

Abstract
We present a new method for multiple sequence alignment (MSA), which we call MSACSA. The method is based on the direct application of a global optimization method called the conformational space annealing (CSA) to a consistency-based score function constructed from pairwise sequence alignments between constituting sequences. We applied MSACSA to two MSA databases, the 82 families from the BAliBASE reference set 1 and the 366 families from the HOMSTRAD set. In all 450 cases, we obtained well optimized alignments satisfying more pairwise constraints producing, in consequence, more accurate alignments on average compared with a recent alignment method SPEM. One of the advantages of MSACSA is that it provides not just the global minimum alignment but also many distinct low-lying suboptimal alignments for a given objective function. This is due to the fact that conformational space annealing can maintain conformational diversity while searching for the conformations with low energies. This characteristics can help us to alleviate the problem arising from using an inaccurate score function. The method was the key factor for our success in the recent blind protein structure prediction experiment.
INTRODUCTION
Multiple sequence alignment (MSA) is a fundamental problem in computational biology and bioinformatics, where either related proteins from the same organism or similar proteins from different organisms are examined to determine their relationship. Such information can then be used to assess the shared evolutionary origins of the sequences and the extent to which functions of related proteins overlap. In addition, MSA is used to model three-dimensional structures of proteins. However, MSA, especially in the context of protein structure prediction, is a nontrivial problem. Reliable protein modeling via MSA depends on three fundamental elements: proper selection of template proteins, the accuracy of a score function for MSA, and the optimization of the score function (1). To obtain biologically meaningful alignments of multiple sequences, we have to deal with these three problems simultaneously.
When we assume that template sequences are already provided, MSA suffers from the following two major obstacles: First, currently there are no ideal score functions that fulfill biologically meaningful applications. In practice, objective functions based on the sum-of-pair scores (2), and their variants (3) have been used widely in the literature. Recent progress has focused on the design of consistency-based objective score functions based on a library of local segments of matches from pairwise alignments where the goal is to find an alignment maximally satisfying the restraints of the library that was implemented in COFFEE score for the first time (4). Second, even with a perfect objective function to be optimized, finding the optimal alignment of given sequences is known as a nondeterministic polynomial-time (NP)-complete problem (5). Exact optimization by dynamic programming (6–8) requires O((2L)N) time complexity (N is the number of sequences, and L the average sequence length) and O(LN) memory complexity. Therefore, carrying out MSA by dynamic programming becomes practically intractable as the number of sequences increases.
Due to these difficulties of rigorous optimization in MSA, practically all current methods in use, employ heuristic strategies such as the progressive alignment. Popular methods include Pileup based on Feng and Doolittle algorithm (9) and ClustalW (10). Progressive alignments align two sequences at a time, and add these alignments to the set of already-aligned sequences based on a guide tree and a reduced pairwise match score. One of the disadvantages of progressive alignments is that errors possibly occurred at early stages of the alignment cannot be fixed at later stages. Various attempts to improve the quality of progressive alignment have been reported by applying progressive alignments iteratively such as in Prrp (11), DiAlign (12), and MUSCLE (13).
More recently, variant methods based on progressive alignment and the consistency-based score functions are proposed (e.g., T-COFFEE (14), ProbCons (15), and SPEM (16)). For example, SPEM combines a profile-profile alignment method called SP2 (17) using secondary structure information, with a consistency-based refinement for pairwise alignment and a progressive algorithm. SPEM is one of the top MSA methods outperforming other popular methods (16).
Among all these efforts, it would be interesting to attempt more rigorous optimization of the consistency-based score function instead of the heuristic progressive alignment approach. In relation to this, we are particularly interested in exploring the relationship between the optimization of the score functions and the alignment accuracy.
In terms of finding the optimal alignment through global optimization (18–21), there have been some earlier attempts to find the global minimum of an objective function for MSA. For example, simulated annealing (SA) was applied to the sum-of-pair score function (22–24). Similarly a genetic algorithm (SAGA) (25) was also applied to these score functions.
One of the simplest algorithms for unbiased global optimization is the SA method, which has been used most widely. Although the SA is very versatile in that it can be easily applied practically to any problem, the drawback is that its efficiency is usually lower than problem-specific algorithms. This is especially problematic for hard optimization problems. For this reason, it is important to find an algorithm that is as general as SA, and yet competitive with problem-specific ones.
In this study, we apply a global optimization method called conformational space annealing (CSA) (26,27) to MSA for a direct optimization of a consistency-based score function. CSA combines the essential ingredients of the three traditional methods of global optimization methods, i.e., SA (28,29), genetic algorithm (GA) (30,31), and Monte Carlo with minimization (MCM) (32). The unique strength of CSA comes from introducing a distance measure between two conformations that makes it possible to systematically control the diversity of sampling.
We show that, by applying CSA to MSA with a consistency score function, the method can find alignments that are more consistent with the library of pairwise alignments and consequently more accurate alignments, compared with existing progressive methods. It should be emphasized that our approach should be contrasted with those popular heuristic approaches based on the progressive alignment in that our approach searches for the optimal alignment in a direct manner.
In addition, our method can provide alternative alignments that might correspond to more biologically meaningful alignments, therefore possibly alleviating the problem arising from an inaccurate objective function.
METHODS
Fig. 1 illustrates the flow diagram of CSA applied to MSA (MSACSA). In MSACSA, we need a series of new concepts. They are: 1), a local minimizer of a given alignment; 2), ways to combine two parent alignments to generate a daughter alignment; 3), a distance measure between two alignments; and 4), an energy function to minimize. Details on these four concepts and the procedures of CSA are described in the following.
FIGURE 1.

Flow diagram of MSACSA is shown. MSACSA runs until the bank size becomes 100 and seed resetting is carried out twice (see Methods).
Local minimization of a given multiple alignment
Throughout the optimization process, CSA keeps in storage (that we call the bank) only locally minimized alignments (i.e., local minima), and explores the phase space within the neighborhood of existing alignments. Local energy minimization is carried out by stochastic quenching. For a given alignment, a series of new potential alignments are generated by perturbations. Whenever the energy of a new alignment is more favorable than that of the old one, the old is replaced by the new. A way to carry out stochastic quenching is to repeat the above procedure until one fails to find a better solution for N times in a row. Typically, one sets N as a multiple of system size and we set N = 10NLmax. N is the number of sequences to align, Lmax is the largest sequence length. For convenience, the maximum number of attempts is set to 100N. Perturbations are generated by local moves that are either horizontal or vertical ones (23,24) both consisting of random insertion, deletion, and relocation of gap(s). Vertical moves are identical to the horizontal ones other than they are applied simultaneously to two or more sequences that share gap(s) at selected columns.
How to combine two multiple alignments
To explore the phase space in a neighborhood of a parent alignment P1, we generate a daughter alignment A by replacing a part of P1 by the corresponding part of another parent alignment P2 in the bank. First we set A = P1, and randomly remove a number of consecutive columns from A. Denote the set of removed residues by r. Find r in P2 and construct the minimal rectangular-shaped partial alignment p, which contains r in the shape of P2 and a number of gaps to fill up the rectangle. Note that the shape of r in P2 may be rugged. Finally, insert p to complete A.
Distance between two multiple alignments
The distance measure between two multiple alignments is a central notion that enables MSACSA to keep diverse alignments in the bank. We count the number of residue mismatches in all pairwise sequence alignments between two given multiple sequence alignments. More precisely, for a given alignment A with N sequences and M aligned columns, we define the set P(A) of aligned residues including gap-residue matches, but no gap-gap matches for all pairwise matches as follows:
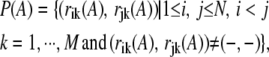 |
(1) |
where rik (A) is the identity of the residue/gap at the ith row and at the kth column of A. Now for two given alignments A and B, the distance is defined as
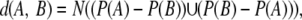 |
(2) |
where N(X) is the number of elements in the set X.
Objective function
To define the objective score function, we need a library that is the set of pairwise aligned residues for all pairs of sequences. Typically, the set of pairwise alignments between constituting sequences is used to generate the library. The idea of a consistency-based score function is to give a higher score to a multiple alignment if more pairwise-aligned residues in it are observed in the library. The score function is designed to give the perfect score to the realization of a multiple alignment if all pairwise restraints in the library are satisfied by the multiple alignment. However, generally speaking, there exist frustrations between pairwise alignment restraints in the library that makes it impossible to satisfy all restraints in the library simultaneously.
For a given third-party alignment method we construct a library of pairwise constraints by performing pairwise alignment for all combinations of sequence pairs. To each aligned residue pair, we assign a weight w, the correlation coefficient between two profiles from the residue pair. Profiles are generated by PSI-BLAST (33). In practice, some w can have negative value, but all matched constraints in the library should contribute positively to an alignment, w is linearly rescaled so that 0.01 ≤ w ≤ 1.0. The small positive value 0.01 maximizes the range of w. The library is now a collection of aligned residue pairs with positive weights. We denote the sum of all weights by 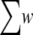 and define the energy of an alignment A with N sequences and M aligned columns as:
and define the energy of an alignment A with N sequences and M aligned columns as:
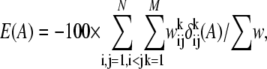 |
(3) |
where, 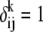 if the aligned residues between the ith and the jth sequences at the kth column are in the library, otherwise
if the aligned residues between the ith and the jth sequences at the kth column are in the library, otherwise 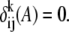
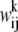 is the corresponding weight of the aligned residue pair from the library. For a position corresponding to a gap where the profile column is not defined, we use
is the corresponding weight of the aligned residue pair from the library. For a position corresponding to a gap where the profile column is not defined, we use 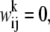 which is equivalent to considering only aligned residues (not including gaps) in the library. The consistency-based energy function (4) of Eq. 3 gives a lower energy to an alignment if more pairwise constraints are satisfied. The energy function gives a perfect score—100 if all constraints are satisfied. However, in general, as mentioned above there are conflicts between constraints or frustration giving an optimization problem on a complex landscape.
which is equivalent to considering only aligned residues (not including gaps) in the library. The consistency-based energy function (4) of Eq. 3 gives a lower energy to an alignment if more pairwise constraints are satisfied. The energy function gives a perfect score—100 if all constraints are satisfied. However, in general, as mentioned above there are conflicts between constraints or frustration giving an optimization problem on a complex landscape.
Conformational space annealing
The CSA method searches the whole conformational space in its early stages and then narrows the search to smaller regions with low energy as the distance cutoff, Dcut, which defines a (varying) threshold of the similarity between two alignments, is reduced. As in genetic algorithms, MSACSA starts with a pre-assigned number (50 in this work) of randomly generated and subsequently energy-minimized alignments. This pool of alignments is called the bank. At the beginning, the bank is a sparse representation of the entire conformational space.
A number of dissimilar alignments (30 in this work) are then selected from the bank, excluding those that have already been used; they are called seeds. Each seed alignment is modified by replacing parts of the seed by the corresponding parts of randomly selected alignment from either the first bank, or the bank. Each alignment is energy-minimized by the stochastic quenching to give a trial alignment. Twenty trial alignments are generated for each seed (a total of 600 alignments). This is the most time-consuming part of the computation, but it is highly suitable for parallel computing, because the local minimizations are independent of each other.
For each trial alignment, α, the closest alignment A from the bank (in terms of the distance d(α, A)) is determined. If d(α, A) ≤ Dcut (Dcut being the current cutoff criterion), α is considered similar to A; in this case α replaces A in the bank, if it is also lower in energy. If α is not similar to A, but its energy is lower than that of the highest-energy alignment in the bank, B, α replaces B. If neither of the above conditions holds, α is rejected. The narrowing of the search regions is accomplished by setting Dcut to a large value initially (usually one-half of the average pair distance, Dave, in the bank), and gradually reducing it as the search progresses. (It is reduced by a fixed ratio after the bank is updated until it becomes Dave/5.)
Special attention is paid to selecting seeds that are far from each other. One round of the procedure is completed when there is no seed to select (i.e., all alignments from the bank have already been used). The round is repeated a predetermined number of times (twice in this work). If necessary, more random alignments (50 in this work) that are subsequently energy-minimized are added to the bank. One resets Dcut to one-half of the average pair distance in the bank and the whole procedure is repeated. In this study, the CSA search stops after complete procedure is finished with 50 + 50 bank conformations. More details can be found elsewhere (26,34–37). The algorithm can be easily parallelized with high parallel efficiency (38).
RESULTS
We applied MSACSA to two manually maintained data sets. One is the BAliBase (39) reference set 1, which contains 82 reference alignments with average sequence identity (ASI) of 31.5%. The other is the HOMSTRAD (40) structural alignment data set of March 1, 2006. There are 368 MSA families with at least three sequences including 129 families with ASI < 30%. We were able to apply MSACSA to 366 families (due to the enormous computational resources necessary, we failed to apply MSACSA to two cases, alpha-amylase and alpha-amylase_NC families). For energy function Eq. 3, pair-wise constraint library of each problem in two data sets is generated by SPEM.
The results are analyzed in three ways: i), the quality of the energy function is assessed by examining energy landscapes; ii), the level of energy optimization is compared with SPEM in terms of average energy and consistency with the pair-wise constraint library; and iii), the alignment accuracy based on the reference alignments are assessed.
Energy landscape and the quality of the energy function
We analyzed the quality of the energy function by examining energy landscapes. Fig. 2 shows two examples of the energy landscape. The first one is the Rhodanese family that exhibits a relatively high correlation between the energy and the alignment accuracy. We found that most of the cases exhibit features similar to this one (see below for more detailed statistics). On average, alignments of lower energies are of higher accuracies. However, the lowest energy alignment is not always the best one because in the case of Rhodanese we find six alignments in the bank that are slightly more accurate. This means that MSACSA provides alternative alignments and, apparently, the energy function used in this study is not perfect.
FIGURE 2.

Two energy landscapes of MSACSA are shown. The lowest-energy alignments are indicated by arrows.
As a rare extreme case of the energy landscape, we can take the DUF170 family for which the energy landscape is shown in Fig. 2 exhibiting a fork-like structure, where we observe four alignments with quite low energies that are separated by a wide gap of alignment accuracy. Although the lowest-energy alignment has the highest accuracy of 85.2%, the accuracy of the second lowest energy alignment is only 61.2%. This means that the two alignments are quite different from each other, with the energy difference of only 0.0008. Therefore, for more decisive differentiation of good alignments from less accurate ones in this case, the energy function needs to be improved.
Fortunately, however, this landscape is one of a few extreme cases. Similar fork-like landscape structure was found in only about four families out of the whole 366 HOMSTRAD families. For further analysis of the energy landscape statistics, we compared the minimum-energy alignments against the alignments with the highest accuracies (in the bank) in terms of the difference in the accuracies. For ∼92% of the 366 HOMSTRAD families, the minimum-energy alignments showed a relatively high correlation between energy and alignment accuracy (similar to the case of Rhodanese family) in which the accuracy of the best alignment did not exceed that of the minimum energy alignment by >4% (0.04). For the remaining cases of ∼7%, the minimum-energy alignments showed lower alignment accuracies by >4% compared with the best alignment, resulting in low correlation between the energy and the accuracy.
From these analyses, we can conclude that, despite some limitation, the consistency-based score function for MSA is quite meaningful in terms of biological applications in the sense that the optimal alignment with respect to the consistency score function is, in almost all cases, also nearly consistent with the manually-constructed reference alignment.
Energy optimization: energy and consistency with the library
Table 1 shows the average energy, the number of satisfied pairwise constraints from the minimum energy alignments of MSACSA and the corresponding values from MSA using SPEM. The energy from Eq. 3 corresponds to the level of consistency of constraints with appropriate weights. The average energies obtained by MSACSA are lower than those by SPEM. Moreover, the energy of the minimum energy alignment by MSACSA is lower than or equal to that by SPEM for all cases. On average, alignment by MSACSA satisfies more pairwise constraints than the SPEM alignment. For BAliBase (HOMSTRAD 366), alignments by MSACSA satisfies 170 (1578) more constraints than those by SPEM.
TABLE 1.
Average energy and number of matches shared in the library
| Constraints in the library
|
|||||
|---|---|---|---|---|---|
| Sets | Methods | Eave | Ntotal | Ncom | Ncomple |
| BAliBase | SPEM | −94.43 | 161,692 | 159,736 | 1956 |
| Reference set 1 | CSA | −95.20 | 161,862 | 2126 | |
| HOMSTRAD | SPEM | −96.28 | 1,206,070 | 1,194,228 | 11,842 |
| (366) | CSA | −96.81 | 1,207,711 | 13,483 | |
Total matches (Ntotal) consist of common (Ncom) and complementary (Ncomple) matches. The total number of constraints for BAliBase (HOMSTRAD) is 169,797 (1,268,069).
Alignment accuracies
Accuracies of MSACSA are compared with those of SPEM. The sum-of-pair score (SPS) provided by BAliBase (39) is used for evaluation. SPS counts all correct matches in an alignment relative to the reference with SPS = 100. Accuracies of SPEM and MSACSA are shown in Table 2. SPS-values are averaged over all families considered. MSACSA (CSAm) according to the lowest energy alignment outperforms SPEM. Moreover, by choosing the best of the alternative alignments in the bank, the results (CSAb) improve significantly.
TABLE 2.
Alignment accuracies
| Sets | Scores | SPEM | CSAm | CSAb | Reference |
|---|---|---|---|---|---|
| BAliBase | SPS | 90.91 | 91.07 | 92.37 | 100.0 |
| Reference set 1 | Nmatches | 111,012 | 111,222 | 111,750 | 117,558 |
| HOMSTRAD | SPS | 86.40 | 86.85 | 88.17 | 100.0 |
| (366) | Nmatches | 1,029,191 | 1,032,783 | 1,040,771 | 1,163,915 |
SPS is the average sum-of-pair score (%). The total number of correctly aligned matches is also shown. CSAm indicates the lowest-energy alignment from MSACSA, and CSAb indicates the best alignment in the bank.
To check whether this kind of improvement in accuracy is statistically meaningful or not, we calculated the so-called P values (41). For BAliBase, we obtain the 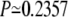 whereas for the case of HOMESTRAD, we get
whereas for the case of HOMESTRAD, we get 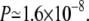 The relatively larger value of P (>0.05) for the case of BAliBase indicates that the better average accuracies of MSACSA over SPEM is not significant in terms of statistics. We note that BAlibase is a relatively easy benchmark set for MSA methods. On the other hand, for the case of HOMSTRAD, we see clearly that MSACSA outperforms SPEM in a statistically meaningful way.
The relatively larger value of P (>0.05) for the case of BAliBase indicates that the better average accuracies of MSACSA over SPEM is not significant in terms of statistics. We note that BAlibase is a relatively easy benchmark set for MSA methods. On the other hand, for the case of HOMSTRAD, we see clearly that MSACSA outperforms SPEM in a statistically meaningful way.
Running time and computational efficiency
We note that there are 368 families in the original HOMSTRAD set. Out of these, we were successful in applying MSACSA to 366 families. In terms of overall running time, 300 families out of the 366 families took <1 day using 128 central processing units (CPUs) (AMD Opteron 2 GHz). The remaining 66 families took ∼1 week. As a typical case, the Rhodanese family, which consists of six sequences with 150 residues on average, took ∼30 s with 128 CPUs. In terms of the number of sequences (in the case of HOMSTRAD set) the maximum number of sequences for which MSACSA was applied was the case of glob family with 41 sequences (average sequence length ∼150) that took 14 h using 128 CPUs.
In terms of the average number of residues per sequence, the maximum average number of residues (for which MSACSA was applied) was the case of rhv family with 854 residues per sequence and Ald_Xan_dh_2 family with 804 residues per sequence and both with six sequences, which took 29 min and 26 min, respectively using 128 CPUs.
It should be noted that, even for families with similar number of sequences as well as similar length of residues per sequence, the running time varied significantly depending on the complexity of the alignments. This is due to the fact that CSA depends greatly on the complexity of the energy landscape rather than the number of the degrees of freedom. This makes it difficult to analyze the running time systematically only based on the number of sequences and the average number of residues per sequence.
To provide an idea about the computational limitation of MSACSA, we sorted the families of HOMSTRAD in terms of the number of sequences (L) times the average number (N) of residues per sequence (i.e., L × N), that is, the total number of residues in a family. In this way, we could see that the two families (alpha-amylase and alpha-amylase_NC) ranked highest (with L × N = 11,615 and L × N = 13,524 respectively). These two families are those to which we failed to apply CSA due to the computational limitation. It seems that L × N ∼ 10,000 is an approximate bound for which MSACSA may take longer than a few days depending on the complexity of the alignment.
Large alignment sets out of the 366 HOMSTRAD families where MSACSA is applied, include the cases of aat (N = 10, L ≈ 421), glob (N = 41, L ≈ 158), and sermam (N = 27, L ≈ 220) families. These families took much more computational resources than the others, ranging from ∼16 min for aat family, 14 h for glob family, to 34 h for sermam family (all with 128 CPUs).
As mentioned above, due to the computational limitation, we were not able to apply MSACSA to two cases, alpha-amylase and alpha-amylase_NC families. The average sequence lengths are L = 402 and L = 486 respectively, and the total number of sequences is N = 23 for both cases. Each of the two families is expected to take longer than 2 weeks with 128 CPUs based on the partial running time for part of the computation.
Although MSACSA has such a limitation due to the heavy computational requirement, we believe, it can be still useful for many practical problems such as protein structure prediction. Moreover, in this work, we are mainly interested in investigating the consequence of rigorous optimization of a score function for the improvement of the alignment accuracies at the cost of the significant computation resources for MSA.
CONCLUSION
In conclusion, we have presented a new method for MSA based on thorough optimization of a consistency based energy function. The method can be applied to any given library of pairwise frustrated constraints provided by a third party alignment method, thus providing additional improvement to existing and future alignment methods. MSACSA finds alignments whose energy values are always less than or equal to those from a progressive method, showing that thorough optimization is accomplished. Consequently, on average, alignment by MSACSA satisfies more consistency conditions of the constraint library. Unlike existing progressive methods, MSACSA can provide alternative alignments as a way to alleviate the problem arising from using an inaccurate energy function. This is important as currently available energy functions for MSA are not perfect and biologically meaningful alignments do not necessarily correspond to the global minimum of current energy functions. Thorough optimization of a consistency-based energy function is shown to provide better alignments on average than the progressive alignment method at the expense of computation time.
The method was implemented in the recent blind protein structure experiment (42) and was a key factor for the most successful protein structure modeling in the high-accuracy template based modeling category (43).
Acknowledgments
We thank J. M. Kosterlitz and S. Gross for useful discussions and critical comments. Computation was carried out using Korea Institute for Advanced Study supercomputers.
Editor: Kathleen B. Hall.
References
- 1.Notredame, C. 2002. Recent progress in multiple sequence alignment: a survey. Pharmacogenomics. 3:131–144. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F. 1989. Gap costs for multiple sequence alignment. J. Theor. Biol. 138:297–309. [DOI] [PubMed] [Google Scholar]
- 3.Altschul, S. F., R. J. Carroll, and D. J. Lipman. 1989. Weights for data related by a tree. J. Mol. Biol. 207:647–653. [DOI] [PubMed] [Google Scholar]
- 4.Notredame, C., L. Holm, and D. G. Higgins. 1998. COFFEE: an objective function for multiple sequence alignments. Bioinformatics. 14:407–422. [DOI] [PubMed] [Google Scholar]
- 5.Wang, L., and T. Jiang. 1994. On the complexity of multiple sequence alignment. J. Comput. Biol. 1:337–348. [DOI] [PubMed] [Google Scholar]
- 6.Carrillo, H., and D. Lipman. 1988. The multiple sequence alignment problem in biology. SIAM J. Appl. Math. 48:1073–1082. [Google Scholar]
- 7.Needleman, S. B., and C. D. Wunsch. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48:443–453. [DOI] [PubMed] [Google Scholar]
- 8.Smith, T. F., and M. S. Waterman. 1981. Identification of common molecular sub sequences. J. Mol. Biol. 147:195–197. [DOI] [PubMed] [Google Scholar]
- 9.Feng, D. F., and R. F. Doolittle. 1987. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol. 25:351–360. [DOI] [PubMed] [Google Scholar]
- 10.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gotoh, O. 1996. Significant improvement in accuracy of multiple protein sequence alignments by iterative refinement as assessed by reference to structural alignments. J. Mol. Biol. 264:823–838. [DOI] [PubMed] [Google Scholar]
- 12.Morgenstern, B., A. Dress, and T. Werner. 1996. Multiple DNA and protein sequence alignment based on segment-to-segment comparison. Proc. Natl. Acad. Sci. USA. 93:12098–12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Notredame, C., D. G. Higgins, and J. Heringa. 2000. T-COFFEE: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. [DOI] [PubMed] [Google Scholar]
- 15.Do, C. B., M. S. Mahabhashyam, M. Brudno, and S. Batzoglou. 2005. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 15:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou, H., and Y. Zhou. 2005. SPEM: improving multiple sequence alignment with sequence profiles and predicted secondary structures. Bioinformatics. 21:3615–3621. [DOI] [PubMed] [Google Scholar]
- 17.Zhou, H., and Y. Zhou. 2005. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins. 58:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajgaria, R., S. R. McAllister, and C. A. Floudas. 2006. A novel high resolution Calpha–Calpha distance dependent force field based on a high quality decoy set. Proteins. 65:726–741. [DOI] [PubMed] [Google Scholar]
- 19.McAllister, S. R., R. Rajgaria, and C. A. Floudas. 2007. Global pairwise sequence alignment through mixed-integer linear programming: a template-free approach. Optimization Methods and Software. 22:127–144. [Google Scholar]
- 20.McAllister, S. R., R. Rajgaria, and C. A. Floudas. 2007. A template-based mixed-integer linear programming sequence alignment model. In Modeling and Algorithms for Global Optimization, Nonconvex Optimization and Its Applications. T. Torn and J. Zilinskas, editors, Springer-Verlag, New York. 343–360.
- 21.McAllister, S. R., R. Rajgaria, and C. A. Floudas. 2008. A path selection approach to global pairwise sequence alignment using integer linear optimization. Optimization. 57:101–111. [Google Scholar]
- 22.Ishikawa, M., T. Toya, M. Hoshida, K. Nitta, A. Ogiwara, and M. Kanehisa. 1993. Multiple sequence alignment by parallel simulated annealing. Comput. Appl. Biosci. 9:267–273. [DOI] [PubMed] [Google Scholar]
- 23.Kim, J., S. Pramanik, and M. J. Chung. 1994. Multiple sequence alignment using simulated annealing. Comput. Appl. Biosci. 10:419–426. [DOI] [PubMed] [Google Scholar]
- 24.Hernández-Guía, M., R. Mulet, and S. Rodríguez-Pérez. 2005. Simulated annealing algorithm for the multiple sequence alignment problem: the approach of polymers in a random medium. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 72:031915. [DOI] [PubMed] [Google Scholar]
- 25.Notredame, C., and D. G. Higgins. 1996. SAGA: sequence alignment by genetic algorithm. Nucleic Acids Res. 24:1515–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, J., H. A. Scheraga, and S. Rackovsky. 1997. New optimization method for conformational energy calculations on polypeptides: conformational space annealing. J. Comput. Chem. 18:1222–1232. [Google Scholar]
- 27.Lee, J., H. A. Scheraga, and S. Rackovsky. 1998. Conformational analysis of the 20-residue membrane-bound portion of melittin by conformational space annealing. Biopolymers. 46:103–116. [DOI] [PubMed] [Google Scholar]
- 28.Kirkpatrick, S., C. D. Gelatt, and M. P. Vecchi. 1983. Optimization by simulated annealing. Science. 220:671–680. [DOI] [PubMed] [Google Scholar]
- 29.Laarhoven, P. J. M., and E. H. L. Aarts. 1992. Simulated Annealing: Theory and Applications. Kluwer, Dordrecht.
- 30.Holland, J. 1973. Genetic algorithms and the optimal allocations of trials. SIAM J. Comput. 2:88–105. [Google Scholar]
- 31.Goldberg, D. E. 1989. Genetic Algorithms in Search, Optimization, and Machine Learning. Addison-Wesley Longman Publishing, Boston, MA.
- 32.Li, Z., and H. A. Scheraga. 1987. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. USA. 84:6611–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee, J., A. Liwo, and H. A. Scheraga. 1999. Energy-based de novo protein folding by conformational space annealing and an off-lattice united-residue force field: application to the 10–55 fragment of staphylococcal protein A and to apo calbindin D9K. Proc. Natl. Acad. Sci. USA. 96:2025–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ko, M. K., S. J. Lee, J. Lee, and B. Kim. 2003. Vortex patterns and infinite degeneracy in the uniformly frustrated XY models and lattice Coulomb gas. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 67:046120. [DOI] [PubMed] [Google Scholar]
- 36.Lee, J., I. H. Lee, and J. Lee. 2003. Unbiased global optimization of Lennard-Jones clusters for N ≤ 201 using the conformational space annealing method. Phys. Rev. Lett. 91:080201. [DOI] [PubMed] [Google Scholar]
- 37.Kim, S. Y., S. J. Lee, and J. Lee. 2007. Ground-state energy and energy landscape of the Sherrington-Kirkpatrick spin glass. Phys. Rev. B. 76:184412. [Google Scholar]
- 38.Lee, J., J. Pillardy, C. Czaplewski, Y. Arnautova, D. R. Ripoll, A. Liwo, K. D. Gibson, R. J. Wawak, H. A. Scheraga. 2000. Efficient parallel algorithms in global optimization of potential energy functions for peptides, proteins, and crystals. Comput. Phys. Commun. 128:399–411. [Google Scholar]
- 39.Thompson, J., F. Plewniak, and O. Poch. 1999. BAliBASE: a benchmark alignment database for the evaluation of multiple alignment programs. Bioinformatics. 15:87–88. [DOI] [PubMed] [Google Scholar]
- 40.Mizuguchi, K., C. M. Deane, T. L. Blundell, and J. P. Overington. 1998. HOMSTRAD: a database of protein structure alignments for homologous families. Protein Sci. 7:2469–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonick, L., and W. Smith. 1993. Cartoon Guide to Statistics. Collins, New York.
- 42.Joo, K., J. Lee, S. Lee, J.-H. Seo, S. J. Lee, and J. Lee. 2007. High accuracy template based modeling by global optimization. Proteins. 69(Suppl 8):83–89. [DOI] [PubMed] [Google Scholar]
- 43.Read, R. J., and G. Chavali. 2007. Assessment of CASP7 predictions in the high accuracy template-based modeling category. Proteins. 69(Suppl 8):27–37. [DOI] [PubMed] [Google Scholar]