

Abstract
Here we investigate the role of rab5 and its cognate exchange factors rabex-5 and hRME-6 in the regulation of AP2 uncoating from endocytic clathrin-coated vesicles (CCVs). In vitro, we show that the rate of AP2 uncoating from CCVs is dependent on the level of functional rab5. In vivo, overexpression of dominant-negative rab5S34N, or small interfering RNA (siRNA)–mediated depletion of hRME-6, but not rabex-5, resulted in increased steady-state levels of AP2 associated with endocytic vesicles, which is consistent with reduced uncoating efficiency. hRME-6 guanine nucleotide exchange factor activity requires hRME-6 binding to α-adaptin ear, which displaces the ear-associated μ2 kinase AAK1. siRNA-mediated depletion of hRME-6 increases phospho-μ2 levels, and expression of a phosphomimetic μ2 mutant increases levels of endocytic vesicle-associated AP2. Depletion of hRME-6 or rab5S35N expression also increases the levels of phosphoinositide 4,5-bisphosphate (PtdIns(4,5)P2) associated with endocytic vesicles. These data are consistent with a model in which hRME-6 and rab5 regulate AP2 uncoating in vivo by coordinately regulating μ2 dephosphorylation and PtdIns(4,5)P2 levels in CCVs.
Introduction
Clathrin-coated pits are a major port of entry into mammalian cells. After assembly of AP2 adaptor complexes and clathrin, other endocytic components including the GTPase dynamin and cargoes, such as transmembrane receptors, become selectively incorporated into coated pits. The coated pits then invaginate and, after scission, form clathrin-coated vesicles (CCVs). Removal (uncoating) of peripheral coat proteins is a prerequisite for the progression of these vesicles through the endocytic pathway (Conner and Schmid, 2003).
Uncoating of clathrin from isolated CCVs in vitro has been extensively characterized and requires the heat shock protein Hsc70 and auxilin, a J domain–containing cofactor (Schlossman et al., 1984; Schmid et al., 1985; Ungewickell et al., 1995; Umeda et al., 2000). However, other investigations demonstrated that AP2 uncoating requires an additional, distinct cytosolic activity (Hannan et al., 1998). Coat disassembly is facilitated by minimizing protein–protein interactions between peripheral coat proteins and transmembrane receptors established during coated pit assembly (Ricotta et al., 2002; Jackson et al., 2003; Honing et al., 2005). Neurons derived from mice lacking synaptojanin, an inositol 5′ phosphatase, display a delay in uncoating. This appears to be because of enhanced AP2 and clathrin association with the plasma membrane in a process that requires phosphoinositide 4,5-bisphosphate (PtdIns(4,5)P2; Cremona et al., 1999).
Assembly of AP2 onto the plasma membrane is mediated by a low affinity interaction between PtdIns(4,5)P2 and a binding site on the α-adaptin subunit of AP2 and is further enhanced by phosphorylation of the μ2 subunit of AP2, which promotes PtdIns(4,5)P2 binding to a distinct site on μ2 (Rohde et al., 2002; Honing et al., 2005). μ2 phosphorylation also specifically enhances its association with YxxΦ motifs within cargoes such as transferrin receptor (TfnR; Fingerhut et al., 2001; Ricotta et al., 2002). There is a μ2 kinase (most likely AAK1 [Conner and Schmid, 2002]) tightly associated with AP2. Previous studies showed that clathrin activates the μ2 kinase (Conner et al., 2003) to promote cargo sequestration into clathrin-coated pits (Jackson et al., 2003). It follows that μ2 dephosphorylation might facilitate uncoating and, indeed, studies using liver CCVs indicated that protein phosphatase 2A (PP2A) is sufficient to mediate AP1 (the adaptor protein complex present in TGN-associated CCVs) and AP2 uncoating from CCVs in vitro (Ghosh and Kornfeld, 2003). However the in vivo significance of PP2A's role has not been explored.
Rab5 is a major regulator of the early endocytic pathway. Through interactions with a variety of effector molecules, it modulates CCV budding, endosomal fusion, motility, and signaling (Zerial and McBride, 2001). Rabex-5 and RME-6 both act as guanine nucleotide exchange factors (GEFs) for rab5. Rabex-5 exists in complex with a rab5 effector, rabaptin5, and this complex appears to be functionally important for rabex-5 recruitment to endosomal membranes (Horiuchi et al., 1997; Lippe et al., 2001). Recent studies in Caenorhabditis elegans have indicated that the rab5 exchange factor RME-6 may act specifically at clathrin-coated pits (Sato et al., 2005). Mammalian orthologues of RME-6, hRME-6 (Sato et al., 2005), also known as RAP6 (Hunker et al., 2006), and GAPex5 (Lodhi et al., 2007) were found to regulate endocytic traffic (Hunker et al., 2006; Su et al., 2006; Lodhi et al., 2007).
Here we demonstrate a novel role for rab5 in specifically regulating AP2 uncoating from CCVs. We demonstrate that rab5 modulates AP2 uncoating via hRME-6 rather than rabex-5. Recruitment of hRME-6 promotes μ2 dephosphorylation. Furthermore, rab5 appears to regulate PtdIns(4,5)P2 levels in endocytic vesicles, thus providing a mechanistic symmetry to AP2 assembly during the disassembly process.
Results
Rab5 regulates uncoating of AP2 from CCVs in vitro
Our previous results indicated that rab5 acts at several steps in the CCV cycle (McLauchlan et al., 1998). We asked whether rab5 might participate in the regulation of CCV uncoating. Using in vitro uncoating assays (Ghosh and Kornfeld, 2003), we examined the effects of cytosols prepared from HEK293T cells overexpressing wild-type rab5 (rab5wt), constitutively active rab5GTP (rab5Q79L), rab5GDP (rab5S34N), and wild-type rab1 (rab1wt) on clathrin and adaptor protein (AP2 and AP1) uncoating. We found that cytosols containing elevated levels of rab5GTP (rab5wt and rab5Q79L) enhanced the extent of AP2 uncoating, monitored by released α-adaptin levels, compared with cytosols containing increased levels of either rab5GDP (rab5S34N) or rab1GTP (rab1wt; Fig. 1 A). Interestingly, the effect of rab5GTP was specific for AP2. Although all of the cytosols contain components that enhance the uncoating of AP1, there was no significant difference in their ability to uncoat either AP1 or clathrin, as assessed by immunoblotting (Fig. 1 A). The comparable effects of rab5wt and rab5Q79L are consistent with previous results showing that cytosol, containing equivalent amounts of posttranslationally modified rab5wt and rab5Q79L, show comparable stimulation of endosomal fusion in vitro (Stenmark et al., 1994a). Because Rab5S34N is presumed to exert its dominant-negative effect in vivo by sequestration of its GEF, the absence of a dominant-negative effect in vitro of cytosol containing overexpressed rab5S34N is most likely because in vitro uncoating assays measure a single round of uncoating, obviating the need for continuous exchange of GDP for GTP on rab5. These data provide the first evidence for a role for rab5 in the specific regulation of AP2 uncoating in vitro.
Figure 1.

Rab5 specifically regulates AP2 uncoating in vitro. (A) Uncoating assays were performed on CCVs isolated from porcine brain incubated with ATP, Hsc70 (1.3 μg), and cytosols (50 μg) prepared from HEK293T cells overexpressing Rab5wt, Rab5Q79L, Rab5S34N, or Rab1wt as indicated. (top) CCVs (P) were separated from released coat proteins (S) and analyzed by Western blotting for clathrin using CHC 5.9, AP2 using anti–α-adaptin mAb 100/2, and AP1 using anti–γ-adaptin mAb 100/3. (bottom) α-Adaptin, clathrin, and γ-adaptin amounts released into supernatant during uncoating quantified by densitometry of Western blots (n = 3). Data expressed as a percentage ± SEM of maximal uncoating seen in the presence of rab5wt or rab5Q79L. AP2 uncoating in the presence of rab5S34N and rab1wt was significantly different from uncoating by rab5wt (**, P < 0.01) and rab5Q79L (***, P < 0.001). (B, top) HEK293T cytosol preincubated with anti-rabaptin5 antibodies to deplete rabex-5 or with nonspecific IgG was assayed for its ability to promote clathrin, AP2, and AP1 uncoating. (bottom) α-Adaptin present in supernatant was quantified and is expressed as a percentage of coat protein uncoating after treatment of cytosol with nonspecific IgG. Results are the mean ± range of duplicate samples. (C, top) Uncoating assays were performed in the presence of HEK293T cytosol or HEK293T cytosol overexpressing rab5wt or hRME-6. (bottom) α-Adaptin present in supernatants was quantified and expressed as a percentage of AP2 uncoating after incubation with cytosol overexpressing HA–hRME-6. Results are means ± range of duplicate samples. (D, top) Cytosol overexpressing hRME-6 was treated with anti-rabaptin5 antibodies or nonspecific IgG and assayed by Western blotting for its ability to uncoat AP2. (bottom) AP2 present in supernatants was quantified and expressed as a percentage of AP2 uncoating after incubation with control cytosol. Results are means ± range of duplicate samples. (E) Uncoating assays were performed in the presence of increasing concentrations of PP2A and supernatants were assayed for release of AP1 and AP2.
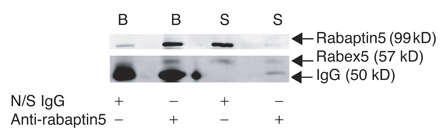
We next examined the effect of altering the levels of two rab5 exchange factors, rabex-5 and RME-6. Because rabex-5 exists in complex with a rab5 effector, rabaptin5 (Horiuchi et al., 1997; Lippe et al., 2001), immunodepletion of rabaptin5 can be used to remove rabex-5 from cytosol (Mattera et al., 2003). Cytosol depleted of rabex-5 (depletion of rabaptin5 was ∼90% and of rabex-5 >65% as assessed by Western blotting; Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200806016/DC1) showed dramatically reduced ability to support the uncoating reaction compared with a mock-depleted control (Fig. 1 B). The effect of depletion of rabaptin5–rabex-5 was specific for AP2 uncoating as AP1 was uncoated to a similar extent by mock- or rabex-5–depleted cytosol (Fig. 1 B).
Similarly, cytosol derived from cells overexpressing hRME-6 enhanced AP2 uncoating compared with control cytosol. The enhancement was comparable to that observed with cytosol overexpressing rab5wt (Fig. 1 C). When the rabaptin5–rabex-5 complex was immunodepleted from cytosol overexpressing hRME-6, there was no significant reduction in the ability of this cytosol to stimulate AP2 uncoating (Fig. 1 D). Collectively, these findings suggest that these two exchange factors are functionally redundant in vitro.
Although Hsc70 and auxilin are necessary and sufficient for clathrin uncoating in vitro, an additional cytosolic factor has been shown to be required for removal of AP2 from CCVs (Hannan et al., 1998). PP2A was shown to be sufficient to remove both AP2 and AP1 from liver-derived CCVs (Ghosh and Kornfeld, 2003). Consistent with these results and using brain-derived CCVs, we also found that PP2A is capable of uncoating AP2 as well as AP1 (Fig. 1 E).
Specific regulation of AP2 uncoating by rab5 in vivo
Having identified a role for rab5 during uncoating in vitro, we wanted to investigate if rab5 also regulates AP2 uncoating in living cells. We established a system to measure differences in the extent of colocalization of AP2 with endocytic vesicles as a relative measure of uncoating. Cells were incubated for 5 min at 37°C with Texas red or Cy5-transferrin (Tfn) and then surface Tfn was removed by acid stripping before antibody staining. Control experiments where cells were incubated on ice revealed that acid stripping effectively removed 95% of the surface fluorescence (unpublished data). The degree of colocalization of AP2 with Tfn (Fig. 2 A) was then measured as described in Materials and methods. In mock-treated cells, no more than 10% of the AP2-positive spots colocalized with Tfn after acid stripping. We compared the extent of AP2 colocalization with endocytic vesicles labeled with Texas red Tfn in mock-transfected cells and those overexpressing rab5S34N, which has a dominant-negative effect on endocytosis (Li and Stahl, 1993). We expressed the extent of overlap of AP2 and Tfn in mock-transfected cells as 1 and found a significant increase in the degree of colocalization of AP2 with endocytic vesicles (1.46 ± 0.13 vs. 1 ± 0.06 fold) in cells with elevated levels of rab5GDP (Fig. 2 B).
Figure 2.

Rab5 regulates AP2 uncoating in vivo. HEK293T cells, either mock transfected or transfected with rab5S34N, were incubated with Texas red Tfn for 5 min at 37°C. After fixation, the surface Texas red Tfn was removed by acid stripping and AP2 was visualized using the mAb AP.6. (A) Representative images of mock-transfected cells or cells overexpressing rab5S34N labeled with Texas red Tfn and AP.6. The arrowheads indicate an overlap between the two shown enlarged in the inset. Bars, 6 μm. (B) Quantification of the degree of overlap of Texas red Tfn with AP.6 in mock-transfected cells and those overexpressing rab5S34N. The degree of overlap between the two markers was set at 1 in mock-transfected cells. Results are expressed as the fold change in overlap compared with mock-transfected cells and are the mean ± SEM of three experiments where at least 25 cells were analyzed for each condition in each experiment. Values are significantly different at P < 0.01 (**). (C) Electron micrograph of a clathrin-coated pit labeled with 10-nm gold–conjugated B3/25 antibodies recognizing the ectodomain of TfnR. Bar, 50 nm. (D) The number of gold-labeled particles per pit was counted in untransfected HEK293T cells or those transfected with rab5wt, rab5Q79L, and rab5S34N. Results are expressed as particles per pit as a percentage of the total number of pits counted. 100 coated pits were counted in each sample. (E) Quantitation of the number of coated pits per profile in untransfected HEK293T cells and those transfected with rab5wt, rab5Q79L, and rab5S34N. Results are expressed as a percentage of pit number per profile in untransfected cells. For each population, at least 60 cell profiles were counted. (F) Representative image of mock-transfected HEK293T cells and those transfected with rab5S34N, which were incubated with Texas red Tfn, acid stripped, and then stained for clathrin using mAb X22. The arrowheads indicate an overlap between the two shown enlarged in the inset. Bars: (top) 7 μm; (bottom) 6 μm. (G) The degree of overlap of clathrin, measured by immunofluorescence using mAb X22, with Texas red Tfn was measured in mock-transfected HEK293T cells or those transfected with rab5S34N. The degree of overlap between X22 and Texas red Tfn was set at 1 in mock-transfected cells and the results are expressed as the fold change in overlap compared with mock-transfected cells and are the mean ± SEM of two experiments where at least 25 cells were analyzed in each experiment.
One possible explanation for these data could be that rab5GDP decreases cargo recruitment into coated pits. If this was the case, then the enhanced association of AP2 with Tfn-positive endocytic, observed after rab5S34N overexpression, could result from lower concentrations of cargo molecules per individual vesicle. To address this, we used gold-labeled B3/25 antibodies that recognize the TfnR ectodomain to measure the density of TfnR per coated pit in mock-transfected cells and in cells overexpressing rab5 mutants (Fig. 2 C). Quantification of gold particles revealed no significant difference in the density of cargo per pit in control cells versus cells overexpressing rab5wt, rab5Q79L, or rab5S34N (Fig. 2 D). However, there were fewer coated pits per profile in cells overexpressing rab5Q79L (Fig. 2 E).
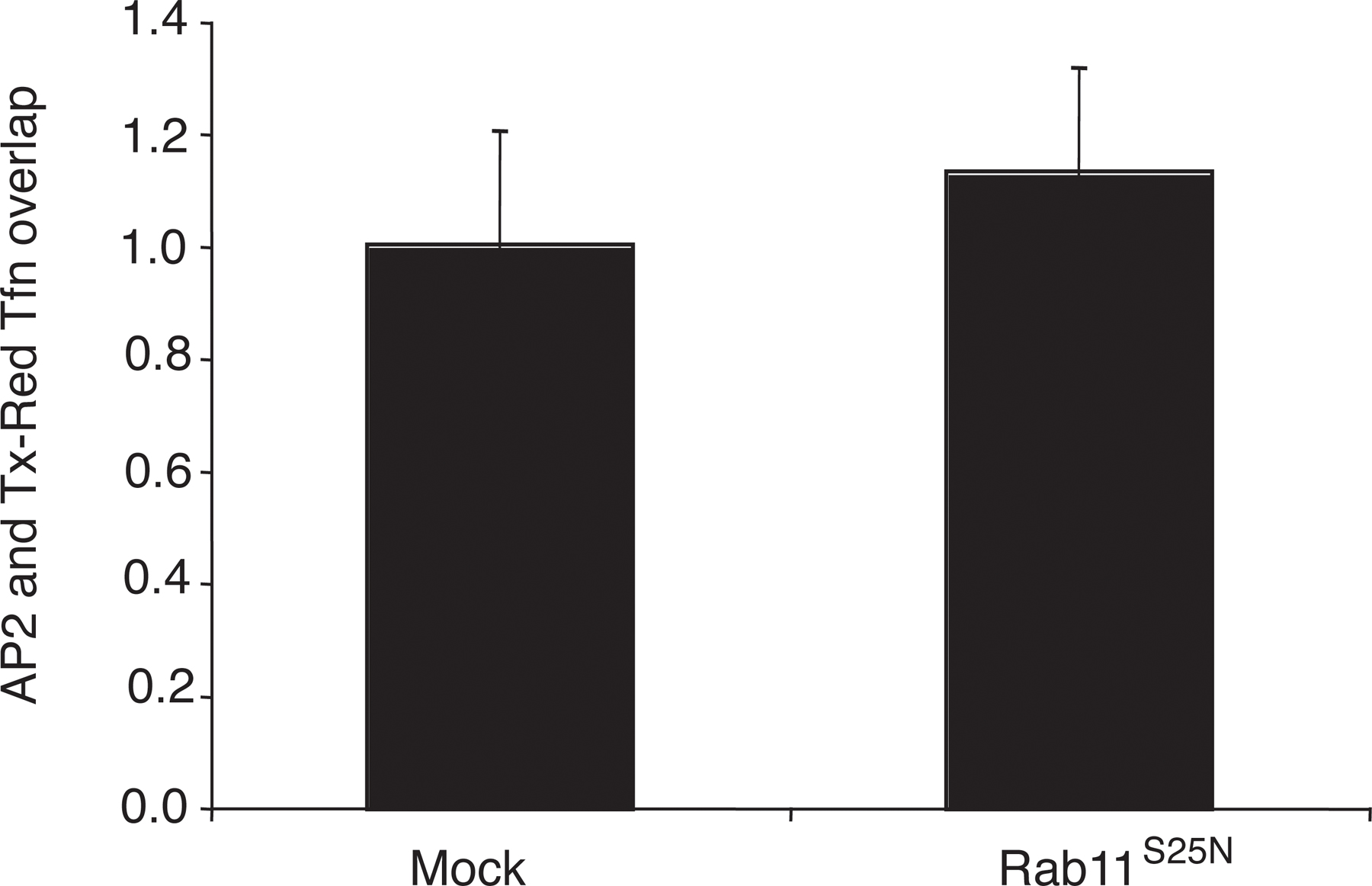
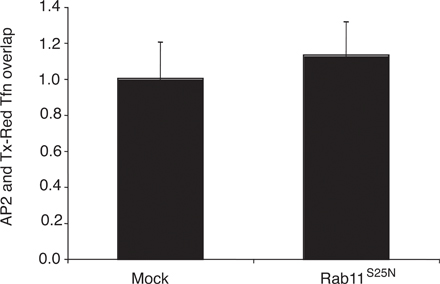
In contrast to our observations on AP2, we found no change in clathrin association with endocytic vesicles in mock-transfected cells versus those where rab5S34N was overexpressed (Fig. 2, F and G). Furthermore, overexpression of dominant-negative rab11 (rab11S25N), which inhibits the recycling pathway, did not significantly affect the degree of colocalization of AP2 with endocytic vesicles compared with mock-treated cells (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200806016/DC1). These data therefore are fully consistent with the hypothesis that rab5 regulates AP2 uncoating in vivo, mirroring our in vitro results showing that rab5 specifically regulates AP2 uncoating.
Recruitment of rab5 to clathrin-coated pits
Because RME-6 acts as a GEF for rab5 at clathrin-coated pits in C. elegans (Sato et al., 2005), we wished to investigate potential mechanisms of hRME-6 recruitment to CCVs to address how rab5 participates in uncoating. GST pull-down experiments indicated that, like its nematode orthologue (Sato et al., 2005), hRME-6 binds to the α-adaptin ear (Fig. 3 A). AAK1 and hRME-6 have been suggested to bind to the same site on α-adaptin, via WxxF motifs (Mishra et al., 2004). In support of this, we found that hRME-6 competes with AAK1 for binding to α-adaptin ear (Fig. 3 A). This suggests a possible mechanism whereby rab5 recruitment would result in net μ2 dephosphorylation as a result of displacement of AAK1 from AP2 by hRME-6. We identified a WxxF motif in the C terminus of hRME-6, 1484WMQF. Mutation of phenylalanine1487 to alanine hRME-6F1487A resulted in an approximately fivefold reduced binding of hRME-6 to α-adaptin ear (Fig. 3 B), confirming that this motif promotes interaction between the two proteins. Binding of hRME-6 to α-adaptin appears to be regulated. hRME-6 derived from cytosol preincubated with MgATP and phosphatase inhibitors bound less efficiently (at least twofold) to α-adaptin compared with hRME-6 from mock-treated cytosol (Fig. 3 C). Phosphorylation may therefore negatively regulate binding of hRME-6 to AP2 early in the CCV cycle when AAK1 activity is required for cargo recruitment.
Figure 3.

hRME-6 acts as a rab5GEF through interactions with clathrin-coated pit components. (A, top) Immobilized GST–α-adaptin ear was incubated with HEK293T cytosol and increasing amounts of HEK293T cytosol prepared from cells overexpressing HA–hRME-6wt as indicated with the total amount of cytosol being maintained constant (120 μg). Bound proteins were detected by Western blotting using anti-HA and anti-AAK1 antibodies. (bottom) Quantitation of HA–hRME-6 versus AAK1 bound proteins. Results are the mean ± the range of three experiments where the values are expressed as a percentage of the maximum HA–hRME-6 and AAK1 bound proteins. (B) Immobilized GST–α-adaptin ear was incubated with cytosol prepared from HEK293T cells overexpressing HA-tagged hRME-6wt or hRME-6F1487A. The specific binding of both proteins to GST–α-adaptin ear was quantified from Western blots using anti-HA antibodies. Results are the mean and standard deviation of four experiments where the values are expressed as a percentage of the maximum binding of HA–hRME-6wt. Asterisk indicates that values are significant at P < 0.05. (C) Immobilized GST–α-adaptin ear was incubated with brain-purified AAK1 (0.75 μg), HEK293T cytosol, or HEK293T cytosol prepared from cells overexpressing HA–hRME-6wt, preincubated for 15 min at 30°C in the presence (phospho conditions) or absence (dephospho conditions) of 2 mM MgATP, 1 μM microcystin, and 1 mM sodium vanadate. Bound proteins were detected using anti-AAK1 and anti-HA antibodies. (D) SDS gel stained with Sypro Ruby of HA–hRME-6wt and HA–hRME-6F1487A affinity purified from HEK293T cells. (E) GEF assays were performed in the presence of 1 mM ATP (regenerating system), 2 μM GST–α-adaptin ear, 8.8 nM HA–hRME-6wt or HA–RME-6F1487A, and 120 nM GAPex5 VPS9 domain as indicated. Results are the means ± SD of at least two experiments performed in duplicate.
Because the VPS9 domain of hRME-6/RAP6 has GEF activity in vitro (Hunker et al., 2006), we set out to measure GEF activity (Hama et al., 1999) of the full-length protein. We overexpressed HA-tagged hRME-6wt or hRME-6F1487A in HEK293T cells and immunopurified both proteins using anti-HA antibodies (Fig. 3 D). Surprisingly, full-length hRME-6 showed no intrinsic GEF activity. However, robust activity was observed in the presence of both ATP and GST α-adaptin ear domain. The measured activity was comparable to that observed at >10-fold higher concentration of the GAPex5 VPS9 domain, which notably was not activated by the presence of ATP or ear (unpublished data). Importantly, addition of α-adaptin ear and ATP had no effect on hRME-6F1487A exchange activity, which lacks the α-adaptin ear binding motif (Fig. 3 E). These data suggest that hRME-6 is inactive outside of coated pits and only activates rab5 upon binding to α-adaptin during or after coated pit formation.
hRME-6 is the rab5 GEF that regulates AP2 uncoating
Our results indicated that hRME-6 might be the relevant GEF for rab5-mediated uncoating but that hRME-6 and rabex-5 are functionally interchangeable in in vitro uncoating assays. To address which GEF is necessary in vivo, we used siRNAs to knockdown either hRME-6 or rabex-5 in HEK293T cells, resulting in 78 and 63% knockdown, respectively (Fig. 4 A). We then asked what effect knockdown of these GEFs had on association of AP2 with endocytic vesicles as described for Fig. 2. As before, we expressed degree of overlap of AP2 with Tfn as 1 in control-treated cells. We found that hRME-6 knockdown resulted in significant enhancement in AP2 colocalization with endocytic vesicles compared with cells transfected with control siRNA (2 ± 0.14-fold compared with 1 ± 0.1-fold for control-treated and 0.89 ± 0.11-fold for rabex-5-treated cells; Fig. 4, B and C). This was comparable to, or greater than, the effect seen upon overexpression of rab5S34N and contrasted with the lack of a significant effect seen upon rabex-5 knockdown. To eliminate the possibility that the effect of hRME-6 on AP2 uncoating was caused by an indirect effect of rab5 acting later in the endocytic pathway, we compared overexpression of HA–hRME-6 and rabaptin5. Overexpression of rabaptin5 (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200806016/DC1; Stenmark et al., 1995), rab5Q79L (Fig. S3; Stenmark et al., 1994b), or rabex-5 (Zhu et al., 2007) resulted in enlarged endosome formation. In contrast, there was no apparent endosomal enlargement in the presence of overexpressed HA–hRME-6 (Fig. S3). These results support the hypothesis that hRME-6 rather than rabex-5 is the relevant GEF for rab5-mediated AP2 uncoating.
Figure 4.

hRME-6 acts as a GEF for rab5 during AP2 uncoating. (A) Western blots of HEK293T cells treated with control siRNA and siRNAs directed against hRME-6 and rabex-5. (B) Representative images of siRNA-treated cells incubated with Texas red Tfn and, after acid stripping, stained for AP2 using AP.6 mAb. The arrowheads indicate an overlap between the two shown enlarged in the inset. Bar, 10 μm. (C) Quantitation of the degree of overlap between AP2 and Texas red Tfn in cells treated with control siRNA and siRNA against hRME-6 and rabex-5. The degree of overlap between AP.6 and Texas red Tfn was set at 1 in control cells. Results are expressed as the fold change in overlap compared with control cells ± SEM and are the results of three experiments where at least 25 cells were analyzed. Values are significant at P < 0.01 (**) for control versus hRME-6 knockdown and rabex-5 versus hRME-6 knockdown.
hRME-6 recruitment results in μ2 dephosphorylation
μ2 phosphorylation is maintained by a balance of kinase (AAK1) and phosphatase activities (Jackson et al., 2003). Although hRME-6 can displace AAK1 from the ear of α-adaptin in vitro, this would be physiologically relevant only if the activity of AAK1 is continuously required during coated pit assembly and if μ2 phosphorylation affects the rate of AP2 uncoating. We first asked whether AAK1 continues to be active in a CCV. CCVs were incubated in the presence of MgATP and the level of phospho-μ2 measured using phosphospecific antibodies (Jackson et al., 2003). As expected, isolated CCVs contain significant amounts of phospho-μ2, which increased in a time-dependent manner upon incubation (Fig. 5 A), indicating that AAK1 continues to be active in CCVs.
Figure 5.

μ2 dephosphorylation is required for efficient AP2 uncoating. (A) AAK1 is active in CCVs. CCVs were incubated in the presence of ATP for various times as indicated. Samples were centrifuged and pellets (containing CCVs) and supernatants were assayed for phosphorylated μ2 using phosphospecific antibodies. (B and C) HEK293T cells were either mock transfected or transfected with HA-μ2wt, HA-μ2T165A, or HA-μ2T156D. After fixation, cells were colabeled with 10-nm gold–conjugated anti-TfnR antibodies (B3/25) and 5-nm gold–labeled anti-HA antibodies. (B) Representative example of a labeled coated pit showing the anti-HA antibodies labeling the inner surface of the plasma membrane (arrow) and the anti-TfnR antibodies (arrowhead) marking the extracellular leaflet. Bar, 50 nm. (C) Anti-TfnR gold particles were counted in HA-positive pits. More than 50 pits were counted for each condition. Results are expressed as the number of pits counted containing zero, one, two, or three gold particles. (D) Cells were transfected with either HA-μ2wt or HA-μ2T156D and incubated with Texas red Tfn for 5 min at 37°C. After fixation and acid stripping of the surface Tfn, cells were labeled with anti-HA antibodies (green). Bar, 7 μm. (E) Quantitation of the overlap of HA with Texas red Tfn. The degree of overlap between HA and Texas red Tfn was set at 1 in HA-μ2wt cells. Results are expressed as the fold change in overlap compared with HA-μ2wt cells and are from two experiments and are significant at P < 0.05 (*). (F) Western blot comparing the levels of μ2 and phospho-μ2 in mock-treated cells and those treated with siRNA against hRME-6. Histogram shows the quantification of the ratio of phospho-μ2/μ2 ± the range of two experiments. (G) Western blot comparing the levels of μ2 and phospho-μ2 in cells overexpressing HA–hRME-6wt and HA–RME-6F1487A compared with mock-transfected cells (Con). Histogram showing the quantification of the ratio of phospho-μ2/μ2 ± SEM from three experiments.
Our previous studies demonstrated that replacement of endogenous μ2 by overexpression of a mutant form of μ2 that cannot be phosphorylated (HAμ2T156A) results in inhibition of Tfn internalization (Olusanya et al., 2001). Overexpression of a phosphomimetic form of μ2 (HAμ2T156D) results in similar inhibition (unpublished data). To investigate how these mutants inhibit internalization, we analyzed cargo recruitment using the electron microscopy assay described in Fig. 2 (C and D). The amount of cargo per coated pit was estimated by labeling the TfnR ectodomain with 10-nm gold–conjugated B3/25. Cells expressing HA-tagged proteins were identified using 5-nm gold–conjugated anti-HA antibodies (Fig. 5 B). Expression of HAμ2T156A resulted in a dramatic reduction in cargo recruitment into clathrin-coated pits, demonstrating in vivo that μ2 phosphorylation does indeed enhance recruitment of cargo such as TfnR as suggested by in vitro studies (Olusanya et al., 2001; Ricotta et al., 2002; Collins et al., 2002; Honing et al., 2005). In contrast, expression of HAμ2T156D had no significant effect on the ability of coated pits to recruit TfnR compared with cells expressing HAμ2wt or untransfected cells (Fig. 5 C). These data indicate that, although unphosphorylatable and phosphomimetic μ2 mutants both inhibit Tfn internalization, the underlying mechanisms are distinct, and that inhibition of endocytosis in the presence of HAμ2T156D must occur after cargo recruitment during the CCV cycle. To test this, we measured the degree of colocalization of AP2 with endocytic vesicles labeled with Texas red Tfn in cells overexpressing either HAμ2wt or HAμ2T156D (Fig. 5, D and E). Expression of the phosphomimetic form resulted in an enhancement in colocalization of AP2 with endocytic vesicles (1.55 ± 0.19-fold vs. 1 ± 0.11-fold), indicating that dephosphorylation of μ2 is indeed a prerequisite for efficient AP2 uncoating.
If hRME-6 acts to displace AAK1, modulation of hRME-6 levels should affect the steady-state levels of phospho-μ2. We compared phospho-μ2 levels in cells where hRME-6 had been depleted using siRNA with mock-depleted cells and found a significant increase in the amount of phospho-μ2 in depleted cells (Fig. 5 F). Similarly, cells overexpressing hRME-6wt showed a significant reduction in the amount of phospho-μ2 compared with control cells and those expressing hRME-6F1487A (Fig. 5 G). These data provide in vivo support for our in vitro data, strongly suggesting that μ2 dephosphorylation is regulated by recruitment of hRME-6.
Modulation of rab5 activity affects PtdIns(4,5)P2 levels during AP2 uncoating
PtdIns(4,5)P2 is the primary determinant for recruitment of AP2 to the plasma membrane (Honing et al., 2005; Motley et al., 2006). Given the specificity of rab5 for the regulation of AP2 uncoating compared with clathrin, we investigated whether PtdIns(4,5)P2 levels were altered in CCVs in cells overexpressing rab5S34N. Using the approach described in Fig. 2, we asked whether overexpression of rab5S34N altered the degree of colocalization of PtdIns(4,5)P2 with endocytic vesicles labeled with a Tfn pulse. To measure PtdIns(4,5)P2 levels, we used 2C11, an antibody specific for PtdIns(4,5)P2 (Thomas et al., 1999; Hammond et al., 2006). We further confirmed its specificity by labeling cells with 2C11 in the presence of an excess of a GST fusion protein including 2xFYVE domains (recognizing PtdIns3P; Pattni et al., 2001) or the pleckstrin homology domain of PLCδ (specific for PtdIns(4,5)P2; Balla, 2005). We found that the latter, but not the former, was capable of competing out the immunofluorescence signal seen with 2C11 (Fig. 6 A). In control cells, <20% of 2C11-positive puncta colocalized with internalized Tfn. In cells overexpressing rab5wt and rab5S34N, there was significantly more colocalization of PtdIns(4,5)P2 with Tfn-positive endocytic vesicles in the presence of overexpressed rab5S34N compared with cells overexpressing rab5wt (1.38 ± 0.08-fold vs. 1 ± 0.07-fold; Fig. 6, B and C). These data indicate that interfering with rab5 activity in vivo affects PtdIns(4,5)P2 turnover in endocytic vesicles.
Figure 6.

Rab5 regulates AP2 uncoating via modulation of PtdIns(4,5)P2 levels. (A) Immunofluorescence images of HEK293T cells labeled with the anti-PtdIns(4,5)P2 antibody 2C11 (left) or with 2C11 in the presence of excess GST-2xFYVE (middle) or GST pleckstrin homology domain of PLCδ (right). Bar, 7 μm. (B) Representative image of HEK293T cells transfected with rab5wt or rab5S34N, which were incubated with Cy5-Tfn, acid-stripped, and then stained for PtdIns(4,5)P2 using mAb 2C11. The arrowheads indicate an overlap between the two shown enlarged in the inset. Bar, 9 μm. (C) Quantitation of the overlap between PtdIns(4,5)P2 and Cy5-Tfn in HEK293T cells overexpressing rab5wt or rab5S34N. The degree of overlap between 2C11 and Cy5-Tfn was set at 1 in rab5wt-transfected cells. Results are expressed as the fold change in overlap compared with rab5wt-transfected cells and are the mean ± of three experiments where at least 25 cells were counted for each condition and are significant at P < 0.01 (**). (D) Representative images of siRNA-treated cells incubated with Cy5-Tfn and, after acid stripping, stained using 2C11. The arrowheads indicate an overlap between the two shown enlarged in the inset. Bar, 10 μm. (E) Quantitation of the degree of overlap between PtdIns(4,5)P2 and Cy5-Tfn in cells treated with control siRNA and siRNA against hRME-6 and rabex-5. The degree of overlap between 2C11 and Cy5-Tfn was set at 1 in control-treated cells and the results are expressed as the fold change in overlap compared with control and are the mean of three experiments ± SEM where at least 25 cells were analyzed. Values are significant at P < 0.01 (**) for control versus hRME-6 knockdown and P < 0.05 (*) for rabex 5 versus hRME-6 knockdown.
We also tested whether hRME-6– or rabex-5–depleted cells showed altered PtdIns(4,5)P2 levels. Cells treated with siRNA against hRME-6, but not rabex-5, showed enhanced colocalization of PtdIns(4,5)P2 with internalized TfnR (1.45 ± 0.09-fold compared with 1 ± 0.09-fold for control siRNA and 1.1 ± 0.06-fold for rabex-5 siRNA; Fig. 6, D and E). Thus, interfering with rab5 activity, using either dominant-negative rab5S34N or hRME-6 depletion, results in alterations in PtdIns(4,5)P2 turnover and increased steady-state levels of AP2 associated with endocytic vesicles. Collectively, these data strongly suggest that hRME-6/rab5 regulates AP2 uncoating by modulating PtdIns(4,5)P2 turnover.
Discussion
Rab5 is a key regulator of the early endocytic pathway and controls multiple steps of the CCV cycle, endosomal fusion, motility, and signaling (Zerial and McBride, 2001). In this study we have identified a further novel role for rab5 and its GEF, hRME-6, in the specific regulation of AP2 uncoating from CCVs. In vitro, we show that the extent of AP2 uncoating from CCVs is dependent on the level of functional rab5. Overexpression of rab5S34N, or siRNA-mediated depletion of hRME-6, but not rabex-5, in intact cells resulted in increased steady-state levels of AP2 associated with endocytic vesicles, which is consistent with reduced efficiency of uncoating. We demonstrated in vitro that GEF activity of full-length hRME-6 requires hRME-6 binding to α-adaptin ear, which displaces the ear-associated μ2 kinase AAK1 and that, in intact cells, depletion of hRME-6 increases phospho-μ2 levels whereas overexpression of hRME-6 reduces phospho-μ2 levels. The contribution of μ2 dephosphorylation to efficient uncoating was shown by expression of a phosphomimetic mutant of μ2, which increases levels of endocytic vesicle-associated AP2. Depletion of hRME-6 or rab5S35N expression also increases the levels of PtdIns(4,5)P2 associated with endocytic vesicles. These data are consistent with a model in which hRME-6 and rab5 regulate AP2 uncoating in vivo by coordinately regulating μ2 dephosphorylation and PtdIns(4,5)P2 levels in CCVs (Fig. 7).
Figure 7.

Model for the role of rab5 in AP2 uncoating. Rab5 acts at multiple steps of the clathrin-coated vesicle cycle (red arrows). PtdIns(4,5)P2 is the primary determinant for the recruitment of AP2 to the plasma membrane during coated pit assembly and this process is enhanced by μ2 phosphorylation, effected by AAK1 (1). Clathrin activates AAK1 to maximize cargo recruitment (2). After coated vesicle scission (3), clathrin is removed by the combined action of Hsc70 and auxilin. Recruitment of hRME-6 displaces AAK1, resulting in net μ2 dephosphorylation. Rab5 mediates PtdIns(4,5)P2 turnover, resulting in efficient removal of AP2 and generation of an uncoated endocytic vesicle (4). There is therefore a mechanistic symmetry between AP2 assembly that requires PtdIns(4,5)P2 and μ2 phosphorylation and disassembly that is mediated by μ2 dephosphorylation and a reduction in PtdIns(4,5)P2. Both the latter processes are modulated by hRME-6/rab5.
A novel role for rab5 in AP2 uncoating
Rab5 has been shown to generate specific membrane domains that are important for its cellular functions, such as endosome fusion and signaling (Zerial and McBride, 2001). The novel role of rab5 in mediating AP2 uncoating may be a prerequisite for the generation of a fusion-competent domain on endocytic vesicles. Although our evidence strongly supports hRME-6 as the relevant GEF for regulation of AP2 uncoating, earlier studies have demonstrated that rabaptin5, and presumably rabex-5, can be recruited to CCVs to promote efficient fusion in vitro (Rubino et al., 2000). Recruitment of rabaptin5–rabex-5 and generation of a “fusion domain” may occur downstream of hRME-6 activity. Therefore, functionally distinct pools of rab5 may be sequentially activated on endocytic vesicles by the coordinated action of different GEFs.
Our in vivo data demonstrate that rab5S34N expression causes a 50% increase in AP2 steady-state levels associated with endocytic vesicles. Previous experiments demonstrated ∼50% reduction in Tfn uptake in cells expressing another dominant-negative form of rab5 (rab5N133I; Bucci et al., 1992). If these results suggest that rab5 is limiting in clathrin-mediated uptake, subsequent in vivo studies have proven controversial with cases in which rab5 is clearly limiting and others in which it is not, suggesting that rab5 may play a regulatory role in the early steps of endocytosis in a cell type–specific manner (Barbieri et al., 2000; Dinneen and Ceresa, 2004). Because rab5 is likely to modulate the rate of AP2 uncoating via interactions with downstream effectors, including lipid and protein kinases and phosphatases, regulation of the activities of these effectors may be dependent on cellular context.
Because rab5 acts at multiple stages on the endocytic pathway, it can be difficult to assign direct rather than indirect effects to a particular event in vivo. For example, enhanced association of AP2 with endocytic vesicles in the presence of rab5S34N could also result from reduced cargo recruitment into coated pits. Quantitative electron microscopy allowed us to eliminate this possibility by demonstrating that neither overexpression of rab5wt, rab5Q79L, nor rab5S34N affected cargo recruitment into clathrin-coated pits. Although it is possible that observed effects on μ2 dephosphorylation and PtdIns(4,5)P2 could be a consequence of a generalized delay in endocytosis when rab5 activity is inhibited, several lines of evidence support the model whereby increased μ2 phosphorylation and PtdIns(4,5)P2 directly contribute to reduced AP2 uncoating. First, overexpression of rab5S34N did not affect clathrin uncoating, which argues against a global disruption of the endocytic pathway. Second, expression of a μ2 phosphomimetic mutant also resulted in delayed AP2 uncoating, thus providing independent evidence for μ2 dephosphorylation promoting efficient AP2 uncoating. Third, in contrast to overexpression of rab5Q79L or rabaptin5, overexpression of hRME-6 did not result in enlarged endosomes, a phenomenon that is also observed on overexpression of rabex-5 (Zhu et al., 2007). Disruption of endosomal dynamics by modulation of rabex-5 levels might be predicted to have indirect effects on the extent of recycling of components required at the plasma membrane. Rabex-5 knockdown did not have any appreciable effect on AP2 uncoating or PtdIns(4,5)P2 levels on endocytic vesicles although we cannot rule out the possibility that the knockdown was not sufficiently complete. However, the effects observed after hRME-6 knockdown are comparable to those seen on expression of rab5S34N, which argues against redundancy of rabex-5 and hRME-6. Fourthly, rab11S25N overexpression, which inhibits recycling, had no significant effect on AP2 uncoating. Finally, because PtdIns(4,5)P2 and μ2 phosphorylation promote AP2 assembly, it is reasonable that the disassembly process might occur by a reversal of these processes.
Although live cell imaging has revealed that clathrin and AP2 are apparently removed from endocytic vesicles with similar kinetics (Ehrlich et al., 2004), a variety of in vitro studies have indicated that uncoating of each has distinct requirements (Chappell et al., 1986; Ungewickell et al., 1995; Hannan et al., 1998; Ghosh and Kornfeld, 2003). Consistent with these observations, we found that expression of rab5S34N interfered with AP2 but not clathrin uncoating in intact cells, suggesting that in wild-type cells, despite identical kinetics, there is no interdependence of clathrin and AP2 uncoating. It is unclear why clathrin and AP2 uncoating are independently regulated although there are at least two possible explanations. First, clathrin participates in several trafficking steps, including formation of TGN-derived CCVs for lysosomal enzyme transport and coated vesicles that bud from recycling endosomes. hRME-6/Rab5–mediated regulation of AP2 uncoating could be a reflection of rab5 specificity in the early endocytic pathway. A second possibility, consistent with previous suggestions (Traub, 2003), is that although clathrin is a component of all CCVs, AP2 acts as the major adaptor complex for a subset of cargo only in HEK293T cells. It follows that distinct mechanisms would therefore operate to uncoat clathrin and specific adaptor complexes and the role of rab5 is restricted to AP2.
hRME-6 is an exchange factor for rab5 in CCVs
The recruitment of rab5 to CCVs is affected by its plasma membrane–specific GEF, hRME-6. Knockdown of hRME-6 but not rabex-5 resulted in enhanced colocalization of AP2 and PtdIns(4,5)P2 with endocytic vesicles. In contrast to rabex-5 (Mattera et al., 2003), hRME-6 binds α-adaptin ear. Although the C-terminal VPS9 domain has been shown to have specific GEF activity against rab5 (Hunker et al., 2006), we were unable to detect any significant GEF activity in the purified full-length protein. However, inclusion of both ATP and α-adaptin ear domain resulted in enhancement of hRME-6 GEF activity in the wild-type protein but not in hRME-6F1487A, which is defective in AP2 binding, strongly suggesting that GEF activity requires a conformational change, perhaps to relieve autoinhibition. Rabex-5 shows a similar requirement to fully activate its GEF activity (Delprato et al., 2004) and precedents for ATP-dependent conformational changes in other GEFs have been reported (Amarasinghe and Rosen, 2005). Our demonstration that α-adaptin ear is required for hRME-6 activation suggests that, in vivo, hRME-6 would only be active after its recruitment to coated pits by AP2, thus providing tight spatiotemporal control of rab5 function.
hRME-6/Rab5 modulates μ2 dephosphorylation and affects PtdIns(4,5)P2 levels during AP2 uncoating
Previous studies have indicated that AP2 disassembly requires an additional cytosolic factor, distinct from Hsc70 and auxilin (Hannan et al., 1998). Consistent with studies using liver CCVs (Ghosh and Kornfeld, 2003), we found that the phosphatase PP2A was sufficient to uncoat both AP2 and AP1 from brain CCVs. Thus, PP2A is likely to antagonize the activity of AAK1, which is active in CCVs, to regulate μ2 phosphorylation during cargo recruitment into newly formed clathrin-coated pits (Jackson et al., 2003). To reduce the affinity of YxxΦ sorting motifs for AP2 during uncoating, the equilibrium of kinase and phosphatase activity would need to be shifted in favor of the latter. In vitro, addition of excess PP2A will therefore be sufficient to uncoat AP2, as found experimentally (Ghosh and Kornfeld, 2003). Moreover, the protein phosphatase inhibitor okadaic acid has been shown to inhibit Tfn internalization but only if cells are preincubated with the inhibitor for at least 10 min (Beauchamp and Woodman, 1994), which is significantly longer than the CCV cycle (30–90 s). Because okadaic acid is a lipophilic drug that would be expected to have immediate access to its target (Haystead et al., 1989), these data argue against phosphatase activity being the driving force for uncoating. This is further supported by the observation that whereas PP2A can uncoat AP1 as well as AP2 in vitro, the effect of rab5 is specific for AP2 only.
Our study demonstrates that AAK1 and hRME-6 show mutually exclusive binding to α-adaptin ear. Recruitment of hRME-6 therefore provides an effective mechanism to favor μ2 dephosphorylation by dissociation of AAK1. A prediction of this model is that modulation of hRME-6 levels will affect the levels of μ2 phosphorylation, and, indeed, hRME-6 appears to be a limiting factor on the endocytic pathway. Its overexpression resulted in reduced levels of phospho-μ2, whereas overexpression of hRME-6F1487A had no effect on μ2 phosphorylation. Knockdown of hRME-6 resulted in increased levels of phospho-μ2. These data provide a strong in vivo correlation for our in vitro data. The recruitment of hRME-6 must be temporally regulated because the activity of AAK1 is required early in coated pit formation during cargo recruitment. In this regard, we have observed that recruitment of hRME-6 to α-adaptin ear is negatively regulated by phosphorylation.
Here we present evidence that overexpression of rab5S34N or depletion of hRME-6 enhances the steady-state levels of PtdIns(4,5)P2 in endocytic vesicles. The primary determinant for the recruitment of AP2 to YxxΦ-containing liposome membranes is PtdIns(4,5)P2 and this is significantly enhanced by μ2 phosphorylation (Honing et al., 2005). In vivo studies using mutant forms of AP2 lacking the PtdIns(4,5)P2 binding site on the α-adaptin subunit have confirmed the importance of this interaction and demonstrated that μ2 phosphorylation facilitates AP2 binding (Motley et al., 2006). We suggest that AP2 dissociation from CCVs occurs by a reversal of this process, i.e., by modulation of μ2 dephosphorylation and by reduction of PtdIns(4,5)P2 levels in CCVs in a rab5-dependent manner. These data are consistent with studies in knockout mice lacking synaptojanin. These mice show an increase in PtdIns(4,5)P2 levels and in the number of CCVs in neurons (Cremona et al., 1999). Live-cell imaging has revealed that the longer synaptojanin isoform SJ1-170, which has clathrin, and AP2 interaction domains appear to be recruited early during coated pit formation. In contrast, a shorter form (SJ1-145) only binds during CCV scission (Perera et al., 2006). The mass action effect of this latter recruitment might be expected to promote PtdIns(4,5)P2 dephosphorylation and consequent productive uncoating (di Paolo and de Camilli, 2006; Perera et al., 2006). Reduction in PtdIns(4,5)P2 levels can occur either as a result of the action of lipid kinases or phosphatases (Shin et al., 2005). Interestingly, in this context, rab5 interacts with several lipid kinases and phosphatases (Simonsen et al., 1998; Christoforidis et al., 1999; Shin et al., 2005; Hyvola et al., 2006). Future studies will be required to establish which, if any, of these are relevant for AP2 uncoating.
In conclusion, we have presented data whereby the uncoating of AP2 is modulated by rab5 and its exchange factor hRME-6 and we provide evidence for a mechanism that displays an elegant symmetry to that involved in assembly of AP2 onto the plasma membrane during coated pit formation (Fig. 7).
Materials and methods
Antibodies, constructs, and proteins
AP.6, X22, B3/25, and 9E10 hybridoma cells were obtained from American Type Culture Collection and secreted antibodies were harvested by centrifugation. Phospho-μ2 antibodies were produced as previously described (Jackson et al., 2003). Anti-AAK1 antibodies were generated in sheep by injection of two peptides corresponding to amino acids 2–18 and 36–55 of the human protein. Antibodies were affinity purified on peptide columns. The anti-PtdIns(4,5)P2 mAb 2C11 was prepared and used as previously described (Thomas et al., 1999; Hammond et al., 2006). An IgG fraction of anti–hRME-6 antibodies was prepared from serum isolated from rabbits inoculated with a synthetic peptide (CMQFTAAVEFIKTIDDRK) conjugated to KLH.
The following commercially available reagents were used in this study: anti–α-adaptin mAb clone 100/2 and anti–γ-adaptin mAb clone 100/3 (Sigma-Aldrich); anti-HA mAb 16B12 (Abcam); anti-rabaptin5, anti–rabex-5, and anti-μ2 (BD Biosciences); mouse anti-clathrin heavy chain and clone CHC 5.9 (MP Biomedicals); Immuno-Pure rabbit anti–mouse IgM (μ chain specific), HRP-conjugated anti–rabbit and anti–mouse IgG (Thermo Fisher Scientific); Texas red Tfn, Cy5-Tfn, Alex Fluor 488 goat anti–mouse IgM (μ chain specific), and ProLong Gold antifade reagent (Invitrogen); FITC-conjugated goat anti–mouse (Jackson ImmunoResearch Laboratories); PP2A (Millipore).
eGFP–rabaptin5 and eGFP-rab11S25N were gifts from M. Zerial (The Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany) and M. McCaffrey (University College Cork, Cork, Ireland). Hsc70 was prepared from porcine brain as described previously (Schlossman et al., 1984). AAK1 was purified from porcine brain as described previously (Pauloin and Thurieau, 1993). HA–hRME-6 was prepared by large scale transfection of HEK293T cells, followed by immunoprecipitation using 16B12 anti-HA antibody and elution of the protein using the HA peptide as previously described (Zhou et al., 1992). A GST fusion protein including the VPS9 domain of GAPex5 (a gift from A. Saltiel, University of Michigan, Ann Arbor, MI) was purified on glutathione agarose.
Cloning of hRME-6
A full-length human RME-6 cDNA was cloned into the Gateway cloning system (Invitrogen) after fusion PCR of two overlapping but incomplete cDNAs from the IMAGE consortium (3355777 and 3451647). The resulting clone, pDONR201-hRME-6, was sequenced and found to contain four nucleotides differing from the genomic sequence of hRME-6, produced by the human genome project, that were predicted to alter the coding region. Each of these reverse transcription and/or PCR errors was corrected by site-directed mutagenesis using the QuikChange kit according to the manufacturer's instructions (Stratagene). The final cDNA was fully verified by DNA sequencing and transferred by Gateway LR reaction to mammalian expression vectors based on pCDNA3.1 modified in house with 2X-HA or 3X-Flag epitope tags and Gateway attR1/attR2 cassettes.
Preparation of cytosols
Cytosol from HEK293T cells and from HEK293T cells transfected with myc-tagged rab5wt, rab5Q79L, rab5S34N, rab1wt, and HA-tagged hRME-6 was prepared by freeze-thawing cells resuspended in 20 mM Hepes, NaOH, pH 7.4, 150 mM KCl, and 1 mM MgCl2, containing EDTA-free protease inhibitor cocktail. Transfections were performed using calcium phosphate as described previously (Sambrook et al., 1989). The levels of overexpression were determined by Western blotting. Rab5wt and Rab5Q79L were overexpressed by approximately ninefold whereas Rab5S34N was overexpressed by approximately three- to ninefold relative to the endogenous proteins. hRME-6wt and hRME-6F1487A were overexpressed by four- to fivefold. For additions to uncoating assays, cytosols were preloaded with 0.6 mM of the relevant nucleotide (GDP or GTP) for 1 h at 4°C. Control experiments indicated that addition of GTP or GDP alone did not affect the extent of uncoating. Cytosols were normalized for the level of expression of the relevant construct using cytosol prepared from untransfected HEK293T cells to balance protein levels.
Preparation of CCVs
CCVs were isolated from porcine brain that had been frozen on dry ice as soon as possible after slaughter. A crude CCV fraction was prepared by differential centrifugation as described previously (Campbell et al., 1984). The CCVs were collected by centrifugation at 85,000 g for 45 min and resuspended in a small volume of homogenization buffer (100 mM MES, pH 6.5, 0.5 mM MgCl2, 1 mM EDTA, 1 mM DTT, and 0.1 mM PMSF). 5–6 ml were layered over 6–7 ml of 8% sucrose-D2O solution in homogenization buffer and centrifuged at 80,000 g in a rotor (SW40; Beckman Coulter) for 2 h at 20°C as described previously (Nandi et al., 1982). The pellet containing CCVs was collected, washed, and resuspended in homogenization buffer. The suspension was spun at 20,000 g for 10 min and the supernatant containing CCVs was snap frozen in aliquots and stored at –80°C.
GEF assays
GEF assays were performed as described previously (Hama et al., 1999).
Uncoating assays
Uncoating assays were performed essentially as previously described (Ghosh and Kornfeld, 2003). Typically 5–6 μg of purified CCVs were incubated with different cytosols in the presence of an ATP-regenerating system (0.8 mM ATP, 5 mM of creatine phosphate, and 0.2 IU of creatine phosphokinase) in a final volume of 50 μl of the following buffer: 10 mM ammonium sulfate, 20 mM Hepes, pH 7.0, 2 mM magnesium acetate, and 25 mM KCl. Assay mixtures were supplemented with 1.3 μg Hsc70 and were incubated for 10 min at 25°C. The reaction was stopped by transferring the assay mixtures to ice and centrifuging them at 100,000 g for 10 min at 4°C in a rotor (TLA 100; Beckman Coulter). Supernatant and pellet were resolved by SDS-PAGE and Western blotting was performed according to standard procedures.
Microscopy and quantitative analysis
HEK293T cells seeded on coverslips coated with poly-l-lysine were transfected using Superfect transfection reagent (QIAGEN) according to the manufacturer's protocol. Superfect was used for microscopy experiments rather than calcium phosphate as it resulted in a consistently higher level (∼90%) of transfection. 16 h after transfection the cells were fixed according to one of the following methods.
Labeling AP2 with AP.6 antibody
Cells were incubated for 20 min in serum-free DME. Subsequently 5 μg/ml of Texas red Tfn was internalized at 37°C for 5 min. After transfer to ice, surface TfnR was acid stripped by washing twice with ice cold PBS, followed by addition of 50 mM glycine-HCl, pH 3.0, 2 M urea, and 100 mM NaCl (ice cold) for 5 min. This procedure was repeated three times to ensure complete removal of surface-bound Tfn. Subsequently, the cells were fixed in 4% PFA/PBS, pH 7.4, at 37°C for 20 min. PFA was quenched by washing three times in 50 mM NH4Cl in PBS. Cells were then permeabilized by incubation in 0.1% Triton X-100 in 0.1% BSA/PBS for 5 min at RT. After blocking with 2% fish skin gelatin in 0.1% BSA/PBS and incubation with primary and secondary antibodies as indicated, the coverslips were mounted on slides in ProLong Gold antifade reagent.
Labeling of PtdIns(4,5)P2 with 2C11 antibody
Cells were incubated with 25 μg/ml Cy5-Tfn for 5 min at 37°C and acid stripped as described in the previous paragraph. Cy5-Tfn was used in this series of experiments because its fluorescent signal was more strongly maintained compared with Texas red Tfn throughout the fixation procedures. All subsequent procedures were carried at 4°C as described previously (Hammond et al., 2006). In competition experiments with GST fusion proteins including the pleckstrin homology domain of PLCδ or 2xFYVE domain, the relevant fusion protein was included in the blocking solution at a concentration of 400 μM.
Specimens were imaged using the DeltaVision RT system based on a microscope (IX71; Olympus) at 100× UPlan Apo NA 1.4 oil objective. Stacks of images were collected at room temperature with 0.2-μm z-step from the bottom to the top of the cells, using a camera (Coolsnap-HQ version I; Roper Scientific). The images were then deconvolved using the conservative ratio of deconvolution of the in-built algorithm of the DeltaVision system. Deconvolved images were transferred to the Volocity 3.7.1 software (ImproVision). Two classifiers were applied selecting either red- or green-labeled objects. The lower limit of fluorescence intensity was based on values taken for at least five different pixels in an area of the cell with background staining. The high limit of fluorescence intensity was set at virtual infinity. Colocalization was determined using Volocity software. Statistical analysis was performed using either Microsoft Excel or SPSS11 for Mac OS X and applying the independent samples t test. The degree of colocalization of AP2 and Tfn or PtdIns(4,5)P2 and Tfn in control cells (mock treated) was set at 1. Representative images have been included in the manuscript but it is important to note that the differences caused by modulation of rab5 activity are not obvious in images from single planes and require quantitation of the extent of overlap over the whole cell.
siRNA procedures
siRNA was performed in HEK293T cells essentially according to the method of Motley et al. (2003), with the following modifications. Cells were plated overnight so that they reached 30% confluency on the day of transfection (day 1). siRNAs against hRME-6, rabex-5, or luciferase (as a control) were added to the cells at a concentration of 0.2 μM. The following day (day 2) the cells were split so that again they would be 30% confluent on day 3 when the same concentration of siRNA was added to the cells. Cells were processed for microscopy or Western blotting on day 4. siRNAs (Thermo Fisher Scientific) had the following sequences: HRME-6, 5′-P.UAUCGAUUGGCCACUUCUUUU-3′, 5′-P.UUAGAAUAGUGGUGCGUUAUU-3′, 5′-UAAUCGUAGAGCACAUUCUUU-3′, 5′-P.AUCAUCGCUAUCUUUGCGUUU-3′; RABEX-5, 5′-P.UGAUUCGGAGCUUAAUUUUU-3′, 5′-P.GAUUGAUCCACAUGAAUUCUU-3′, 5′-P.CACGAGUUUGCAUCCUUUCUU-3′, 5′-P.UCUUUCUGGAGGCACUUUCUU-3′.
Electron microscopy
B3/25 and 16B12 antibodies were conjugated to colloidal gold according to standard procedures (Griffiths, 1993). Samples were coded and analyzed “blind” until all of the data were collected. HEK293T, grown in 6-well dishes to a confluency of 70–80%, were prefixed at RT in 4% PFA for 30 min. After three washes in 50 mM (NH4)2SO4 in PBS, cells were incubated with gold-labeled antibodies for 45 min at room temperature. After three washes in PBS, cells were scraped from the dish and collected by centrifugation in a rotor (GH-3.8 swing-out; Beckman Coulter) at 1,500 g. The supernatant was removed and 1% glutaraldehyde containing 2% tannic acid was layered gently on the top of the pellet. Samples were incubated at room temperature for 30 min. After dehydration in a graded series of ethanol solutions, pellets were embedded in epon and sectioned using an Ultracut E microtome (Reichert-Jung). Specimens were visualized at 80 kV and images were captured using a transmission electron microscope (CM10; Philips). Negative images were converted into positive digital images by scanning into Photoshop (version 7.0; Adobe). For experiments with rab5 constructs, cells were cotransfected with CD8 and selected by FACs cell sorting using a MoFlo cell sorter (Dako). Roughly 106 cells were recovered per transfection and replated on a 6-well dish. Cells were allowed to recover overnight and were then labeled with B3/25 gold.
Online supplemental material
Fig. S1 is a Western blot to show the extent of depletion of rabex-5 when rabaptin5 is immunoprecipitated from cytosol. Fig. S2 is a histogram of the overlap of AP2 with Texas red Tfn containing endocytic vesicles in cells overexpressing rab11S25N compared with mock-treated cells. Fig. S3 is immunofluorescent images showing the effect of overexpression of rab5Q79L, rabaptin5, and hRME-6. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200806016/DC1.
Supplementary Material
Acknowledgments
We thank Matt Child and Chris Hill for excellent technical assistance. We are grateful to Geoff Cope for valuable electron microscopy advice and to Carl Smythe for insightful discussion.
Microscopy was performed in the Sheffield BioImaging Facility (Wellcome grant GR077544AIA). Work in the E. Smythe laboratory is supported by the Medical Research Council (MRC; G0300452) and Biotechnology and Biological Sciences Research Council (BBS/B/04072). B. Shortt was supported by a British Heart Foundation PhD studentship (FS/2001053) and S. Singh by an MRC studentship. B.D. Grant was supported by National Institutes of Health (grant GM67237) and G. Schiavo by Cancer Research UK.
Abbreviations used in the paper: CCV, clathrin-coated vesicle; GEF, guanine nucleotide exchange factor; PP2A, protein phosphatase 2A; PtdIns(4,5)P2, phosphoinositide 4,5-bisphosphate; Tfn, transferrin; TfnR, tranferrin receptor.
References
- Amarasinghe, G.K., and M.K. Rosen. 2005. Acidic region tyrosines provide access points for allosteric activation of the autoinhibited Vav1 Dbl homology domain. Biochemistry. 44:15257–15268. [DOI] [PubMed] [Google Scholar]
- Balla, T. 2005. Inositol-lipid binding motifs: signal integrators through protein-lipid and protein-protein interactions. J. Cell Sci. 118:2093–2104. [DOI] [PubMed] [Google Scholar]
- Barbieri, M.A., R.L. Roberts, A. Gumusboga, H. Highfield, C. Alvarez-Dominguez, A. Wells, and P.D. Stahl. 2000. Epidermal growth factor and membrane trafficking. EGF receptor activation of endocytosis requires Rab5a. J. Cell Biol. 151:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp, J.R., and P.G. Woodman. 1994. Regulation of transferrin receptor recycling by protein phosphorylation. Biochem. J. 303:647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci, C., R.G. Parton, I.H. Mather, H. Stunnenberg, K. Simons, B. Hoflack, and M. Zerial. 1992. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 70:715–728. [DOI] [PubMed] [Google Scholar]
- Campbell, C., J. Squicciarini, M. Shia, P.F. Pilch, and R.E. Fine. 1984. Identification of a protein kinase as an intrinsic component of rat liver coated vesicles. Biochemistry. 23:4420–4426. [DOI] [PubMed] [Google Scholar]
- Chappell, T.G., W.J. Welch, D.M. Schlossman, K.B. Palter, M.J. Schlesinger, and J.E. Rothman. 1986. Uncoating ATPase is a member of the 70 kilodalton family of stress proteins. Cell. 45:3–13. [DOI] [PubMed] [Google Scholar]
- Christoforidis, S., M. Miaczynska, K. Ashman, M. Wilm, L. Zhao, S.C. Yip, M.D. Waterfield, J.M. Backer, and M. Zerial. 1999. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1:249–252. [DOI] [PubMed] [Google Scholar]
- Collins, B.M., A.J. McCoy, H.M. Kent, P.R. Evans, and D.J. Owen. 2002. Molecular architecture and functional model of the endocytic AP2 complex. Cell. 109:523–535. [DOI] [PubMed] [Google Scholar]
- Conner, S.D., and S.L. Schmid. 2002. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J. Cell Biol. 156:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner, S.D., and S.L. Schmid. 2003. Regulated portals of entry into the cell. Nature. 422:37–44. [DOI] [PubMed] [Google Scholar]
- Conner, S.D., T. Schroter, and S.L. Schmid. 2003. AAK1-mediated micro2 phosphorylation is stimulated by assembled clathrin. Traffic. 4:885–890. [DOI] [PubMed] [Google Scholar]
- Cremona, O., P.G. Di, M.R. Wenk, A. Luthi, W.T. Kim, K. Takei, L. Daniell, Y. Nemoto, S.B. Shears, R.A. Flavell, et al. 1999. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 99:179–188. [DOI] [PubMed] [Google Scholar]
- Delprato, A., E. Merithew, and D.G. Lambright. 2004. Structure, exchange determinants, and family-wide rab specificity of the tandem helical bundle and Vps9 domains of Rabex-5. Cell. 118:607–617. [DOI] [PubMed] [Google Scholar]
- Di Paolo, G., and P. de Camilli. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature. 443:651–657. [DOI] [PubMed] [Google Scholar]
- Dinneen, J.L., and B.P. Ceresa. 2004. Expression of dominant negative rab5 in HeLa cells regulates endocytic trafficking distal from the plasma membrane. Exp. Cell Res. 294:509–522. [DOI] [PubMed] [Google Scholar]
- Ehrlich, M., W. Boll, A. Van Oijen, R. Hariharan, K. Chandran, M.L. Nibert, and T. Kirchhausen. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 118:591–605. [DOI] [PubMed] [Google Scholar]
- Fingerhut, A., K. von Figura, and S. Honing. 2001. Binding of AP2 to sorting signals is modulated by AP2 phosphorylation. J. Biol. Chem. 276:5476–5482. [DOI] [PubMed] [Google Scholar]
- Ghosh, P., and S. Kornfeld. 2003. AP-1 binding to sorting signals and release from clathrin-coated vesicles is regulated by phosphorylation. J. Cell Biol. 160:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, G. 1993. Fine Structure Immunocytochemistry. Springer Verlag, Heidelberg, Germany. 495 pp.
- Hama, H., G.G. Tall, and B.F. Horazdovsky. 1999. Vps9p is a guanine nucleotide exchange factor involved in vesicle-mediated vacuolar protein transport. J. Biol. Chem. 274:15284–15291. [DOI] [PubMed] [Google Scholar]
- Hammond, G.R., S.K. Dove, A. Nicol, J.A. Pinxteren, D. Zicha, and G. Schiavo. 2006. Elimination of plasma membrane phosphatidylinositol (4,5)-bisphosphate is required for exocytosis from mast cells. J. Cell Sci. 119:2084–2094. [DOI] [PubMed] [Google Scholar]
- Hannan, L.A., S.L. Newmyer, and S.L. Schmid. 1998. ATP- and cytosol-dependent release of adaptor proteins from clathrin-coated vesicles: a dual role for Hsc70. Mol. Biol. Cell. 9:2217–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haystead, T.A., A.T. Sim, D. Carling, R.C. Honnor, Y. Tsukitani, P. Cohen, and D.G. Hardie. 1989. Effects of the tumour promoter okadaic acid on intracellular protein phosphorylation and metabolism. Nature. 337:78–81. [DOI] [PubMed] [Google Scholar]
- Honing, S., D. Ricotta, M. Krauss, K. Spate, B. Spolaore, A. Motley, M. Robinson, C. Robinson, V. Haucke, and D.J. Owen. 2005. Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol. Cell. 18:519–531. [DOI] [PubMed] [Google Scholar]
- Horiuchi, H., R. Lippe, H.M. McBride, M. Rubino, P. Woodman, H. Stenmark, V. Rybin, M. Wilm, K. Ashman, M. Mann, and M. Zerial. 1997. A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell. 90:1149–1159. [DOI] [PubMed] [Google Scholar]
- Hunker, C.M., A. Galvis, I. Kruk, H. Giambini, M.L. Veisaga, and M.A. Barbieri. 2006. Rab5-activating protein 6, a novel endosomal protein with a role in endocytosis. Biochem. Biophys. Res. Commun. 340:967–975. [DOI] [PubMed] [Google Scholar]
- Hyvola, N., A. Diao, E. McKenzie, A. Skippen, S. Cockcroft, and M. Lowe. 2006. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. 25:3750–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, A.P., A. Flett, C. Smythe, L. Hufton, F.R. Wettey, and E. Smythe. 2003. Clathrin promotes incorporation of cargo into coated pits by activation of the AP2 adaptor μ2 kinase. J. Cell Biol. 163:231–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G., and P.D. Stahl. 1993. Structure-function relationship of the small GTPase rab5. J. Biol. Chem. 268:24475–24480. [PubMed] [Google Scholar]
- Lippe, R., M. Miaczynska, V. Rybin, A. Runge, and M. Zerial. 2001. Functional synergy between Rab5 effector Rabaptin-5 and exchange factor Rabex-5 when physically associated in a complex. Mol. Biol. Cell. 12:2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi, I.J., S.H. Chiang, L. Chang, D. Vollenweider, R.T. Watson, M. Inoue, J.E. Pessin, and A.R. Saltiel. 2007. Gapex-5, a Rab31 guanine nucleotide exchange factor that regulates Glut4 trafficking in adipocytes. Cell Metab. 5:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera, R., C.N. Arighi, R. Lodge, M. Zerial, and J.S. Bonifacino. 2003. Divalent interaction of the GGAs with the Rabaptin-5-Rabex-5 complex. EMBO J. 22:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLauchlan, H., J. Newell, N. Morrice, A. Osborne, M. West, and E. Smythe. 1998. A novel role for rab5-GDI in ligand sequestration into clathrin-coated pits. Curr. Biol. 8:34–45. [DOI] [PubMed] [Google Scholar]
- Mishra, S.K., M.J. Hawryluk, T.J. Brett, P.A. Keyel, A.L. Dupin, A. Jha, J.E. Heuser, D.H. Fremont, and L.M. Traub. 2004. Dual engagement regulation of protein interactions with the AP-2 adaptor alpha appendage. J. Biol. Chem. 279:46191–46203. [DOI] [PubMed] [Google Scholar]
- Motley, A.M., N.A. Bright, M.N. Seaman, and M.S. Robinson. 2003. Clathrin mediated endocytosis in AP2-depleted cells. J. Cell Biol. 162:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley, A.M., N. Berg, M.J. Taylor, D.A. Sahlender, J. Hirst, D.J. Owen, and M.S. Robinson. 2006. Functional analysis of AP-2 alpha and mu2 subunits. Mol. Biol. Cell. 17:5298–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi, P.K., G. Irace, P.P. Van Jaarsveld, R.E. Lippodt, and H. Edelhoch. 1982. Instability of coated vesicle in concentrated sucrose solutions. Proc. Natl. Acad. Sci. USA. 79:5881–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olusanya, O., P.D. Andrews, J.R. Swedlow, and E. Smythe. 2001. Phosphorylation of threonine156 of the μ2 subunit of the AP2 complex is essential for endocytosis in vitro and in vivo. Curr. Biol. 11:896–900. [DOI] [PubMed] [Google Scholar]
- Pattni, K., M. Jepson, H. Stenmark, and G. Banting. 2001. A PtdIns(3)P-specific probe cycles on and off host cell membranes during Salmonella invasion of mammalian cells. Curr. Biol. 11:1636–1642. [DOI] [PubMed] [Google Scholar]
- Pauloin, A., and C. Thurieau. 1993. The 50 kDa protein subunit of assembly polypeptide (ap) AP-2 adaptor from clathrin-coated vesicles is phosphorylated on threonine-156 by AP-1 and a soluble AP50 kinase which co-purifies with the assembly polypeptides. Biochem. J. 296:409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera, R.M., R. Zoncu, L. Lucast, P. De Camilli, and D. Toomre. 2006. Two synaptojanin 1 isoforms are recruited to clathrin-coated pits at different stages. Proc. Natl. Acad. Sci. USA. 103:19332–19337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricotta, D., S.D. Conner, S.L. Schmid, K. von Figura, and S. Honing. 2002. Phosphorylation of the AP2 μ subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J. Cell Biol. 156:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde, G., D. Wenzel, and V. Haucke. 2002. A phosphatidylinositol (4,5)-bisphosphate binding site within μ2-adaptin regulates clathrin-mediated endocytosis. J. Cell Biol. 158:209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino, M., M. Miaczynska, R. Lippe, and M. Zerial. 2000. Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J. Biol. Chem. 275:3745–3748. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Standard protocol for calcium phosphate-mediated transfection of adherent cells. In Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 16.33–16.36
- Sato, M., K. Sato, P. Fonarev, C.J. Huang, W. Liou, and B.D. Grant. 2005. Caenorhabditis elegans RME-6 is a novel regulator of RAB-5 at the clathrin-coated pit. Nat. Cell Biol. 7:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlossman, D.M., S.L. Schmid, W.A. Braell, and J.E. Rothman. 1984. An enzyme that removes clathrin coats: purification of an uncoating ATPase. J. Cell Biol. 99:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, S.L., W.A. Braell, and J.E. Rothman. 1985. ATP catalyzes the sequestration of clathrin during enzymatic uncoating. J. Biol. Chem. 260:10057–10062. [PubMed] [Google Scholar]
- Shin, H.W., M. Hayashi, S. Christoforidis, S. Lacas-Gervais, S. Hoepfner, M.R. Wenk, J. Modregger, S. Uttenweiler-Joseph, M. Wilm, A. Nystuen, et al. 2005. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J. Cell Biol. 170:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.M. Gaullier, A. Brech, J. Callaghan, B.H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Stenmark, H., R.G. Parton, O. Steele-Mortimer, A. Lutcke, J. Gruenberg, and M. Zerial. 1994. a. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 13:1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark, H., A. Valencia, O. Martinez, O. Ullrich, B. Goud, and M. Zerial. 1994. b. Distinct structural elements of rab5 define its functional specificity. EMBO J. 13:575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark, H., G. Vitale, O. Ullrich, and M. Zerial. 1995. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell. 83:423–432. [DOI] [PubMed] [Google Scholar]
- Su, X., I.J. Lodhi, A.R. Saltiel, and P.D. Stahl. 2006. Insulin-stimulated interaction between insulin receptor substrate 1 and p85alpha and activation of protein kinase B/Akt require Rab5. J. Biol. Chem. 281:27982–27990. [DOI] [PubMed] [Google Scholar]
- Thomas, C.L., J. Steel, G.D. Prestwich, and G. Schiavo. 1999. Generation of phosphatidylinositol-specific antibodies and their characterization. Biochem. Soc. Trans. 27:648–652. [DOI] [PubMed] [Google Scholar]
- Traub, L.M. 2003. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J. Cell Biol. 163:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda, A., A. Meyerholz, and E. Ungewickell. 2000. Identification of the universal cofactor (auxilin 2) in clathrin coat dissociation. Eur. J. Cell Biol. 79:336–342. [DOI] [PubMed] [Google Scholar]
- Ungewickell, E., H. Ungewickell, S.E. Holstein, R. Lindner, K. Prasad, W. Barouch, B. Martin, L.E. Greene, and E. Eisenberg. 1995. Role of auxilin in uncoating clathrin-coated vesicles. Nature. 378:632–635. [DOI] [PubMed] [Google Scholar]
- Zerial, M., and H. McBride. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2:107–117. [DOI] [PubMed] [Google Scholar]
- Zhou, Q., P.M. Lieberman, T.G. Boyer, and A.J. Berk. 1992. Holo-TFIID supports transcriptional stimulation by diverse activators and from a TATA-less promoter. Genes Dev. 6:1964–1974. [DOI] [PubMed] [Google Scholar]
- Zhu, H., G. Zhu, J. Liu, Z. Liang, X.C. Zhang, and G. Li. 2007. Rabaptin-5-independent membrane targeting and Rab5 activation by Rabex-5 in the cell. Mol. Biol. Cell. 18:4119–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}