

Abstract
Despite the fact that many genomes have been decoded, proteome chips comprising individually purified proteins have been reported only for budding yeast, mainly because of the complexity and difficulty of high-throughput protein purification. To facilitate proteomics studies in prokaryotes, we have developed a high-throughput protein purification protocol that allowed us to purify 4,256 proteins encoded by the Escherichia coli K12 strain within 10 h. The purified proteins were then spotted onto glass slides to create E. coli proteome chips. We used these chips to develop assays for identifying proteins involved in the recognition of potential base damage in DNA. By using a group of DNA probes, each containing a mismatched base pair or an abasic site, we found a small number of proteins that could recognize each type of probe with high affinity and specificity. We further evaluated two of these proteins, YbaZ and YbcN, by biochemical analyses. The assembly of libraries containing DNA probes with specific modifications and the availability of E. coli proteome chips have the potential to reveal important interactions between proteins and nucleic acids that are time-consuming and difficult to detect using other techniques.
Protein chips, also known as protein microarrays, are miniaturized, parallel assay systems that contain small amounts of purified proteins in a high-density format1,2. Individually purified proteins, synthesized polypeptides or protein fragments (such as protein domains) are immobilized to derivatized glass surfaces3, allowing simultaneous screening with a variety of analytes in small-volume samples within a single experiment. When all or most (for example, >80%) of the individually purified proteins in a given proteome are present on such a microarray, a proteome chip is created1,4. Despite the fact that many genomes have been decoded, only yeast proteome chips, in both N- and C-terminally tagged form, have been reported1,5. Because of the complexity and difficulty associated with protein chip fabrication, the other protein chips that have been described have usually contained only a particular family of proteins, a collection of known members of a certain domain6,7, a small fraction of the proteome of a higher eukaryote8,9 or even an unpurified cell extract10.
We and others have shown that proteome microarrays are useful for analyzing the biochemical activities of proteins at the proteome level1,2,11–13. In an effort to promote high-throughput studies in prokaryotes, Mori and colleagues14 have constructed an open reading frame (ORF) collection that carries 4,256 of 4,288 genes in the E. coli K12 genome. These ORFs are cloned into a bacterial expression vector that allows over-production of proteins fused with N-terminal polyhistidine (His6) tags under the control of an isopropyl-α-d-thiogalactoside (IPTG)-inducible promoter. This work opens the door for high-throughput production and purification of E. coli proteins.
DNA stores genetic information and performs many other processes that are essential for life. The four DNA bases are constantly subjected to various kinds of damage and modification15. In the past two decades, many proteins have been identified that detect and repair base lesions. Almost all of these base-repair proteins recognize and repair the base damage by flipping the lesioned base out of the DNA duplex into an extrahelical conformation16,17. Despite the fact that the detection and repair of DNA base damage have been extensively studied, a comprehensive picture of DNA damage repair, and of damage sensing in particular, is still lacking. Therefore, the use of a proteome-wide approach would be a particularly fruitful strategy for exploring the repair and modification of DNA bases18,19.
We and others have been developing tools that make it possible to identify new functions and to characterize new base-repair and base-modification proteins. In the course of synthesizing unique DNA probes to perform pull-down experiments, we realized that many DNA repair proteins occur in low copy numbers and are masked by other abundant proteins in cell extracts. To circumvent this problem, we performed a genome-wide screen for new DNA repair activities using synthetic inhibitors of glycosylases and an in vitro expression cloning protein library and identified a new glycosylase using this approach20.
We also took a different and less tedious approach by probing the interactions of specific DNA with proteins presented on proteome chips. This strategy has been successfully used to reveal new DNA-binding proteins and to identify interactions between transcription factors and DNA motifs21,22. These studies have suggested that an E. coli proteome protein chip of this type could serve as an efficient tool for profiling potential DNA base damage recognition and repair events within a particular host's genome.
Here, we report the development of an extremely high-throughput protocol for the purification of ∼4,200 E. coli proteins, the fabrication of a prokaryotic proteome chip, and the application of a proteome-wide screening approach to detect undescribed activities involved in DNA base-damage recognition.
Results
A protocol for high-throughput protein purification
To individually purify 4,256 His6-tagged fusion proteins encoded by the E. coli K12 genome, we developed a high-throughput protein purification protocol that allowed us to purify all 4,256 proteins at once. Bacterial strains from the Mori collection were inoculated from glycerol stocks into 96-well dishes. After overnight incubation, the cultures were inoculated into fresh medium in 96-well deep-well plates, and the cells were induced for 3.5 h to produce His-tagged fusion proteins after the optical density (OD)600 reached 0.7. The cell pellets were then collected and stored in a freezer at −80 °C.
To reduce manual pipetting and improve the throughput of the protein purification, we decided to perform the cell lysis, affinity capture and resin washing in the same plate. We added lysis buffer and the Ni-NTA resin to the frozen cell pellets and transferred them to a bottom-sealed filter plate after thawing (Fig. 1). We chose a particular filter plate with a large pore size that was big enough to allow the cell debris to pass through easily but small enough to retain the resin. After incubation in a cold room, the filter plate was unsealed to remove the cell debris. The retained resin was washed under high- and low-salt conditions in the filter plate, and elution buffer was added. Because only 25 μl of the elution buffer was added to each well, we observed no leakage from the filter plate during elution. Finally, the eluted proteins could be conveniently collected into a 96-well receiver plate by centrifugation. Using this protocol, we could purify all 4,256 individual proteins from 1.6 ml of bacterial culture within 10 h.
Figure 1.
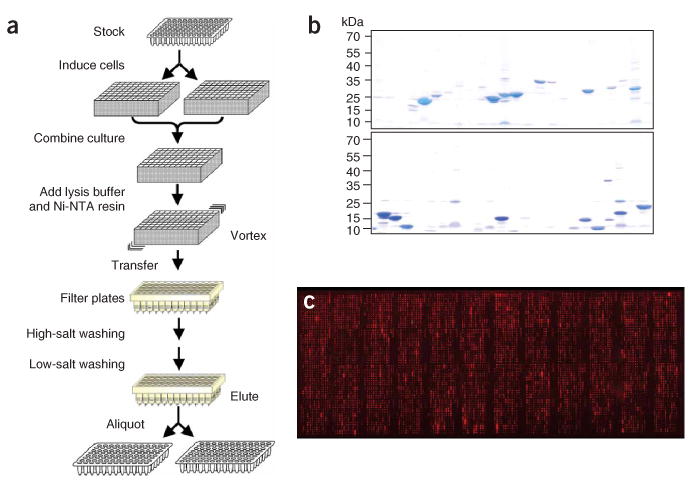
An E. coli proteome chip. (a) Protocol for high-throughput E. coli protein expression and purification. (b) The quality and quantities of the purified E. coli proteins were determined by Coomassie staining. (c) Proteins immobilized on a FullMoon slide were visualized by probing the slide with anti-His6 monoclonal antibody followed by Cy5-labeled secondary antibody.
To ensure the purity of these proteins, we monitored the quality and quantity of the purified proteins by using electrophoresis and Coomassie staining to test random samples (Fig. 1b). We estimated that approximately 88% of the proteins were purified at the expected molecular weight with yields of more than 0.2 μg/ml, of which around 50% were seen as the predominant band.
Fabrication of E. coli proteome chips
To prepare the proteome chips, we printed the purified proteins in duplicate onto glass slides using a ChipWriter Pro (Bio-Rad) with 48 pins. Different surface chemistries were tested, including the FullMoon, aldehyde-derivatized, hydrogel- and nitrocellulose-coated (FAST slides) surfaces. All of the tests produced satisfactory immobilization results, with the FullMoon surfaces performing the best. To visualize the immobilization of the proteins on the glass surfaces, we labeled the proteins on the chip with either an anti-His6 monoclonal antibody or DyLight 547 NHS ester. Data analysis indicated that more than 95% of the proteins showed substantial signals above background (Fig. 1c), consistent with the Coomassie analysis. Our results also show that the DyLight 547 NHS ester could efficiently label proteins immobilized on selected surfaces—for example, the FullMoon and aldehyde-derivatized surfaces—with minimum background.
Screening for proteins that recognize DNA damage
To demonstrate the power of this E. coli proteome chip, we probed the chip with probes exemplifying two types of potential damage, mismatches and abasic sites, as a means of identifying responses to DNA base damage. Previous work by our group and others has indicated that DNA base-flipping and base-repair proteins preferentially recognize unstable base pairs such as mismatches or abasic sites in double-stranded DNA (dsDNA)16,20,23–27. We hypothesized that for many repair proteins, locating unstable regions in a duplex DNA constitutes the first step in finding lesions27. Thus, using DNA probes containing mismatches or an abasic site to conduct proteome-wide screening would be expected to provide valuable insight into known and unknown DNA repair-related functions.
We therefore prepared seven 19-mer dsDNA probes with Cy3 labels incorporated at the 5′-ends (Fig. 2). Probe 1, a perfectly matched dsDNA, served as a control. Probes 2 and 3, respectively, contained mismatches of A:C and G:T in their sequences. Probes 4–7 each contained an abasic site mimic-complementary to G, A, C and T, respectively. To optimize the binding conditions, we carried out a series of pilot assays and determined that the FullMoon surface produced the best signal-to-noise ratio with a stringent high-salt wash. The seven Cy3-labeled DNA probes were then individually used to probe separate E. coli proteome chips under the optimized conditions. The resulting binding signals were acquired with a microarray scanner. The signals obtained from each experiment using probes 2–7 (either a mismatch or abasic site) were divided by the signal for probe 1 to normalize the data. To identify potential ‘hits’, we ranked the bacterial proteins on the basis of these ratios.
Figure 2.

Design of the DNA probes. Probe 1 is an unmodified, 19-mer duplex DNA. Probes 2 and 3 contain A:C and G:T mismatches, respectively. Probes 4–7 contain an abasic site complementary to G, A, C or T. All probes were labeled with Cy3 dye.
The 20 top-ranking proteins identified by probes 3 and 4 are listed in Tables 1 and 2. (The top 20 rankings obtained with the other probes are shown in Supplementary Table 1 online.) Only a small number of proteins showed intensity ratios substantially higher than those of the rest of the proteins. For example, CspE and YbcN showed severalfold intensity ratios (Table 1). CspE is a cold-shock DNA-binding protein28. It has been proposed that CspE destabilizes nucleic acid secondary structures and induces nucleic acid melting. Its recognition of the G:Tmismatch in probe 3 is not altogether unexpected. Notably, CspE showed a high preference for the G:T over the A:C mismatch (probe 2) and the abasic sites in probes 4–7 (Supplementary Fig. 1 online). The other hit, YbcN, is a protein of unknown function. As shown in Figure 3a and in Supplementary Figure 2a online, YbcN selectively bound the G:T-mismatched DNA over the perfectly matched and A:C-mismatched DNA. It could also recognize abasic sites, but much less efficiently. The experimental results with probe 2 revealed that the protein YicP, a putative adenine deaminase29, was ranked at the top of the list (Supplementary Table 1), with its signal ratio being at least threefold higher than those of the other hits.
Table 1.
The top 20 proteins specifically recognized by Probe 3 with a G:T mismatch
| Rank | Ratioa | Protein | Function or proposed function |
|---|---|---|---|
| 1 | 12.17 ± 0.24 | cspE | Cold shock–like protein |
| 2 | 6.97 ± 1.35 | ybcN | Hypothetical protein |
| 3 | 4.49 ± 0.40 | ybfN | Predicted lipoprotein |
| 4 | 4.28 ± 0.36 | cspC | Cold shock–like protein |
| 5 | 4.12 ± 0.17 | dgt | Deoxyguanosine triphosphate triphosphohydrolase |
| 6 | 3.57 ± 0.07 | cspA | Cold shock–like protein |
| 7 | 3.53 ± 0.26 | yeaO | Conserved hypothetical protein |
| 8 | 2.55 ± 0.10 | dsbG | Thiol:disulfide interchange protein dsbG precursor |
| 9 | 2.41 ± 0.50 | ybdB | Esterase |
| 10 | 2.16 ± 1.22 | yfeH | Putative cytochrome oxidase |
| 11 | 2.16 ± 0.14 | polA | DNA polymerase I, 3′ → 5′ polymerase, 5′ → 3′ and 3′ → 5′ exonuclease |
| 12 | 2.10 ± 0.02 | pldB | Lysophospholipase L2 |
| 13 | 2.01 ± 0.12 | yhfS | Hypothetical protein |
| 14 | 2.00 ± 0.05 | modC | Molybdenum import ATP-binding protein |
| 15 | 1.95 ± 0.17 | yjiV | Hypothetical protein |
| 16 | 1.84 ± 0.09 | ygbT | Conserved protein |
| 17 | 1.83 ± 0.25 | treR | HTH-type transcriptional regulator |
| 18 | 1.75 ± 0.02 | secD | Protein-export membrane protein |
| 19 | 1.74 ± 0.04 | ycaJ | Recombination protein |
| 20 | 1.74 ± 0.03 | yfeP | MntH manganese ion NRAMP transporter |
The signals obtained from probing experiments with probe 3 were normalized against those from probe 1 as the control. The ranked ratios are shown.
Table 2.
The top 20 proteins that preferentially bound to Probe 4
| Rank | Ratioa | Protein | Function or proposed function |
|---|---|---|---|
| 1 | 10.43 ± 0.33 | ybaZ | Predicted methylated DNA-protein cysteine methyltransferase |
| 2 | 3.24 ± 0.24 | polA | DNA polymerase I, 3′ → 5′ polymerase, 5′ → 3′ and 3′ → 5′ exonuclease |
| 3 | 2.65 ± 0.15 | lysR | Transcriptional activator protein |
| 4 | 2.22 ± 0.05 | rfaF | ADP-heptose:lipopolysaccharide heptosyltransferase II |
| 5 | 2.15 ± 0.10 | ybbS | Putative transcriptional regulator |
| 6 | 1.95 ± 0.37 | ybcN | Hypothetical protein |
| 7 | 1.86 ± 0.12 | betA | Choline dehydrogenase |
| 8 | 1.83 ± 0.03 | ygbT | Conserved protein |
| 9 | 1.82 ± 0.03 | smtA | S-adenosylmethionine–dependent methyltransferase |
| 10 | 1.82 ± 0.09 | fadD | Long-chain fatty acid CoA ligase |
| 11 | 1.78 ± 0.02 | ycbG | Hypothetical protein |
| 12 | 1.76 ± 0.02 | rfaS | Lipopolysaccharide core biosynthesis protein |
| 13 | 1.74 ± 0.02 | yqhH | Predicted outer membrane lipoprotein |
| 14 | 1.73 ± 0.06 | ybhA | Pyridoxal phosphatase/fructose 1,6-bisphosphatase |
| 15 | 1.72 ± 0.06 | ygcJ | Predicted protein |
| 16 | 1.71 ± 0.05 | msbA | Lipid A export ATP-binding/permease protein |
| 17 | 1.71 ± 0.02 | acpS | Holo-[acyl-carrier-protein] synthase |
| 18 | 1.70 ± 0.15 | bisZ | Trimethylamine N-oxide reductase III, TorZ subunit |
| 19 | 1.70 ± 0.06 | talC | Fructose 6-phosphate aldolase 2 |
| 20 | 1.68 ± 0.03 | yhaR | Predicted L-PSP (mRNA) endoribonuclease |
The signals obtained from probing experiments with probe 4 were normalized against those from probe 1 as the control. The ranked ratios are shown.
Figure 3.

Identification of YbcN and YbaZ. (a) Images from experiments with seven probes show that YbcN preferentially binds to probe 3. It also recognizes probes 4–7, each of which contains an abasic site in duplex DNA. (b) YbaZ binds to DNA probes that contain an abasic site. In all images, the first panel is the control, and the rectangle defines the target protein (in duplicate).
When probes 4–7, each containing an abasic site, were applied to the proteome chip, the YbaZ protein was predominant. This protein preferentially recognized dsDNA that contained an abasic site (Fig. 3b, Supplementary Fig. 2b, Table 2 and Supplementary Table 1). Its signal ratios were manyfold higher than those for other hits with these probes. However, it did not seem to recognize mismatched base pairs or to bind to probe 1. Gel-shift experiments verified our conclusion from the chip assays that YbaZ preferentially binds to DNA that contains an abasic site (Supplementary Fig. 3 online). We also measured the binding affinities of YbaZ and YbcN to abasic DNA (probe 5) and G:T-mismatched DNA (probe 3), respectively, with a chip-based method30,31. The dissociation constant (Kd) of YbaZ with probe 5 was determined to be 3.5 × 10−12 M and that for YbcN with probe 3 to be 1.1 × 10−12 M, indicating high affinity in both cases (Supplementary Fig. 4 online).
Sequence alignment indicated that YbaZ is a putative alkyltransferase-like protein (ATL)32; however, its exact function is unknown. YbaZ resembles the well-known O6-methylguanine-DNA methyltransferases such as Ada and Ogt in E. coli and MGMT (or AGT) in humans. These proteins directly remove alkyl adducts to the O6-position of guanine through an irreversible, suicidal transfer of the damage to an activated cysteine residue33. However, the reactive cysteine residue is not conserved in YbaZ. Our preliminary in vitro tests showed no repair activity of YbaZ toward alkylated base lesions.
Biochemical characterization of YbcN and YbaZ
We next investigated the biochemical activities of YbcN and YbaZ that are relevant to DNA damage recognition and repair. To test whether these proteins are DNA base-flipping proteins16,20,23–27, we used 2-aminopyrine (Ap)-modified DNA and studied base-flipping by following the fluorescence intensity of Ap. The fluorescence of 2-Ap is typically quenched in duplex DNA, but it shows enhanced fluorescence intensity when it is flipped into an extrahelical conformation. YbaZ effectively induced a marked increase in fluorescence from 2-Ap in duplex DNA (Fig. 4a). With a less stable Ap:A base pair incorporated into the DNA, an increase in fluorescence intensity of more than 1,000-fold was observed, confirming that YbaZ is a base-flipping protein. The same results were obtained with YbcN, indicating that this protein is also a DNA base-flipping protein (Fig. 4b).
Figure 4.
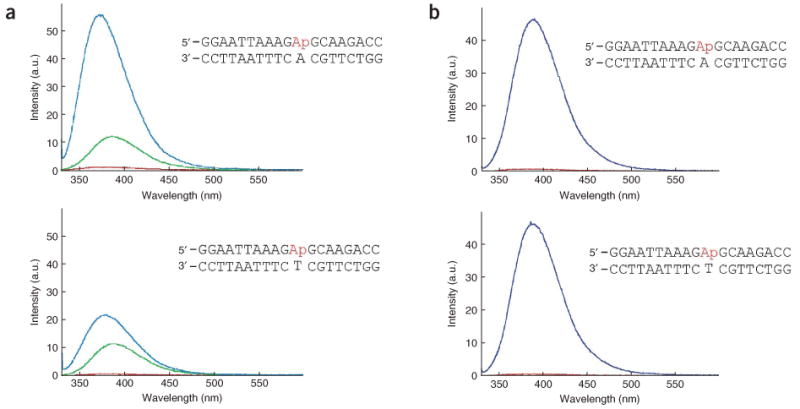
Base-flipping assay of YbcN and YbaZ. (a) Base-flipping by YbaZ. Two probes with 2-aminopyrine (Ap) opposite A (upper panel, an unstable base pair) or T (lower panel, a relatively stable but non–Watson-Crick base pair) were used to probe base-flipping by YbaZ in a fluorescence assay. All spectra were obtained after subtraction of the buffer background obtained under the same conditions. Red, DNA probe alone; green, spectrum taken immediately after mixing YbaZ with DNA; blue, spectrum taken 14 min after mixing. The fluorescence intensity increase indicates that the 2-aminopyrine base is in an extrahelical conformation after mixing with YbaZ. (b) Base-flipping by YbcN. YbcN efficiently flips out Ap from the two DNA probes. The base-flipping by YbcN was much faster than that by YbaZ; there was no difference between the signal immediately after mixing and that taken 14 min after mixing in the case of YbcN. Red, DNA probe alone; dark blue, spectrum taken after mixing YbcN with DNA.
Potential binding partners of YbaZ
Given the preference of YbaZ for various abasic site–containing DNA probes and its sequence homology to DNA repair alkyltransferases, we were interested in investigating its physiological roles. We therefore used labeled YbaZ to identify its potential partners on the E. coli proteome chips. On the basis of the normalized binding signal intensities, HelD showed the highest level of interaction with YbaZ (Supplementary Table 2 online). HelD is a type IV helicase in E. coli and is known to be involved in conjugational recombination and the repair of methylation-based DNA damage34. As biological systems are known to use multiple redundant pathways to insure the proper repair of various types of DNA damage, it is possible that YbaZ and helicase IV are involved in locating DNA damage and in the subsequent recruitment of repair machinery to complement other repair pathways. YbaZ has been shown to recognize O6-meG, which might subsequently recruit helicase IV for further processing of this type of lesion. Thus, the results described here might point to new strategies in DNA alkylation repair that can be further tested in E. coli.
Discussion
An important hurdle in fabricating proteome chips is the need to produce and purify a large number of proteins in a timely manner. In recent years there has been rapid progress in improving the throughput process and implementing alternative approaches. For example, several companies now sell reagents and devices that are adapted for high-throughput protein expression and purification of proteins in E. coli using standard 96-well plates, and automation of the entire process is also underway.
In vitro transcription-translation systems offer an alternative approach for producing proteins. Our group35 and Labaer's36 have taken this process one step further by directly applying in vitro transcription-translation systems to protein chip fabrication. The obvious advantage is that the expensive and time-consuming steps of ORF cloning, protein expression and purification can be eliminated. However, because of inherent problems with the in vitro system, the size of proteins that can be produced is limited, and the reproducibility of the system is not high.
Here we describe the development of a high-throughput protein purification protocol based on protein expression in E. coli that allowed us to purify more than 4,200 proteins within 10 h from prepared cultures. By combining the steps of cell lysis and protein capture on affinity resins in sealed filter plates, we have reduced the number of pipetting steps and therefore lowered the potential for problems resulting from human error. Our current throughput compares favorably with dedicated commercial systems. Because no special reagents were required, the cost of purifying a single protein was much less than the cost of using a robotic liquid handling system. In addition, because we chose unsophisticated and inexpensive liquid handling systems (for example, Q-Fill2 and Apricot) for protein purification and rearraying, our protocol can readily be transferred to and adopted by other research laboratories. With the use of this protocol and the ORF collection that carries 4,256 of 4,288 genes in the E. coli K12 genome, we fabricated an E. coli proteome chip incorporating 99.3% of the genome.
We then used this E. coli proteome chip to identify proteins that might be involved in DNA damage recognition. To our surprise, instead of observing multiple proteins showing a range of gradually decreasing affinities for these probes, we found one or two proteins that had much higher affinities for the modified probe than did the other proteins. These results underscore our contention that proteome chips provide a suitable platform for performing studies of this type—for example, identifying proteins with low copy numbers inside cells—in a high-throughput manner.
With our DNA probes, we have been able to identify several interesting protein targets, and we have corroborated these findings from the chip experiments in biochemical assays. Thus, our results indicate that both YbcN and YbaZ are base-flipping proteins. Applying labeled YbaZ to the E. coli proteome chip allowed further exploration of the potential networks involved in DNA repair. One possible limitation of this approach is that problems with protein folding and the integrity of the proteins printed on the chips might sometimes produce false-positives or missed ‘hits’. However, the possibility of using a range of different DNA probes, each with a specific modification, to profile cellular proteins on the whole-proteome level is exciting. If, as shown here, one or two proteins can be identified with each probe that is applied, a substantial database can be built for further biological validation and reference.
Methods
Protein expression and purification in the 96-well format
E. coli cells were first activated from frozen stocks by inoculation using a 96-pin prong into 0.5 ml of 2 × LB medium containing 30 μg/ml chloramphenicol in deep-well 96-well plates The cells were allowed to grow overnight at 37 °C, and each culture was then divided into two aliquots, diluting each with 0.8 ml of 2 × LB medium containing 30 μg/ml chloramphenicol to give a final OD600 of ∼0.1 in deep-well 96-well plates. The cells were grown at 37 °C to an OD600 of 0.7–0.9, with shaking at 250 r.p.m., and then induced with 1 mM IPTG for ∼3.5 h at 37 °C with shaking at 250 r.p.m. The liquid cultures were then harvested by centrifugation at 2,547g for 5 min at 4 °C. The pellets were stored in deep-well 96-well plates at −80 °C for future protein purification.
To purify the fusion proteins, frozen cell pellets were thawed at room temperature and resuspended at 4 °C in 80 μl of lysis buffer containing 50 mM NaH2PO4, pH 8, with 300 mM NaCl, 20 mM imidazole, CelLytic B (Sigma), lysozyme at 1 mg/ml, benzonase at 50 units/ml, proteinase inhibitor cocktail (Sigma) and 1 mM PMSF. The mixtures were transferred to bottom-sealed filter plates (Multiscreen Nylon Mesh) together with 25 μl pre-washed Ni-NTA Superflow (QIAGEN) per well. The plates were sealed with plate seals and incubated for 1.5 h at 4 °C with vigorous shaking. The seals were then removed, and the resin-protein complexes in the filter plates were washed three times with 250 μl/well of wash buffer I (50 mM NaH2PO4 with 300 mM NaCl, 10% glycerol, 20 mM imidazole and 0.01% Triton X-100, pH 8) and three times with wash buffer II (50 mM NaH2PO4 with 150 mM NaCl, 25% glycerol, 20 mM imidazole and 0.01% Triton X-100, pH 8) using a Q-fill2 (Gentix). After the filter plates were spun at 300g to completely remove the leftover wash buffer, the fusion protein in each well was eluted twice with 25 μl of elution buffer (50 mM NaH2PO4, 150 mM NaCl, 25% glycerol, 250 mM imidazole and 0.01% Triton X-100, pH 7.5). The eluted proteins were collected in 96-well PCR plates by centrifugation at 1,872g for 5 min.
Printing of the E. coli K12 proteome chips
The purified proteins were re-arrayed and aliquotted from 96-well plates into 384-well plates in a cold room using an Apricot system (PerkinElmer). The re-arrayed proteins were printed in duplicate onto glass slides, including FullMoon (FullMoon Biosystem), aldehyde-derivatized (Telechem), hydrogel (Schott) and FAST (Whatman) slides, using a ChipWriter Pro (Bio-Rad). After being left in the cold room for at least 8 h to ensure proper immobilization, the protein chips were stored at −80 °C.
Additional methods
The methods for E. coli ORF collection, oligonucleotide synthesis, DNA probing, YbaZ probing, fluorescence assays measurement of Kd and the detailed step-by-step protocols for the fabrication of the E. coli K12 proteome chips are available online in Supplementary Methods online.
Supplementary Material
Note: Supplementary information is available on the Nature Methods website.
Acknowledgments
We thank H. Mori (Nara Institute of Science and Technology, Japan) for providing the E. coli ORF collection, A. Osterman for help, C.L. Woodard for reviewing this manuscript, and D. McClellan for editorial assistance. This work was supported in part by the National Institutes of Health (grant GM071440 to C.H.; U54 RR020839 to H.Z.), the W. M. Keck Foundation (to C.H.), the Arnold and Mabel Beckman Foundation (to C.H.) and the Research Corporation (to C.H.).
Footnotes
Author Contributions: C.-S.C. developed the high-throughput protein purification protocol, printed chips, performed chip assays, analyzed chip assay data and wrote the manuscript. E.K. made the DNA probes, analyzed chip assay data, and performed the base-flipping assays and electrophoretic mobility shift assays with the help of H.C. and X.J. J.Z. measured the Kd and helped purify proteins. S.-C.T. helped purify proteins and print chips. C.H. and H.Z. planned the project and wrote the manuscript.
References
- 1.Zhu H, et al. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 2.MacBeath G, Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 3.Zhu H, Snyder M. Protein chip technology. Curr Opin Chem Biol. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 4.Michaud GA, et al. Analyzing antibody specificity with whole proteome microarrays. Nat Biotechnol. 2003;21:1509–1512. doi: 10.1038/nbt910. [DOI] [PubMed] [Google Scholar]
- 5.Gelperin DM, et al. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carinci F, et al. P-15 cell-binding domain derived from collagen: analysis of MG63 osteoblastic-cell response by means of a microarray technology. J Periodontol. 2004;75:66–83. doi: 10.1902/jop.2004.75.1.66. [DOI] [PubMed] [Google Scholar]
- 7.Espejo A, Cote J, Bednarek A, Richard S, Bedford MT. A protein-domain microarray identifies novel protein-protein interactions. Biochem J. 2002;367:697–702. doi: 10.1042/BJ20020860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lueking A, et al. A nonredundant human protein chip for antibody screening and serum profiling. Mol Cell Proteomics. 2003;2:1342–1349. doi: 10.1074/mcp.T300001-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Ge H. UPA, a universal protein array system for quantitative detection of protein-protein, protein-DNA, protein-RNA and protein-ligand interactions. Nucleic Acids Res. 2000;28:e3. doi: 10.1093/nar/28.2.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies DH, et al. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc Natl Acad Sci USA. 2005;102:547–552. doi: 10.1073/pnas.0408782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall DA, et al. Regulation of gene expression by a metabolic enzyme. Science. 2004;306:482–484. doi: 10.1126/science.1096773. [DOI] [PubMed] [Google Scholar]
- 12.Huang J, et al. Finding new components of the target of rapamycin (TOR) signaling network through chemical genetics and proteome chips. Proc Natl Acad Sci USA. 2004;101:16594–16599. doi: 10.1073/pnas.0407117101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ptacek J, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 14.Kitagawa M, et al. Complete set of ORF clones of Escherichia coli ASKA library (A Complete Set of E. coli K-12 ORF Archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 15.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 16.Roberts RJ, Cheng X. Base flipping. Annu Rev Biochem. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 17.Verdine GL, Bruner SD. How do DNA repair proteins locate damaged bases in the genome? Chem Biol. 1997;4:329–334. doi: 10.1016/s1074-5521(97)90123-x. [DOI] [PubMed] [Google Scholar]
- 18.Phizicky E, Bastiaens PI, Zhu H, Snyder M, Fields S. Protein analysis on a proteomic scale. Nature. 2003;422:208–215. doi: 10.1038/nature01512. [DOI] [PubMed] [Google Scholar]
- 19.Templin MF, et al. Protein microarrays: promising tools for proteomic research. Proteomics. 2003;3:2155–2166. doi: 10.1002/pmic.200300600. [DOI] [PubMed] [Google Scholar]
- 20.Haushalter KA, Stukenberg PT, Kirschner MW, Verdine GL. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr Biol. 1999;9:174–185. doi: 10.1016/s0960-9822(99)80087-6. [DOI] [PubMed] [Google Scholar]
- 21.MacBeath G. Protein microarrays and proteomics. Nat Genet. 2002;32(Suppl):526–532. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- 22.Arenkov P, et al. Protein microchips: use for immunoassay and enzymatic reactions. Anal Biochem. 2000;278:123–131. doi: 10.1006/abio.1999.4363. [DOI] [PubMed] [Google Scholar]
- 23.Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001;29:3784–3795. doi: 10.1093/nar/29.18.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verdine GL. The flip side of DNA methylation. Cell. 1994;76:197–200. doi: 10.1016/0092-8674(94)90326-3. [DOI] [PubMed] [Google Scholar]
- 25.Hornby DP, Ford GC. Protein-mediated base flipping. Curr Opin Biotechnol. 1998;9:354–358. doi: 10.1016/s0958-1669(98)80007-4. [DOI] [PubMed] [Google Scholar]
- 26.Labahn J, et al. Structural basis for the excision repair of alkylation-damaged DNA. Cell. 1996;86:321–329. doi: 10.1016/s0092-8674(00)80103-8. [DOI] [PubMed] [Google Scholar]
- 27.Duguid EM, Mishina Y, He C. How do DNA repair proteins locate potential base lesions? A chemical crosslinking method to investigate O6-alkylguanine-DNA alkyltransferases. Chem Biol. 2003;10:827–835. doi: 10.1016/j.chembiol.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Phadtare S, Severinov K. Nucleic acid melting by Escherichia coli CspE. Nucleic Acids Res. 2005;33:5583–5590. doi: 10.1093/nar/gki859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsui H, Shimaoka M, Kawasaki H, Takenaka Y, Kurahashi O. Adenine deaminase activity of the yicP gene product of Escherichia coli. Biosci Biotechnol Biochem. 2001;65:1112–1118. doi: 10.1271/bbb.65.1112. [DOI] [PubMed] [Google Scholar]
- 30.Jones RB, Gordus A, Krall JA, MacBeath G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 31.Zhu J, et al. RNA-binding proteins that inhibit RNA virus infection. Proc Natl Acad Sci USA. 2007;104:3129–3134. doi: 10.1073/pnas.0611617104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearson SJ, Ferguson J, Santibanez-Koref M, Margison GP. Inhibition of O6-methylguanine-DNA methyltransferase by an alkyltransferase-like protein from Escherichia coli. Nucleic Acids Res. 2005;33:3837–3844. doi: 10.1093/nar/gki696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mishina Y, Duguid EM, He C. Direct reversal of DNA alkylation damage. Chem Rev. 2006;106:215–232. doi: 10.1021/cr0404702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendonca VM, Kaiser-Rogers K, Matson SW. Double helicase II (uvrD)-helicase IV (helD) deletion mutants are defective in the recombination pathways of Escherichia coli. J Bacteriol. 1993;175:4641–4651. doi: 10.1128/jb.175.15.4641-4651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao SC, Zhu H. Protein chip fabrication by capture of nascent polypeptides. Nat Biotechnol. 2006;24:1253–1254. doi: 10.1038/nbt1249. [DOI] [PubMed] [Google Scholar]
- 36.Ramachandran N, et al. Self-assembling protein microarrays. Science. 2004;305:86–90. doi: 10.1126/science.1097639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Methods website.