

Abstract
The initial concentration of monomeric amyloidogenic proteins is a crucial factor in the in vitro formation of amyloid fibrils. We use quantitative atomic force microscopy to study the effect of the initial concentration of human α-synuclein on the mean length of mature α-synuclein fibrils, which are associated with Parkinson's disease. We determine that the critical initial concentration, below which low-molecular-weight species dominate and above which fibrils are the dominant species, lies at ∼15 μM, in good agreement with earlier measurements using biochemical methods. In the concentration regime where fibrils dominate, we find that their mean length increases with initial concentration. These results correspond well to the qualitative predictions of a recent statistical-mechanical model of amyloid fibril formation. In addition, good quantitative agreement of the statistical-mechanical model with the measured mean fibril length as a function of initial protein concentration, as well as with the fibril length distributions for several protein concentrations, is found for reasonable values of the relevant model parameters. The comparison between theory and experiment yields, for the first time to our knowledge, an estimate of the magnitude of the free energies associated with the intermolecular interactions that govern α-synuclein fibril formation.
INTRODUCTION
The aggregation of proteins into amyloid or amyloid-like fibrils is a process of crucial importance in many neurological disorders (1–3). In the case of Parkinson's disease (PD), the hallmark pathological features are Lewy bodies: intracellular neuronal inclusions consisting mainly of misfolded and aggregated α-synuclein (4,5). Nanoscale knowledge of the morphology of the protein aggregates in these inclusions may help increase understanding of the etiology of the disease. Earlier biophysical research has shown that aggregates of various amyloidogenic proteins typically take the form of thread-like fibrils. These fibrils may assemble hierarchically, that is, by protofilaments winding together (6), but also by lateral association without formation of a helical structure (7,8). Similar fibrillar structures have been observed in vitro formed by various disease-related and nondisease-related proteins, such as α-synuclein (6,8–10), glucagon (11,12), insulin (6,7), amyloid-β (Aβ) peptide 1–40 (13), prion protein (14), and others.
The morphology of the resulting aggregates depends on such diverse factors as solution conditions (15), the shape of any preformed aggregates which may serve as nuclei (7), and mutations in the amino acid sequence of the protein (10). Based on these observations, structural models describing the assembly of amyloid fibrils have been proposed. The dominant structural model for α-synuclein fibrillization (6) proposes that two protofilaments (linear chains of β-sheet-folded monomers) wind together to form an intermediate fibril, and two of these intermediate fibrils in turn wind together to form a so-called “mature fibril”. Note that various authors use different terminologies to describe the various species during the aggregation process. We follow the definitions of Kodali and Wetzel (13).
The aggregation of proteins into amyloid fibrils is considered to be a nucleation-polymerization process (16). As such, the initial concentration of α-synuclein is expected to have a profound effect on the fibril length. The concentration of α-synuclein present in neural cells is a factor relevant to the etiology of PD, since triplication of the α-synuclein gene (and subsequent overexpression of the protein) is associated with familial PD (17). Another clue to the significance of α-synuclein concentration for the etiology of the disease is that in pathological conditions, α-synuclein aggregates are also found in glial cells, where in vitro overexpression of α-synuclein leads to cell death (18). Apart from overexpression, the “effective concentration” of α-synuclein can also change from its normal value (which we estimate to be 70–140 μM in healthy neural cells, see the Materials and Methods section) due to reduced degradation or unspecific molecular crowding. These effects have been found to significantly reduce the aggregation lag time in in vitro experiments (19,20).
To capture the essential factors that allow many different proteins to form similar fibrils, a general statistical-mechanical model of amyloid fibril formation was recently outlined (21). This model couples a theory describing self-assembly and conformational transition to a description of the association of linear chains. The model focuses on the formation of linear, unbranched amyloid fibrils commonly observed in studies of protein aggregation and does not address amorphous aggregation. The model predicts the effect of protein concentration on the properties of a dilute solution of fibrillogenic protein molecules, given the free energies associated with various intermolecular interactions (Fig. 1). For reasonable values of these free-energy parameters, the model predicts that there exists a critical concentration below which most protein molecules are present as free monomers. It also predicts fibril formation above this concentration, with the fibril length increasing with protein concentration. The existence of a critical concentration is consistent with the notion of amyloid fibril formation being a nucleation-polymerization process (1,16).
FIGURE 1.

(A) Schematic and simplified representation of relevant species in α-synuclein fibrillization. Intrinsically disordered monomers (left) misfold and aggregate to form protofilaments (middle). Mature fibrils (right) can consist of up to four laterally interacting protofilaments. In this cartoon, molecules represented by disks possess the β-strand conformation that characterizes amyloid fibrils, whereas those represented by blobs do not. In this work we assume that the fibril ends can be in either a β- or a non-β-conformation. The experimentally observed helical twist in the mature fibril is not represented in this model. All processes are assumed to be reversible; (B) all interactions between protein molecules in the fibril have a free energy associated with them. P is the free energy for the interaction between β-folded monomers due to cross-β-sheet formation; E is the free energy for the interaction between a β-folded molecule and a molecule that is in a non-β-conformation, which is taken to be equal to the free energy between two non-β monomers; R is the free energy penalty for a transition between a region along the fibril axis in which the molecules have a β-conformation and one in which they are in a non-β-state; F is the lateral interaction free energy. For a full description of the statistical-mechanical model, see van Gestel and de Leeuw (21).
In this investigation, we test the predictions of the statistical-mechanical model introduced in van Gestel and de Leeuw (21), both qualitatively and quantitatively, using atomic force microscopy (AFM) to image mature α-synuclein fibrils formed in vitro at various initial protein concentrations. Our experimental results demonstrate that α-synuclein fibril formation is well described by the model. We determine the critical concentration for α-synuclein fibrillization to be 15 μM. From the measured dependence of the average fibril length on the protein concentration, we extract values for the free energies of interactions in the fibrils: the free energy of interaction between adjacent β-folded monomers is found to lie between −16.2 and −15.4 kJ/mol, and the lateral interaction between protofilaments in the fibril has a bond energy between −11.0 and −7.4 kJ/mol.
MATERIALS AND METHODS
Expression, purification, and aggregation of recombinant human α-synuclein
Wild-type α-synuclein (140 aa, M = 14,460 Da) was expressed and purified as described before (10).
Protein solutions taken from stock at −80°C were defrosted and centrifuged for 1 h at 21,000 × g to remove any preformed aggregates or contaminating particles. Native gradient polyacrylamide gel electrophoresis confirmed the presence of only monomeric α-synuclein. The initial protein concentration was determined by measuring the absorbance at 275 nm with a NanoDrop ND-1000 absorption spectrophotometer (NanoDrop Technologies, Wilmington, DE) and using an extinction coefficient ɛ(275 nm) = 5600 M−1cm−1 (1400 M−1cm−1 per tyrosine residue). Monomeric α-synuclein was diluted to the desired initial protein concentrations in the 5–250 μM range in a buffer containing 10 mM HEPES and 50 mM NaCl at pH 7.4. We estimate the concentration uncertainty to be 6% for all initial concentrations based on analysis of absorption measurements and pipetting accuracy.
Aggregation was performed in a temperature-controlled shaking incubator (ThermoMixer, Eppendorf, Hamburg, Germany) at 37°C while shaking at 500 rpm. This shaking frequency speeds up the aggregation process to a manageable timescale but is considered “gentle”. Any shear-force-induced fibril breakage would occur to the same extent for all concentration conditions in this experiment since incubation conditions were equal. The aggregation of amyloidogenic proteins is not an artifact of agitation: aggregation occurs in undisturbed solutions, only much slower (22). Each vial contained 400 μl of protein solution, and all aggregations were performed in duplicate. Before samples were taken out of the aggregation vessels, they were rotated at an angle and aspirated to maximize homogeneity of the sample without disrupting any aggregates.
Samples were taken for detailed AFM analysis after 20–28 days, when the aggregation reactions had reached their final equilibrium state as verified from fibril morphology measurements. The presence of monomeric protein in equilibrium with fibrils was confirmed not only by measuring the 275 nm absorbance of the supernatant after pelleting the fibrils by centrifugation but also from native gradient polyacrylamide gel electrophoresis.
Acquisition of AFM images
We deposited 4 μl aliquots of aggregated protein solutions on freshly cleaved mica and incubated them for 2 min in a humid environment to avoid drying of the droplet and salt crystal formation. The samples were then gently rinsed with 200 μl of MilliQ water (resistivity > 18 MΩ·cm−1; Millipore, Bedford, MA) and blown dry with a gentle flow of N2(g). Aggregates of all sizes were found to adhere equally well to freshly cleaved mica without further surface modification (see, for example, Hoyer et al. (9)).
AFM images were acquired on a custom-built standalone AFM instrument (23) and on a Multimode AFM with a Nanoscope IV controller (Veeco, Santa Barbara, CA) in tapping mode under ambient conditions. The drying of protein aggregate samples for imaging in air influences their morphology (especially height and periodicity) but does not affect the observed fibril length. We used Veeco Probes MSCT-AU tip F (Si3N4), nominal tip radius 10 nm, spring constant k = 0.5 N/m; and MikroMasch (Tallinn, Estonia) NSC36/Cr-Au tip B (Si), nominal tip radius < 10 nm, spring constant k = 1.75 N/m. Tapping amplitude was between 50 and 100 nm, depending on tip-sample adhesion assessed on a measurement-by-measurement basis. For the aggregates formed by 5 and 10 μM α-synuclein solutions, images were taken at a pixel resolution of 4 nm/pixel (image size 4 μm) for the 20–30 μM aggregates at 20 nm/pixel (image size 20 μm) and for the 50–250 μM aggregates at 40 nm/pixel (image size 20 μm).
Measurement of fibril length distributions
Raw AFM height images were processed using Scanning Probe Image Processor (Image Metrology, Hørsholm, Denmark) to remove sample tilt and scanner bow. Sample tilt was removed using manual tilt correction while monitoring x and y cross sections until both cross sections were horizontal. Then, any scanner bow artifacts were corrected using a second or third order average profile fit. To minimize distortion of apparent morphology of the objects in the image, the fit was calculated excluding these objects by setting limits on the z color scale. Finally, any line-to-line scanner jumps were corrected by a zeroth order linewise fit.
Lengths of individual fibrils were measured using segmented line profiles in ImageJ (24). To minimize observer bias, all fibrils that fit the following criteria were included in the analysis:
The fibril lies completely within the image.
The fibril can be unambiguously distinguished from any overlapping fibrils.
The fibril appears in the image as larger than four pixels.
We estimate the accuracy of the individual fibril length measurements to be 40 nm (20 and 30 μM concentrations) and 80 nm (50–250 μM concentrations), mainly limited by tip-sample convolution and pixel resolution. The analysis procedure is demonstrated in Fig. 2.
FIGURE 2.

AFM image illustrating the length measurement procedure. Using the plane-corrected height images (A), lengths were measured manually for all fibrils that could be resolved individually, did not fall off the edge of the image, and were larger than four pixels (B). Scale bar 2 μm.
The aggregates formed by initial concentrations of 5 and 10 μM were amorphous with reported sizes on the order of 20 nm, which corresponds to the “tip-sample-convolution resolution”. The size of these aggregates was characterized by their height instead of their length.
Modeling α-synuclein fibril length as a function of initial concentration
We have adapted a recently outlined statistical-mechanical model of protein aggregation in dilute solution (21) to the specific case of α-synuclein fibrillization. The model assumes that only two conformational states of the protein molecules are sufficiently populated to have an effect on the aggregation behavior: either proteins can be in a β-strand conformation or they can be in a less ordered conformation. The model then describes the properties of mature fibrils in an equilibrium situation. One key prediction is the distribution of the lengths of mature fibrils as a function of initial concentration. The statistical-mechanical model does not attempt to model the early stages of aggregation. It would in principle be possible to extend the model to include parameters that represent monomer conformation. However, for every conformation taken into account, we need an extra free energy parameter.
A model with an infinite number of adjustable parameters may be complete but will not be very informative. It will also be next to impossible to independently determine the appropriate values for these parameters from experimental data. It would also in principle be possible to include an activation step into the model. However, the concentration of “activated” monomers (that are in a conformation capable of adding to a fibril) will be extremely small since they would be incorporated into the fibrils immediately. The equilibrium model then simplifies to one without the activation step. The kinetics of the process would be influenced significantly by an activation step, but since our interest lies in the morphology of the resulting species, that does not pose a problem. Any conformational changes in the monomers will likely involve such small free energy changes that it would not significantly influence the predictions of our model if we took them into account.
The model accounts for three species that participate in the aggregation process: monomers, protofilaments, and fibrils (Fig. 1). Monomers are defined as single protein molecules that possess a non-β-conformation. Protofilaments are linear chains of interacting monomers, each of which can be in a non-β-state or in a β-strand state. Mature fibrils are defined as rod-like aggregates, which in the case of α-synuclein contain up to four protofilaments.
Because proteins that possess different conformations interact differently, we introduce two free-energy parameters: one that accounts for the interaction between two proteins that are both in a β-strand conformation (labeled P in Fig. 1) and one for the interaction of two proteins that are not both in this conformation (E). Furthermore, we introduce an interaction free energy for lateral protein-protein contacts (F) and a free-energy penalty that is applied whenever an ordered region and a disordered one meet (R). (Note that the symbols for some of the free energies are different than those in van Gestel and de Leeuw (21). E replaces M, to avoid confusion with the molar mass, and P replaces P*, because in the current context it is not necessary to distinguish between the β-bond free energy and the excess β-bond free energy.) According to the current structural model of α-synuclein fibrillization, mature α-synuclein fibrils consist of two intertwined intermediate fibrils, which in turn consist of two intertwined protofilaments (6). This is reflected in the theory by taking into account only fibrils that consist of four or fewer protofilaments. Each fibril contains (p − 1) × m lateral protein-protein contacts, with p the number of protofilaments making up the fibril and m the length of each protofilament expressed in the number of protein molecules. Combining the model with self-assembly theory allows us to obtain values for the mean aggregate size, the distribution of fibril lengths, and the mean fibril length.
The temperature at which aggregation is performed (or modeled) affects the kinetics of amyloidogenesis, but not so much the fibril morphology or the equilibrium concentrations. In the model, as in the experiment, temperature was kept constant at the physiologically relevant value of 37°C.
Although a full description of the model has been given in van Gestel and de Leeuw (21), it is appropriate to summarize the theory and present the key equations here. To describe a polydisperse system of dissolved protein aggregates, two characteristics are of vital importance. The first is the number density ρ, which gives the total number of particles (aggregates and monomers) that are present in solution, and the second is the volume fraction φ of protein molecules, which in effect counts the total number of protein molecules present. In van Gestel and de Leeuw (21), these parameters were determined to equal
 |
(1) |
and
 |
(2) |
respectively. In the above equations, x, y, λ1, and λ2 are prefactors depending on the description of the protofilament ends (21). These prefactors depend only on Boltzmann factors σ and s (defined below). The equations thus contain five variables: the fugacity 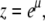 with μ the chemical potential of protein molecules given in units of the thermal energy (kBT with kB Boltzmann's constant and T the absolute temperature), and the Boltzmann factors
with μ the chemical potential of protein molecules given in units of the thermal energy (kBT with kB Boltzmann's constant and T the absolute temperature), and the Boltzmann factors 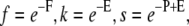 and
and  F, E, P, and R are the free energies introduced above (Fig. 1) and are also given in terms of the thermal energy.
F, E, P, and R are the free energies introduced above (Fig. 1) and are also given in terms of the thermal energy.
In Eqs. 1 and 2 the number density and volume fraction have each been split into five terms that can be used separately if required. The first term in each equation gives the number density or volume fraction of monomers, the second term that of dimers, the (combined) third and fourth terms of protofilaments of all lengths, and the final term for fibrils of all lengths, containing p protofilaments. These latter terms equal
 |
(3) |
and
 |
(4) |
If φ and ρ are known, the mean number of protein molecules per particle can be calculated as
 |
(5) |
By taking the last term from Eqs. 1 and 2, we can calculate the mean aggregation number for fibrils only in a similar way:
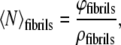 |
(6) |
or alternatively, for all fibrils containing p = 4 protofilaments,
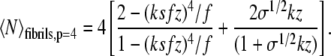 |
(7) |
To calculate the mean length (expressed in number of monomers) of such fibrils, one then needs only to divide the mean aggregation number by the number of protofilaments, p:
 |
(8) |
To compare theory and experiment, we need to convert experimental units to those reflected in the theoretical model. The volume fraction of (initially monomeric) protein ϕ is calculated as 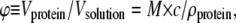 since
since  where Vprotein is the volume occupied by the protein molecules, Vsolution is the total volume, mprotein is the mass of the dissolved protein, c is the protein molar concentration, M is the protein molar mass, and ρprotein is the protein mass density. The mass density of α-synuclein was estimated according to Fischer ((25), Eq. 2), inserting M = 14,460 kDa for the molar mass, giving ρ = 1.46 × 103 mg/ml. The mass density is assumed to be constant upon folding and aggregation of the protein. We realize that this assumption may be an oversimplification. However, to our knowledge there are currently no exact values for the mass density of α-synuclein molecules inside a fibril. Detailed structural information about the fibril architecture is necessary to reach a more accurate estimate of the mass density.
where Vprotein is the volume occupied by the protein molecules, Vsolution is the total volume, mprotein is the mass of the dissolved protein, c is the protein molar concentration, M is the protein molar mass, and ρprotein is the protein mass density. The mass density of α-synuclein was estimated according to Fischer ((25), Eq. 2), inserting M = 14,460 kDa for the molar mass, giving ρ = 1.46 × 103 mg/ml. The mass density is assumed to be constant upon folding and aggregation of the protein. We realize that this assumption may be an oversimplification. However, to our knowledge there are currently no exact values for the mass density of α-synuclein molecules inside a fibril. Detailed structural information about the fibril architecture is necessary to reach a more accurate estimate of the mass density.
A second conversion is that between the units in which length is measured. In the theory, the length of a fibril is given as the degree of polymerization divided by the number of protofilaments per fibril, i.e., in terms of a number of molecules, rather than in nanometers. The “length of one protein molecule” along the fibril long axis equals one inter-β-strand distance of 0.47 nm (26). A fibril that contains four protofilaments and has a length of 1 μm would thus contain ∼8.5 × 103 monomers.
Finally, the theory requires that the conformation of the end monomers of the fibrils be specified. This can be done in three ways: we can force all fibril ends to be in a non-β-conformation, we can fix them in a β-conformation, or we can allow them to attain either of these conformations (21). The first of these boundary conditions causes the model to predict the formation of unrealistically long fibrils for reasonable values of the free-energy parameters. The other two descriptions of fibril end conformation yield realistic, and equivalent, results. We chose to allow both conformations since the actual conformation of fibril ends is not known.
Estimation of concentration of α-synuclein in a neural cell
To relate the critical concentration for α-synuclein fibrillization found in our theory and experiments to the in vivo situation, we estimate the concentration of α-synuclein in a neural cell. It is estimated that α-synuclein (Ma = 14,460 g/mol) comprises a fraction of fa ≅ 0.5%–1.0% % of brain cytosolic protein (5). If we assume that proteins make up fp = 20% of a cell's weight and the average mass density of a cell is ρc = 1.03 g/ml (both estimates from Lodish et al. (27)), we can approximate the concentration of α-synuclein in a neuron as
 |
using na = ma/Ma, ma = fa fp mc, and mc = ρc Vc, where ca is the molar concentration of α-synuclein in the cell, na is the number of moles of α-synuclein in the cell, ma is the total mass of the α-synuclein, and Vc and mc are the cell volume and mass, respectively, which cancel in the equation.
RESULTS
The extent of α-synuclein fibrillization depends on the initial protein concentration
Under the conditions employed in this investigation, α-synuclein forms no fibrillar aggregates when the initial concentration is 5 or 10 μM (see representative images in Fig. 3, A and B). The apparent lateral dimensions of these aggregates are exaggerated by tip-sample convolution (∼20 nm). Their height is in the order of a few nanometers (see also Fig. 4).
FIGURE 3.

AFM height images of wild-type α-synuclein aggregates formed at initial protein concentrations increasing from 5 to 250 μM. All scale bars 1 μm.
FIGURE 4.

Fibril length distributions for wild-type α-synuclein fibrils formed in vitro from a range of initial protein concentrations (5, 10, 20, 30, 50, 100, 250 μM). For 5 and 10 μM samples, the aggregate height instead of length was measured as the indicator of aggregate size, since no significant fibrillization occurred. The solid lines in the distributions for 20–250 μM are theoretical predictions using the same free-energy parameters as in Fig. 5. The reported n is the number of fibrils measured at each concentration. Bin sizes are 0.4 nm (for 5 and 10 μM concentrations) and 100 nm (20–250 μM).
The aggregation with an initial concentration of 20 μM produced many small aggregates similar to those observed for 5 and 10 μM and a small amount of short fibrils, most of them under 1 μm in length (Fig. 3 C). For higher concentrations (30 μM up to 250 μM), progressively more and longer fibrils were observed (Fig. 3, D–F).
The critical α-synuclein concentration for fibril formation is ∼15 μM
The fibril length measurements for each initial concentration condition are summarized in the length distribution histograms shown in Fig. 4. The distribution of mature α-synuclein fibril lengths depends on the initial protein concentration: the distributions display a tail toward longer fibril lengths that becomes more pronounced at higher concentrations (Fig. 4). The shortest fibrils (<100 nm in length) at each aggregation condition are underrepresented in the histograms because the tip-sample convolution and pixel resolution require a minimum length for identification as a fibril in the AFM images, in this case 40–80 nm.
To quantify the effect of increasing protein concentration, the numerical average of the fibril lengths was calculated from each length distribution. Under our experimental conditions, there is no significant fibrillization below a threshold initial concentration of ∼15 μM; above this concentration progressively longer fibrils form (Fig. 5). See the next section for a discussion of the error on the critical concentration estimate. The vertical error bars at the 5 and 10 μM data points in Fig. 5 are set to 20 nm because that is the “tip-sample convolution resolution”: the lateral size (“length”) of the aggregates appears as 20 nm due to the finite AFM tip size, but the real length is smaller. The resolution of the length measurements is taken as the uncertainty on the means of the other concentrations and is 40 nm for 20, 30, and 50 μM and 80 nm for the higher concentrations.
FIGURE 5.

Concentration dependence of mean α-synuclein fibril length. The data points are the mean lengths from the fibril length distributions (Fig. 4), the solid line is the concentration dependence predicted by the statistical-mechanical model (lateral binding free energy F = − 3.8 kBT, binding free energy between β-folded monomers P = −6.2 kBT).
Comparison of the statistical-mechanical model with mean fibril lengths yields free energy parameters of α-synuclein fibrillization
We use the experimentally determined mean fibril lengths to establish the free energies of the interactions in α-synuclein fibrils as modeled by the statistical-mechanical model. Although the original model contains four free-energy parameters, only two of these parameters influence the predicted concentration dependence of the mean fibril length. These are the free energy of an interaction between β-folded monomers P and the lateral-interaction free energy F (Fig. 1). The other two parameters, which describe the interaction between disordered protein molecules and the transition between a disordered and an ordered regime along the fibril axis, turn out to have a negligible effect. This observation indicates that the mature fibrils that dominate the system at high enough protein concentrations contain very few disordered protein molecules. A similar result was found by Nyrkova and co-workers in their analysis of protein fibril formation by a synthetic peptide (28).
To quantitatively compare the experimental results to the theory, we fixed the values of all four free-energy parameters and calculated the unknown fugacity z from the protein concentration using Eq. 2. Subsequently, Eq. 8 was used to calculate the mean fibril length for the fixed values of the free-energy parameters and at the specified concentration. By repeating this process for different values of ϕ, we obtain the dependence between the protein concentration and the mean fibril length (Fig. 5). By systematically varying the free-energy parameters P and F, we conclude that the mean fibril length at high concentrations depends only on the β-bond free energy P and not on F, whereas the predicted critical concentration depends on both free-energy parameters. Observing the experimental results (Fig. 5), we set the limits between which the critical concentration must fall at 10 and 20 μM and use this as a criterion to determine which values of the free energy parameters yield good agreement between theory and experiment. We then specify the error margin of the critical concentration as ccrit = 15 ± 5 μM. The constant slope of the curve (Fig. 5) at high concentrations is virtually independent of the value of either parameter.
Good agreement between theory and experiment is found when the β-interaction free energy P lies between −6.3 and −6.0 times kBT and the lateral-association free energy F is between −4.3 and −2.9 times kBT, with kB the Boltzmann's constant and T the absolute temperature. The solid line in Fig. 5 shows the predicted concentration dependence of the mean fibril length for P = −6.2 kBT and F = −3.8 kBT. These values correspond to P = −15.9 kJ/mol and F = −9.8 kJ/mol at the fibrillization temperature of 37°C.
Using the same values for the free-energy parameters, we compared theoretically calculated length distributions to those measured by AFM. The statistical-mechanical model reproduced the experimental results semiquantitatively for concentrations above 10 μM (Fig. 4). We plotted the number density (proportional to the number of fibrils per unit volume) of fibrils with a length that falls within each specified interval (expressed as a number of monomers m), divided by the total fibril number density. The total fibril number density was calculated by summation of the fibril number density over all fibril lengths. This is summarized in Eq. 9.
Only fibrils consisting of four protofilaments were taken into account in the calculation, because fibrils that contain fewer than the maximum allowed number of protofilaments tend to stay very short (21):
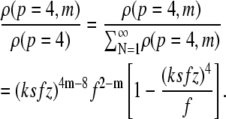 |
(9) |
The theory predicts an exponential decay of the number of fibrils of a given degree of polymerization with their length (solid lines in Fig. 4). The theory provides an excellent prediction for the fractions of long fibrils, but the agreement is less obvious for short fibrils. In particular, fewer short fibrils (with lengths below a few hundred nanometers) are found experimentally than would be expected from the model prediction. This may be due to the finite resolution of the AFM imaging and length measurement procedure. Both the theory and the AFM measurements indicate that virtually no fibrils are formed in aggregations with initial protein concentrations of 5 and 10 μM.
DISCUSSION
The critical concentration for α-synuclein fibrillization of 15 ± 5 μM is in the same order of magnitude as an earlier reported critical concentration for α-synuclein of 28 μM, determined by quantitative amino acid analysis (16). The critical concentration is well below the estimated in vivo concentration of α-synuclein in neural cells (70–140 μM). This indicates the possibility of amyloid fibril formation at normal physiological conditions, not necessarily involving overexpression of α-synuclein. The fibril lengths we find are also realistic: fibrils up to several micrometers do not have to fold upon themselves to fit in a Lewy body with an approximate diameter of 10 μm.
The use of a single critical concentration for a polymerization process requires two assumptions, as explained in Frieden (29). One assumption is that the equilibrium of the conformational changes must be rapid relative to the equilibrium of the monomers with the fibrils. This assumption certainly holds: monomer folding conversions typically take place on the order of microseconds to milliseconds, and the aggregation process is in the order of hours, even weeks.
The other implicit assumption one makes when using a single critical concentration is that all monomeric conformers interact with the fibril. In the polymerization process discussed here, where it is likely that multiple conformations exist (30), the case may be that there are monomeric conformers that do not add to the fibrils. However, it is mainly the kinetics of growth, not the morphology of the resulting fibrils, that would be affected. Sandal et al. report the detection of various α-synuclein conformers based on force spectroscopy data but also indicate that it is at present impossible to prove spectroscopically what conformation the conformers actually possess (30). This being the case, and given that the conformer equilibrium is much more rapid than the fibril formation process, the assumption that all monomers are available for fibril formation seems justified.
We determined the free energies of two types of bonds within an α-synuclein amyloid fibril: the free-energy difference between bound and unbound states of the monomers, reflected by the parameter P, which we determined to lie between −16.2 and −15.4 kJ/mol, and the free energy of the lateral bond between two protofilaments in a fibril, reflected by the parameter F, which is between −11.0 and −7.4 kJ/mol. These values imply that the bonds that make up the fibrils are ∼2–4 times as strong as a hydrogen bond in a protein (∼4.2–8.4 kJ/mol), four times as strong as a typical Van der Waals interaction (∼4.2 kJ/mol), and ∼20 times weaker than a single covalent C-C bond (∼347 kJ/mol).
The bond between the monomers in a protofilament is ∼1.5 times as strong as the bond between protofilaments. Considering nanodeformation experiments using AFM tips, one would expect “unzipping” of protofilaments to occur rather than breakage of fibrils perpendicular to the long axis. This prediction is consistent with results on Aβ reported by Kellermayer et al., who used an AFM tip as a nanomanipulation tool (31). Our value for the lateral association free energy F is very similar to their free energy of lateral binding of ∼9.6 kJ/mol determined using mechanical unzipping of β-sheets from Aβ fibrils (31).
The statistical-mechanical model we employ holds under two conditions. The first is that the protein solution is dilute enough that the effects of interaggregate interactions (such as those of the excluded-volume type) may be neglected. This condition is likely satisfied, as the α-synuclein concentrations used in this investigation are in the micromolar range, and the concentration of fibrils is much lower still. The second condition is that the processes described by the model are reversible, where the observed species can be reasonably assumed to be in thermodynamic equilibrium with the surroundings. Fibrillization of α-synuclein, as of Alzheimer's Aβ protein (32), can be considered to be reversible: fibrils were shown to dissociate under high hydrostatic pressure (33).
The application of the statistical-mechanical theory to actual morphological data allows us to look in a new way at the interactions involved in α-synuclein fibrillization and to quantify the strength of the bonds involved. The model is equally applicable to other amyloid-forming proteins, provided that the maximum number of protofilaments per fibril and the intermolecular distance along the fibril long axis are known. Its predictions of the mean fibril length, the critical concentration, and the fractions of fibrils with a given length will be valuable in establishing quantitative insights into the biophysics of fibril formation in other proteins. Detailed analysis of the kinetics and energetics of the aggregation process are essential to map the energy landscape for fibrillization and to fill in the gaps in suitable theoretical models consistent with the physics of these complex biopolymer systems. The existence of a critical concentration for aggregation is particularly interesting because critical monomer/nucleus concentration fluctuations may trigger the onset of nucleation (34), a process analogous to protein crystallization (35). Since there is a growing consensus that early aggregate species are likely responsible for disease etiology, detailed morphological studies of the intermediate species around the critical concentration for fibrillization will yield key insights into potentially cytotoxic intermediates on the pathway to fibrillization and to the development of intervention strategies for inhibition of aggregation or for fibril dissolution.
Acknowledgments
M.v.R. thanks Kirsten van Leijenhorst for protein expression and purification, Martijn Stopel and Robert van der Meer for performing protein aggregation and initial AFM experiments instrumental to this research, Kees van der Werf for expert advice on AFM, Hetty ten Hoopen and the Polymer Chemistry and Biomaterials group for the use of their Multimode AFM, and Prof. Wim Briels of the Computational Biophysics group for helpful discussions on statistical mechanics. J.v.G. and S.W.d.L. thank Maarten Wolf, Jaap Jongejan, and Jon Laman for stimulating discussions.
The work of M.v.R. is part of the research program of the Stichting voor Fundamenteel Onderzoek der Materie (FOM), which is financially supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO). J.v.G. and S.W.d.L. thank NWO for funding (grant No. 635.100.012, program for computational life sciences).
Editor: Peter Hinterdorfer.
References
- 1.Chiti, F., and C. M. Dobson. 2006. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75:333–366. [DOI] [PubMed] [Google Scholar]
- 2.Rochet, J. C., and P. T. Lansbury Jr. 2000. Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 10:60–68. [DOI] [PubMed] [Google Scholar]
- 3.Tycko, R. 2004. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr. Opin. Struct. Biol. 14:96–103. [DOI] [PubMed] [Google Scholar]
- 4.Goedert, M. 2001. α-Synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2:492–501. [DOI] [PubMed] [Google Scholar]
- 5.Shults, C. W. 2006. Lewy bodies. Proc. Natl. Acad. Sci. USA. 103:1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khurana, R., C. Ionescu-Zanetti, M. Pope, J. Li, L. Nielson, M. Ramirez-Alvarado, L. Regan, A. L. Fink, and S. A. Carter. 2003. A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophys. J. 85:1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jansen, R., W. Dzwolak, and R. Winter. 2005. Amyloidogenic self-assembly of insulin aggregates probed by high resolution atomic force microscopy. Biophys. J. 88:1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segers-Nolten, I., K. van der Werf, M. van Raaij, and V. Subramaniam. 2007. Quantitative characterization of protein nanostructures using atomic force microscopy. Conf. Proc. IEEE Eng. Med. Biol. Soc. 6609–6612. [DOI] [PubMed]
- 9.Hoyer, W., D. Cherny, V. Subramaniam, and T. Jovin. 2004. Rapid self-assembly of α-synuclein observed by in situ atomic force microscopy. J. Mol. Biol. 340:127–139. [DOI] [PubMed] [Google Scholar]
- 10.Van Raaij, M. E., I. M. J. Segers-Nolten, and V. Subramaniam. 2006. Quantitative morphological analysis reveals ultrastructural diversity of amyloid fibrils from α-synuclein mutants. Biophys. J. 91:L96–L98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Jong, K. L., B. Incledon, C. M. Yip, and M. R. DeFelippis. 2006. Amyloid fibrils of glucagon characterized by high-resolution atomic force microscopy. Biophys. J. 91:1905–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong, M., M. B. Hovgaard, S. Xu, D. E. Otzen, and F. Besenbacher. 2006. AFM study of glucagon fibrillation via oligomeric structures resulting in interwoven fibrils. Nanotechnology. 17:4003–4009. [DOI] [PubMed] [Google Scholar]
- 13.Kodali, R., and R. Wetzel. 2007. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 17:48–57. [DOI] [PubMed] [Google Scholar]
- 14.Anderson, M., O. V. Bocharova, N. Makarava, L. Breydo, V. V. Salnikov, and I. V. Baskakov. 2006. Polymorphism and ultrastructural organization of prion protein amyloid fibrils: an insight from high resolution atomic force microscopy. J. Mol. Biol. 358:580–596. [DOI] [PubMed] [Google Scholar]
- 15.Hoyer, W., T. Antony, D. Cherny, G. Heim, T. M. Jovin, and V. Subramaniam. 2002. Dependence of α-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 322:383–393. [DOI] [PubMed] [Google Scholar]
- 16.Wood, S. J., J. Wypych, S. Steavenson, J. C. Louis, M. Citron, and A. L. Biere. 1999. α-Synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J. Biol. Chem. 274:19509–19512. [DOI] [PubMed] [Google Scholar]
- 17.Singleton, A. B., M. Farrer, J. Johnson, A. Singleton, S. Hague, J. Kachergus, M. Hulihan, T. Peuralinna, A. Dutra, R. Nussbaum, S. Lincoln, A. Crawley, M. Hanson, D. Maraganore, C. Adler, M. R. Cookson, M. Muenter, M. Baptista, D. Miller, J. Blancato, J. Hardy, and K. Gwinn-Hardy. 2003. α-Synuclein locus triplication causes Parkinson's disease. Science. 302:841. [DOI] [PubMed] [Google Scholar]
- 18.Stefanova, N., L. Klimaschewski, W. Poewe, G. K. Wenning, and M. Reindl. 2001. Glial cell death induced by overexpression of α-synuclein. J. Neurosci. Res. 65:432–438. [DOI] [PubMed] [Google Scholar]
- 19.Shtilerman, M. D., T. T. Ding, and P. T. Lansbury Jr. 2002. Molecular crowding accelerates fibrillization of α-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry. 41:3855–3860. [DOI] [PubMed] [Google Scholar]
- 20.Munishkina, L. A., E. M. Cooper, V. N. Uversky, and A. L. Fink. 2004. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 17:456–464. [DOI] [PubMed] [Google Scholar]
- 21.van Gestel, J., and S. W. de Leeuw. 2006. A statistical-mechanical theory of fibril formation in dilute protein solutions. Biophys. J. 90:3134–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramirez-Alvarado, M., J. S. Merkel, and L. Regan. 2000. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. USA. 97:8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Werf, K. O., C. A. J. Putman, B. G. de Grooth, F. B. Segerink, E. H. Schipper, N. F. van Hulst, and J. Greve. 1993. Compact stand-alone atomic force microscope. Rev. Sci. Instrum. 64:2892–2897. [Google Scholar]
- 24.Rasband, W. S. 1997. –2006. ImageJ, http://rsb.Info.Nih.Gov/ij/.
- 25.Fischer, H., I. Polikarpov, and A. F. Craievich. 2004. Average protein density is a molecular-weight-dependent function. Protein Sci. 13:2825–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serpell, L. C., J. Berriman, R. Jakes, M. Goedert, and R. A. Crowther. 2000. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc. Natl. Acad. Sci. USA. 97:4897–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lodish, H., A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore, and J. Darnell. 2000. Molecular Cell Biology, 4th ed. W. H. Freeman, New York.
- 28.Nyrkova, I. A., A. N. Semenov, A. Aggeli, and N. Boden. 2000. Fibril stability in solutions of twisted β-sheet peptides: a new kind of micellization in chiral systems. Eur. Phys. J. B. 17:481–497. [Google Scholar]
- 29.Frieden, C. 2007. Protein aggregation processes: in search of the mechanism. Protein Sci. 16:2334–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sandal, M., F. Valle, I. Tessari, S. Mammi, E. Bergantino, F. Musiani, M. Brucale, L. Bubacco, and B. Samori. 2008. Conformational equilibria in monomeric α-synuclein at the single-molecule level. PLoS Biol. 6:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kellermayer, M. S. Z., L. Grama, A. Karsai, A. Nagy, A. Kahn, Z. L. Datki, and B. Penke. 2005. Reversible mechanical unzipping of amyloid β-fibrils. J. Biol. Chem. 280:8464–8470. [DOI] [PubMed] [Google Scholar]
- 32.Wetzel, R. 2006. Kinetics and thermodynamics of amyloid fibril assembly. Acc. Chem. Res. 39:671–679. [DOI] [PubMed] [Google Scholar]
- 33.Foguel, D., M. C. Suarez, A. D. Ferrao-Gonzales, T. C. Porto, L. Palmieri, C. M. Einsiedler, L. R. Andrade, H. A. Lashuel, P. T. Lansbury, J. W. Kelly, and J. L. Silva. 2003. Dissociation of amyloid fibrils of α-synuclein and transthyretin by pressure reveals their reversible nature and the formation of water-excluded cavities. Proc. Natl. Acad. Sci. USA. 100:9831–9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Podesta, A., G. Tiana, P. Milani, and M. Manno. 2006. Early events in insulin fibrillization studied by time-lapse atomic force microscopy. Biophys. J. 90:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.ten Wolde, P. R., and D. Frenkel. 1997. Enhancement of protein crystal nucleation by critical density fluctuations. Science. 277:1975–1978. [DOI] [PubMed] [Google Scholar]