by treatment of cells with low doses of MNNG requires the ataxia telangiectasia and Rad3-related (ATR) kinase, whose downstream target is the checkpoint kinase Chk1 (Jiricny, 2006). Interestingly, cell cycle arrest only ensues in the second cell cycle after exposure to MNNG (Kaina, 2004; Stojic et al., 2004).
So how does alkylation damage kill cells in a MMR-dependent manner? A longstanding explanation is that since O 6-meG resides in the parental strand, it is not excised by the MMR system that targets only the nascent strand. Repeated rounds of excision by MMR or “futile cycles” eventually result in lethal DSBs. In vitro studies of MMR using mammalian proteins are consistent with iterative rounds of excision repair providing support for this scheme (York and Modrich, 2006). In E. coli, homologous recombination is required for resistance to methylating agents that give rise to MMR-dependent DSBs. In dam recB(Ts) ada ogt cells exposed to MNNG, a subset of these DSBs are replication-dependent, and likely to be the primary contributor to cytotoxicity (Nowosielska and Marinus, 2008). Rapidly dividing cells were more sensitive to killing by MNNG than slowly growing cells consistent with a futile cycling model.
A second possibility based in part on studies in budding yeast is that MMR in MNNG-treated cells is thwarted in some way leaving toxic repair intermediates that block replication or yield a DSB in the second round of replication (Cejka et al., 2005). In this scheme, homologous recombination performs a vital rescue function. The Jiricny lab has recently reported that treatment of mammalian cells with MNNG gives rise to MMR-dependent single-stranded gaps in newly replicated DNA (Mojas et al., 2007). These findings lend support for a “processing model” whereby MMR targets O 6-meG mismatches but fails to complete excision repair leaving single-strand gaps that persist unrepaired into the second cell cycle where they cause replication fork collapse. In addition, deletion of Exo1 in the mouse conferred partial resistance of ear fibroblasts to 6-thioguanine (6-TG) (Schaetzlein et al., 2007). 6-TG is a purine analogue that gives rise to cytotoxic S6meG adducts that are recognized by the MMR system (Karran, 2007). EXO1 is therefore required to elicit an apoptotic response to DNA damaging agents consistent with a processing model for ATR activation. Genetic interaction studies in S. cerevisiae point to a structural or scaffolding role for Exo1 in the formation of MMR complexes distinct from its nuclease activity (Amin et al., 2001). It remains to be determined whether this structural function of Exo1 plays any role in damage signalling.
A third possibility is one in which MMR proteins function as direct sensors of DNA damage and recruit either directly or indirectly the ATR-ATRIP complex to sites of damage. In support of this idea, two separation of function alleles in murine Msh2G674A and Msh6T1217D were identified in which MMR function is lost but the apoptotic response to DNA damaging agents like MNNG is intact (Lin et al., 2004; Yang et al., 2004a). The knock-in mice die from cancer and cells are MSI+ and fail to support an in vitro MMR reaction. Nevertheless, MEFs from the knock-in exhibit the same sensitivity to cell killing by MNNG and cisplatin as do MEFs from wild-type mice. These results suggest that MMR and damage signalling involve overlapping but distinct pathways and argue against a futile cycle model. Recently, we demonstrated that ATR-ATRIP preferentially localizes to O 6-meG/T mismatches in vitro, but only when MMR proteins are present (Yoshioka et al., 2006). Similarly, phosphorylation of Chk1 in vitro by ATR is observed only in the presence of O 6-meG/T and also requires MutSα and MutLα. These results support the direct recognition of O 6-meG/T mismatches by MMR and subsequent activation of ATR. However, they do not address to what extent MMR processes O 6-meG/T mismatches leading up to ATR activation.
The question of how ATR is activated in response to DNA damage remains to be determined, e.g. does it involve ssDNA-RPA as has been posited for replication arrest (Zou and Elledge, 2003)? Recent findings alluded to above would seem to support this idea, however, alternative pathways may exist. Mutant human and Xenopus ATRIP proteins lacking an N-terminal region required for interaction with RPA and stable DNA binding nevertheless support ATR-mediated phosphorylation of Chk1 in response to DNA damage (Ball et al., 2005; Kim et al., 2005), and TopBP1 has been postulated to be a direct activator of ATR via specific protein-protein interactions (Kumagai et al., 2006). Understanding why O6-meG poses a block to gap repair in the MMR pathway is an important unanswered question as it does not appear to pose a block to replication per se (Mojas et al., 2007).
MMR has been implicated in cell killing by the fluoropyrimidine antimetabolite 5-fluoro-2′-uridine (FU) though the mechanism of cell killing is unclear. FU is widely used as an adjuvant therapy for advanced colorectal cancer and has pleiotropic effects on cell metabolism. It is incorporated into both RNA and DNA, and FdUMP inhibits thymidylate synthase (TS), the enzyme that utilizes dUMP as substrate in the sole biosynthetic pathway leading to the creation of dTMP. Correspondingly, cells treated with FU exhibit elevated dUTP levels and deoxynucleoside triphosphate precursor pool imbalances.
The MMR system has been implicated in cell killing by fluoropyrimidines (FPs) as MMR-deficient cells exhibit increased survival after treatment with FdU (Meyers et al., 2004). Arrest in G2 is observed within the first cell cycle in MMR-proficient cells, but not in MMR-deficient cells (Carethers et al., 1999; Meyers et al., 2003, 2005). Inhibition of TS without FU treatment elicits a G2 arrest regardless of MMR status suggesting that the contribution of MMR to DNA damage signalling is via incorporation of FPs into DNA as opposed to the inhibition of TS and the resulting imbalance in nucleotide pools. MutSα recognizes FdU mispairs in vitro and is a substrate for in vitro MMR assays (Fischer et al., 2007; York and Modrich, 2006). In the clinic, resistance to FU treatment occurs frequently. Since 15% of colorectal tumours exhibit microsatellite instability (MSI) and are presumed to be MMR-deficient, there is concern that this subset of patients might be resistant to FU. Clinical data to test this possibility are equivocal due to several confounding factors including the fact that MSI+ individuals have a better prognosis than MSI− individuals independent of FU treatment.
The role of MMR proteins in DNA damage signalling presents additional interesting problems related to the convergence of multiple DNA repair pathways on a single class of DNA damage. Since DNA damaging agents like the alkylating agent temozolomide, cisplatin, and fluoropyrimidines are widely used in chemotherapy often times with the development of drug resistance in patients, understanding how these various repair pathways intersect, enhance, or inhibit each other has important practical implications (see Kovtun and McMurray, 2007). For example, Pani et al. (2007) have shown that the MMR pathway contributes to the cytoxicity of cisplatin in human cells; however, other pathways are also critical in mediating the response to cisplatin particularly homologous recombination. In the case of FdU, widely used as an adjuvant therapy in advanced colorectal cancer, MMR-mediated apoptosis (Meyers et al., 2005) may be one of several contributors to the overall cytotoxicity of FdU as base excision repair (BER) enzymes also target FdU (Meyers et al., 2004). The BER enzyme SMUG1 removes FdU from DNA and confers protection from cell killing (An et al., 2007). MED1 (MBD4), a BER N-glycosylase also targets halogenated pyrimidines, and by analogy to MMR, loss of MED1 confers resistance to FdU (Bellacosa et al., 1999). Interestingly, MED1 was isolated in a two-hybrid screen using MLH1 as bait (Cortellino et al., 2003). Finally, in addition to SMUG1 and MED1, two other BER enzymes have been implicated in the processing of FdU, thymine-DNA glycosylase (TDG) and uracil-DNA glycosylase (UNG) (Fischer et al., 2007). Clearly the interactions between MMR and BER pathways are critical in modulating the DNA damage response (Kovtun and McMurray, 2007).
Cross-talk between MMR and NER, the predominant pathway for the repair of UV damage, may also operate. Msh2−/− mice exhibit an increased predisposition to skin cancer in response to UVB exposure that is enhanced when mice are additionally defective for the NER Xpc gene (Meira et al., 2002). Interactions between scMsh2 and the NER proteins Rad2, Rad10, and Rad14 have been observed in co-immunoprecipitation experiments (Bertrand et al., 1998), and loss of MMR in human cells leads to a deficiency in transcription-coupled NER (Mellon et al., 1996). These and other studies point to a still poorly defined interaction between elements of these two excision repair pathways.
Wang and colleagues have described a p73-dependent, p53-independent apoptotic pathway utilized in an MLH1-dependent damage response to cisplatin (Gong et al., 1999; Shimodaira et al., 2003). Exposure of cells to cisplatin results in an increase in p73 levels that requires both MLH1 and PMS2 and induces a physical interaction between PMS2 and p73 that appears to stabilize the latter.
In conclusion, MMR functions as a damage sensor recognizing a variety of structurally diverse DNA lesions and triggering DNA damage signalling involving multiple cellular signalling pathways. A number of interactions between MMR proteins and DNA damage signalling proteins have been reported including interactions between MSH2 and ATR, Chk1 and Chk2 (Wang and Qin, 2003; Yoshioka et al., 2006); MLH1 and ATM (Adamson et al., 2005; Brown et al., 2003); PMS2 and p73 (Shimodaira et al., 2003); the three MutL homologues, MLH1, PMS2, and PMS1 with a large number of proteins involved in cell cycle/signalling/apoptosis functions (Cannavo et al., 2007). The challenge is to determine the functional significance of these interactions and the biological context in which they operate.
1.3. Other functions of MMR
MMR proteins participate in a number of other cellular processes including recombination processes that have been most extensively studied in fungi. Mismatches are present in heteroduplex DNA formed during homologous recombination between closely related but not identical sequences derived from the genetic exchange between two parental chromosomes. The repair of these mismatches can, depending on which DNA strand is targeted, result in restoration of the parental genotypes (so-called 2:2 Mendelian ratio of alleles), or can yield a non-Mendelian segregation (6:2 or 2:6 segregation) commonly known as a gene conversion event. If the mismatch is not repaired, an aberrant 5:3 segregation of alleles known as post-meiotic segregation (PMS) occurs, so called because the two strands of the heteroduplex separate in the first mitotic division after meiosis. In MMR-deficient cells, PMS is greatly elevated at the expense of gene conversion events. MMR proteins also influence homologous recombination events in mammalian cells; for example, MutSα has been observed to reduce recombination at DSBs possibly via an interaction with the BLM helicase so as to suppress inappropriate recombination (Smith et al., 2007; Yang et al., 2004b).
A second function of MMR in recombination is its “antirecombination” role in which recombination between related but distinct sequences (known as homologous recombination) is inhibited by the MMR system. In prokaryotes, the antirecombination function of MMR is thought to be a critical barrier to the creation of new species by blocking the transfer of large amounts of genetic material between two divergent cells. Although, the molecular mechanism by which MMR proteins inhibit homologous recombination is unclear, recent studies in S. cerevisiae examining the Msh6-PCNA interaction reinforce an earlier proposal that MMR-mediated antirecombination may involve two steps, an early fidelity check perhaps involving the RecQ. helicase Sgs1 that is largely independent of Msh6-PCNA interactions and a more canonical spellchecker step operating on recombination intermediates containing newly synthesized DNA (see, Stone et al. (2008) and references cited therein). A practical consequence of MMR-mediated antirecombination is the now widespread use of isogenic targeting constructs in the production of gene-targeted mice (te Riele et al., 1992). By reducing the density of potential mismatches formed between the targeting vector and the endogenous chromosomal locus, the efficiency of targeting via a homologous recombination mechanism is greatly enhanced. In a related scenario, unconstrained recombination between repeated sequences that share homology would give rise to potentially deleterious chromosomal rearrangements. That these are relatively rare is, in part, attributable to the antirecombination activity of MMR. For example, loss of MMR in human cells increases the rate of gene duplication 50–100-fold and may predispose these cells to cancer (Chen et al., 2001).
A third function of MMR in recombination involves two MutS homologues, MSH2 and MSH3. MSH2-MSH3 acts in concert with Rad 1 –Rad 10 endonuclease to trim nonhomologous DNA ends from a recombination pairing intermediate formed by annealing at a DSB between directly repeated sequences (reviewed in Schofield and Hsieh, 2003).
MMR proteins also figure prominently in meiosis where they play essential roles in mismatch correction of heteroduplex DNA intermediates of meiotic recombination and the formation of crossovers required for the proper pairing or synapsis of homologous chromosomes and their subsequent separation in the first reductional division (reviewed in Cohen et al., 2006; Harfe and Jinks Robertson, 2000; Hoffmann and Borts, 2004). In fungi and in mice, the loss of MMR proteins that function in meiosis results in arrested or aberrant meiosis and, in many cases, sterility. MSH4 and MSH5 function as a heterodimer that binds Holliday junctions in vitro though not in a mismatch-specific fashion as these meiosis-specific MutS homologues are missing the N-terminal mismatch binding motif (Snowden et al., 2004,2008). MLH1–MLH3, MutLγ, is required for stable crossovers and chiasmata formation and for normal meiotic progression (see Kan et al., 2008). Despite their central importance in meiosis, much remains to be learned concerning their function and mode of action.
MSH2–MSH3 also plays an important role in trinucleotide repeat expansion, the process that underlies several hereditary and progressive neurodegenerative diseases such as Huntington disease, myotonic dystrophy, and fragile-X syndrome (Cummings and Zoghbi, 2000). In the mouse, somatic and germline expansions of the CAG-CTG repeat within the Huntington’s gene requires MSH2 (Kovtun and McMurray, 2001; Manley et al., 1999; Wheeler et al., 2003). Repeat instability is reduced in MSH3-null mice but elevated in MSH6-null mice pointing to a role for MSH3 in promoting repeat instability (van den Broek et al., 2002). MSH2–MSH3 has been shown in vitro to target CAG-hairpin DNA and to be modulated by an A-A mismatch in the hairpin stem (Owen et al., 2005).
MMR is involved in the generation of immunoglobulin diversity. Antibody diversity requires V(D)J recombination, class isotype switching, and somatic hypermutation in regions encoding the variable regions of immunoglobulin genes in B lymphocytes. Inactivation of MSH2, MSH6, PMS2, MLH1, or EXO1 reduces the recovery of somatic mutations that occur specifically at A-T base pairs at immunoglobulin loci. A two-stage model has been invoked in which deamination of cytosines at G-C base pairs by activation-induced cystidine deaminse (AID) leads to G-U mispairs (reviewed in Di Noia and Neuberger, 2007). These are targets for base excision repair (BER). However, they are also potential targets for the MSH2–MSH6 MMR protein, and errors at A-T base pairs in the ensuing excision and resynthesis steps of MMR result in the observed mutation spectrum. Inactivation of polη also reduces mutations at A-T base pairs implicating it in somatic hypermutation. Interestingly, MSH2–MSH6 stimulates the catalytic activity of polη in vitro and associates with polriη in cell extracts (Wilson et al., 2005). Unexpectedly, Mlh3−/− mice exhibited an increased frequency of mutations in Ig variable regions suggesting that MLH3 normally inhibits the accumulation of mutations in this region (Li et al., 2006).
1.4. MMR defects and human cancer
Each year approximately 150,000 people in the United States and half a million worldwide are diagnosed with colon cancer. Of these, approximately 3–4% occur in familial cancer syndromes of which Hereditary Nonpolyposis Colorectal Cancer (HNPCC) or Lynch Syndrome is the most common (Lynch et al., 1985). HNPCC is characterized by an increased risk of colorectal cancer and other cancers. It is a common, autosomal dominant syndrome characterized by early onset (average age <45 years), and the occurrence of neoplastic lesions in a variety of tissues including endometrial, skin, ovarian, gastric, and renal. In the HNPCC population (up to 70 years of age), the cancer risks are 80% in colon, 20–60% in endometrium, 11–19% in stomach, and 9–11% in ovary, while, in the general population, the risks are 5.5% in colon, 2.7% in endometrium, <1% in stomach, and 1.6% in ovary (Kohlmann and Gruber, 2006; Watson et al., 2001). The diagnosis of HNPCC can be determined using the Amsterdam Criteria I, II (Vasen et al., 1999) and then by molecular genetic testing for germline mutations in mismatch repair genes. Bethesda guidelines can be used for identification of those patients who should undergo more detailed laboratory investigation (Silva et al., 2005).
A hallmark of HNPCC tumour cells is MSI. Microsatellites are repetitive DNA sequence of 1–4 base nucleotides that are particularly susceptible to DNA replication errors when the MMR system is absent. Five markers (BAT26, BAT25, D5S346, D2S123, and D17S250) are recommended by the National Cancer Institute to assess microsatellite instability. A tumour is classified as MSI-high if two or more of the five markers show instability, as MSI-low if 1 of the markers shows instability, or as MS-stable if no marker does (Boland et al., 1998). In HNPCC tumours, more than 90% exhibit MSI (Soreide, 2007). Early attempts at identifying the genetic basis for HNPCC revealed frequent insertions and deletions at di- and trinucleotide repeats or microsatellite regions at a putative HNPCC locus on human chromosome 2 as well as throughout the genome (see Li, 2003). These mutations were called replication error positive (RER+).
Work on DNA repair in bacteria and fungi had already revealed that loss of MMR conferred a mutator phenotype in which base substitutions as well as frameshift mutations were greatly elevated. Petes and colleagues tested the stability of poly(dGT) tracts in S. cerevisiae cells harbouring single and double mutations in MMR genes MSH2, MLH1, and PMS1 (Strand et al., 1993). Loss of any one MMR gene was sufficient to elevate the frequency of tract instability two orders of magnitude.
Using degenerate PCR primers homologous to highly conserved regions of bacterial mutS and fungal MSH2 genes, two groups independently cloned the human MSH2 gene and located it on chromosome 2 (Fishel et al., 1993; Leach et al., 1993). Germline mutations in the MSH2 gene were identified in HNPCC families. In another approach, tumour cells from HNPCC and sporadic tumours with MSI were tested in in vitro MMR assays and found to be deficient (Parsons et al., 1993; Umar et al., 1994). In 1994, the human MLH1 gene was identified by a search of expressed sequence tags or a degenerate PCR method. The human MLH1 gene resides on chromosome 3p21, close to markers previously linked to cancer susceptibility in HNPCC kindreds. Mutations of MLH1 were identified in such kindreds (Bronner et al., 1994; Papadopoulos et al., 1994). To date, the bulk of germline HNPCC mutations, roughly 90%, reside in two MMR genes, MSH2 and MLH1, with mutations in MSH6 (7–10%) associated with atypical HNPCC and PMS2 mutations being quite rare (Peltomaki, 2001).
Biochemical studies led to the restoration of MMR in nuclear extracts derived from colorectal tumour cell lines by the addition of purified hMLH1/hPMS2 or hMSH2/hMSH6 (Drummond et al., 1995; Li and Modrich, 1995). In another line of experimentation, chromosome or gene transfer experiments in MMR-deficient tumour cells demonstrated that restoration of MMR also rescued the MSI phenotype. Thus, Boland and colleagues restored MMR to an hMLH1 -deficient colorectal tumour cell line by the transfer of chromosome 3 harbouring a wild type copy of hMLH1 (Koi et al., 1994). Similar experiments were carried out for tumour cell lines deficient in MSH6, PMS2, and MLH1 (reviewed in Li, 2003).
The defects by HNPCC mutations (http://www.insight-group.org/) in MutSα can hypothetically fall into at least six classes: interference with DNA binding, loss of ATPase activity, loss of allosteric communication between DNA and ATP binding sites, loss of protein-protein interactions with downstream effectors, loss of MSH2–MSH6 interaction, and general loss of protein stability. HNPCC mutations have been mapped onto the three-dimensional structure of hMutSα (Fig. 3; Warren et al., 2007). Clearly, mutations are located throughout the protein structure; the challenge is to identify informative mutations that can shed light on molecular mechanism.
Approximately, 15% of spontaneous colorectal cancers are MSI+; the vast majority of these are caused by epigenetic silencing of MLH1 (Jenkins et al., 2007; Samowitz et al., 2001a). That hypermethylation is likely a common mechanism for MMR inactivation in tumour cells was demonstrated in several sporadic colon tumours and cell lines that are free of any mutations in MLH1 (Kane et al., 1997).
Microsatellite instability (MSI) and chromosomal instability (CIN) are two major pathways in the formation of colorectal cancers (Soreide, 2007). tumours with CIN have mutations in p53 and APC, including chromosomal abnormalities. In contrast, tumours with MSI have frameshift mutations in specific target genes such as β-catenin and TGFβRII genes (Kim et al., 2003), and fewer mutations are found in K-Ras and p53 (Samowitz et al., 2001 b). Actually, frameshift mutations of the microsatellite repeats in the TGFβRII coding region were found in ~90% of HNPCC tumours (Markowitz et al., 1995). It is important that MSI can mutate specific genes, probably forming multi-step carcinogenesis. Additionally, it is also possible that MSI and CIN pathways interact with each other. Recently, BRCA1 is shown to regulate an MMR-induced G2/M checkpoint, which is critical for inhibition of CIN (Yamane et al., 2007). The observation of alterations of key growth regulatory genes in MMR-deficient cells such as NF1, APC, p53, K-Ras may suggest that even in the presence of MSI, tumour progression is mainly driven by a process of natural selection (Bertholon et al., 2006). MSI+ colorectal carcinoma has been associated with a more favourable clinical outcome (Chung and Rustgi, 2003) that has confounded clinical studies assessing drug efficacy among colorectal cancer patients. Compared with MMR-proficient tumours, MMR-deficient tumours (MSI+) tend to be proximal to splenic flexure, poorly differentiated, mucinous, characterized by marked lymphocyte infiltration, and frequently large in size (Wheeler et al., 2000). MSI+ was also strongly associated with a decreased likelihood of lymph node and distant organ metastases (Malesci et al., 2007).
1.5. Animal models of MMR deficiency
Animal models for MMR deficiency in which each MMR gene has been knocked out confirm that loss of MMR confers a mutator phenotype (MSI+), increased incidence of cancer and decreased lifespan (see Table 2; reviewed in Edelmann and Edelmann, 2004). Surprisingly, however, these mice do not develop colorectal cancer, but have an array of other tumours, particularly lymphoma. Gene disruption experiments indicate that some MMR proteins including MSH2 appear to be tumour suppressors though their role is in mutation avoidance. While mice-deficient in MSH2 appear normal at birth and are fertile, they develop T cell lymphoma, gastrointestinal, skin, or other tumours (de Wind et al., 1995, 1998; Reitmair et al., 1995). Mice lacking MSH2 show an inability of G-T mismatch binding in cell lysates, a mutator phenotype, and resistance to a mismatch-inducing reagent in isolated cells. Half of the MSH2−/− mice die by 6 months of age, and all of them succumb by 12 months.
Table 2.
Mouse lines with mutations in MMR genes
| Genotype | Tumour
|
MSI incidencea |
Fertility Male/female | Reference | ||
|---|---|---|---|---|---|---|
| Spectrumb | Incidence | Mononucleotide | Dinucleotide | |||
| MutS homologues | ||||||
| MSH2−/− | Lymphoma, GI, skin and other tumours | High | High | High | +/+ | de Wind et al. (1995), Reitmair et al. (1995, 1997), Smits et al. (2000) |
| MSH2 C674A/C674A | Lymphoma, GI, skin | High | High | High | +/+ | Lin et al. (2004) |
| MSH3−/− | GI tumours | Low | Moderate | High | +/+ | Edelmann et al. (2000), de Wind et al. (1999) |
| MSH6−/− | Lymphoma, GI, skin and | High | None | Low | +/+ | Edelmann et al. (1997) |
| MSH6 TI217D/T1217D | Lymphoma, GI, skin | High | High | High | +/+ | Yang et al. (2004a,b) |
| MSH3−/− MSH6−/− | Lymphoma, GI, skin and other tumours | High | High | High | +/+ | Edelmann et al. (2000), de Wind et al. (1999) |
| MSH4−/− | None | None | N/A | N/A | −/− | Kneitz et al. (2000) |
| MSH5−/− | None | None | N/A | N/A | −/− | Edelmann et al. (1999a), de Vries et al. (1999) |
| MutL homologues | ||||||
| MLH1−/− | Lymphoma, GI, skin and other tumours | High | High | High | −/− | Baker et al. (1996), Edelmann et al. (1996), Prolla et al. (1998), Edelmann et al. (1999b) |
| PMS1−/− | None | None | Low | Low | +/+ | Prolla et al. (1998) |
| PMS2−/− | Lymphoma and sarcoma | High | High | High | −/+ | Baker et al. (1995), Prolla et al. (1998) |
| MLH3−/− | N/A | N/A | Low | Low | −/−; | Lipkin et al. (2002) |
| Exonuclease | ||||||
| EXO1−/− | Lymphoma | Moderate | High | Low | −/− | Wei et al. (2003) |
GI tumours: Adenoma, adenocarcinoma, and flat adenoma in gastrointestinal organs. Skin tumours: Squamous cell carcinoma, keratoacanthoma, sebaceous (adenoma, epithelioma, carcinoma), papilloma, haemangioma, and pylomatricoma. Other tumours include: Tumours of the uterus, brain, lung, liver, and mammary gland. Sarcoma: Leiomyosarcoma, myxoid sarcoma, and fibroid sarcoma. Adapted from Edelmann and Edelmann (2004) and used with permission.
MSI was analysed in tumour samples, normal tissue or culture cells.
Tumour spectrum includes: Lymphoma: B and T cell lymphoma (NHL, non-Hodgkin’s lymphoma).
MSH3−/− mice do not have a significant phenotype, although a small subset develop gastrointestinal tumours (de Wind et al., 1999; Edelmann et al, 2000). This correlates with the fact that MSH3 mutations are very rare in human HNPCC, and to the fact that the MSH2–MSH6 complex is partially redundant for repair of a single base insertion. MSH6−/− mice have reduced survival (50% survival at 11 months) with a tumour spectrum similar to that of MSH2−/−. In agreement with their substrate specificity (primarily base-base mispairs and +1 IDLs), mutations in human MSH6 are associated with MSI-low tumours. The cancers in families with MSH6 mutations are late onset, and endometrial cancer is commonly associated with these mutations (Berends et al., 2002; Wu et al., 1999).
Slightly lower risks for colon cancer and higher risks for endometrial cancer have been reported in families with MSH6 alterations. It should be noted that double-mutant MSH3−/− MSH6−/− mice have phenotypes very similar to those of MSH2−/− (de Wind et al., 1999; Edelmann et al., 2000). This reflects the fact that MSH2 is a common subunit for both MutSα and MutSβ, the two MutS homologues that carry out MMR on base-base and insertion-deletion mismatches, respectively. An MSH6 missense mutation (Thr 1217 to Asp) in mice causes loss of MMR function, and shows increased mutation rates and cancer susceptibility, while having no effect on the apoptotic response to DNA damaging agents (Yang et al., 2004a).
MLH1−/− mice (Baker et al., 1996; Edelmann et al., 1996) have similar lifespans to MSH2−/− mice (50% survival at 6 months; all succumb by 12 months) and a similar tumour spectrum including T cell lymphoma, gastrointestinal or skin tumours. MLH1−/− fibroblaste also are MSI-high and deficient in MMR. In contrast to the MSH2−/− mice, MLH1−/− males and females are sterile (Edelmann et al., 1996; Woods et al., 1999). Germ line cells in MLH1−/− male mice enter meiotic prophase, arrest at pachytene phase, and undergo apoptosis. PMS2−/− mice develop lymphomas and sarcomas but not gastrointestinal tumours, and display 50% survival at 9–10 month ages. PMS2−/− males are sterile possibly due to abnormal chromosomal pairing in pachytene phase while PMS2−/− females are fertile.
Gene disruption of EXO1 in mice results in MMR defects, increased cancer susceptibility, and male and female sterility (Wei et al., 2003). The repair defect in EXO1−/−cells also caused elevated microsatellite instability at a mononucleotide repeat marker and a significant increase in mutation rate. EXO1−/− mice displayed reduced survival, increased susceptibility to lymphomas and meiotic defects.
The mouse models have confirmed that defects in MMR are the underlying cause of HNPCC and give rise to a mutator phenotype, cancer predisposition, and in some cases, sterility reflecting the role of some MMR proteins, like MLH1, in meiosis. They also support the idea articulated by Loeb (2001) that cancers exhibit a mutator phenotype early in their evolution (see discussion in Yang et al., 2004a). The mouse studies have yielded some unexpected differences from HNPCC. HNPCC patients are heterozygous for the germline mutation whereas mice heterozygous for the null allele are healthy. In addition, the tumour spectrum of the knock-out mice differs from their human counterparts.
The link between MMR-deficiency, a mutator phenotype, and the development of cancer has been firmly established. Work proceeds now to understand how a mutator phenotype gives rise to cancer, to mine mechanistic information from HNPCC alleles, and to develop better diagnostic tools in the clinic and more effective treatments. With respect to clinical outcomes, continuous treatment with nitric-oxide-donating aspirin derivatives suppresses MSI in MMR-deficient and HNPCC cancer cells (Mcllhatton et al., 2007). These compounds appear to enhance acquisition of a microsatellite-stable phenotype by an as yet undefined genetic selection process and may offer promise in the future as a chemopreventive for HNPCC carriesrs.
1.6. MMR and ageing
Since the accumulation of DNA damage and resulting genomic instability is thought to be an important initiating event in ageing as in cancer, which is itself an age-associated human disease, much attention has been directed at elucidating the role of DNA repair DNA damage signalling pathways in the ageing process (see this issue). Several mechanisms of age-dependent genome instability have been proposed including the accumulation of oxidative DNA damage, mitochondrial dysfunction that contributes to oxidative stress, and defects in genome maintenance pathways including DNA repair, replication, checkpoint signalling, and telomere maintenance. Defects in several genes encoding proteins involved in DNA repair or damage signalling, e.g. members of the RecQ. helicase family, give rise to human segmental progeroid syndromes characterized by short telomeres and premature ageing or early onset of age-related disease such as cancer (reviewed in Blasco, 2007; Brosh and Bohr, 2007; Hanada and Hickson, 2007).
The role of the MMR pathway in ageing is less clearly discernible. HNPCC patients do not possess characteristics of premature ageing, and MEFs from PMS2−/− mice have normal telomeres (Siegl-Cachedenier et al., 2007b). Attempts to monitor the effects of caloric restriction, known to prolong lifespan in rodents, on cancer incidence and survival in MMR-deficient Mlh1−/− mice reveal only modest effects (Tsao et al., 2002). However, as the authors note, the marked inherent carcinogenic potential of the mutator phenotype in these MMR-deficient mice may mask effects of caloric restriction. Several studies suggest that the capacity for MMR as judged by MSI or mismatch levels is diminished as a function of ageing possibly due to the loss of key MMR proteins (see, e.g. Annett et al., 2005; Ben Yehuda et al., 2000; Krichevsky et al., 2004; Neri et al., 2005). Although we do not know whether correlations between ageing and loss of MMR reflect a direct role for MMR in preventing ageing or are indirect consequences of the ageing process, these studies raise intriguing possibilities. Finally, oxidative damage of DNA has been correlated with the ageing process. MutSα can target 8-oxo-guanine mispairs for repair (Mazurek et al., 2002; Ni et al., 1999), and treatment of HEL cells with H2O2 is associated with a reduction in MMR and with decreased levels of some MMR proteins (see, e.g. Chang et al., 2002). Although BER remains the primary repair pathway for oxidative DNA damage (Wilson and Bohr, 2007), the possible link between MMR and oxidative damage and ageing remains a fertile area of investigation.
A landmark finding is the observation in budding yeast that loss of MMR function promotes cell proliferation in cells missing telomerase (Rizki and Lundblad, 2001). In S. cerevisiae, an est-2Δ strain missing the catalytic subunit of telomerase exhibits healthy growth initially, but subsequently undergoes a rapid decline in colony formation. The est-2Δ msh2Δ strain, on the other hand, exhibits improved growth and increased cell proliferation. Mutations in MLH1 or PMS1 confer a similar improvement in growth. The enhanced growth phenotype is dependent on RAD52 implicating homologous recombination. In the absence of telomerase, telomeres can undergo recombination-based alternative elongation of telomeres, or ALT mechanisms (Blasco, 2005). Given the antirecombination function of MMR, the logical conclusion is that the loss of MMR is rescuing telomere function by allowing homologous recombination between sister telomeres. In support of this notion, HCT 15 human colon cancer cells that are MSHG −/− and expressing a dominant-negative hTERT exhibit telomere lengthening via an ALT-like mechanism (Bechter et al., 2004).
Recently, Siegl-Cachedenier et al. (2007a) have reported that loss of MMR prolongs the mean lifespan and median survival of Terc−/− mice. Interestingly, the Terc−/− PMS2−/− mouse exhibit a progressive reduction in the number of tumours normally seen in PMS2−/−mice consistent with a tumour suppressor function for short telomeres in the absence of MMR. Further analyses reveal that the increased survival conferred by loss of MMR function in telomerase-deficient mice or cells derived from these mice is not due to an amelioration of telomerase dysfunction or apoptosis; nor is it attributable to increased recombination between sister telomeres. Instead, the loss of MMR stimulates the proliferative capacity of cells through an attenuation of p21, a cyclin-dependent kinase inhibitor that is a downstream effector of p53-mediated G1/S checkpoints; p21 also interacts with PCNA causing inactivation of PCNA-mediated DNA replication (reviewed in Ju et al, 2007). In fact, the Terc−/− PMS2−/− mouse phenocopies in several important aspects a Terc−/− p21−/− mouse in which loss of p21 confers improves cell proliferation and organ maintenance in telomere-dysfunctional mice (Choudhury et al., 2007). These findings point to a surprising new role for PMS2 in signalling cell cycle arrest in response to telomere dysfunction. Loss of EXO1, the exonuclease that functions in MMR, rescues organ maintenance and lifespan in telomerase-dysfunctional Terc−/− mice (Schaetzlein et al., 2007). Whether EXO1 and PMS2 function under conditions of telomere shortening in the same p21 repression pathway remains to be determined as does the molecular pathway by which PMS2 regulates cell proliferation via p21.
2. Summary
Studies of MMR continue to yield unexpected findings and intriguing connections to many essential biological processes. Although the broad outlines of MMR function have been identified, issues regarding DNA target accessibility, spatial and temporal regulation of MMR protein activity, and interactions with proteins that clearly operate in pathways quite distinct from DNA repair remain ill defined. Given the seemingly ubiquitous presence of MMR proteins in a diverse array of important cellular functions, we can be confident that many important and challenging problems remain to be solved.
Acknowledgments
We apologize to our many colleagues whose work we only cite indirectly. We thank Tom Kunkel and Dorothy Erie for permission to reproduce Table 1, Lorena Beese for Fig. 3, Winfried Edelmann for Table 2. We thank Galina Obmolova for Fig. 2. This work was supported by the Intramural Research Program of NIDDK.
References
- Acharya S, Foster PL, Brooks PJ, Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Molecular Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- Adamson AW, Beardsley DI, Kim WJ, Gao Y, Baskaran R, Brown KD. Methylator-induced, mismatch repair-dependent G2 arrest is activated through Chk1 and Chk2. Molecular Biology of the Cell. 2005;16:1513–1526. doi: 10.1091/mbc.E04-02-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin NS, Nguyen MN, Oh S, Kolodner RD. exo1-dependent mutator mutations: a model system for studying functional interactions in mismatch repair. Molecular and Cellular Biology. 2001;21:5142–5155. doi: 10.1128/MCB.21.15.5142-5155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Q, Robins P, Lindahl T, Barnes DE. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Research. 2007;67:940–945. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- Annett K, Duggan O, Freeburn R, Hyland P, Pawelec G, Barnett Y. An investigation of DNA mismatch repair capacity under normal culture conditions and under conditions of supra-physiological challenge in human CD4+T cell clones from donors of different ages. Experimental Gerontology. 2005;40:976–981. doi: 10.1016/j.exger.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Antony E, Hingorani MM. Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry. 2003;42:7682–7693. doi: 10.1021/bi034602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony E, Hingorani MM. Asymmetric ATP binding and hydrolysis activity of the Thermus aquaticus MutS dimer is key to modulation of its interactions with mismatched DNA. Biochemistry. 2004;43:13115–13128. doi: 10.1021/bi049010t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony E, Khubchandani S, Chen S, Hingorani MM. Contribution of Msh2 and Msh6 subunits to the asymmetric ATPase and DNA mismatch binding activities of Saccharomyces cerevisiae Msh2-Msh6 mismatch repair protein. DNA Repair. 2006;5:153–162. doi: 10.1016/j.dnarep.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, Elliott EA, Yu J, Ashley T, Arnheim N, Flavell RA, Liskay RM. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A, Ashley T, Liskay RM. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nature Genetics. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Ball HL, Myers JS, Cortez D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATR1P localization but is dispensable for Chk1 phosphorylation. Molecular Biology of the Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Flores-Rozas H. Cadmium inhibits mismatch repair by blocking the ATPase activity of the MSH2-MSH6 complex. Nucleic Acids Research. 2005;33:1410–1419. doi: 10.1093/nar/gki291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechter OE, Zou Y, Walker W, Wright WE, Shay JW. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Research. 2004;64:3444–3451. doi: 10.1158/0008-5472.CAN-04-0323. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3969–3974. doi: 10.1073/pnas.96.7.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Yehuda A, Globerson A, Krichevsky S, Bar On H, Kidron M, Friedlander Y, Friedman G, Ben Yehuda D. Ageing and the mismatch repair system. Mechanisms of Ageing and Development. 2000;121:173–179. doi: 10.1016/s0047-6374(00)00208-6. [DOI] [PubMed] [Google Scholar]
- Berardini M, Mazurek A, Fishel R. The effect of O6-methylguanine DNA adducts on the adenosine nucleotide switch functions of hMSH2-hMSH6 and hMSH2-hMSH3. The Journal of Biological Chemistry. 2000;275:27851–27857. doi: 10.1074/jbc.M003589200. [in process citation] [DOI] [PubMed] [Google Scholar]
- Berends MJ, Wu Y, Sijmons RH, Mensink RG, van der Sluis T, Hordijk-Hos JM, de Vries EG, Hollema H, Karrenbeld A, Buys CH, van der Zee AG, Hofstra RM, Kleibeuker JH. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. American Journal of Human Genetics. 2002;70:26–37. doi: 10.1086/337944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholon J, Wang Q, Galmarini CM, Puisieux A. Mutational targets in colorectal cancer cells with microsatellite instability. Family Cancer. 2006;5:29–34. doi: 10.1007/s10689-005-2573-5. [DOI] [PubMed] [Google Scholar]
- Bertrand P, Tishkoff DX, Filosi N, Dasgupta R, Kolodner RD. Physical interaction between components of DNA mismatch repair and nucleotide excision repair. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14278–14283. doi: 10.1073/pnas.95.24.14278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas I, Obmolova G, Takahashi M, Herr A, Newman MA, Yang W, Hsieh P. Disruption of the helix-u-turn-helix motif of MutS protein: loss of subunit dimerization, mismatch binding and ATP hydrolysis. Journal of Molecular Biology. 2001;305:805–816. doi: 10.1006/jmbi.2000.4367. [DOI] [PubMed] [Google Scholar]
- Bjornson KP, Modrich P. Differential and simultaneous adenosine di- and triphosphate binding by MutS. The Journal of Biological Chemistry. 2003;278:18557–18562. doi: 10.1074/jbc.M301101200. [DOI] [PubMed] [Google Scholar]
- Blackwell LJ, Wang S, Modrich P. DNA chain length dependence of formation and dynamics of hMutSalpha-hMutLalpha-heteroduplex complexes. The Journal of Biological Chemistry. 2001;276:33233–33240. doi: 10.1074/jbc.M105076200. [DOI] [PubMed] [Google Scholar]
- Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nature Reviews Genetics. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- Blasco MA. Telomere length, stem cells and aging. Nature Chemical Biology. 2007;3:640–649. doi: 10.1038/nchembio.2007.38. [DOI] [PubMed] [Google Scholar]
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Research. 1998;58:5248–5257. [PubMed] [Google Scholar]
- Bowers J, Sokolsky T, Quach T, Alani E. A mutation in the MSH6 subunit of the Saccharomyces cerevisiae MSH2-MSH6 complex disrupts mismatch recognition. The Journal of Biological Chemistry. 1999;274:16115–16125. doi: 10.1074/jbc.274.23.16115. [DOI] [PubMed] [Google Scholar]
- Bowers J, Tran PT, Joshi A, Liskay RM, Alani E. MSH-MLH complexes formed at a DNA mismatch are disrupted by the PCNA sliding clamp. Journal of Molecular Biology. 2001;306:957–968. doi: 10.1006/jmbi.2001.4467. [DOI] [PubMed] [Google Scholar]
- Brieger A, Plotz G, Raedle J, Weber N, Baum W, Caspary WF, Zeuzem S, Trojan J. Characterization of the nuclear import of human MutLalpha. Molecular Carcinogenesis. 2005;43:51–58. doi: 10.1002/mc.20081. [DOI] [PubMed] [Google Scholar]
- Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Research. 2007;35:7527–7544. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R. The mismatch repair system is required for S-phase checkpoint activation. Nature Genetics. 2003;33:80–84. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P. In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6765–6770. doi: 10.1073/pnas.121183298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmann MA, Nowosielska A, Marinus MG. Separation of mutation avoidance and antirecombination functions in an Escherichia coli mutS mutant. Nucleic Acids Research. 2005a;33:1193–1200. doi: 10.1093/nar/gki263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmann MA, Nowosielska A, Marinus MG. The MutS C terminus is essential for mismatch repair activity in vivo. Journal of Bacteriology. 2005b;187:6577–6579. doi: 10.1128/JB.187.18.6577-6579.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Gerrits B, Marra G, Schlapbach R, Jiricny J. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. The Journal of Biological Chemistry. 2007;282:2976–2986. doi: 10.1074/jbc.M609989200. [DOI] [PubMed] [Google Scholar]
- Cannavo E, Marra G, Sabates-Bellver J, Menigatti M, Lipkin SM, Fischer F, Cejka P, Jiricny J. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Research. 2005;65:10759–10766. doi: 10.1158/0008-5472.CAN-05-2528. [DOI] [PubMed] [Google Scholar]
- Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Mojas N, Gillet L, Schar P, Jiricny J. Homologous recombination rescues mismatch-repair-dependent cytotoxicity of S(N)1-type methylating agents in S. cerevisiae. Current Biology. 2005;15:1395–1400. doi: 10.1016/j.cub.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Chang CL, Marra G, Chauhan DP, Ha HT, Chang DK, Ricciardiello L, Randolph A, Carethers JM, Boland CR. Oxidative stress inactivates the human DNA mismatch repair system. American Journal of Physiology Cell Physiology. 2002;283:C148–C154. doi: 10.1152/ajpcell.00422.2001. [DOI] [PubMed] [Google Scholar]
- Chen S, Bigner SH, Modrich P. High rate of CAD gene amplification in human cells deficient in MLH1 or MSH6. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13802–13807. doi: 10.1073/pnas.241508098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J, Lee HW, von Zglinicki T, Ganser A, Schirmacher P, Nakauchi H, Rudolph KL. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nature Genetics. 2007;39:99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- Chung DC, Rustgi AK. The hereditary nonpolyposis colorectal cancer syndrome: genetics and clinical implications. Annals of Internal Medicine. 2003;138:560–570. doi: 10.7326/0003-4819-138-7-200304010-00012. [DOI] [PubMed] [Google Scholar]
- Clark AB, Deterding L, Tomer KB, Kunkel TA. Multiple functions for the N-terminal region of Msh6. Nucleic Acids Research. 2007;35:4114–4123. doi: 10.1093/nar/gkm409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AB, Kunkel TA. Cadmium inhibits the functions of eukaryotic MutS complexes. The Journal of Biological Chemistry. 2004;279:53903–53906. doi: 10.1074/jbc.C400495200. [DOI] [PubMed] [Google Scholar]
- Clark AB, Valle F, Drotschmann K, Gary RK, Kunkel TA. Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. The Journal of Biological Chemistry. 2000;275:36498–36501. doi: 10.1074/jbc.C000513200. [DOI] [PubMed] [Google Scholar]
- Cohen PE, Pollack SE, Pollard JW. Genetic analysis of chromosome pairing, recombination, and cell cycle control during first meiotic prophase in mammals. Endocrine Reviews. 2006;27:398–426. doi: 10.1210/er.2005-0017. [DOI] [PubMed] [Google Scholar]
- Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. The Journal of Biological Chemistry. 2005;280:39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L, Bellacosa A. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15071–15076. doi: 10.1073/pnas.2334585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings CJ, Zoghbi HY. Fourteen and counting: unraveling trinucleotide repeat diseases. Human Molecular Genetics. 2000;9:909–916. doi: 10.1093/hmg/9.6.909. [DOI] [PubMed] [Google Scholar]
- Das Gupta R, Kolodner RD. Novel dominant mutations in Saccharomyces cerevisiae MSH6. Nature Genetics. 2000;24:53–56. doi: 10.1038/71684. [DOI] [PubMed] [Google Scholar]
- de Vries SS, Baart EB, Dekker M, Siezen A, de Rooij DG, de Boer P, te Riele H. Mouse MutS-like protein Msh5 is required for proper chromosome synapsis in male and female meiosis. Genes & Development. 1999;13:523–531. doi: 10.1101/gad.13.5.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Claij N, Jansen L, van Klink Y, Radman M, Riggins G, van der Valk M, van’t Wout K, te Riele H. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nature Genetics. 1999;23:359–362. doi: 10.1038/15544. [DOI] [PubMed] [Google Scholar]
- de Wind N, Dekker M, van Rossum A, van der Valk M, te Riele H. Mouse models for hereditary nonpolyposis colorectal cancer. Cancer Research. 1998;58:248–255. [PubMed] [Google Scholar]
- Deschenes SM, Tomer G, Nguyen M, Erdeniz N, Juba NC, Sepulveda N, Pisani JE, Liskay RM. The E705K mutation in hPMS2 exerts recessive, not dominant, effects on mismatch repair. Cancer Letters. 2007;249:148–156. doi: 10.1016/j.canlet.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annual Review of Biochemistry. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- Drotschmann K, Yang W, Brownewell FE, Kool ET, Kunkel TA. Asymmetric recognition of DNA local distortion: structure-based functional studies of eukaryotic Msh2-Msh6. The Journal of Biological Chemistry. 2001;276:46225–46229. doi: 10.1074/jbc.C100450200. [DOI] [PubMed] [Google Scholar]
- Drummond JT, Li GM, Longley MJ, Modrich P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science (New York, NY) 1995;268:1909–1912. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- Duckett DR, Drummond JT, Murchie AIH, Reardon JT, Sancar A, Lilley DMJ, Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6443–6447. doi: 10.1073/pnas.93.13.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufner P, Marra G, Raschle M, Jiricny J. Mismatch recognition and DNA-dependent stimulation of the ATPase activity of hMutSα abolished by a single mutation in the hMSH6 subunit. The Journal of Biological Chemistry. 2000;275:36550–36555. doi: 10.1074/jbc.M005987200. [DOI] [PubMed] [Google Scholar]
- Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P. A defined human system that supports bidirectional mismatch-provoked excision. Molecular Cell. 2004;15:31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Yang K, Umar A, Heyer J, Lau K, Fan K, Liedtke W, Cohen PE, Kane MF, Lipford JR, Yu N, Crouse GF, Pollard JW, Kunkel T, Lipkin M, Kolodner R, Kucherlapati R. Mutation in the Mismatch Repair Gene Msh6 Causes Cancer Susceptibility. Cell. 1997;91:467–477. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Edelmann W. Loss of DNA mismatch repair function and cancer predisposition in the mouse: animal models for human hereditary nonpolyposis colorectal cancer. American Journal of Medical Genetics C: Seminar Medical Genetics. 2004;129:91–99. doi: 10.1002/ajmg.c.30021. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Cohen PE, Kane M, Lau K, Morrow B, Bennett S, Umar A, Kunkel T, Cattoretti G, Chaganti R, Pollard JW, Kolodner RD, Kucherlapati R. Meiotic pachytene arrest in MLH1 -deficient mice. Cell. 1996;85:1125–1134. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Cohen PE, Kneitz B, Winand N, Lia M, Heyer J, Kolodner R, Pollard JW, Kucherlapati R. Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nature Genetics. 1999a;21:123–127. doi: 10.1038/5075. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Umar A, Yang K, Heyer J, Kucherlapati M, Lia M, Kneitz B, Avdievich E, Fan K, Wong E, Crouse G, Kunkel T, Lipkin M, Kolodner RD, Kucherlapati R. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Research. 2000;60:803–807. [PubMed] [Google Scholar]
- Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, Fan K, Brown AM, Lipkin M, Kucherlapati R. Tumorigenesis in Mlh1 and Mlh1/ Apc1638N mutant mice. Cancer Research. 1999b;59:1301–1307. [PubMed] [Google Scholar]
- Erdeniz N, Nguyen M, Deschenes SM, Liskay RM. Mutations affecting a putative MutLalpha endonuclease motif impact multiple mismatch repair functions. DNA Repair. 2007;6:1463–1470. doi: 10.1016/j.dnarep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Baerenfaller K, Jiricny J. 5-Fluorouracil is efficiently removed from DNA by the base excision and mismatch repair systems. Gastroenterology. 2007;133:1858–1868. doi: 10.1053/j.gastro.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [published erratum appears in Cell 1994 Apr 8;77(1):167] [DOI] [PubMed] [Google Scholar]
- Flores-Rozas H, Clark D, Kolodner RD. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nature Genetics. 2000;26:375–378. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- Genschel J, Bazemore LR, Modrich P. Human exonuclease I is required for 5′ and 3′ mismatch repair. The Journal of Biological Chemistry. 2002;277:13302–13311. doi: 10.1074/jbc.M111854200. [DOI] [PubMed] [Google Scholar]
- Genschel J, Modrich P. Mechanism of 5′-directed excision in human mismatch repair. Molecular Cell. 2003;12:1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr, Levrero M, Wang JY. The tyrosine kinase c-Ab1 regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- Gorbalenya AE, Koonin EV. Superfamily of UvrA-related NTP-binding proteins: implications for rational classification of recombination/repair systems. Journal of Molecular Biology. 1990;213:583–591. doi: 10.1016/S0022-2836(05)80243-8. [DOI] [PubMed] [Google Scholar]
- Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Molecular Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilley M, Welsh KM, Su SS, Modrich P. Isolation and characterization of the Escherichia coli mutL gene product. The Journal of Biological Chemistry. 1989;264:1000–1004. [PubMed] [Google Scholar]
- Gu L, Hong Y, McCulloch S, Watanabe H, Li GM. ATP-dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Research. 1998;26:1173–1178. doi: 10.1093/nar/26.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarne A, Junop MS, Yang W. Structure and function of the N-terminal 40 kDa fragment of human PMS2: a monomeric GHL ATPase. The EMBO Journal. 2001;20:5521–5531. doi: 10.1093/emboj/20.19.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarne A, Ramon-Maiques S, Wolff EM, Ghirlando R, Hu X, Miller JH, Yang W. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. The EMBO Journal. 2004;23:4134–4145. doi: 10.1038/sj.emboj.7600412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber LT, Pang PP, Sobell DI, Mankovich JA, Walker GC. Nucleotide sequence of the Salmonella typhimurium mutS gene required for mismatch repair: homology of MutS and HexA of Streptococcus pneumoniae. Journal of Bacteriology. 1988;170:197–202. doi: 10.1128/jb.170.1.197-202.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MC, Shcherbakova PV, Kunkel TA. Differential ATP binding and intrinsic ATP hydrolysis by amino-terminal domains of the yeast Mlh1 and PMS1 proteins. The Journal of Biological Chemistry. 2002;277:3673–3679. doi: 10.1074/jbc.M106120200. [DOI] [PubMed] [Google Scholar]
- Hanada K, Hickson ID. Molecular genetics of RecQ helicase disorders. Cellular and Molecular Life Sciences. 2007;64:2306–2322. doi: 10.1007/s00018-007-7121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annual Review of Genetics. 2000;34:359–399. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]
- Harrington JM, Kolodner RD. Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Molecular and Cellular Biology. 2007;27:6546–6554. doi: 10.1128/MCB.00855-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawk JD, Stefanovic L, Boyer JC, Petes TD, Farber RA. Variation in efficiency of DNA mismatch repair at different sites in the yeast genome. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8639–8643. doi: 10.1073/pnas.0503415102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess MT, Mendillo ML, Mazur DJ, Kolodner RD. Biochemical basis for dominant mutations in the Saccharomyces cerevisiae MSH6 gene. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:558–563. doi: 10.1073/pnas.0510078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann ER, Borts RH. Meiotic recombination intermediates and mismatch repair proteins. Cytogenetic and Genome Research. 2004;107:232–248. doi: 10.1159/000080601. [DOI] [PubMed] [Google Scholar]
- Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chemical Review. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- Jacobs-Palmer E, Hingorani MM. The effects of nucleotides on MutS-DNA binding kinetics clarify the role of MutS ATPase activity in mismatch repair. Journal of Molecular Biology. 2007;366:1087–1098. doi: 10.1016/j.jmb.2006.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MA, Hayashi S, O’Shea AM, Burgart LJ, Smyrk TC, Shimizu D, Waring PM, Ruszkiewicz AR, Pollett AF, Redston M, Barker MA, Baron JA, Casey GR, Dowty JG, Giles GG, Limburg P, Newcomb P, Young JP, Walsh MD, Thibodeau SN, Lindor NM, Lemarchand L, Gallinger S, Haile RW, Potter JD, Hopper JL, Jass JR. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology. 2007;133:48–56. doi: 10.1053/j.gastro.2007.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Clark AB, Slebos RJ, Al-Refai H, Taylor JA, Kunkel TA, Resnick MA, Gordenin DA. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nature Genetics. 2003;34:326–329. doi: 10.1038/ng1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J. The multifaceted mismatch-repair system. Nature Reviews. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Kovvali GK, Guzder SN, Amin NS, Holm C, Habraken Y, Sung P, Prakash L, Prakash S. Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair. The Journal of Biological Chemistry. 1996;71:27987–27990. doi: 10.1074/jbc.271.45.27987. [DOI] [PubMed] [Google Scholar]
- Ju Z, Choudhury AR, Rudolph KL. A dual role of p21 in stem cell aging. Annals of the New York Academy of Sciences. 2007;1100:333–344. doi: 10.1196/annals.1395.036. [DOI] [PubMed] [Google Scholar]
- Junop MS, Obmolova G, Rausch K, Hsieh P, Yang W. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Molecular Cell. 2001;7:1–12. doi: 10.1016/s1097-2765(01)00149-6. [DOI] [PubMed] [Google Scholar]
- Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- Kadyrov FA, Holmes SF, Arana ME, Lukianova OA, O’Donnell M, Kunkel TA, Modrich P. Saccharomyces cerevisiae MutLa is a mismatch repair endonuclease. The Journal of Biological Chemistry. 2007;282:37181–37190. doi: 10.1074/jbc.M707617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaina B. Mechanisms and consequences of methylating agent-induced SCEs and chromosomal aberrations: a long road traveled and still a far way to go. Cytogenetic and Genome Research. 2004;104:77–86. doi: 10.1159/000077469. [DOI] [PubMed] [Google Scholar]
- Kan R, Sun X, Kolas NK, Avdievich E, Kneitz B, Edelmann W, Cohen PE. Comparative analysis of meiotic progression in female mice bearing mutations in genes of the DNA mismatch repair pathway. Biology of Reproduction. 2008;78:462–471. doi: 10.1095/biolreprod.107.065771. [DOI] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors. Cancer Research. 1997;57:808–811. [PubMed] [Google Scholar]
- Karran P. Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis. 2001;22:1931–1937. doi: 10.1093/carcin/22.12.1931. [DOI] [PubMed] [Google Scholar]
- Karran P. Thiopurines, DNA damage, DNA Repair and therapy-related cancer. British Medical Bulletin. 2007;79:153–170. doi: 10.1093/bmb/ldl020. [DOI] [PubMed] [Google Scholar]
- Kim IJ, Kang HC, Park JH, Shin Y, Ku JL, Lim SB, Park SY, Jung SY, Kim HK, Park JG. Development and applications of a beta-catenin oligonucleotide microarray: beta-catenin mutations are dominantly found in the proximal colon cancers with microsatellite instability. Clinical Cancer Research. 2003;9:2920–2925. [PubMed] [Google Scholar]
- Kim SM, Kumagai A, Lee J, Dunphy WG. Phosphorylation of Chk1 by ATM- and Rad3-related (ATR) in Xenopus egg extracts requires binding of ATRIP to ATR but not the stable DNA-binding or coiled-coil domains of ATRIP. The Journal of Biological Chemistry. 2005;280:38355–38364. doi: 10.1074/jbc.M508673200. [DOI] [PubMed] [Google Scholar]
- Kleczkowska HE, Marra G, Lettieri T, Jiricny J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes & Development. 2001;15:724–736. doi: 10.1101/gad.191201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneitz B, Cohen PE, Avdievich E, Zhu L, Kane MF, Hou H, Jr, Kolodner RD, Kucherlapati R, Pollard JW, Edelmann W. MutS homolog4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes & Development. 2000;14:1085–1097. [PMC free article] [PubMed] [Google Scholar]
- Knudsen NO, Nielsen FC, Vinther L, Bertelsen R, Holten-Andersen S, Liberti SE, Hofstra R, Kooi K, Rasmussen LJ. Nuclear localization of human DNA mismatch repair protein exonuclease 1 (hEXO1) Nucleic Acids Research. 2007;35:2609–2619. doi: 10.1093/nar/gkl1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann W, Gruber SB. Hereditary non-polyposis colon cancer. 2006 http://www.geneclinics.org/profiles/hnpcc.
- Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Research. 1994;54:4308–4312. [PubMed] [Google Scholar]
- Kolodner RD, Mendillo ML, Putnam CD. Coupling distant sites in DNA during DNA mismatch repair. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12953–12954. doi: 10.1073/pnas.0705698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, McMurray CT. Trinucleotide expansion in haploid germ cells by gap repair. Nature Genetics. 2001;27:407–411. doi: 10.1038/86906. [DOI] [PubMed] [Google Scholar]
- Kovtun IV, McMurray CT. Crosstalk of DNA glycosylases with pathways other than base excision repair. DNA Repair. 2007;6:517–529. doi: 10.1016/j.dnarep.2006.10.015. [DOI] [PubMed] [Google Scholar]
- Krichevsky S, Pawelec G, Gural A, Effros RB, Globerson A, Yehuda DB, Yehuda AB. Age related microsatellite instability in T cells from healthy individuals. Experimental Gerontology. 2004;39:507–515. doi: 10.1016/j.exger.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annual Review of Biochemistry. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Lamers MH, Georgijevic D, Lebbink JH, Winterwerp HH, Agianian B, de Wind N, Sixma TK. ATP increases the affinity between MutS ATPase domains. Implications for ATP hydrolysis and conformational changes. The Journal of Biological Chemistry. 2004;279:43879–43885. doi: 10.1074/jbc.M406380200. [DOI] [PubMed] [Google Scholar]
- Lamers MH, Perrakis A, Enzlin JH, Winterwerp HHK, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G:T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- Lau PJ, Kolodner RD. Transfer of the MSH2{middle dot}MSH6 complex from proliferating cell nuclear antigen to mispaired bases in DNA. The Journal of Biological Chemistry. 2003;278:14–17. doi: 10.1074/jbc.C200627200. [DOI] [PubMed] [Google Scholar]
- Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen LA, Mystrom-Lahti M, Guan XY, Zhang J, Meltzer PS, Yu JW, Kao FT, Chen DJ, Cerosaletti KM, Fournier REK, Todd S, Lewis T, Leach RJ, Naylor SL, Weissenbach J, Mecklin JP, Jarvinen H, Petersen GM, Hamilton SR, Green J, Jass J, Watson P, Lynch HT, Trent JM, de la Chappelle A, Kinzler KW, Vogelstein B. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- Lebbink JH, Georgijevic D, Natrajan G, Fish A, Winterwerp HH, Sixma TK, de Wind N. Dual role of MutS glutamate 38 in DNA mismatch discrimination and in the authorization of repair. The EMBO Journal. 2006;25:409–419. doi: 10.1038/sj.emboj.7600936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SD, Alani E. Analysis of interactions between mismatch repair initiation factors and the replication processivity factor PCNA. Journal of Molecular Biology. 2006;355:175–184. doi: 10.1016/j.jmb.2005.10.059. [DOI] [PubMed] [Google Scholar]
- Li GM. DNA mismatch repair and cancer. Frontier in Biosciences. 2003;8:d997–d1017. doi: 10.2741/1121. [DOI] [PubMed] [Google Scholar]
- Li GM, Modrich P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:1950–1954. doi: 10.1073/pnas.92.6.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Peled JU, Zhao C, Svetlanov A, Ronai D, Cohen PE, Scharff MD. A role for Mlh3 in somatic hypermutation. DNA Repair. 2006;5:675–682. doi: 10.1016/j.dnarep.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, Umar A, Kucherlapati R, Lipkin M, Kunkel TA, Edelmann W. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Research. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- Lipkin SM, Moens PB, Wang V, Lenz M, Shanmugarajah D, Gilgeous A, Thomas J, Cheng J, Touchman JW, Green ED, Schwartzberg P, Collins FS, Cohen PE. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nature Genetics. 2002;31:385–390. doi: 10.1038/ng931. [DOI] [PubMed] [Google Scholar]
- Loeb LA. A mutator phenotype in cancer. Cancer Research. 2001;61:3230–3239. [PubMed] [Google Scholar]
- Lopez de Saro FJ, Marinus MG, Modrich P, O’Donnell M. The beta sliding clamp binds to multiple sites within MutL and MutS. The Journal of Biological Chemistry. 2006;281:14340–14349. doi: 10.1074/jbc.M601264200. [DOI] [PubMed] [Google Scholar]
- Lopez de Saro FJ, O’Donnell M. Interaction of the β sliding clamp with MutS, ligase and DNA Polymerase I. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8376–8380. doi: 10.1073/pnas.121009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzen A, Liberti SE, Rasmussen LJ. Cadmium inhibits human DNA mismatch repair in vivo. Biochemical and Biophysical Research Communications. 2004;321:21–25. doi: 10.1016/j.bbrc.2004.06.102. [DOI] [PubMed] [Google Scholar]
- Lynch HT, Schuelke GS, Kimberling WJ, Albano WA, Lynch JF, Biscone KA, Lipkin ML, Deschner EE, Mikol YB, Sandberg AA, et al. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). II. Biomarker studies. Cancer. 1985;56:939–951. doi: 10.1002/1097-0142(19850815)56:4<939::aid-cncr2820560440>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Malesci A, Laghi L, Bianchi P, Delconte G, Randolph A, Torri V, Carnaghi C, Doci R, Rosati R, Montorsi M, Roncalli M, Gennari L, Santoro A. Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clinical Cancer Research. 2007;13:3831–3839. doi: 10.1158/1078-0432.CCR-07-0366. [DOI] [PubMed] [Google Scholar]
- Malkov VA, Biswas L, Camerini-Otero RD, Hsieh P. Photocross-linking of the NH2-terminal region of Taq MutS protein to the major groove of a hetero-duplex DNA. The Journal of Biological Chemistry. 1997;272:23811–23817. doi: 10.1074/jbc.272.38.23811. [DOI] [PubMed] [Google Scholar]
- Manelyte L, Urbanke C, Giron-Monzon L, Friedhoff P. Structural and functional analysis of the MutS C-terminal tetramerization domain. Nucleic Acids Research. 2006;34:5270–5279. doi: 10.1093/nar/gkl489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nature Genetics. 1999;23:471–473. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science (New York, NY) 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- Marsischky GT, Kolodner RD. Biochemical characterization of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 complex and mispaired bases in DNA. The Journal of Biological Chemistry. 1999;274:26668–26682. doi: 10.1074/jbc.274.38.26668. [DOI] [PubMed] [Google Scholar]
- Martik D, Baitinger C, Modrich P. Differential specificities and simultaneous occupancy of human MutSalpha nucleotide binding sites. The Journal of Biological Chemistry. 2004;279:28402–28410. doi: 10.1074/jbc.M312108200. [DOI] [PubMed] [Google Scholar]
- Matson SW, Robertson AB. The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucleic Acids Research. 2006;34:4089–4097. doi: 10.1093/nar/gkl450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur DJ, Mendillo ML, Kolodner RD. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Molecular Cell. 2006;22:39–49. doi: 10.1016/j.molcel.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Mazurek A, Berardini M, Fishel R. Activation of human MutS homologs by 8-oxo-guanine DNA damage. The Journal of Biological Chemistry. 2002;277:8260–8266. doi: 10.1074/jbc.M111269200. [DOI] [PubMed] [Google Scholar]
- Mcllhatton MA, Tyler J, Burkholder S, Ruschoff J, Rigas B, Kopelovich L, Fishel R. Nitric oxide-donating aspirin derivatives suppress microsatellite instability in mismatch repair-deficient and hereditary nonpolyposis colorectal cancer cells. Cancer Research. 2007;67:10966–10975. doi: 10.1158/0008-5472.CAN-07-2562. [DOI] [PubMed] [Google Scholar]
- Meira LB, Cheo DL, Reis AM, Claij N, Burns DK, te Riele H, Friedberg EC. Mice defective in the mismatch repair gene Msh2 show increased predisposition to UVB radiation-induced skin cancer. DNA Repair. 2002;1:929–934. doi: 10.1016/s1568-7864(02)00143-x. [DOI] [PubMed] [Google Scholar]
- Mellon I, Rajpal DK, Koi M, Boland CR, Champe GN. Transcription-coupled repair deficiency and mutations in human mismatch repair genes. Science (New York, NY) 1996;272:557–560. doi: 10.1126/science.272.5261.557. [DOI] [PubMed] [Google Scholar]
- Mendillo ML, Mazur DJ, Kolodner RD. Analysis of the interaction between the Saccharomyces cerevisiae MSH2–MSH6 and MLH1–PMS1 complexes with DNA using a reversible DNA end-blocking system. The Journal of Biological Chemistry. 2005;280:22245–22257. doi: 10.1074/jbc.M407545200. [DOI] [PubMed] [Google Scholar]
- Mendillo ML, Putnam CD, Kolodner RD. Escherichia coli MutS tetramerization domain structure reveals that stable dimers but not tetramers are essential for DNA mismatch repair in vivo. The Journal of Biological Chemistry. 2007;282:16345–16354. doi: 10.1074/jbc.M700858200. [DOI] [PubMed] [Google Scholar]
- Meyers M, Hwang A, Wagner MW, Boothman DA. Role of DNA mismatch repair in apoptotic responses to therapeutic agents. Environmental and Molecular Mutagenesis. 2004;44:249–264. doi: 10.1002/em.20056. [DOI] [PubMed] [Google Scholar]
- Meyers M, Hwang A, Wagner MW, Bruening AJ, Veigl ML, Sedwick WD, Boothman DA. A role for DNA mismatch repair in sensing and responding to fluoropyrimidine damage. Oncogene. 2003;22:7376–7388. doi: 10.1038/sj.onc.1206941. [DOI] [PubMed] [Google Scholar]
- Meyers M, Wagner MW, Mazurek A, Schmutte C, Fishel R, Boothman DA. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. The Journal of Biological Chemistry. 2005;280:5516–5526. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
- Modrich P. Mechanisms in eukaryotic mismatch repair. The Journal of Biological Chemistry. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojas N, Lopes M, Jiricny J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes & Development. 2007;21:3342–3355. doi: 10.1101/gad.455407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natrajan G, Lamers MH, Enzlin JH, Winterwerp HH, Perrakis A, Sixma TK. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Research. 2003;31:4814–4821. doi: 10.1093/nar/gkg677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri S, Gardini A, Facchini A, Olivieri F, Franceschi C, Ravaglia G, Mariani E. Mismatch repair system and aging: microsatellite instability in peripheral blood cells from differently aged participants. The Journal of Gerontology. 2005;60:285–292. doi: 10.1093/gerona/60.3.285. [DOI] [PubMed] [Google Scholar]
- Ni TT, Marsischky GT, Kolodner RD. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Molecular Cell. 1999;4:439–444. doi: 10.1016/s1097-2765(00)80346-9. [DOI] [PubMed] [Google Scholar]
- Nielsen FC, Jager AC, Lutzen A, Bundgaard JR, Rasmussen LJ. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene. 2004;23:1457–1468. doi: 10.1038/sj.onc.1207265. [DOI] [PubMed] [Google Scholar]
- Nowosielska A, Marinus MG. DNA mismatch repair-induced double-strand breaks. DNA Repair. 2008;7:48–56. doi: 10.1016/j.dnarep.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- Owen BA, Yang Z, Lai M, Gajek M, Badger JD, 2nd, Hayes JJ, Edelmann W, Kucherlapati R, Wilson TM, McMurray CT. (CAG)(n)-hairpin DNA binds to Msh2–Msh3 and changes properties of mismatch recognition. Nature Structural & Molecular Biology. 2005;12:663–670. doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- Pani E, Stojic L, El-Shemerly M, Jiricny J, Ferrari S. Mismatch repair status and the response of human cells to cisplatin. Cell Cycle (Georgetown, TX) 2007;6:1796–1802. doi: 10.4161/cc.6.14.4472. [DOI] [PubMed] [Google Scholar]
- Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Hseltine WA, Flesichmann RD, Fraser CM, Adams MD, Venter JC, Hamilton SR, Petersen GM, Watson P, Lynch HT, Peltomaki P, Mecklin JP, de la Chapelle A, Kinzler KW, Vogelstein B. Mutation of a mutL homolog in hereditary colon cancer. Science (New York, NY) 1994;268:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, Modrich P. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- Pavlov YI, Mian IM, Kunkel TA. Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Current Biology. 2003;13:744–748. doi: 10.1016/s0960-9822(03)00284-7. [DOI] [PubMed] [Google Scholar]
- Peltomaki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Human Molecular Genetics. 2001;10:735–740. doi: 10.1093/hmg/10.7.735. [DOI] [PubMed] [Google Scholar]
- Plotz G, Piiper A, Wormek M, Zeuzem S, Raedle J. Analysis of the human MutLalpha. MutSalpha complex. Biochemical and Biophysical Research Communications. 2006;340:852–859. doi: 10.1016/j.bbrc.2005.12.096. [DOI] [PubMed] [Google Scholar]
- Plotz G, Raedle J, Brieger A, Trojan J, Zeuzem S. hMutSalpha forms an ATP-dependent complex with hMutLalpha and hMutLbeta on DNA. Nucleic Acids Research. 2002;30:711–718. doi: 10.1093/nar/30.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluciennik A, Modrich P. Protein roadblocks and helix discontinuities are barriers to the initiation of mismatch repair. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12709–12713. doi: 10.1073/pnas.0705129104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, PMS1 and Pms2 DNA mismatch repair. Nature Genetics. 1998;18:276–279. doi: 10.1038/ng0398-276. [DOI] [PubMed] [Google Scholar]
- Räschle M, Dufner P, Marra G, Jiricny J. Mutations within the hMLH1 and hPMS2 subunits of the human MutLα mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSα. The Journal of Biological Chemistry. 2002;277:21810–21820. doi: 10.1074/jbc.M108787200. [DOI] [PubMed] [Google Scholar]
- Reitmair AH, Risley R, Bristow RG, Wilson T, Ganesh A, Jang A, Peacock J, Benchimol S, Hill RP, Mak TW, Fishel R, Meuth M. Mutator phenotype in Msh2-deficient murine embryonic fibroblasts. Cancer Research. 1997;57:3765–3771. [PubMed] [Google Scholar]
- Reitmair AH, Schmits R, Ewel A, Bapat B, Redston M, Mitri A, Waterhouse P, Mittrucker HW, Wakeham A, Liu B, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nature Genetics. 1995;11:64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- Rizki A, Lundblad V. Defects in mismatch repair promote telomerase-independent proliferation. Nature. 2001;411:713–716. doi: 10.1038/35079641. [DOI] [PubMed] [Google Scholar]
- Robertson A, Pattishall SR, Matson SW. The DNA binding activity of MutL is required for methyl-directed mismatch repair in Escherichia coli. The Journal of Biological Chemistry. 2006a;281:8399–8408. doi: 10.1074/jbc.M509184200. [DOI] [PubMed] [Google Scholar]
- Robertson AB, Pattishall SR, Gibbons EA, Matson SW. MutL-catalyzed ATP hydrolysis is required at a post-UvrD loading step in methyl-directed mismatch repair. The Journal of Biological Chemistry. 2006b;281:19949–19959. doi: 10.1074/jbc.M601604200. [DOI] [PubMed] [Google Scholar]
- Sacho EJ, Kadyrov FA, Modrich P, Kunkel TA, Erie DA. Direct visualization of asymmetric adenine nucleotide-induced conformational changes in MutL alpha. Molecular Cell. 2008;29:112–121. doi: 10.1016/j.molcel.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samowitz WS, Curtin K, Lin HH, Robertson MA, Schaffer D, Nichols M, Gruenthal K, Leppert MF, Slattery ML. The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer. Gastroenterology. 2001a;121:830–838. doi: 10.1053/gast.2001.27996. [DOI] [PubMed] [Google Scholar]
- Samowitz WS, Holden JA, Curtin K, Edwards SL, Walker AR, Lin HA, Robertson MA, Nichols MF, Gruenthal KM, Lynch BJ, Leppert MF, Slattery ML. Inverse relationship between microsatellite instability and K-ras and p53 gene alterations in colon cancer. American Journal of Pathology. 2001b;158:1517–1524. doi: 10.1016/S0002-9440(10)64102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaetzlein S, Kodandaramireddy NR, Ju Z, Lechel A, Stepczynska A, Lilli DR, Clark AB, Rudolph C, Kuhnel F, Wei K, Schlegelberger B, Schirmacher P, Kunkel TA, Greenberg RA, Edelmann W, Rudolph KL. Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell. 2007;130:863–877. doi: 10.1016/j.cell.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield MJ, Brownewell FE, Nayak S, Du C, Kool ET, Hsieh P. The Phe-X-Glu DNA binding motif of MutS. The Journal of Biological Chemistry. 2001a;276:45505–45508. doi: 10.1074/jbc.C100449200. [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annual Review of Microbiology. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P. Interaction of Escherichia coli MutS and MutL at a DNA mismatch. Thejournal of Biological Chemistry. 2001b;276:28291–28299. doi: 10.1074/jbc.M103148200. [DOI] [PubMed] [Google Scholar]
- Selmane T, Schofield MJ, Nayak S, Du C, Hsieh P. Formation of a DNA mismatch repair complex mediated by ATP. Journal of Molecular Biology. 2003;334:949–965. doi: 10.1016/j.jmb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Shell SS, Putnam CD, Kolodner RD. Chimeric Saccharomyces cerevisiae Msh6 protein with an Msh3 mispair-binding domain combines properties of both proteins. Proceedings of the National Academy of Sciences of the United States of America. 2007a;104:10956–10961. doi: 10.1073/pnas.0704148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell SS, Putnam CD, Kolodner RD. The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA. Molecular Cell. 2007b;26:565–578. doi: 10.1016/j.molcel.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H, Yoshioka-Yamashita A, Kolodner RD, Wang JY. Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2420–2425. doi: 10.1073/pnas.0438031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegl-Cachedenier I, Flores I, Klatt P, Blasco MA. Telomerase reverses epidermal hair follicle stem cell defects and loss of long-term survival associated with critically short telomeres. Thejournal of Cell Biology. 2007a;179:277–290. doi: 10.1083/jcb.200704141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegl-Cachedenier I, Munoz P, Flores JM, Klatt P, Blasco MA. Deficient mismatch repair improves organismal fitness and survival of mice with dysfunctional telomeres. Genes & Development. 2007b;21:2234–2247. doi: 10.1101/gad.430107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva RV, Garicochea B, Cotti G, Maranho IC, Cutait R. Hereditary nonpolyposis colorectal cancer identification and surveillance of high-risk families. Clinics. 2005;60:251–256. doi: 10.1590/s1807-59322005000300011. [DOI] [PubMed] [Google Scholar]
- Sixma T. DNA mismatch repair: MutS structures bound to mismatches. Current Opinion in Structural Biology. 2001;11:47–52. doi: 10.1016/s0959-440x(00)00169-x. [DOI] [PubMed] [Google Scholar]
- Smith BT, Grossman AD, Walker GC. Visualization of mismatch repair in bacterial cells. Molecular Cell. 2001;8:1197–1206. doi: 10.1016/s1097-2765(01)00402-6. [DOI] [PubMed] [Google Scholar]
- Smith JA, Bannister LA, Bhattacharjee V, Wang Y, Waldman BC, Waldman AS. Accurate homologous recombination is a prominent double-strand break repair pathway in mammalian chromosomes and is modulated by mismatch repair protein Msh2. Molecular and Cellular Biology. 2007;27:7816–7827. doi: 10.1128/MCB.00455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits R, Hofland N, Edelmann W, Geugien M, Jagmohan-Changur S, Albuquerque C, Breukel C, Kucherlapati R, Kielman MF, Fodde R. Somatic Apc mutations are selected upon their capacity to inactivate the beta-catenin downregulating activity. Genes, Chromosomes & Cancer. 2000;29:229–239. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1033>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Snowden T, Acharya S, Butz C, Berardini M, Fishel R. hMSH4–hMSH5 recognizes Holliday Junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Molecular Cell. 2004;15:437–451. doi: 10.1016/j.molcel.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Snowden T, Shim KS, Schmutte C, Acharya S, Fishel R. hMSH4–hMSH5 adenosine nucleotide processing and interactions with homologous recombination machinery. Thejournal of Biological Chemistry. 2008;283:145–154. doi: 10.1074/jbc.M704060200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreide K. Molecular testing for microsatellite instability and DNA mismatch repair defects in hereditary and sporadic colorectal cancers–ready for prime time? Tumour Biology. 2007;28:290–300. doi: 10.1159/000110427. [DOI] [PubMed] [Google Scholar]
- Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes & Development. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J, Gealy R, Petes T, Jinks-Robertson S. Role of PCNA interactions in the mismatch repair-dependent processing of mitotic and meiotic recombination intermediates in yeast. Genetics. 2008 doi: 10.1534/genetics.107.085415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand M, Prolla TA, Liskay RM, Petes TD. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature. 1993;365:274–276. doi: 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- te Riele H, Maandag ER, Berns A. Highly efficient gene targeting in embryonic stem cells through homologous recombination with isogenic DNA constructs. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:5128–5132. doi: 10.1073/pnas.89.11.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, Kane MF, Kolodner RD. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:7487–7492. doi: 10.1073/pnas.94.14.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Degtyareva NP, Gordenin DA, Resnick MA. Genetic factors affecting the impact of DNA polymerase delta proofreading activity on mutation avoidance in yeast. Genetics. 1999a;152:47–59. doi: 10.1093/genetics/152.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Gordenin DA, Resnick MA. The 3′-5′ exonucleases of DNA polymerases delta and epsilon and the 5′-3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Molecular Biology of the Cell. 1999b;19:2000–2007. doi: 10.1128/mcb.19.3.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PT, Fey JP, Erdeniz N, Gellon L, Boiteux S, Liskay RM. A mutation in EXO1 defines separable roles in DNA mismatch repair and post-replication repair. DNA Repair. 2007;6:1572–1583. doi: 10.1016/j.dnarep.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao JL, Dudley S, Kwok B, Nickel AE, Laird PW, Siegmund KD, Liskay RM, Shibata D. Diet, cancer and aging in DNA mismatch repair deficient mice. Carcinogenesis. 2002;23:1807–1810. doi: 10.1093/carcin/23.11.1807. [DOI] [PubMed] [Google Scholar]
- Tsurimoto T. PCNA binding proteins. Frontier in Biosciences. 1999;4:D849–D858. doi: 10.2741/tsurimoto. [DOI] [PubMed] [Google Scholar]
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. Journal of the National Cancer Institute. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar A, Boyer JC, Thomas DC, Nguyen DC, Risinger JI, Boyd J, Ionov Y, Perucho M, Kunkel TA. Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. The Journal of Biological Chemistry. 1994;269:14367–14370. [PubMed] [Google Scholar]
- Umar A, Buermeyer AB, Simon JA, Thomas DC, Clark AB, Liskay RM, Kunkel TA. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996;87:65–73. doi: 10.1016/s0092-8674(00)81323-9. [DOI] [PubMed] [Google Scholar]
- van den Broek WJ, Nelen MR, Wansink DG, Coerwinkel MM, te Riele H, Groenen PJ, Wieringa B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Human Molecular Genetics. 2002;11:191–198. doi: 10.1093/hmg/11.2.191. [DOI] [PubMed] [Google Scholar]
- Varlet L, Canard B, Brooks P, Cerovic G, Radman M. Mismatch repair in Xenopus egg extracts: DNA strand breaks act as signals rather than excision points. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10156–10161. doi: 10.1073/pnas.93.19.10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- Vo AT, Zhu F, Wu X, Yuan F, Gao Y, Gu L, Li GM, Lee TH, Her C. hMRE11 deficiency leads to microsatellite instability and defective DNA mismatch repair. EMBO Reports. 2005;6:438–444. doi: 10.1038/sj.embor.7400392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hays JB. Mismatch repair in human nuclear extracts: effects of internal DNA-hairpin structures between mismatches and excision-initiation nicks on mismatch correction and mismatch-provoked excision. The Journal of Biological Chemistry. 2003;278:28686–28693. doi: 10.1074/jbc.M302844200. [DOI] [PubMed] [Google Scholar]
- Wang H, Hays JB. Signaling from DNA mispairs to mismatch-repair excision sites despite intervening blockades. The EMBO Journal. 2004;23:2126–2133. doi: 10.1038/sj.emboj.7600153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hays JB. Human DNA mismatch repair: coupling of mismatch recognition to strand-specific excision. Nucleic Acids Research. 2007;35:6727–6739. doi: 10.1093/nar/gkm734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hoffman PO, Lawrence C, Hays JB. Testing excision models for responses of mismatch-repair systems to UV photoproducts in DNA. Environmental and Molecular Mutagenesis. 2006;47:296–306. doi: 10.1002/em.20206. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang Y, Schofield MJ, Du C, Fridman Y, Lee SD, Larson ED, Drummond JT, Alani E, Hsieh P, Erie DA. DNA bending and unbending by MutS govern mismatch recognition and specificity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:14822–14827. doi: 10.1073/pnas.2433654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qjn J. MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15387–15392. doi: 10.1073/pnas.2536810100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JJ, Forsberg LJ, Beese LS. The structural basis for the mutagenicity of O(6)-methyl-guanine lesions. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19701–19706. doi: 10.1073/pnas.0609580103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Molecular Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Watson P, Butzow R, Lynch HT, Mecklin JP, Jarvinen HJ, Vasen HF, Madlensky L, Fidalgo P, Bernstein L. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecologic Oncology. 2001;82:223–228. doi: 10.1006/gyno.2001.6279. [DOI] [PubMed] [Google Scholar]
- Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, Russell R, Hou H, Kneitz B, Kunkel TA, Kolodner RK, Cohen PE, Edelmann W. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility and male and female sterility. Genes & Development. 2003;17:603–614. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JM, Bodmer W, Mortensen NJ. DNA mismatch repair genes and colorectal cancer. Gut. 2000;47:148–153. doi: 10.1136/gut.47.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, Lebel LA, Vrbanac V, Teed A, te Riele H, MacDonald ME. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Human Molecular Genetics. 2003;12:273–281. doi: 10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- Wilson DM, 3rd, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair. 2007;6:544–559. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, Yasui A, Woodgate R, Gearhart PJ. MSH2–MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. The Journal of Experimental Medicine. 2005;201:637–645. doi: 10.1084/jem.20042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods LM, Hodges CA, Baart E, Baker SM, Liskay M, Hunt PA. Chromosomal influence on meiotic spindle assembly: abnormal meiosis I in female Mlh1 mutant mice. The Journal of Cell Biology. 1999;145:1395–1406. doi: 10.1083/jcb.145.7.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhu BB, Yu J, Zhu H, Qju L, Kindy MS, Gu L, Seidel A, Li GM. In vitro and in vivo modulations of benzo[c]phenanthrene-DNA adducts by DNA mismatch repair system. Nucleic Acids Research. 2003a;31:6428–6434. doi: 10.1093/nar/gkg875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Platt JL, Cascalho M. Dimerization of MLH1 and PMS2 limits nuclear localization of MutLalpha. Molecular and Cellular Biology. 2003b;23:3320–3328. doi: 10.1128/MCB.23.9.3320-3328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Berends MJ, Mensink RG, Kempinga C, Sijmons RH, van Der Zee AG, Hollema H, Kleibeuker JH, Buys CH, Hofstra RM. Association of hereditary nonpolyposis colorectal cancer-related tumors displaying low microsatellite instability with MSH6 germline mutations. American Journal of Human Genetics. 1999;65:1291–1298. doi: 10.1086/302612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Schofield M, Biswas L, Hsieh P. Requirement for Phe36 for DNA binding and mismatch repair by Escherichia coli MutS protein. Nucleic Acids Research. 2000;28:3564–3569. doi: 10.1093/nar/28.18.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Schupp JE, Kinsella TJ. BRCA1 activates a G2-M cell cycle checkpoint following 6-thioguanine-induced DNA mismatch damage. Cancer Research. 2007;67:6286–6292. doi: 10.1158/0008-5472.CAN-06-2205. [DOI] [PubMed] [Google Scholar]
- Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, Kucherlapati R, Kolodner RD, Edelmann W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004a;6:139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Yang Q, Zhang R, Wang XW, Linke SP, Sengupta S, Hickson ID, Pedrazzi G, Perrera C, Stagljar I, Littman SJ, Modrich P, Harris CC. The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene. 2004b;23:3749–3756. doi: 10.1038/sj.onc.1207462. [DOI] [PubMed] [Google Scholar]
- Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair. 2006;5:654–666. doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- York SJ, Modrich P. Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. The Journal of Biological Chemistry. 2006;281:22674–22683. doi: 10.1074/jbc.M603667200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Molecular Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, Gu L, Guo S, Wang C, Li GM. Evidence for involvement of HMGB1 protein in human DNA mismatch repair. The Journal of Biological Chemistry. 2004;279:20935–20940. doi: 10.1074/jbc.M401931200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science (New York, NY) 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]