

Abstract
Viruses of the DNA tumor virus family share the ability to transform vertebrate cells through the action of virus-encoded tumor antigens that interfere with normal cell physiology. They accomplish this very efficiently by inhibiting endogenous tumor suppressor proteins that control cell proliferation and apoptosis. Simian virus 40 (SV40) encodes two oncoproteins, large tumor antigen, which directly inhibits the tumor suppressors p53 and Rb, and small tumor antigen (ST), which interferes with serine/threonine protein phosphatase protein phosphatase 2A (PP2A). We have constructed a Drosophila model for SV40 ST expression and show that ST induces super-numerary centrosomes, an activity we also demonstrate in human cells. In early Drosophila embryos, ST also caused increased microtubule stability, chromosome segregation errors, defective assembly of actin into cleavage furrows, cleavage failure, a rise in cyclin E levels and embryonic lethality. Using ST mutants and genetic interaction experiments between ST and PP2A subunit mutations, we show that all of these phenotypes are dependent on ST’s interaction with PP2A. These analyses demonstrate the validity and utility of Drosophila as a model for viral oncoprotein function in vivo.
Keywords: SV40 ST, centriole, centrosome, PP2A, aneuploidy, cyclin E
Introduction
Viruses of the DNA tumor virus family induce cell proliferation to promote the replication of the viral genome. Extensively investigated members of this family include human papillomavirus (HPV), adenovirus, polyomavirus and simian virus 40 (SV40). By hijacking cell-cycle and apoptosis regulation, viral oncoproteins transform cells. Investigation of DNA tumor virus oncoproteins has led to the identification of many fundamental mechanisms of tumor suppression (Lavia et al., 2003; Ahuja et al., 2005). By altering the activity of p53, retinoblastoma protein and serine/threonine protein phosphatase 2A (PP2A), three key tumor suppressors, SV40 can cause tumor formation in transgenic mouse models (Ahuja et al., 2005; Arroyo and Hahn, 2005).
A single transcript expressed from the early region of the SV40 genome encodes three proteins by alternative splicing: large tumor antigen (LT), small tumor antigen (ST) and 17kT (Sullivan and Pipas, 2002). ST cooperates with LT to transform cells (Skoczylas et al., 2004). ST has two domains and two known binding partners: a domain with homology to dnaJ proteins, or ‘J domain’ that binds Hsc70, and a PP2A-binding domain. The oncogenic activity of ST requires PP2A binding, but not the J domain (Mungre et al., 1994; Saenz-Robles et al., 2001; Skoczylas et al., 2004). Because of this property, ST has been used as a tool to assess PP2A’s role in transformation (Chen et al., 2004; Skoczylas et al., 2004). By inhibiting PP2A, ST disrupts dephosphorylation of targets for transformation, including c-myc and RalA (Mumby, 2007). The PP2A holoenzyme consists of three subunits, a scaffolding subunit (A), a regulatory subunit (B) and a catalytic subunit (C). In addition, substrate specificity is conferred by four classes of regulatory subunits: B, B’, B” and B”’ (Janssens and Goris, 2001; Sontag, 2001). ST binds directly to the PP2A A subunit (PP2AA), displacing B subunits from the holoenzyme but retaining the C subunit (Yang et al., 1991; Chen et al., 2007b). The structure of PP2AA bound to ST revealed a direct association between several PP2AA HEAT repeats with the second of two zincbinding domains in ST, an association that is similar to PP2AB’ bound to PP2AA (Xu et al., 2006; Chen et al., 2007b; Cho and Xu, 2007; Cho et al., 2007). Thus, ST might function as a PP2A B subunit (Cho et al., 2007). Therefore, in addition to inhibiting activity toward normal substrates through competition with B subunits, ST may confer new substrate specificities to PP2A.
Although DNA tumor virus oncoproteins disrupt pathways controlling the cell cycle and apoptosis, they also compromise cell division, potentially leading to aneuploidy (Lavia et al., 2003). For example, HPV E7 and adenovirus E1A induce centrosome overduplication, impacting genomic stability by increasing the incidence of multipolar mitotic spindles (De Luca et al., 2003; Duensing and Munger, 2003; Duensing et al., 2004). Although the transforming properties of ST have been extensively investigated, there is little understanding of the impact that disruption of normal signaling caused by ST and other viral oncoproteins have on animal development. To address this question, we have constructed a Drosophila melanogaster model for SV40 ST pathogenesis; the first Drosophila model for expression of a viral oncoprotein. We show that ST causes increased centrosome numbers, chromosome segregation defects, aberrant cytoskeleton assembly, cytokinesis failures and a rise in cyclin E levels. Using ST mutations and Drosophila mutant analysis, we show that disruption of embryogenesis by ST requires PP2A subunits, confirming the ST-PP2A connection from vertebrate studies.
Results
ST induces centrosome overduplication, and cytoskeletal and cleavage defects that lead to embryonic lethality
Expression of the SV40 early region, which encodes the LT, ST and 17kT proteins through alternative splicing (Figure 1a)(Sullivan and Pipas, 2002), in Drosophila early embryos caused an increase in centrosome numbers and lethality (Figures 1c and d; Supplementary Results; Supplementary Figure S1). ST was expressed exclusively from this construct (Figure 1e). To test whether ST indeed caused the increased centrosome numbers and other phenotypes (see below) observed with the early region construct, and to eliminate a potential contribution from undetectable expression of LT and 17kT, transgenic flies that express an ST cDNA fused to a 3 × FLAG tag at the C terminus were generated (ST-FLAG). As a control, transgenic flies containing the FLAG tag vector were generated (vector). Comparison among several ST-FLAG transgenic lines showed that independent lines varied in expression levels, with embryonic lethality directly correlating with the level of ST-FLAG (Supplementary Figure S1).
Figure 1.

Centrosome abnormalities in Drosophila embryos expressing Simian virus 40 (SV40) small tumor antigen (ST). (a) Schematic diagram of the SV40 early region gene products large tumor antigen (LT), ST and 17kT, produced by alternative splicing. Black lines indicate introns. Size of each protein (in amino acids) and region used to raise antibodies (Ab) are indicated. (b) ST has two domains: a J domain and a PP2A-binding domain. The mutations used in this study are indicated. (c, d) SV40 ST-expressing embryos exhibit supernumerary centrosomes and mitotic spindle abnormalities. Early cleavage stage embryos were stained for centrosomin (red), α-tubulin (green), and DNA (blue). Note some of the spindle poles are located outside of the image stack shown. Bar: 25 μm. (e) SV40 early region-expressing embryos produce ST protein, but no detectable LT. Lysate from mouse liver expressing SV40 early region (Comerford et al., 2003) is included as a positive control.
We next examined embryos that express ST-FLAG (hereafter referred to as ST embryos) for centrosome duplication, cytoskeleton, chromosome organization and cleavage defects by immunostaining embryos with antibodies to centrosomin (CNN) and α-tubulin to label centrosomes and microtubules, respectively, and with dyes for DNA and filamentous actin (Figures 2a-d). Normally, centrosomes duplicate with high fidelity once each cleavage cycle, ensuring a single centrosome at each spindle pole at mitosis (Figure 2a). ST not only caused centrosome overduplication by the first mitotic cycle (Figure 2b, arrow), but also caused other spindle assembly defects (for example, Figure 1d) including a failure to organize or maintain chromosomes within the spindle (Figure 2b). Often, the extra centrosomes in ST embryos were free, and not associated with a mitotic spindle (for example, Figure 1d). Thus, ST disrupts centrosome duplication and chromosome segregation from the earliest embryonic cleavage cycles.
Figure 2.
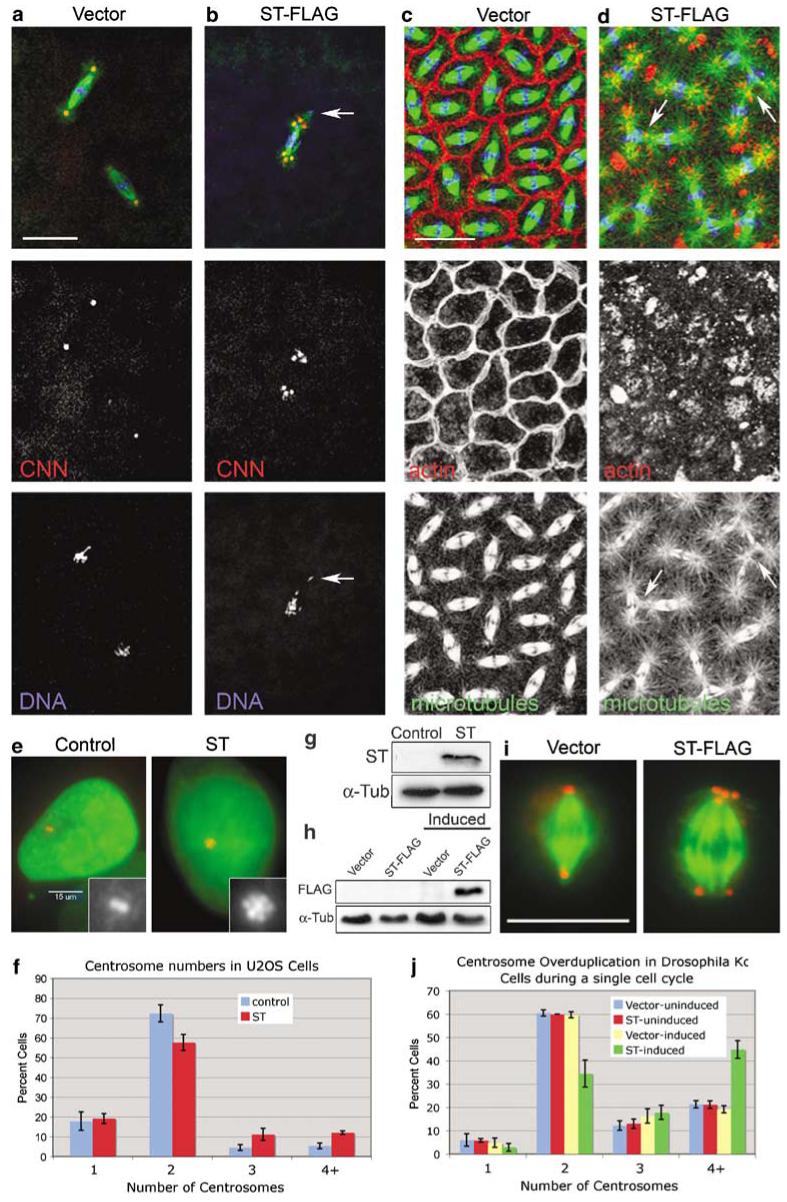
Small tumor antigen (ST) induces centrosome overduplication and cytoskeleton disruption. (a, b) Expression of FLAG-tagged ST causes centrosome overduplication and chromosome segregation errors from the earliest embryonic divisions. Spindles in ST-FLAG embryos have extra centrosomes at spindle poles and scattered arrangement of chromosomes, some displaced completely from the spindle (arrow in (b)). Embryos were stained for α-tubulin (green), centrosomin (CNN) (red) and DNA (blue). At cortical cleavage stages (c, d) actin is not assembled properly into furrows, astral microtubules are larger and interact with neighboring spindles in ST embryos (arrows in (d)). (c, d) Embryos were stained for α-tubulin (green), filamentous actin (red) and DNA (blue). (e) In human U2OS cells, ST expression increases centrosome numbers. Cells transfected with pGFP-histone (a marker for transfected cells) with or without the ST expression plasmid pCMV5/Smt (Sontag et al., 1993)were stained for γ-tubulin (red) and green fluorescent protein (GFP) (green). Insets show the centrosomes; note the flower-like arrangement of the multiple centrosomes in ST-expressing cells. (f) Quantitation of supernumerary centrosomes in human U2OS cells. Mean±s.d. (23.2±3.9%) of ST-expressing cells had >2 centrosomes, versus 9.8±2.9% in control cells. n = 200 cells per sample for three independent experiments. (g) Western blot for ST expression in U2OS cells. (h) Western blot for ST-FLAG expression in Drosophila Kc cells. Lanes labeled ‘Induced’ were exposed to 1.0 mM CuSO4 for 20 h. Detection of α-tubulin in each blot was used as a loading control. (i) Supernumerary centrosomes in Kc cells following induction of ST-FLAG. Cells were stained for α-tubulin (green) and CNN (red). (j) Quantitation of centrosome overduplication in Kc cells. Mitotic cells were counted following a 20 h induction period. Four or more centrosomes were found in 44.8±3.8% (mean±s.d.)of ST-induced cells versus 21.2±1.6% for uninduced ST cells; n = 200 cells per sample for three independent experiments. Bars: 25 μm in (a, b), 15 μm in (c, d, e) and 10 μm in (i).
As induction of supernumerary centrosomes is a novel phenotype associated with ST, we asked whether ST elicited this activity in mammalian cells. To test for centrosome duplication effects of ST in human cells, we transiently transfected U2OS cells with a plasmid that expresses native ST protein. Figures 2e-g show that ST-expressing U2OS cells also have increased centrosome numbers, with 23.2±3.9% (mean±s.d.) having >2 centrosomes, compared to 9.8±2.9% of control cells.
To examine the effects of ST on centrosome duplication within the time frame of a single cell cycle, we established a stable Drosophila cell line that expresses ST-FLAG under inducible control from the metallothionein promoter (Figure 2h). The doubling time of Kc cells is approximately 24 h at 25 °C, yet after only 20 h of induction, ST-FLAG caused a significant increase in centrosome number within a single cell cycle from 21.2±1.6% (mean±s.d.) (uninduced)to 44.8±3.8% of cells that contain ≥4 centrosomes (Figures 2i and j). The excess centrosomes coalesced at the spindle poles in mitotic cells, resulting in bipolar spindles (Figure 2i). For this analysis, we restricted centrosome counts to mitotic cells, making it unlikely that tallied cells experienced cytokinesis during the 20-h induction period. Flow cytometry was used to examine cell-cycle effects of ST expression in Kc cells, yet no significant differences in cell-cycle parameters or in the polyploid cell population were seen at 20 and 42 h of induction of ST compared to the controls (Supplementary Figure S2). Therefore, centrosome overduplication did not arise as an indirect perturbation of the cell cycle by ST. Moreover, double staining of Kc cells for Cid, a kinetochore marker, and CNN showed that there was no increase in chromosome number coinciding with increased centrosome numbers (Supplementary Figure S3). Thus, supernumerary centrosomes induced by ST are achieved by overduplication rather than through a failure of cytokinesis in Kc cells.
Although centrosomes function primarily as microtubule-organizing centers (MTOCs), they also have a critical role in organizing actin at the cortex and cleavage furrow assembly in syncytial Drosophila embryos during cleavage cycles 10-14 (Raff and Glover, 1989; Rothwell and Sullivan, 2000). We find that during cortical cleavage divisions the MTOC activity of centrosomes is enhanced in ST embryos, resulting in larger astral microtubule arrays containing longer microtubules (Figure 2d). Moreover, the organization of actin into cleavage furrows is severely aberrant in ST embryos, frequently failing to assemble into furrows, but rather organizing into clumps nearby the centrosomes (Figure 2c). Neighboring spindles, normally separated by cleavage furrows, come into direct contact with each other in ST embryos, with astral microtubules from one spindle integrating with the spindle microtubules of their neighbors (arrows in Figure 2d), aprobable consequence of cleavage furrow failure (Rothwell and Sullivan, 2000). Thus, in late syncytial embryonic cleavage cycles, ST enhances microtubule stability and disrupts assembly of actin into cleavage furrows.
The disruption of actin assembly into cleavage furrows is predicted to have a severe impact on proper progression of cleavage cycles. To examine cleavage dynamics live, we imaged ST embryos using eGFP-tagged CNN and mRFP-tagged histone to label centrosomes and chromosomes, respectively (Schuh et al., 2007; Zhang and Megraw, 2007). The fluorescent signals were too weak to image early cleavage deep within the embryo; however, cortical cycles 10-13 were recorded by time-lapse confocal imaging. Control centrosomes split late in telophase and the nuclei divided synchronously as normal for blastoderm divisions (Figure 3a; Supplementary movie S1)(Callaini and Riparbelli, 1990; Rothwell and Sullivan, 2000). In ST embryos, cleavage cycles retained synchrony and progressed on a similar timescale as control embryos, demonstrating no delay in cycle completion. However, some nuclei failed to complete cleavage, causing daughter chromosomes to collapse back together after anaphase and producing polyploid nuclei associated with four centrosomes that fail to enter the next cleavage cycle (Figure 3b; Supplementary movie S2). From this analysis we conclude that ST causes cytokinesis failure in blastoderm embryos.
Figure 3.
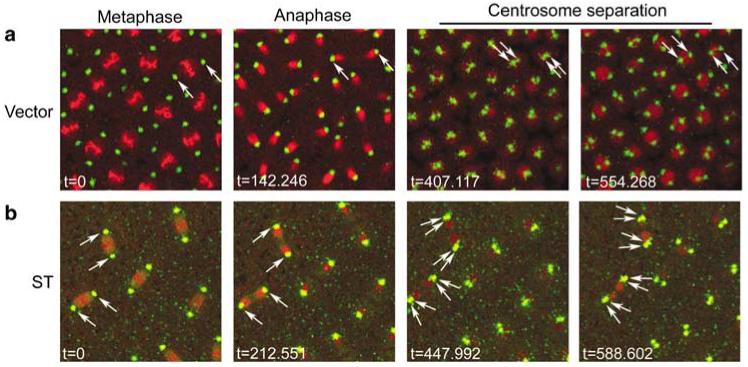
Live imaging of small tumor antigen (ST) embryos shows cytokinesis failures. Syncytial blastoderm embryos that express eGFP-centrosomin (CNN) to label centrosomes (green) and His2Av-mRFP to label chromosomes (red) were imaged by time-lapse confocal microscopy in (a) vector control embryos, and (b) ST embryos. Movies corresponding to these still images are found in Supplementary materials (movies 1 and 2). In the vector time series (a), a pair of centrosomes is followed (arrows) through mitosis and centrosome splitting until cleavage is completed. In the ST series (b), two pairs of centrosomes (arrows) are followed through a cleavage cycle in which the nuclei collapse back onto each other and fuse during centrosome separation. The following cleavage cycle for this series shows that the two indicated polyploid nuclei, each associated with four centrosomes, fail to proceed into the next mitosis (see Supplementary movie 2).
ST activities in the embryo depend on PP2A binding
To determine the relative contributions of PP2A and Hsc70 binding to the ST-associated embryonic phenotypes, we introduced independent mutations in the J domain and PP2A-binding domains (Figure 1b). To disrupt the J domain, we mutated the aspartic acid at amino acid 44 in the conserved HPD loop to asparagine (STD44N). This mutation, when introduced into LT, disrupts its interaction with Hsc70 (Sullivan et al., 2000; Sullivan and Pipas, 2002). To disrupt ST’s ability to bind PP2A, we introduced two independent mutations: a cysteine to serine amino-acid substitution at position 103 (STC103S) and a nonsense mutation at 111 (STΔPP2A). These mutations block PP2A binding and inhibition completely (STΔPP2A) (Mateer et al., 1998) or by approximately 50% (STC103S) (Mungre et al., 1994). Transgenic lines that express ST-FLAG and these mutants at similar levels (Figure 4a) were selected for phenotype analysis.
Figure 4.
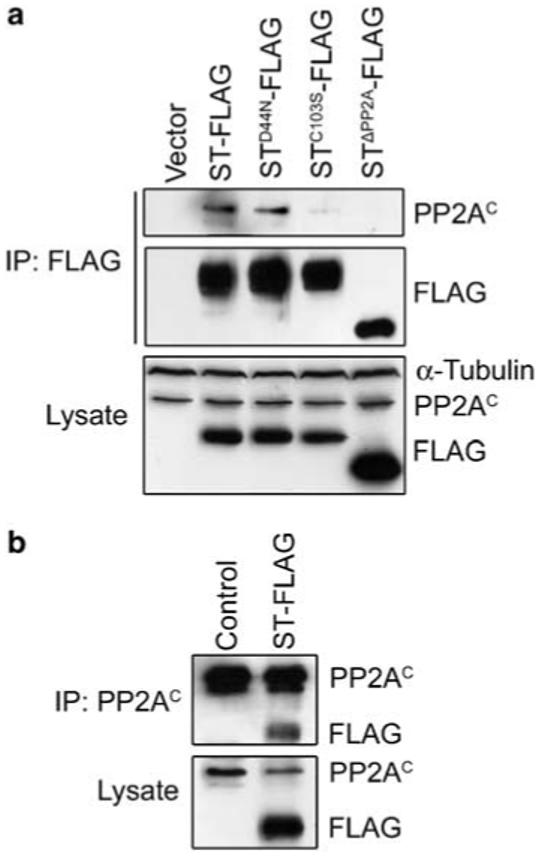
Small tumor antigen (ST) interacts with PP2A in Drosophila embryos. (a) PP2AC co-immunoprecipitates (co-IP) with ST-FLAG, but poorly with ST PP2A-binding mutants STC103S and STΔPP2A. Western blotting of lysates shows that ST-FLAG and mutant derivatives are expressed at similar levels, except for STΔPP2A-FLAG, which is approximately 1.8-fold higher than the others. α-Tubulin was used as a loading control. PP2AC co-IPs with ST-FLAG and STD44N-FLAG. A reduced amount of PP2AC co-IPs with STC103S-FLAG, and none is detected with STΔPP2A-FLAG. (b) ST-FLAG co-IPs with PP2AC from embryo lysates.
As ST interacts directly with PP2AAC (Yang et al., 1991), association of ST and ST mutant proteins with PP2A was assayed in embryo extracts by immunoprecipitation (IP). The PP2A catalytic subunit (PP2AC)coimmunoprecipitates (co-IP) with ST and STD44N efficiently from embryo lysates (Figure 4a). In reciprocal binding assays, ST co-IP with PP2AC (Figure 4b). However, IP of the two ST PP2A-binding mutants showed either reduced (STC103S), or no binding to PP2A (STΔPP2A) (Figure 4a). These data show that the association of ST with PP2A is conserved between mammals and Drosophila.
We next examined the effects of expression of ST mutants in embryos to compare their contribution to the phenotypes associated with ‘wild-type’ ST expression. The first phenotype we quantified, lethality, was dependent on PP2A binding, but not on the J domain: embryonic expression of ST and STD44N was deleterious, with ∼70% of embryos failing to hatch. Expression of STC103S and STΔPP2A, on the other hand, had little effect on embryonic survival (Figure 5a). These results indicate that binding/inhibition of PP2A, but not association with Hsc70, is required for ST-induced lethality in early embryos.
Figure 5.

Small tumor antigen (ST) phenotypes are dependent on the PP2A-binding domain. (a) Approximately 70% of ST-FLAG (ST) and STD44N-FLAG (STD44N) embryos fail to hatch (y axis is the percent of embryos that fail to hatch) at 29 °C. (b) The relative phases of embryogenesis among 3-h collections of embryos from the indicated cross. Fewer ST and STD44N embryos progress to later embryonic stages compared to control and PP2A-binding mutant ST embryos. (c) Supernumerary centrosomes occur in 55-60% of ST and STD44N embryos, whereas STC103S and STΔPP2A embryos are similar to control. (d-h) Multiple centrosomes are associated with spindle poles in ST and STD44N embryos. Control (d), ST (e) and ST mutant (f-h) embryos were stained for α-tubulin (green), centrosomin (CNN) (red) and DNA (blue). (i) Larger astral microtubules and disruption of actin assembly into cleavage furrows occurs during cortical cycles with ST and STD44N embryos, but less so in STC103S and STΔPP2A embryos. (j-n) Assembly of actin into cleavage furrows is disrupted by ST in a PP2A-dependent manner. Embryos were stained from control (j), ST (k) and ST mutants (l-n) for α-tubulin (green), actin (red) and DNA (blue). In ST and STD44N embryos, spindles collide with each other because of defective furrow assembly (k, l). The embryo in (n) is at a later cycle than those in (j-m), which is why the spindles are smaller. Bars: 25 μm in (d-h), 15 μm in (j-n).
To examine developmental delay or arrest as a possible cause for lethality, we compared embryonic stages between ST and ST mutants from a 3-h embryo collection and found that most ST and STD44N embryos failed to progress beyond the early syncytial and blastoderm cleavage cycles of embryogenesis, whereas significantly greater numbers of the vector control, STC103S and STΔPP2A embryos progressed farther to cellularization and gastrulation stages (Figure 5b). These data indicate that ST blocks or delays embryonic development during the early cleavage cycles, a stage in development that is dependent on rapid division and dynamic cytoskeletal rearrangements (Rothwell and Sullivan, 2000).
Next, we compared ST embryos to ST mutant embryos to determine which phenotypes—increased centrosome numbers, microtubule aster size and actin organization—are affected by ST mutations. We find that ST and STD44N cause supernumerary centrosomes in 55-60% of early embryos (Figure 5c). At mitosis, vector control embryos had normal bipolar spindles with one centrosome at each pole (Figure 5d), whereas ST and STD44N embryos assembled spindles with multiple centrosomes at spindle poles (Figures 5e and f). Supernumerary centrosomes occur rarely in STC103S and STΔPP2A embryos (Figures 5c, g and h). In addition, large astral microtubule arrays and disorganized cleavage furrows formed in ST and STD44N embryos compared to control embryos (Figures 5i-l). However, these effects were reduced or absent when the PP2A-binding mutants STC103S and STΔPP2A were expressed (Figures 5i, m and n). These results demonstrate that the centrosomal and cytoskeleton defects caused by ST are due to PP2A binding.
Genetic enhancement of ST by heterozygous PP2A subunit mutants
Although our results show that PP2A binding is required for ST embryonic phenotypes, we turned to genetic analysis to examine potential modifying effects that mutations in PP2A subunit genes have on ST embryos. Drosophila have single genes encoding PP2AC, PP2AA and PP2AB’ subunits, and two genes that encode PP2AB subunits. PP2AC is encoded by the microtubule star (mts)gene, PP2AA by PP2A-29B, PP2AB by twins (tws), PP2AB’-1 by well rounded (wrd)and PP2AB’-2 by widerborst (wdb). Mutations in all of these PP2A subunit genes were available or, in the case of wdb generated here.
To generate a mutation in wdb in which PP2AB’-2 expression is eliminated, we mobilized a P element transposon located at the 5′ end of the wdb gene, resulting in a 1.9-kb deletion that removes both promoters (wdb12-1; see Supplementary Figure S4). Disruption of PP2AB’-1 and PP2AB’-2 expression in these mutants was confirmed using antibodies specific to each B’ subunit (Supplementary Figure S4). The wrdKG01108 mutant was viable, producing fertile adults. On the other hand, the wdb12-1 mutation was lethal, arresting development at late pupal stages. These observations are consistent with published reports of wdb and wrd mutants (Hannus et al., 2002; Viquez et al., 2006). We also generated a wrd wdb double mutant to test for genetic redundancy between these two PP2AB’-encoding genes. The wrdKG01108 wdb12-1 double mutant arrests development at an earlier stage than the single wdb12-1 mutant, dying as third instar larvae. This novel finding indicates that the two PP2AB’ subunits are at least partially redundant in function.
With the exception of the wrdKG01108 mutant, the PP2A subunit mutations are homozygous lethal. In the heterozygous state, all the PP2A subunit mutants are viable and healthy and are maintained as heterozygous stocks.
To test for genetic interactions between ST and PP2A subunit genes, we crossed PP2A subunit mutants with ST-expressing flies to generate embryos that express ST and are heterozygous for each PP2A subunit mutation being tested. At 25 °C, a temperature that results in lower ST expression and therefore increased embryo survival, approximately 20% of ST embryos fail to hatch into larvae (Figure 6a; compare to 68% lethality at 29°C in Figure 5a). However, the addition of one mutant copy of mts, PP2A-29B or tws enhanced STembryonic lethality to >90%, whereas the wrd wdb double mutant enhanced ST-dependent lethality to 78% (Figure 6a). Although the wrd wdb double mutant enhanced ST lethality, the wrd and wdb single mutants exhibited little or no enhancement of ST-dependent lethality, again indicating redundancy between the two PP2AB’ subunits.
Figure 6.

Small tumor antigen (ST) embryonic phenotypes are enhanced by PP2A subunit-encoding mutations. Heterozygous microtubule star (mts)(PP2AC), PP2A-29B (PP2AA), tws (PP2AB), well-rounded (wrd)(PP2AB’-1), widerborst (wdb) (PP2AB’-2)and the wrd wdb double mutant were introduced into ST embryos at 25 °C. (a) All mutations enhanced lethality except for PP2AB’-1.(b) Inhibition of early embryonic development by PP2A subunit mutants. (c) The PP2AC, PP2AA and PP2AB’ double mutant increased the mitotic index. In some embryos, cleavage was disrupted to such a degree that cell-cycle stage could not be determined (‘undetermined’). (d) Supernumerary centrosomes were enhanced by all mutations except for the PP2AB’ single mutants. Representative phenotypes include multiple centrosomes (e-g), ST + PP2AA + / - showing a large spindle and scattered chromosomes (e), ST PP2AB + / - demonstrating linked spindles with aberrant microtubules and lagging chromosomes (f, arrow marks a lagging chromosome), ST + PP2AB +/- and ST + PP2AB’-1,B’-2 + / - showing loss of chromosomes from the spindle or localized near centrosomes (f, g, arrowheads) and ST PP2AC +/- demonstrating large microtubule asters associated with clumps ofactin filaments near the cortex (h). Bars: 25 μmin (e-g), 15 μm in (h).
To determine any developmental stage, cell-cycle phase, and PP2A subunit bias associated with the enhancement of ST pathology, we examined ST + PP2A+/- mutant embryos by immunostaining for centrosomes, microtubules, actin and DNA. By scoring embryonic stages among a 3-h collection, we determined that ST + PP2A+/- mutations showed a delay or arrest of embryos at early cleavage cycles, including the wrd wdb double mutant, but less so for the single mutants of either PP2AB’ gene (Figure 6b). A small increase in mitotic index occurs in ST embryos (Figure 6c). This mitotic delay is limited to the very early cleavage cycles (cycles 1-9), before the cortical cycles (10-13) (data not shown). This accounts for our observation, by live imaging, of no difference in cortical cleavage cycle duration between control and ST embryos (Figure 3; Supplementary movies S1 and S2). Nevertheless, mutations in PP2AC, PP2AA and the PP2AB’ double mutant all enhanced the mitotic index (Figure 6c).
Centrosome number increase by ST was enhanced by PP2AC, PP2AA, PP2AB and the PP2AB’ double mutant, but not by the individual PP2AB’ mutants (Figures 6d-g). In addition to increased centrosome replication, PP2A subunit mutations also enhanced spindle abnormalities, including aberrant microtubule assembly (Figure 6f), large spindles (Figure 6e) and chromosome scattering or loss from spindles (Figures 6e-g). At the cortex, the abundant centrosomes organize long microtubule asters associated with clusters of actin filaments (Figure 6h). Interestingly, although the centrosome replication, spindle microtubule and chromosome arrangement defects were associated with PP2AC, PP2AA, PP2AB and the PP2AB’ double mutants, enhancement of long astral microtubules at the cortex were seen with the PP2AC, PP2AA and PP2AB mutants, but not with the PP2AB’ double or single mutants. This suggests that the microtubule stabilization imposed by ST is mediated through competition with the PP2A-AB’C complex and not PP2A-AB’C. These strong genetic interactions between ST and PP2A subunit mutants unequivocally support a primary role for PP2A in ST-induced pathology in vivo.
Downstream effects of ST expression
PP2A has approximately 75 reported substrates, with likely more yet undiscovered (Janssens and Goris, 2001). Although ST binds PP2A and disrupts normal PP2A activity in Drosophila embryos, it did not affect the expression levels of PP2A subunits PP2AC (Figure 4)or PP2AB’-2 (data not shown). One downstream target of ST action is increased cyclin A expression (Porras et al., 1996; Schuchner and Wintersberger, 1999; Goetz et al., 2001; Skoczylas et al., 2005). We found that cyclin A levels were not altered in ST embryos (data not shown). However, the levels of another G1/S cyclin and Cdk2 activator, cyclin E, were elevated in response to ST expression by 2.3±0.58 fold (mean±s.d.) at 29 °C (Figure 7).
Figure 7.
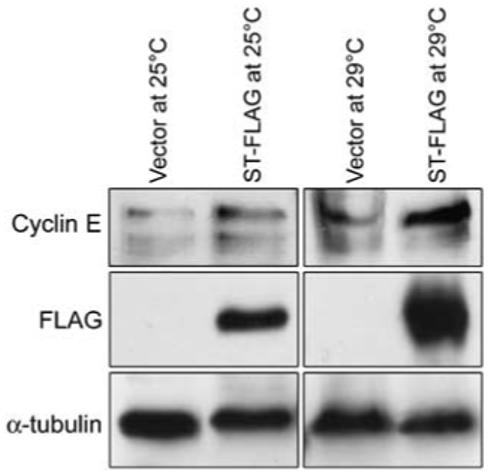
Cyclin E levels are elevated in small tumor antigen (ST) embryos. Western blot of embryo lysates from ST embryos show that cyclin E levels are elevated in ST embryos at 25 °C, and more so at 29 °C when ST levels are also increased.
Discussion
SV40 ST, together with LT, promotes cell transformation through disruption of key regulators of cell proliferation and apoptosis (Saenz-Robles et al., 2001; Lavia et al., 2003; Skoczylas et al., 2004; Arroyo and Hahn, 2005; Sontag and Sontag, 2006). Here, we show that SV40 ST causes lethality when expressed in early Drosophila embryos. In addition, ST induces increased centrosome numbers in human and Drosophila cultured cells as well as in Drosophila embryos. Moreover, in Drosophila embryos, ST expression induces multiple defects in cytoskeleton organization and in cleavage divisions, including longer astral microtubules, defective microtubule organization into mitotic spindles, chromosome segregation defects, aberrant actin assembly into cleavage furrows and cytokinesis failure. The phenotypes elicited by ST are due to disruption of PP2A, since mutations in the PP2A-binding domain, but not the J domain of ST inhibit disruption of embryo development. Moreover, mutations in the genes that encode the PP2A C, A B and B’ subunits are strong enhancers of ST phenotypes, consistent with PP2A as the primary effector of ST pathogenesis in Drosophila embryos. In addition, the levels of cyclin E, a regulator of cell-cycle entry and centrosome duplication, are elevated in ST embryos. These data establish the first Drosophila model for a viral oncoprotein, and show the efficacy of this model for genetic manipulation and for the potential to discover novel features of viral oncoprotein biology.
ST causes centrosome overduplication
Centrosomes have a dominant role in microtubule assembly into the bipolar spindle apparatus at mitosis. Therefore, centrosome duplication must be tightly regulated to ensure spindle bipolarity and accurate chromosome segregation at cell division (Nigg, 2002; Sluder and Nordberg, 2004). Chromosome missegregation leads to aneuploidy, a common feature of cancer cells. Supernumerary centrosomes are another common feature of cancer cells, and centrosomal proteins have been implicated as tumor suppressors or oncogenes (Brinkley, 2001; Pihan et al., 2001; Lingle et al., 2002; Nigg, 2002; Fukasawa, 2007).
The ability of ST to induce supernumerary centrosomes in Drosophila and human cells is a novel finding, and indicates conservation of this activity. Expression of ST in human cell culture was previously reported to block centrosome assembly and function (Gaillard et al., 2001). This discrepancy with our findings may be attributed to differences in the levels of ST expressed and/or the cell type used, suggesting context-dependent effects of ST expression. We found that centrosome overduplication was potent in ST-naïve Drosophila Kc cells: a 20-h induction of ST increased the incidence of supernumerary centrosomes from approximately 21 to 45%. Although centrosome numbers can increase through overduplication or can arise as a consequence of failed cytokinesis, ST expression in Kc cells specifically affected centrosome duplication with little or no effect on cytokinesis. Interestingly, multiple centrioles induced by ST in U2OS cells appear to assemble around a single parent centriole, similar to that seen with overexpression of Plk4, a key regulator of centriole duplication (Duensing et al., 2007; Kleylein-Sohn et al., 2007). Increased centrosome numbers are elicited by ST through PP2A perturbation, as this activity is abolished by ST mutations that block PP2A binding, and enhanced by PP2A subunit mutations. Other viral oncoproteins, including E7 from HPV-16, E1A from adenovirus-5, HBx from Hepatitis-B and Tax from HTLV-1, also induce supernumerary centrosomes (De Luca et al., 2003; Duensing and Munger, 2003; Fujii et al., 2006; Nitta et al., 2006). As E7, E1A and Tax bind to PP2A and affect its phosphatase activity (Liao and Hung, 2004; Pim et al., 2005; Hong et al., 2007), PP2A disruption may represent a shared mechanism for deregulating centrosome replication by viral oncoproteins.
Mutations in PP2AC (mts) cause uncoupling of centrosome and nuclear divisions, and longer astral microtubules that fail to attach to kinetochores (Snaith et al., 1996). Although mts mutant embryos show excess centrosomes, increased centrosome numbers were not observed in PP2AB (tws) mutant cells (Mayer-Jaekel et al., 1993; Snaith et al., 1996). RNAi depletion of PP2AC and PP2AA in Drosophila S2 cells also showed elongated microtubules in mitotic cells (Chen et al., 2007a). However, depletion of PP2AA, PP2AC or PP2AB did not increase centrosome numbers in S2 cells, and even resulted in decreased centrosome numbers (Chen et al., 2007a), a result that differs from ST expression presented here. These data suggest that, in addition to inhibiting activities toward normal PP2A substrates, ST may direct PP2A toward novel targets that regulate centrosome duplication. The idea that ST confers new target specificities to PP2A is consistent with reports of ST increasing PP2A substrate specificity for histone H1, and for novel targeting of PP2A to the androgen receptor (Yang et al., 1991, 2005). Moreover, the crystal structures of PP2A AB’C heterotrimer and the A-ST dimer are consistent with ST acting as a viral B-type PP2A subunit, perhaps conferring a new range of substrate specificities (Xu et al., 2006; Chen et al., 2007b; Cho et al., 2007; Cho and Xu, 2007). Thus, ST inhibits endogenous PP2A activity while bestowing novel target specificities.
Although the targets of PP2A-ST that contribute to the phenotypes presented here are unknown, one downstream effector of ST is cyclin E, whose levels are elevated in ST embryos. In other systems, ST stimulates AP-1 to promote transcription of cyclin D (Frost et al., 1994; Watanabe et al., 1996), which is expected to then induce cyclin E expression, driving G1 cells into S phase (Reed, 1997). Cyclin E/Cdk2 activity is required for centrosome duplication, and cyclin E + Cdk2 overexpression induces centrosome amplification (Tsou and Stearns, 2006; Duensing et al., 2007; Nigg, 2007). However, although Cdk2 + cyclin E overexpression can act synergistically with Plk4 overexpression, HPV-16 E7 expression or proteasome inhibitors to induce centriole overduplication, cyclin E or Cdk2 + cyclin E overexpression alone has only a minor effect on centriole replication (Duensing et al., 2004, 2007). Thus, the elevation of cyclin E levels induced by ST expression is unlikely sufficient to account for the increased centrosome replication we observed, but may be a contributing factor.
Microtubule stability and actin organization
The major pool of cellular PP2A is associated with microtubules and centrosomes, showing the highest degree of association at S phase, when centrosome duplication occurs (Sontag et al., 1995). Previous studies showed that PP2AC mutant Drosophila embryos display longer, more stabilized microtubules, which is why the gene encoding PP2AC was named ‘mts’ (Snaith et al., 1996). Consistent with a role for PP2A in promoting shorter microtubules, ST embryos have longer and more abundant astral microtubules. Moreover, this phenotype is dependent on ST-PP2A binding and is strongly enhanced by PP2A subunit mutants, sometimes producing astral microtubules >100-μm long. Interestingly, although the PP2A A, C and B subunit mutants enhanced microtubule stability and actin disruption at cleavage furrows, the B’ and double B’ mutants did not. This suggests that the targets for PP2A that regulate these cytoskeleton dynamics are dependent more on the B subunit than on B’. Microtubule assembly at centrosomes requires γ-tubulin for nucleation and regulators such as Aurora A kinase to promote microtubule polymerization or stability (Wiese and Zheng, 2006). PP2A regulates Aurora A levels (Horn et al., 2007) and the activity of the microtubuledestabilizing phosphoprotein Op18/Stathmin (Tournebize et al., 1997). The large asters in ST embryos may therefore be due to alterations in levels or activities of these factors. Elucidating the role of ST in microtubule stability will require the identification of PP2A substrates that regulate microtubule dynamics.
Actin assembly into cleavage furrows is severely aberrant in ST embryos, showing thicker bundles of actin cables and abnormal clumps of actin at the cortex. Enhancement of actin defects in ST embryos by PP2A A, C and B subunit mutants resulted in clumping of actin in the proximity of centrosomes, with furrow assembly severely inhibited. The conserved small G-protein regulators of actin, Cdc42 and Rac are activated by ST in MDCK cells, whereas Rho is inhibited (Nunbhakdi-Craig et al., 2003). Consistent with this, actin assembly into Drosophila cleavage furrows is regulated differentially by the active or inactive forms of Rho and Cdc42, but not Rac (Crawford et al., 1998). Thus, Cdc42 and Rho are potential ST targets affecting cleavage in Drosophila embryos. In addition, proteins that regulate actin at cytokinetic furrows are targets of PP2A (Sontag and Sontag, 2006), and therefore may be affected in ST embryos causing cytokinesis failure.
We have created the first Drosophila model for expression of a viral oncoprotein. Compared to mammalian models, some unique advantages this in vivo model for ST expression has are the molecular genetic tools available for Drosophila research, and the relative rapidity of experimental approaches to investigate the determinants of ST pathology. Establishment of this model has revealed novel ST functions, including stimulation of centrosome duplication, stabilization of microtubules, chromosomal instability (Supplementary Discussion) and inhibition of cytokinesis, all of which could reflect fundamental mechanisms by which ST contributes to transformation. Furthermore, Drosophila is a useful model for genetic screens targeting tumorigenesis and metastasis (Brumby and Richardson, 2003; Pagliarini and Xu, 2003). Future experiments based on ST expression in somatic tissues will test tumor-promoting capabilities of ST and possibly pave the way for genetic dissection of ST pathways.
Materials and methods
Antibodies
Soluble GST-ST fusion protein (amino acids 1-110) from Escherichia. coli was purified by glutathione agarose chromatography. 6XHis-tagged B56-1 (amino acids 543-656) and B56-2 (amino acids 425-524) were purified from E. coli using immobilized Ni+ + on chelating sepharose fast flow (Amersham Biosciences). Antisera were raised in rabbits by Cocalico Biologicals Inc.. For immunostaining, the following antibodies were used: rabbit anti-CNN (Zhang and Megraw, 2007) 1:2000, α-tubulin DM1a 1:1000 (Sigma), Alexa546-phalloidin 1:200 (Molecular Probes), γ-tubulin GTU-88 1:500 (Sigma), green fluorescent protein (GFP) 1:1000 (Invitrogen), rabbit anti-Cid 1:100 (Abcam), Draq5 1:1000 (Axxora). Secondary antibodies included Alexa 488 and 546 coupled goat antibodies used at 1:400 (Invitrogen). For western blotting, the following antibodies were used: FLAG (M2 monoclonal from Sigma) 1:10 000, cyclin E (a gift from H Richardson) 1:20, cyclin A clone A12 (Developmental Studies Hybridoma Bank (DSHB)) 1:5, cyclin B clone F2F4 (DSHB) 1:1000, anti-B56-1 1:10 000, anti-B56-2 1:10 000, anti-PP2AC clone 1D6 (Upstate Cell Signaling) 1:5000, anti-ST (UT450) 1:10 000, pAb280 1:1000 (Oncogene), pAb419 1:1000 (Oncogene), α-tubulin DM1a 1:10 000 (Sigma). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies were diluted 1:20 000 (Jackson IRL Inc.). Western blots were processed with SuperSignal West Pico chemiluminescent substrate (Pierce).
Additional ‘Materials and methods’ are provided in the Supplementary information.
Supplementary Material
Acknowledgements
We thank Estelle Sontag, Hui Zou, Helena Richardson, Stefan Heidmann and Kristen Johansen for reagents and Marc Mumby for a critical reading of our paper. This work was supported by grants from the National Institutes of Health (GM068756) and the Welch Foundation (I-1610).
References
- Ahuja D, Saenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–7745. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- Brumby AM, Richardson HE. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769–5779. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaini G, Riparbelli MG. Centriole and centrosome cycle in the early Drosophila embryo. J Cell Sci. 1990;97(Part 3):539–543. doi: 10.1242/jcs.97.3.539. [DOI] [PubMed] [Google Scholar]
- Chen F, Archambault V, Kar A, Lio P, D'Avino PP, Sinka R, et al. Multiple protein phosphatases are required for mitosis in Drosophila. Curr Biol. 2007a;17:293–303. doi: 10.1016/j.cub.2007.01.068. [DOI] [PubMed] [Google Scholar]
- Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004;5:127–136. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- Chen Y, Xu Y, Bao Q, Xing Y, Li Z, Lin Z, et al. Structural and biochemical insights into the regulation of protein phosphatase 2A by small t antigen of SV40. Nat Struct Mol Biol. 2007b;14:527–534. doi: 10.1038/nsmb1254. [DOI] [PubMed] [Google Scholar]
- Cho US, Morrone S, Sablina AA, Arroyo JD, Hahn WC, Xu W. Structural basis of PP2A inhibition by small t antigen. PLoS Biol. 2007;5:e202. doi: 10.1371/journal.pbio.0050202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445:53–57. doi: 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- Comerford SA, Clouthier DE, Hinnant EA, Hammer RE. Induction of hepatocyte proliferation and death by modulation of T-Antigen expression. Oncogene. 2003;22:2515–2530. doi: 10.1038/sj.onc.1206259. [DOI] [PubMed] [Google Scholar]
- Crawford JM, Harden N, Leung T, Lim L, Kiehart DP. Cellularization in Drosophila melanogaster is disrupted by the inhibition of rho activity and the activation of Cdc42 function. Dev Biol. 1998;204:151–164. doi: 10.1006/dbio.1998.9061. [DOI] [PubMed] [Google Scholar]
- De Luca A, Mangiacasale R, Severino A, Malquori L, Baldi A, Palena A, et al. E1A deregulates the centrosome cycle in a Ran GTPase-dependent manner. Cancer Res. 2003;63:1430–1437. [PubMed] [Google Scholar]
- Duensing A, Liu Y, Perdreau SA, Kleylein-Sohn J, Nigg EA, Duensing S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene. 2007;26:6280–6288. doi: 10.1038/sj.onc.1210456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Duensing A, Lee DC, Edwards KM, Piboonniyom SO, Manuel E, et al. Cyclin-dependent kinase inhibitor indirubin-3′-oxime selectively inhibits human papillomavirus type 16 E7-induced numerical centrosome anomalies. Oncogene. 2004;23:8206–8215. doi: 10.1038/sj.onc.1208012. [DOI] [PubMed] [Google Scholar]
- Duensing S, Munger K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J Virol. 2003;77:12331–12335. doi: 10.1128/JVI.77.22.12331-12335.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JA, Alberts AS, Sontag E, Guan K, Mumby MC, Feramisco JR. Simian virus 40 small t antigen cooperates with mitogen-activated kinases to stimulate AP-1 activity. Mol Cell Biol. 1994;14:6244–6252. doi: 10.1128/mcb.14.9.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii R, Zhu C, Wen Y, Marusawa H, Bailly-Maitre B, Matsuzawa S, et al. HBXIP, cellular target of hepatitis B virus oncoprotein, is a regulator of centrosome dynamics and cytokinesis. Cancer Res. 2006;66:9099–9107. doi: 10.1158/0008-5472.CAN-06-1886. [DOI] [PubMed] [Google Scholar]
- Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer. 2007;7:911–924. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- Gaillard S, Fahrbach KM, Parkati R, Rundell K. Over-expression of simian virus 40 small-T antigen blocks centrosome function and mitotic progression in human fibroblasts. J Virol. 2001;75:9799–9807. doi: 10.1128/JVI.75.20.9799-9807.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz F, Tzeng YJ, Guhl E, Merker J, Graessmann M, Graessmann A. The SV40 small t-antigen prevents mammary gland differentiation and induces breast cancer formation in transgenic mice; truncated large T-antigen molecules harboring the intact p53 and pRb binding region do not have this effect. Oncogene. 2001;20:2325–2332. doi: 10.1038/sj.onc.1204355. [DOI] [PubMed] [Google Scholar]
- Hannus M, Feiguin F, Heisenberg CP, Eaton S. Planar cell polarization requires Widerborst, a B’ regulatory subunit of protein phosphatase 2A. Development. 2002;129:3493–3503. doi: 10.1242/dev.129.14.3493. [DOI] [PubMed] [Google Scholar]
- Hong S, Wang LC, Gao X, Kuo YL, Liu B, Merling R, et al. Heptad repeats regulate protein phosphatase 2a recruitment to I-kappaB kinase gamma/NF-kappaB essential modulator and are targeted by human T-lymphotropic virus type 1 tax. J Biol Chem. 2007;282:12119–12126. doi: 10.1074/jbc.M610392200. [DOI] [PubMed] [Google Scholar]
- Horn V, Thelu J, Garcia A, Albiges-Rizo C, Block MR, Viallet J. Functional interaction of Aurora-A and PP2A during mitosis. Mol Biol Cell. 2007;18:1233–1241. doi: 10.1091/mbc.E06-12-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. Plk4-induced centriole biogenesis in human cells. Dev Cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Lavia P, Mileo AM, Giordano A, Paggi MG. Emerging roles of DNA tumor viruses in cell proliferation: new insights into genomic instability. Oncogene. 2003;22:6508–6516. doi: 10.1038/sj.onc.1206861. [DOI] [PubMed] [Google Scholar]
- Liao Y, Hung MC. A new role of protein phosphatase 2a in adenoviral E1A protein-mediated sensitization to anticancer drug-induced apoptosis in human breast cancer cells. Cancer Res. 2004;64:5938–5942. doi: 10.1158/0008-5472.CAN-04-1533. [DOI] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA. 2002;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateer SC, Fedorov SA, Mumby MC. Identification of structural elements involved in the interaction of simian virus 40 small tumor antigen with protein phosphatase 2A. J Biol Chem. 1998;273:35339–35346. doi: 10.1074/jbc.273.52.35339. [DOI] [PubMed] [Google Scholar]
- Mayer-Jaekel RE, Ohkura H, Gomes R, Sunkel CE, Baumgartner S, Hemmings BA, et al. The 55 kd regulatory subunit of Drosophila protein phosphatase 2A is required for anaphase. Cell. 1993;72:621–633. doi: 10.1016/0092-8674(93)90080-a. [DOI] [PubMed] [Google Scholar]
- Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell. 2007;130:21–24. doi: 10.1016/j.cell.2007.06.034. [DOI] [PubMed] [Google Scholar]
- Mungre S, Enderle K, Turk B, Porras A, Wu YQ, Mumby MC, et al. Mutations which affect the inhibition of protein phosphatase 2A by simian virus 40 small-t antigen in vitro decrease viral transformation. J Virol. 1994;68:1675–1681. doi: 10.1128/jvi.68.3.1675-1681.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer. 2002;2:815–825. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosome duplication: of rules and licenses. Trends Cell Biol. 2007;17:215–221. doi: 10.1016/j.tcb.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Nitta T, Kanai M, Sugihara E, Tanaka M, Sun B, Nagasawa T, et al. Centrosome amplification in adult T-cell leukemia and human T-cell leukemia virus type 1 Tax-induced human T cells. Cancer Sci. 2006;97:836–841. doi: 10.1111/j.1349-7006.2006.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunbhakdi-Craig V, Craig L, Machleidt T, Sontag E. Simian virus 40 small tumor antigen induces deregulation of the actin cytoskeleton and tight junctions in kidney epithelial cells. J Virol. 2003;77:2807–2818. doi: 10.1128/JVI.77.5.2807-2818.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302:1227–1231. doi: 10.1126/science.1088474. [DOI] [PubMed] [Google Scholar]
- Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res. 2001;61:2212–2219. [PubMed] [Google Scholar]
- Pim D, Massimi P, Dilworth SM, Banks L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene. 2005;24:7830–7838. doi: 10.1038/sj.onc.1208935. [DOI] [PubMed] [Google Scholar]
- Porras A, Bennett J, Howe A, Tokos K, Bouck N, Henglein B, et al. A novel simian virus 40 early-region domain mediates transactivation of the cyclin A promoter by small-t antigen and is required for transformation in small-t antigen-dependent assays. J Virol. 1996;70:6902–6908. doi: 10.1128/jvi.70.10.6902-6908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff JW, Glover DM. Centrosomes, and not nuclei, initiate pole cell formation in Drosophila embryos. Cell. 1989;57:611–619. doi: 10.1016/0092-8674(89)90130-x. [DOI] [PubMed] [Google Scholar]
- Reed SI. Control of the G1/S transition. Cancer Surv. 1997;29:7–23. [PubMed] [Google Scholar]
- Rothwell WF, Sullivan W. The centrosome in early Drosophila embryogenesis. Curr Top Dev Biol. 2000;49:409–447. doi: 10.1016/s0070-2153(99)49020-9. [DOI] [PubMed] [Google Scholar]
- Saenz-Robles MT, Sullivan CS, Pipas JM. Transforming functions of simian virus 40. Oncogene. 2001;20:7899–7907. doi: 10.1038/sj.onc.1204936. [DOI] [PubMed] [Google Scholar]
- Schuchner S, Wintersberger E. Binding of polyomavirus small T antigen to protein phosphatase 2A is required for elimination of p27 and support of S-phase induction in concert with large T antigen. J Virol. 1999;73:9266–9273. doi: 10.1128/jvi.73.11.9266-9273.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh M, Lehner CF, Heidmann S. Incorporation of Drosophila CID/CENP-A and CENP-C into centromeres during early embryonic anaphase. Curr Biol. 2007;17:237–243. doi: 10.1016/j.cub.2006.11.051. [DOI] [PubMed] [Google Scholar]
- Skoczylas C, Fahrbach KM, Rundell K. Cellular targets of the SV40 small-t antigen in human cell transformation. Cell Cycle. 2004;3:606–610. [PubMed] [Google Scholar]
- Skoczylas C, Henglein B, Rundell K. PP2A-dependent transactivation of the cyclin A promoter by SV40 ST is mediated by a cell cycle-regulated E2F site. Virology. 2005;332:596–601. doi: 10.1016/j.virol.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Sluder G, Nordberg JJ. The good, the bad and the ugly: the practical consequences of centrosome amplification. Curr Opin Cell Biol. 2004;16:49–54. doi: 10.1016/j.ceb.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Snaith HA, Armstrong CG, Guo Y, Kaiser K, Cohen PT. Deficiency of protein phosphatase 2A uncouples the nuclear and centrosome cycles and prevents attachment of microtubules to the kinetochore in Drosophila microtubule star (mts)embryos. J Cell Sci. 1996;109(Part 13):3001–3012. doi: 10.1242/jcs.109.13.3001. [DOI] [PubMed] [Google Scholar]
- Sontag E. Protein phosphatase 2A: the Trojan horse of cellular signaling. Cell Signal. 2001;13:7–16. doi: 10.1016/s0898-6568(00)00123-6. [DOI] [PubMed] [Google Scholar]
- Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell. 1993;75:887–897. doi: 10.1016/0092-8674(93)90533-v. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Bloom GS, Mumby MC. A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J Cell Biol. 1995;128:1131–1144. doi: 10.1083/jcb.128.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag JM, Sontag E. Regulation of cell adhesion by PP2A and SV40 small tumor antigen: an important link to cell transformation. Cell Mol Life Sci. 2006;63:2979–2991. doi: 10.1007/s00018-006-6300-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Cantalupo P, Pipas JM. The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol Cell Biol. 2000;20:6233–6243. doi: 10.1128/mcb.20.17.6233-6243.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Pipas JM. T antigens of simian virus 40: molecular chaperones for viral replication and tumorigenesis. Microbiol Mol Biol Rev. 2002;66:179–202. doi: 10.1128/MMBR.66.2.179-202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournebize R, Andersen SS, Verde F, Doree M, Karsenti E, Hyman AA. Distinct roles of PP1 and PP2A-like phosphatases in control of microtubule dynamics during mitosis. EMBO J. 1997;16:5537–5549. doi: 10.1093/emboj/16.18.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou MF, Stearns T. Controlling centrosome number: licenses and blocks. Curr Opin Cell Biol. 2006;18:74–78. doi: 10.1016/j.ceb.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Viquez NM, Li CR, Wairkar YP, DiAntonio A. The B’ protein phosphatase 2A regulatory subunit well-rounded regulates synaptic growth and cytoskeletal stability at the Drosophila neuromuscular junction. J Neurosci. 2006;26:9293–9303. doi: 10.1523/JNEUROSCI.1740-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe G, Howe A, Lee RJ, Albanese C, Shu IW, Karnezis AN, et al. Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc Natl Acad Sci USA. 1996;93:12861–12866. doi: 10.1073/pnas.93.23.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Zheng Y. Microtubule nucleation: gamma-tubulin and beyond. J Cell Sci. 2006;119:4143–4153. doi: 10.1242/jcs.03226. [DOI] [PubMed] [Google Scholar]
- Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, et al. Structure of the protein phosphatase 2A holoenzyme. Cell. 2006;127:1239–1251. doi: 10.1016/j.cell.2006.11.033. [DOI] [PubMed] [Google Scholar]
- Yang CS, Vitto MJ, Busby SA, Garcia BA, Kesler CT, Gioeli D, et al. Simian virus 40 small t antigen mediates conformation-dependent transfer of protein phosphatase 2A onto the androgen receptor. Mol Cell Biol. 2005;25:1298–1308. doi: 10.1128/MCB.25.4.1298-1308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SI, Lickteig RL, Estes R, Rundell K, Walter G, Mumby MC. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol Cell Biol. 1991;11:1988–1995. doi: 10.1128/mcb.11.4.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Megraw TL. Proper recruitment of gamma-tubulin and D-TACC/Msps to embryonic Drosophila centrosomes requires centrosomin motif 1. Mol Biol Cell. 2007;18:4037–4049. doi: 10.1091/mbc.E07-05-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.