

Abstract
Topoisomerase I (Top1) is an abundant and essential enzyme. Top1 is the selective target of camptothecins, which are effective anticancer agents. Top1-DNA cleavage complexes can also be trapped by various endogenous and exogenous DNA lesions including mismatches, abasic sites and carcinogenic adducts. Tyrosyl-DNA phosphodiesterase (Tdp1) is one of the repair enzymes for Top1-DNA covalent complexes. Tdp1 forms a multiprotein complex that includes poly(ADP) ribose polymerase (PARP). PARP-deficient cells are hypersensitive to camptothecins and functionally deficient for Tdp1. We will review recent developments in several pathways involved in the repair of Top1 cleavage complexes and the role of Chk1 and Chk2 checkpoint kinases in the cellular responses to Top1 inhibitors. The genes conferring camptothecin hypersensitivity are compiled for humans, budding yeast and fission yeast.
A. Introduction: Mammalian Topoisomerase Families, Top1 Functions and Catalytic Mechanisms
Seven topoisomerase genes are encoded in the human nuclear genome [1]. The enzymes (abbreviated Topo or Top) have been numbered in the order of their discovery except for the most recent enzyme, mitochondrial topoisomerase I (Top1mt) [2, 3]. Vertebrate cells contain two Top1 (Top1 for the nuclear genome and Top1mt for the mitochondrial genome), two Top2 (Top2α and β) and two Top3 (Top3α and β). The seventh topoisomerase is Spo11, whose expression is restricted to germ cells. Top3α forms heterodimers with BLM (the gene product deficient in Bloom syndrome) and is functionally related to the resolution of post-replicative hemicatenanes and recombination intermediates [4, 5]. Top1 proteins belong to the family of the tyrosine recombinases (which includes λ-integrase, Flip and Cre recombinases), and Top2 is related to bacterial gyrase and Topo IV, which are the targets of quinolone antibiotics.
Topoisomerases and tyrosine recombinases nick and religate DNA by forming a covalent enzyme-DNA intermediate between an enzyme catalytic tyrosine residue and the end of the broken DNA (Fig. 1). These covalent intermediates are generally referred to as “cleavage (or cleavable) complexes” (Fig. 2). Topoisomerases have also been classified in two groups depending whether they cleave and religate one strand (type I) or both strands (type II) of the DNA duplex. Type I enzymes include Top1 (nuclear), Top1mt, Top3α and β and type II enzymes include Top2α and β and Spo11.
Fig. 1. Topoisomerase cleavage complexes.

Topoisomerases (abbreviated Topo in panel A) utilize a catalytic tyrosine residue for nucleophilic attack and breakage of a DNA phosphoester bond. A. The polarity depends on the Topo (only human enzymes are considered here). B. Topoisomerases I (nuclear and mitochondrial Top1) form a covalent bond with the 3′-DNA end and generate a 5′-hydroxyl-end. This cleavage intermediate allow controlled rotation of the 5′-end around the intact DNA strand (see Fig. 3B). Under normal conditions, the reaction is reversible. Religation (back arrow from B -> A) is favored over cleavage and requires the alignment of the 5′-hydroxyl-end with the phosphoester tyrosyl-DNA bond for nucleophilic attack. C. All other human Topo enzymes (Top2 and Top3) have an opposite polarity compared to Top1 (see Fig. 2). They form covalent bonds with the 5′-end of the break and generate 3′-hydroxyl ends.
Fig. 2. Schematic architecture of the topoisomerase cleavage complexes.
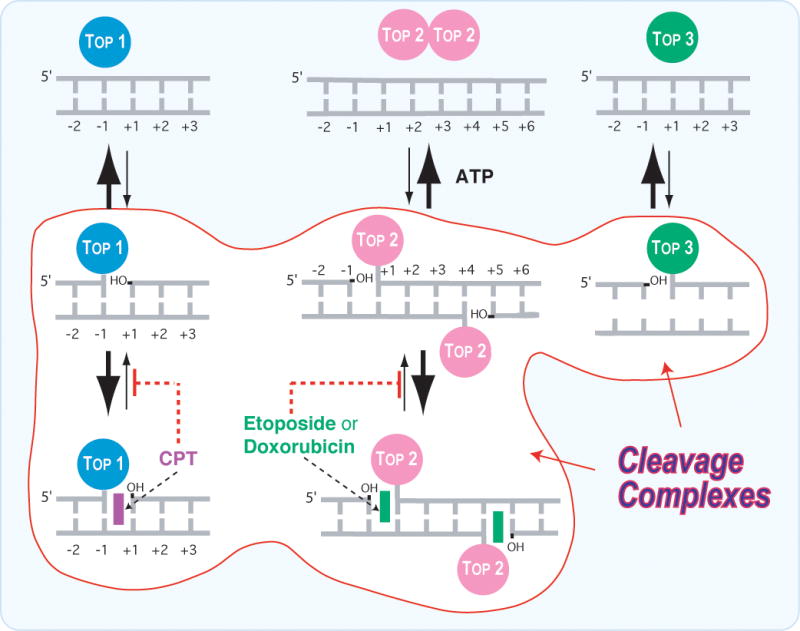
A. Topoisomerases I (Top1 nuclear and Top1mt) bind to double-stranded DNA and form covalent complexes at the 3′-end of the breaks. All other topoisomerases form covalent complexes at the 5′-end of the breaks. Top1 cleavage complexes are selectively stabilized by the natural alkaloid camptothecin (CPT). B. Topoisomerase II homodimers (Top2α and Top2β) bind to double-stranded DNA and form cleavage complexes with a canonical 4-base pair overhang. Top2 binds and hydrolyze ATP during catalysis. Top2 inhibitors stabilize the Top2 cleavage complexes and are potent anticancer drugs. C. Topoisomerases III (Top3α and Top3β) bind as monomers to non-canonical DNA structures (single-stranded DNA) [194] in association with a RecQ helicase (BLM in humans, Sgs-1 in budding yeast, Rhq1 in fission yeast). Top3 has been proposed to resolve double-holiday junctions arising from stalled replication forks (see Fig. 5A and corresponding text). Top3 inhibitors have not been reported.
Top1 is essential in vertebrates and flies but not in yeast. Knocking out the TOP1 gene results in early embryonic lethality in mouse [6] and fly [7]. By contrast, yeast survives in the absence of TOP1 [8]. Top1 is expressed constitutively throughout the cell cycle [9] and is concentrated in the nucleolus [10, 11]. Its main function is to relieve both positive and negative DNA supercoiling generated by transcription and replication, and possibly DNA repair and chromatin remodeling [1, 12–14]. The mechanistic similarities between Top1 and other tyrosine recombinases suggest that Top1 may also play a role in DNA recombinations [15, 16]. The Top1 recombinase activity has been proposed for the replication of vaccinia [17] and hepadnaviruses [16]. Top1 probably contributes also to RNA splicing by phosphorylating SR proteins [18, 19].
Top1 relaxes DNA supercoiling in the absence of energy cofactor by nicking the DNA and allowing rotation of the broken strand around the Top1-bound DNA strand (Fig. 3B – curved arrow). Crystal structures of Top1 [20–22] show the enzyme encircling the DNA tightly like a clamp (Fig. 3D), which accounts for the fact that Top1 controls the processive relaxation of supercoiled DNA [20, 23–25]. Once the DNA is relaxed, Top1 religates the breaks by reversing its covalent binding. Religation requires the DNA end 5′-hydroxyl-group to be aligned with the tyrosine-DNA phosphodiester bond. Under normal condition, the cleavage intermediates (Figs. 2A and 3B) are transient and religation is favored over cleavage.
Fig. 3. Trapping of Top1 cleavage complexes by camptothecin and non-camptothecin inhibitors.
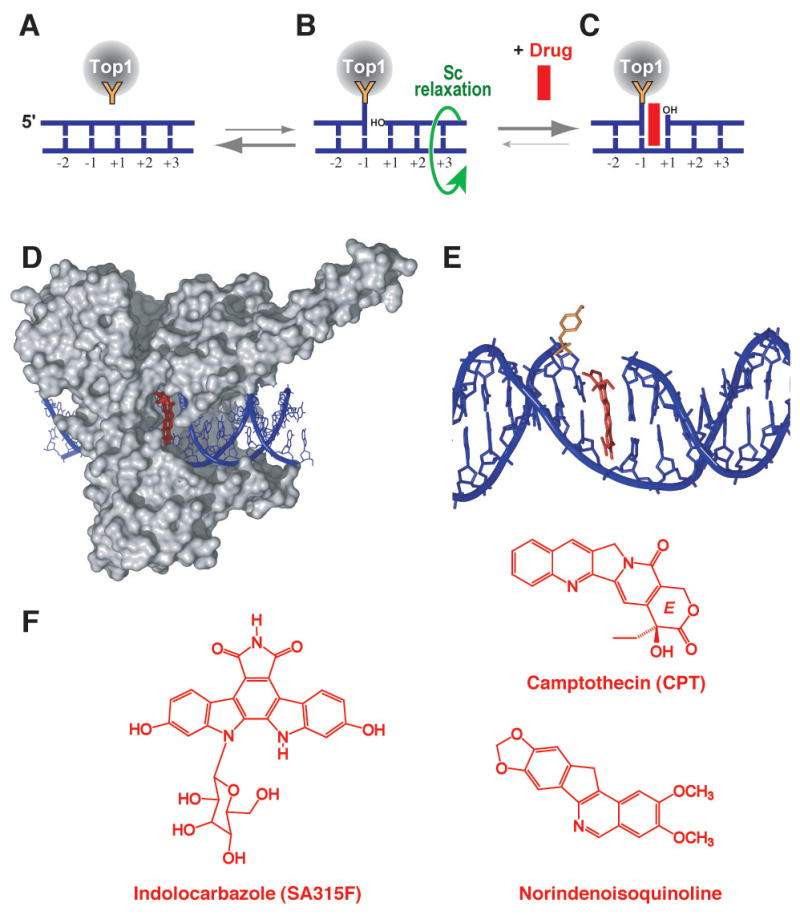
A. Under physiological conditions, Top1 is associated with chromatin in non-covalent complexes. B. A small fraction of Top1 forms cleavage complexes that relax DNA supercoiling by controlled rotation of the cleaved strand around the intact strand (green curved arrow). C. Anticancer drugs such as those shown in panel F reversibly trap the Top1 cleavage complex by inhibiting religation. D. Crystal structure of camptothecin bound to the Top1-DNA cleavage complex [from [29]] showing “interfacial inhibition” [26, 27] of the Top1 cleavage complex by camptothecin. Interfacial inhibition also applies to non-camptothecin Top1 inhibitors shown in panel F [29, 30]. E. Same structure as in panel D. The Top1 has been removed except for the catalytic tyrosine (in orange). Camptothecin is shown intercalated between the base pairs flanking the Top1 cleavage site. F. Structures of three Top1 inhibitors.
B. Induction and stabilization of Top1 cleavage complexes by camptothecin and anticancer drugs and by carcinogens and endogenous DNA lesions
The normally transient Top1 cleavage complexes can be converted into potential DNA lesions. Stabilization of the cleavage complexes generally results from misalignment of the 5′-hydroxyl-DNA end. Misalignments can be generated by drugs bound at the interface of the enzyme and broken DNA [26, 27] and by alterations of the DNA substrate (Table 1 and Fig. 4A).
Table 1.
Exogenous and endogenous factors producing Top1 cleavage complexes
| Drugsa | Mechanismb | Revc | Notes | Refs. |
|---|---|---|---|---|
| Camptothecins | T | r | Derived from the natural alkaloid | [196] |
| Indenoisoquinolines | T | r | Synthetic; in preclinical development | [32] |
| Indolocarbazoles (NB-506) | T | r | Semi-synthetic; in clinical development | [32] |
| Actinomycin D | T | r | Other effects: DNA, RNA polymerase | [196] |
| Hoechst minor groove | T | r | Other effects: DNA | [32] |
| Ecteinascidin 743 (YondelisR) | T | r | Other effects: traps TC-NER complex | [32] |
| Cytosine Arabinoside | T | r | Other effects: blocks DNA synthesis | [197, 198] |
| Gemcitabine | T | r | Other effects: blocks DNA synthesis | [199] |
| Endogenous DNA lesions | [37, 44] | |||
|
| ||||
| Single base mismatches | T | r | Polymerase & mismatch defects | [37, 40] |
| Mismatched loops | T | ir | Mismatch deficiencies | [40] |
| Abasic sites | T | ir | AP sites; base excision repair | [40] |
| 8-oxoguanosine | B | r | Free radicals | [21] |
| 5-hydroxycytosine | ? | r | Free radicals | [21] |
| Single-strand breaks | T | ir | Free radicals; base excision repair | [41] |
| Cytosine methylation | F+T | r | Physiological | [200] |
| Triple helix formation | F+T | r | ? | [201] |
| Apoptotic chromatin fragmentation | B+T | ir | Appears ubiquitous during apoptosis | [42–45] |
| Exogenous DNA lesions | [37] | |||
|
| ||||
| UV lesions | ? | ? | Dimers & 6,4-photoproducts | [202, 203] |
| IR-induced DNA breaks | T | ir | Both single- & double-strand breaks | [41] |
| 06-methylguanine | T | r | Produced by alkylating drugs (MNNG) | [204] |
| O6-dA-benzo[a]pyrene adducts | T | r | Intercalated carcinogenic adducts | [205] |
| N2-dG-benzo[a]pyrene adducts | F | ir | Minor groove carcinogenic adducts | [206, 207] |
| N2-dG-benzo[c]phenanthrene adducts | T | r | Intercalated carcinogenic adducts | [207] |
| N6-Ethenoadenine | T | r | Carcinogenic vinyl adduct | [208] |
| N2-dG-ethyl adducts | T | r | Produced by acetaldehyde (alcohol) | [209] |
For detailed review on non-camptothecin inhibitors see [32].
Mechanism for Top1 cleavage complex production: T: Trapping of the Top1 cleavage complexes (i.e.: inhibition of religation) (see Fig. 3B); B: enhancement of binding; F: enhancement of the forward (cleavage) reaction.
Reversibility of the Top1 cleavage complexes: r: reversible; ir: irreversible.
Fig. 4. Conversion of reversible Top1 cleavage complexes into DNA damage.
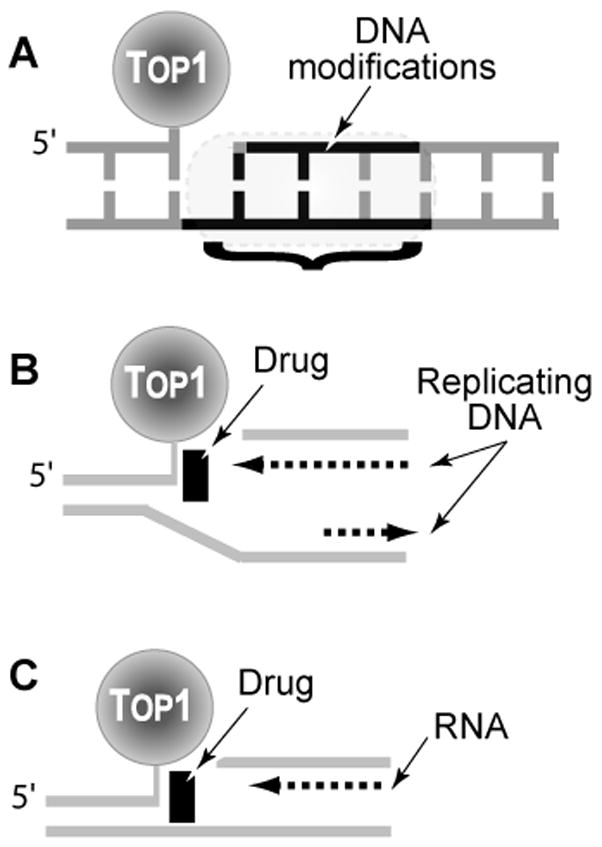
A. Irreversible (“suicide”) Top1 cleavage complexes are produced when Top1 cleaves previously damaged DNA (DNA modifications that trap Top1 are detailed in Table 1). B. Top1 cleavage complexes can be converted to irreversible complexes upon replication fork collision when the Top1 cleavage complex is on the leading strand for DNA synthesis. The drug is shown as the initiating event for the collision. However, once the replication double-strand break (Rep-DSB) is formed, dissociation of the drug has no impact on the irreversible covalent complex. C. Conversion of Top1 cleavage complexes into irreversible covalent complexes by transcription complexes.
Camptothecins and non-camptothecin Top1 inhibitors trap Top1 cleavage complexes by binding at the enzyme-DNA interface (Fig. 3D–E) [22, 28–30]. Hence Top1 inhibitors represent a paradigm for “interfacial inhibitors” [26, 27]. Interfacial inhibition has recently been shown to account for the molecular mechanism of inhibition of many natural products that block specific conformational states of macromolecular complexes. Aphidicolin and Top2 inhibitors have also been proposed to follow the interfacial inhibition paradigm [26, 27].
Two camptothecin derivatives are used in cancer therapy: hycamtin (Topotecan®) and CPT-11 (Irinotecan; Camptosar®) [31]. CPT-11 is an inactive prodrug. It needs to be converted to its active metabolite SN-38. Hence, it is preferable to use SN-38, topotecan, or camptothecin for pharmacological studies. Additional camptothecin derivatives are in preclinical and clinical development [32].
Two key pharmacological properties of camptothecins need to be stressed. First, camptothecins bind reversibly to the Top1 cleavage complexes. Under pharmacological conditions, a rapid equilibrium is established between the ternary drug-enzyme-DNA complex and the dissociated complex. Hence, once camptothecins are diluted out and removed from cell culture, the cleavage complexes reverse rapidly [33]. The equilibrium can be shifted toward religation by increasing the temperature to 65°C and the salt concentration (≥ 0.35 M NaCl) in biochemical reactions. Salt-reversal is commonly used to study the “on” and “off” rates of Top1 cleavage complexes under various conditions [for recent example see [34]]. A second key pharmacological feature of camptothecins is the trapping of only a subset of the existing Top1 cleavage complexes, i.e. those with a guanine at the 5′-end of the break (+1 position – see Fig. 3) [35, 36]. Indenoisoquinolines (Fig. 3F) on the other hand tend to stabilize those cleavage complexes with a cytosine at the 3′-end of the breaks (−1 position) [34]. This sequence selectivity explains why camptothecins are relatively poor Top1 catalytic inhibitors as a fraction of the Top1 cleavage complexes (those not bearing a guanine +1) are immune to the drugs. It also explains why camptothecins only reveal a subset of the Top1 sites and should not be used alone to map all the Top1 cleavage complexes in a given DNA or chromatin segment.
Top1 cleavage complexes can be trapped by endogenous and frequent DNA lesions including abasic sites, mismatches, oxidized bases, nicks and carcinogenic DNA adducts [37] (Table 1 and references therein). For instance, DNA modifications such as those associated with oxidative damage [thousands per cell per day [38]] can produce Top1 cleavage complexes [39]. By contrast to camptothecins and other Top1 inhibitory drugs, these DNA modifications can produce irreversible cleavage complexes when the 5′-end of the DNA is irreversibly misaligned as in the case of abasic sites [40] or DNA breaks [41] (Table 1, Fig. 4A). Finally, we recently found the formation of Top1 cleavage complexes during apoptosis [42–45], which we have been explained by the trapping of Top1 by chromatin modifications (primarily due to reactive oxygen species).
C. Conversion of Top1 cleavage complexes into DNA damage
Cleavage complexes induced by DNA modifications that induce pronounced DNA structural alterations (abasic sites, mismatches, breaks) can be irreversible (Fig. 4A, Table 1). Such irreversible cleavage complexes have been referred to as “suicide complexes” [46, 47]. They constitute composite DNA lesions associating disruption of the DNA backbone (break on one or both strands) in association with a large protein covalently bound to the 3′-end of the broken DNA (Top1 is a 100 kDa protein). Reversible cleavage complexes can also produce irreversible cleavage complexes after processing by DNA and RNA polymerases (Fig. 4B and C). Thus, both DNA and RNA synthesis convert reversible cleavage complexes into DNA lesions. The relative contribution of DNA replication and transcription depends on the camptothecin concentration and the cell type. In highly proliferative cancer cells, replication-induced DNA damage contributes to most of the cytotoxicity at low camptothecin doses, whereas transcription-induced DNA damage contributes to the cytotoxicity of high doses of camptothecin [48, 49]. In non-dividing cells (neurons and lymphocytes) transcription-induced damage can kill cells at pharmacological concentrations [50, 51].
Camptothecin-induced Top1 cleavage complexes can readily be converted into replication double-strand breaks (Rep-DSB) (Fig. 4B) as demonstrated by: i) analyses of the broken ends by ligation-mediated PCR [52] showing the extension of the leading strand up to last nucleotide [leading to blunt-ended DSB by “replication run-off” [52]], and ii) rapid phosphorylation of histone H2AX (referred to as γ-H2AX) [53], which is a hallmark for double-strand breaks [54, 55]. Inhibition of DNA synthesis occurs within minutes following camptothecin treatment. It is intense (≥ 80%) and persists for several hours following drug removal [56]. At least two mechanisms lead to DNA synthesis inhibition: i) direct block of replication forks that have collided with the Top1 cleavage complexes (Fig. 4B), and ii) indirect replication arrest by S-phase checkpoint activation. The checkpoint implication is consistent with the fact that the checkpoint abrogator 7-hydroxystaurosporine (UCN-01) prevents inhibition of DNA synthesis by camptothecins [56–58]. The lethality of the replication double-strand breaks stems from the fact that when DNA synthesis is inhibited by aphidicolin, a specific inhibitor of replicative DNA polymerases [59], cells become immune to camptothecin in spite of their ability to form reversible cleavage complexes [48, 49]. Similarly, aphidicolin prevents the formation of γ-H2AX foci in camptothecin-treated cells [53].
Camptothecin is a potent inhibitor of both nucleoplasmic (mRNA) and nucleolar (rRNA) transcription [60–62]. Although the overall level of transcripts decreases rapidly following Top1 inhibition, specific genes are differentially affected. For example, camptothecin causes a strong holdback of the endogenous c-myc gene at the P2 promoter, whereas it produces minimal effect on an episomal c-myc gene or on the basal transcription of the Hsp70 and Gapdh genes [63]. Camptothecin also enhances the expression of a large number of genes including c-fos [63–66]. Transcription inhibition is primarily due to transcription elongation blocks by trapped Top1 cleavage complexes (Fig. 4C), which is a high probability event considering that Top1 is associated with transcription complexes [12]. Camptothecin has little effect on transcription initiation [11, 67]. It has been proposed that the elongating RNA polymerase collides with trapped Top1 cleavage complexes on the transcribed strand, resulting in the conversion of reversible Top1 cleavage complexes into irreversible strand breaks (Fig. 4C) [68, 69]. Inhibition of Top1 catalytic activity by camptothecins might also inhibit transcription by producing an accumulation of positive supercoils upstream from the elongating RNA polymerase [63, 70] and by compacting chromatin domains [70–72]. The transcriptional effects of camptothecins could also be related to functions of Top1 besides its DNA nicking-closing activity. Top1 regulates transcription initiation by interacting with TATA binding proteins [73, 74], and phosphorylates/activates RNA splicing factors from the SR family [19].
By contrast to replication [56], transcription inhibition recovers rapidly following camptothecin treatment [62, 67, 75]. Recovery of RNA synthesis depends both on degradation of Top1 and functional transcription-coupled nucleotide excision repair (TC-NER) [75]. Tumor cells that are deficient in Top1 degradation following camptothecin treatment, and Cockayne syndrome cells that are deficient in TC-NER are hypersensitive to camptothecin [76, 77], suggesting the importance of transcription-coupled DNA repair for RNA-synthesis recovery and cell survival in response to Top1-mediated DNA damage.
D. Repair of Top1-associated DNA damage
The molecular mechanisms/pathways involved in the repair of Top1-associated DNA damage are better understood than for Top2. Because camptothecin can be readily used in yeast, multiple pathways have been uncovered. We will consider three main repair pathways: i) Reversal of the covalent Top1-DNA complexes by 5′-end religation, ii) Top1 excision by Tdp1, and iii) Top1 excision by endonucleases. In spite of an apparent redundancy, it remains to be determined which pathways are preferred or selective for the Top1-associated DNA damages represented in Figure 4.
I. Reversal of Top1-DNA covalent complexes by 5′ end religation (Fig. 5A)
Fig. 5. Schematic representation of three main pathways for the repair of Top1 covalent complexes.

A. 5′-end-religation requires realignment of the 5′-hydroxyl with the end of the broken DNA bonded to Top1. This would require a “pull-back” (“regression”) of the replication or transcription complexes (see Figs. 4B and 4C). Fork regression and restart require helicase activities (in particular RecQ helicases such as Sgs-1 in budding yeast and BLM or WRN in humans) in association with Top3. B. Top1 excision by Tdp1 requires prior proteolysis of Top1 [106] or denaturation of Top1 [105] to expose the phosphotyrosyl bond to be attacked (see Fig. 6A–C). Tdp1 generates a 3′-phosphate DNA end, which needs to be hydrolyzed by polynucleotide kinase phosphatase (PNKP = hPNK). PNKP also catalyzes the phosphorylation of the 5′-end of the DNA. Tdp1 and PNKP are part of the XRCC1 complex (shown at the bottom). C. Excision of the Top1-DNA covalent complex by 3′-endonucleases. Studies in budding yeast have implicated at least 3 endonuclease families. The human orthologs are listed: Rad1/Rad10, Mus81/Eme1 and Mre11/Rad50. The resulting DNA lesion is probably processed by homologous recombination initiated by the Rad51, Rad52 complexes and by non-homologous end joining (Ku-DNA-PK pathway).
Top1-mediated DNA religation requires that the intact 5′-hydroxyl end be aligned with the 3′-end bonded to Top1 for nucleophilic attack of the tyrosyl-phosphoester bond (see Fig. 1B). Thus, this pathway/mechanism excludes the Top1 suicide complexes generated by DNA lesions affecting the 5′-end of the broken DNA (Fig. 4A and Table 1) unless they are repaired first. Top1 religation can be envisaged for the replication and transcription breaks (Fig. 4B and C) following regression (“pull-back”) of the replication or transcription complexes (Fig. 5A).
Replication fork regression could generate a “chicken foot” [see Fig. 6 in ref. [78]], which is topologically equivalent to a Holiday junction. RecQ helicases (the Bloom syndrome helicase BLM in mammals and Sgs1 in yeast) and Top3 (Top3α in mammals) form helicase-Top3 complexes, which have been implicated in the regression of replication forks and their restart [4, 79]. A plausible intermediate is the conversion of the “chicken foot” into a Double-Holiday junction catalyzed by Rad51, which can then be resolved by Top3 [80, 81]. The role of the RecQ helicases in processing Top1-mediated DNA damage is demonstrated by the hypersensitivity of yeast Sgs1 and Rhq1 mutants (see Tables 3 and 4) and the hypersensitivity of Bloom syndrome cells to camptothecin [79, 82] (see Table 2).
Fig. 6. Tyrosyl-DNA phosphodiesterase (Tdp1)-mediated reactions and substrates.
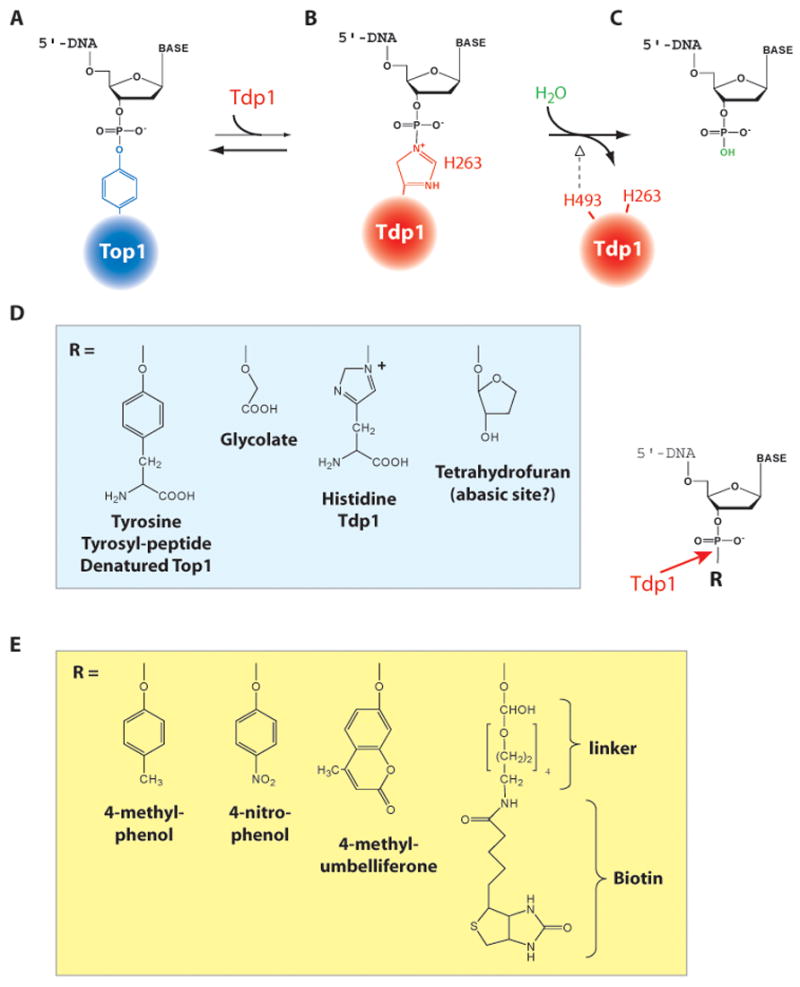
A. Structure of the Top1-DNA covalent intermediate (see Fig. 1B). B. Tdp1 releases the Top1 by forming a covalent bond between its active histidine 263 and the DNA end. C. Histidine 493 from the second HKD motif of Tdp1 (and which is mutated H493R in SCAN1) promotes the hydrolysis of the Tdp1-DNA intermediate, and frees the 3′-phosphate DNA end. Tdp1 is regenerated for another catalytic cycle. D. Physiological substrates for Tdp1. E. Tdp1 substrates used for biochemical assays.
Table 3.
Genetic alterations conferring hypersensitivity to Top1 poisoning in budding yeast:
| Yeast Saccharomyces Cerevisiae |
Humans
|
|||||
|---|---|---|---|---|---|---|
| Gene | Effect | Refs. | Function | Gene | Effect | Refs. |
| RAD52/51 homologous recombination (HR) | ||||||
| RAD52a | HS | [8, 98, 99, 131, 235–237] | Strand annealing | RAD52 | ? | |
| RAD51 | HS | [98, 99, 131, 236] | RecA homolog: strand invasion | RAD51C | HS | [223] |
| RAD55 | HS | [98, 99, 131] | Strand annealing, exchange | XRCC2 | HS | [219, 230, 234] |
| RAD57 | HS | [98, 99, 131] | Strand annealing, exchange | XRCC3 | HS | [219] |
| RAD54 | HS | [98, 99] | ATPase | |||
| MMS1 | S | [99, 238] | Replication repair/epistatic Rad52 | |||
| RAD59 | S | [98, 99] | Rad52-related recombination | |||
|
| ||||||
| MRX (MRN) 3′- nuclease/checkpoint (HR + NHEJ) | ||||||
| SAE2 | HS | [99] | Activates Mre11 endonuclease; meiotic and mitotic recombination | |||
| MRE11 | HS | [97–99, 237] | MRX/N complex; endonuclease | MRE11 | ? | |
| RAD50 | HS | [99, 131, 236] | MRX/N complex; scaffold | RAD50 | ? | |
| XRS2 | HS | [131] | MRX/N complex; signaling | NBS1 | HS | [212, 221] |
|
| ||||||
| Mus81/Mms4 (Mus81/Eme1) 3′-Flap Endonuclease | ||||||
| MUS81 | S | [98, 99, 126, 131] | 3′-flap endonuclease with Mms4 | MUS81 | NS | [137] |
| MMS4 | S | [98, 99, 126] | Partner for Mus81 endonuclease | EME1 | ? | |
|
| ||||||
| Tdp1-PNKP 3″-end processing | ||||||
| TDP1 | CSb | [97–99] | Tyrosyl-DNA phosphodiesterase | TDP1 | ? | [93, 94, 224] |
| TPP1 | CSb,c | [98, 110] | Polynucleotide 3′-phosphatase | PNKPb | HS | [115] |
| APN1 | CSb,c | [97, 98, 236] | AP endonuclease (endo IV family) | |||
| APN2 | CSb,c | [97, 98] | AP endonuclease (exo III family) | APE1 | ? | |
|
| ||||||
| Rad1/Rad10 (XPF/ERCC1) 3″-endonuclease | ||||||
| RAD1 | CSb | [97–99, 236] | 3′-flap endonuclease with Rad10 | XPF | NS | (our observations) |
| RAD10 | CSb | [97, 98] | Partner for Rad1 | ERCC1 | NS | |
|
| ||||||
| Rad27 (FEN1) 5″-endonuclease | ||||||
| RAD27 | MS | [98, 99, 133] | 5′-flap endonuclease | FEN1 | HS | [133] |
| SLX4/SLX1 | MS | [99] | Endonuclease with broad range of activities against various substrates | ? | ||
|
| ||||||
| Mismatch repair | ||||||
| MMS1 | S | [99] | Mismatch repair | |||
|
| ||||||
| RecQ/Top3 helicases/topoisomerase | ||||||
| SGS1 | MS | [126, 131] | Top3-associated helicase | WRN; BLM | HS | [82, 227–229] |
| SRS2 | S | [98, 99] | Rad51-associated helicase | ? | ||
| TOP3 | S | [98, 126] | Replication/recombination topoisomerase | TOP3α TOP3β | ? | |
|
| ||||||
| 9-1-1(“PCNA-like”) Clamp | ||||||
| DDC1 | MS | [98] | Replication/Repair Clamp; “9-1-1” | RAD9 | ? | |
| RAD17 | MS | [131, 236, 239] | Replication/Repair Clamp; “9-1-1” | RAD1 | ? | |
| MEC3 | MS | [131] | Replication/Repair Clamp; “9-1-1” | HUS1 | ? | |
| RAD24 | MS | [237] | Clamp loader for 9-1-1 | RAD17 | ? | |
|
| ||||||
| Replication | ||||||
| CDC45 | S | [134] | Initiation of DNA replication | CDC45L | ? | |
| POL32 | MS | [131] | Small subunit for Polδ | TEX14 | ? | |
| TRF4 | S | [240] | DNA polymerase | POLS | ? | |
| DPB11 | S | [134] | Replication initiation/checkpoint | TOPBP1 | ? | |
| RAD6 | MS | [131, 236, 241] | PRRd; Ub conjug | RAD6A, B | ? | |
| RAD18 | S | [131, 236] | PRRd; loads Rad6 | RAD18 | ? | |
| SLX4 | MS | [99] | DNA replication | ? | ||
| CLB5 | S | [99] | B type cyclin | Cyclin B | ? | |
| Dcc1 | S | [242] | Sister chromatid cohesion | ? | ||
|
| ||||||
| Transcription | ||||||
| HPR1 | S | [131] | Transcription & recombination | MGC5350 | ? | |
| SFP1 | S | [131] | Transcription factor | REQ | ? | |
| CCR4 | S | [131] | Transcription | KIAA1194 | ? | |
| BUR2 | S | [131] | Cyclin partner for Bur1 | Cyclin H | ? | |
| RPB9 | S | [131] | RNA polymerase subunit | POLR21 | ? | |
| HTZ1 | S | [99] | Regulation of transcription from Pol II promoter | |||
| SPT21 | S | [99] | Regulation of transcription from Pol II promoter | |||
| PAT1 | S | [99] | Controls mRNA decay | |||
| LSM1 | S | [99] | Controls mRNA decay | |||
| HMO1 | S | [99] | Involved in rDNA transcription | |||
| MPH1 | MS | [131, 241] | RNA helicase | MPH1 | ? | |
|
| ||||||
| Sensor PI3K-related protein kinases | ||||||
| MEC1 | HS | [236, 237] | PI3LK checkpoint sensor kinase | ATR | HS | [163] |
| DDC2 | ? | Partner for MEC1 | ATRIP | ? | ||
| TEL1 | S | [237] | PI3LK checkpoint sensor kinase | ATM | HS | [211–214] |
| PI3LK checkpoint sensor kinase | DNA-PK | HS | [56, 219] | |||
|
| ||||||
| Transducer protein kinases; BRCT proteins | ||||||
| RAD53 | MS | [236] | Checkpoint effector kinase | CHK2 | S | [162, 218] |
| RAD9 | MS | [236, 237] | Adaptor for checkpoint kinases | MDC1 | ? | |
| BRCA1 | HS | [216] | ||||
|
| ||||||
| Chromatin | ||||||
| HTA1/2 | S | [237] | Histone H2A | H2AX | S | [53] |
| HHF1/2 | S | [243] | Histone H4 | H4 | ? | |
| GCN5 | S | [244] | Histone H3 acetyltransferase | PCAF | ? | |
| YNG2 | S | [244] | Histone H4 acetyltransferase | ING1-5 | ? | |
| ESA1 | S | [243] | Histone H4 acetyltransferase | MYST1/HAT | ? | |
| ASF1 | S | [98, 245] | Chromatin assembly | ASF1B | ? | |
| MCD1 | S | [240] | Chromatin cohesion | RAD21 | ? | |
| CTF4 | MS | [98] | Chromatid cohesion & segregation | AND-1 | ? | |
| TOF1 | S | [99, 242] | Chromatid cohesion | TIM-1 | ? | |
| CSM3 | S | [99, 242] | Meiotic chromosome segregation | TIPIN | ? | |
| FUN30 | S | [99] | DNA-dependent ATPase; chromosome stability | |||
|
| ||||||
| Ubiquitin | ||||||
| UBC9 | S | [246] | Ubiquitin ligase | UBE2I | ? | |
| DOA4 | S | [246] | Ubiquitin hydrolase | |||
| UBC4 | S | [99] | Ubiquitin conjugation; stress response | |||
| RTT101 | S | [99] | Ubiquitin ligase; chromosome stability | |||
| ULA1 | S | [99] | RUB1-protein conjugation | |||
|
| ||||||
| Others | ||||||
| VAC14 | S | [99] | Vacuole inheritance | |||
| NUP60 | S | [99] | Nucleocytoplasmic transport | |||
| PPH3 | S | [99] | Serine/threonine phosphatase | |||
| PSY2 | S | [99] | Unknown | |||
| ILM1 | S | [99] | Unknown | |||
| CLB5 | S | [99] | B type cyclin in G1/S transition | |||
Abbreviations for effects: HS, S, MS, and CS correspond to hypersensitivity, sensitivity, moderate sensitivity, and conditional sensitivity to camptothecin, respectively. NS: no hypersensitivity.
The Rad52 epistasis group includes the RAD 50, 51, 52, 54, 55, 57, 59, MRE11 and XRS2 genes.
Tdp1 deficiency results in HS only in the presence of Rad1/Rad10 deficiency [97, 98]; conversely Rad1 deficiency does not confer hypersensitivity to CPT [126] unless the Tdp1-Apn1 pathway is defective [97]. Tpp1, Apn1+Apn2+Tpp1 need to be inactivated to confer full camptothecin hypersensitivity [111]; see Fig. 3A.
PNKP possesses both 3′-phosphatase and 5′-kinase activities, whereas the yeast ortholog, Tpp1 only possesses 3′-phosphatase activity. Neither Apn1, Apn2 or Tpp1 possess AP endonuclease activity) [111].
PRR: post-replication repair. Deficiency of Rhp6 or Rhp18 (YSP orthologs of Rad6 and Rad18) does not confer CPT hypersensitivity [247].
Table 4.
Genetic alterations conferring hypersensitivity to Top1 poisoning in fission yeast:
| Yeast Saccharomyces Pombe |
Humans
|
|||||
|---|---|---|---|---|---|---|
| Gene | Effect | Refs. | Function | Gene | Effect | Refs. |
| Rhp54 | HS | [129] | Homologous recombination (HR) | RAD52 | ? | |
| Rhp55 | LS | [129] | Homologous recombination (HR) | XRCC2 | HS | [219, 230, 234] |
| Rhp22A | LS | [129] | Homologous recombination (HR) | XRCC3 | HS | [219] |
| Rhp51 | S | [129] | RecA homolog; Rad52 epistasis G. | RAD51C | HS | [223] |
| rad22 | HS | [248] | Rec A homolog; functions with Mus81 | |||
|
| ||||||
| Rad50 | HS | [129] | MRX/N complex; scaffold | RAD50 | ? | |
| Mus81 | S | [129] | 3′-flap endonuclease with Eme1; meiotic recombination | MUS81 | NS | [137] |
| Eme1 | S | [129] | Partner for mus81 nuclease | MUS81 | ? | |
| RusA | RSa | [129] | HJ resolvase | |||
|
| ||||||
| Pnk1 | Sb | [112] | Polynucleotide kinase phosphatase | PNKP | HS | [115] |
|
| ||||||
| Rqh1 | MS | [98] | Top3-associated helicase | WRN | HS | [227–229] |
| BLM | HS | [82] | ||||
|
| ||||||
| Crb2 | S | [249] | Adaptor for checkpoint kinases and checkpoint proteins | 53BP1 | ? | |
| BRCA1 | HS | [216] | ||||
|
| ||||||
| Chk1 | S | [155, 249, 250] | Checkpoint effector kinase | CHK1 | S | [162, 165] |
|
| ||||||
| Swi1 | HS | [251] | Mating-type switching | TIMELESS | ||
Abbreviations for effect: HS, S, MS, and CS correspond to hypersensitivity, sensitivity, moderate sensitivity, and conditional sensitivity to camptothecin, respectively. NS: no hypersensitivity.
rusA suppresses hypersensitivity of Mus81/Eme1− but does not reverse sensitivity of rqh1−; rusA also suppresses the lethality of double mutants for Mus81/Eme1 + rqh1 [129]. RusA expressed in budding yeast partially suppresses hypersensitivity to CPT in Mms4-deficient cells [126].
Pnk1- cells are hypersensitive to CPT in the absence of additional defects, indicating difference from budding (see [a]) and importance of this pathway in fission yeast, which like mammals possesses a gene that has both 3′-phosphatase and 5′-kinase activity [112].
Table 2.
Genetic Alterations sensitizing mammalian cells to Top1 poisons
| Genes | Functions | Refs. |
|---|---|---|
| APTX | Mutated in AOA1; Encodes aprataxin, which associate XRCC1 (Fig. 5B) | [210] |
| ATMa | Mutated in AT; Protein kinase from the PI3K family implicated in DSB response | [211–214] |
| ATR | Protein kinase from the PI3K family; Implicated in replication stress | [163] |
| BCL-2 | Mutated in B-cell lymphoma; Apoptosis | [215] |
| BLM | Mutated in BS; Helicase from the RecQ family involved in genomic stability | [79, 82] |
| BRCA1 | Mutated in familial breast cancers; DNA damage response; TC-NER | [216] |
| BRCA2 | Mutated in familial breast cancers; Rad51 loading; Homologous recombination | [217] |
| CSA/CSB | Mutated in CS; TCR/BER | [76] |
| Chk1 | Checkpoint kinase phosphorylated/activated by PI3K (ATR) | [162, 165] |
| Chk2 | Checkpoint kinase phosphorylated/activated by PI3K (ATM etc…) | [162, 218] |
| DNA-PKcs | Protein kinase from the PI3K family; Implicated in DSB response | [56, 219, 220] |
| FEN-1 | Flap and gap endonuclease; processing of stalled replication forks | [133] |
| H2AX | Core histone; phosphorylated in response to DSB (γ-H2AX foci) | [53] |
| NBS1 | Mutated in NBS; Scaffolding protein forming a complex with Mre11 and Rad50 (MRN complex); DSB repair and recombination pathways | [212, 221] |
| PARP | BER (see Fig. 7) | [118, 222] |
| PNKP/hPNK | Processing of DNA ends: 3′-DNA-phosphatase + 5′-DNA-kinase | [115] |
| Rad51C | One of the five Rad51 paralogs; Implicated in DNA strand exchange/homologous recombination | [223] |
| TDP1 | Mutated in SCAN; Hydrolysis of 3′-phosphodiesters (phosphotyrosyl and phosphoglycolate) and phosphamides (Tdp1 cleavage complex) | [93, 94, 224] |
| TP53 | Mutated in Li-Fraumeni syndrome; encodes p53; Checkpoints; apoptosis | [225, 226] |
| WRN | Mutated in Werner syndrome; RecQ helicase involved in genomic stability | [227–229] |
| XRCC1 | BER; binds to Tdp1, PARP, β-polymerase, ligase III and aprataxin | [116, 117, 119, 230–233] |
| XRCC2 | One of the five Rad51 paralogs: Rad51B, Rad51C, Rad51D, XRCC2 & XRCC3; Implicated in DNA strand exchange/homologous recombination | [219, 230, 234] |
| XRCC3 | One of the five Rad51 paralogs; Implicated in DNA strand exchange/homologous recombination | [167, 219] |
The contribution of ATM has not been found consistently: ATM-siRNA cells are not hypersensitive to camptothecin [162] and we found that AT-complemented cells are not hypersensitive to camptothecin (our unpublished results).
Genes are in alphabetic order.
Abbreviations: AOA1: Ataxia-oculomotor apraxia 1; ATM: Ataxia Telangiectasia Mutant; ATR: Ataxia Telangiectasia and Rad3-related; BER: Base Excision Repair; BLM: Bloom syndrome (BS); CSA/CSB: Cockayne Syndrome (CS) complementation groups A and B; DNA-PKcs: DNA-dependent protein kinase catalytic subunit; DSB: DNA double-strand breaks; NBS: Nijmegen Breakage Syndrome; NER: nucleotide excision repair; PARP: poly(ADP-ribose) polymerase; PI3K: phosphatidyl inositol 3 kinase; PNKP: polynucleotide kinase phosphatase; SCAN: Spino Cerebellar Ataxia Axonal Neuropathy; TCR: transcription-coupled repair; WRN: Werner syndrome.
In the case of transcription, blocked RNA complexes might be displaced (“pull-back” mechanism) from the Top1 cleavage complexes without removal from the transcribed DNA. Transcription elongation restart can finish the incomplete mRNA. Rad26 (the yeast homologue of CSB) and TFIIS have been implicated in the backtracking of RNA polymerase II [83]. RNA polymerase II can also be degraded following camptothecin treatment but this process is limited to some cell types and delayed as compared to RNA-synthesis recovery [75].
Because Top1 is very effective in joining a 5′-hydroxyl end from a non-homologous substrate to the Top1 covalent complex, Top1 cleavage complexes might reverse by religation of a non-homologous end, which leads to DNA recombinations [84]. Camptothecin is indeed a potent inducer of sister chromatid exchanges and chromosomal abnormalities [85–87]. Top1-mediated 5′-end ligation with vaccinia Top1 is commonly used for cloning recombinant genomes (TOPO cloning kit; Invitrogen, Carlsbad, CA).
II. Top1 excision by Tyrosyl-DNA-phoshodiesterase (Tdp1) (Fig. 5B)
Tdp1 was discovered by Nash and coworkers [88] as the enzyme capable of hydrolyzing the covalent bond between the Top1 catalytic tyrosine and the 3′-end of the DNA [89]. Tdp1 generates a 3′-phosphate, which is further processed by a 3′-phosphatase, such as PNKP (hPNK) (Fig. 5B).
Tdp1 belongs to the phospholipase D superfamily [90] of phospholipid hydrolyzing enzymes. It is ubiquitous and highly conserved in eukaryotes (from yeast to humans – see Tables 2–4). Tdp1 is physiologically important since the homozygote mutation H493R causes spinocerebellar ataxia with axonal neuropathy (SCAN1) [91]. SCAN1 cells are hypersensitive to camptothecin [92–94] (Table 2) and ionizing radiation [95], but not to etoposide or bleomycin [94]. The budding yeast TDP1 knock out is viable [88] and hypersensitive to high levels of Top1 cleavage complexes generated by overexpression of a toxic Top1 [96]. It is hypersensitive to camptothecin only when the checkpoint gene Rad9 is simultaneously inactivated [96] or when some endonuclease pathways (Rad1/Rad10 and Slx1/Slx4) are inactive [97–99] (Table 3). Because Tdp1 specifically processes 3′- but not 5′-tyrosyl-DNA complexes [88, 89], Tdp1 cannot hydrolyze the cleavage complexes produced by other topoisomerases besides Top1 (see Fig. 2). However, Tdp1 function is probably not limited to the repair of Top1 cleavage complexes. Tdp1 can remove 3′-phosphoglycolate (Fig. 6D) generated by oxidative DNA damage, which suggests a broader role for Tdp1 in the maintenance of genomic stability [100].
Human Tdp1 acts as a monomer and crystal structures demonstrate the presence of two catalytic domains related by a pseudo-2-fold axis of symmetry [101–103]. Each domain contains 3 conserved HKN residues (H263, K266 and N283 and H493, K495 and N516 (equivalent to the “HKD motifs” of phospholipases) [101, 102, 104] forming a catalytic network with two water molecules critical for Tdp1 activity [90, 91]. Tdp1 hydrolyzes the DNA phosphotyrosine bond in two consecutive SN2 reactions (Fig. 6B–C). In the first reaction, H263 of the first HKD motif releases the Top1 tyrosine from the phosphodiester by forming a transient covalent phosphoamide bond between the Nε2 atom of the nucleophilic H263 and the 3′-end of the DNA (Fig. 6B). Mutating H263 totally abolishes Tdp1 catalytic activity [90]. In the second reaction, H493 from the second HKD motif catalyzes the nucleophilic attack of the phosphoamide bond by a water molecule. This regenerates Tdp1 and produces a 3′-phosphate DNA end [90] (Figs. 6C and 5B). The SCAN1 mutation H493R affects preferentially the second step of the reaction, which leads to an accumulation of Tdp1-DNA covalent intermediate and reduces Tdp1 catalytic activity ≈ 25-fold [94, 105]. Interthal, Champoux and coworkers have recently shown that wild-type Tdp1 can hydrolyze the phosphoamide Tdp1-DNA covalent intermediate (Fig. 6B) [105], and proposed that lack of symptoms in SCAN1 heterozygote carriers might be due to the release of these covalent intermediates by the coexisting wild-type Tdp1 (Fig. 6D). They also proposed that the covalent Tdp1-DNA intermediates rather than deficient Tdp1 catalytic activity might be responsible for DNA damage leading to SCAN [105].
Both the structure of the DNA segment bound to Top1 [88, 106] and the length of the Top1 polypeptide chain determine Tdp1’s activity [106]. Optimum Tdp1 activity requires: 1/ a DNA segment consisting of at least a few nucleotides [106]; 2/ an exposed phosphotyrosyl bond at the Top1-DNA junction [a tyrosyl group linked to the 3′-end of a nick is a poor substrate [96], indicating that Tdp1 acts after the 5′-end of the broken DNA has been either digested or displaced to provide access to the 3′-phosphotyrosyl bond]; and 3/ a short Top1 polypeptide segment, as the effectiveness of Tdp1 decreases with the length of the Top1 polypeptide [106]. In fact, Top1 needs to be proteolyzed or denatured for efficient Tdp1 activity [89, 105, 106]. Top1 ubiquitination and degradation have been observed following camptothecin treatment [107, 108]. The Top1 degradation pathway appears selectively deficient in transformed cells, although not all transformed cells appear equally able to proteolyze Top1 following camptothecin treatment [77]. Such differences have been proposed to contribute to camptothecin resistance [77]. A recent structure of Tdp1 bound to a tyrosine-containing peptide demonstrate that both the DNA and the Top1 polypeptide need to adapt their structure to bind Tdp1 in the crystal structure [103]. The DNA binds in a narrow groove that fits a single-stranded substrate and the short Top1 polypeptide is folded differently from the native Top1 [103]. Recently, an alternative model has been presented for duplex DNA, which can also be processed effectively by Tdp1 [88, 106, 109].
The 3′-phosphate ends generated by Tdp1 need to be hydrolyzed to a 3′-hydroxyl for further processing by DNA polymerases and/or ligases (Fig. 5B). In budding yeast, this 3′-phosphatase activity is carried out by Tpp1 [110] and by the two functionally overlapping multifunctional apurinic (AP) endonucleases, Apn1 and Apn2 [97] (see Fig. 8A). Apn1 is the ortholog of E. coli endonuclease IV and represents the major yeast AP endonuclease. Apn2 (also called Eth1) belongs to the second family of AP endonuclease (the E. coli exonuclease III family), and is the ortholog of Ape1 in humans. Simultaneous inactivation of Tpp1, Apn1 and Apn2 is required to sensitize yeast to camptothecin [110], indicating the functional redundancy of the 3′-phosphatase pathways. Noticeably, the hypersensitivity of the tpp1 apn1 apn2 triple mutant is rescued by inactivation of Tdp1 [111], which indicates that in the absence of Tdp1, budding yeast uses an alternative endonuclease pathway for removal of the Top1 covalent complexes (see Figs. 5C and 8A, and next section). The 3′-phosphatase orthologs of Tpp1 are Pnk1 in fission yeast [112] and PNKP (also referred to as hPNK) in humans [110, 113, 114] (see Tables 2–4). In addition to their 3′-phosphatase activity, Pnk1 [112] and PNKP/hPNK [113, 114] possess 5′-kinase activity, which is missing for Tpp1. Stable down-regulation of human PNKP (hPNK) sensitizes to camptothecin (Table 2), ionizing- and UV-radiation, H2O2 and UV, and increases spontaneous mutation frequency [115].
Fig. 8. Repair involved in the repair of Top1 covalent complexes in budding yeast.
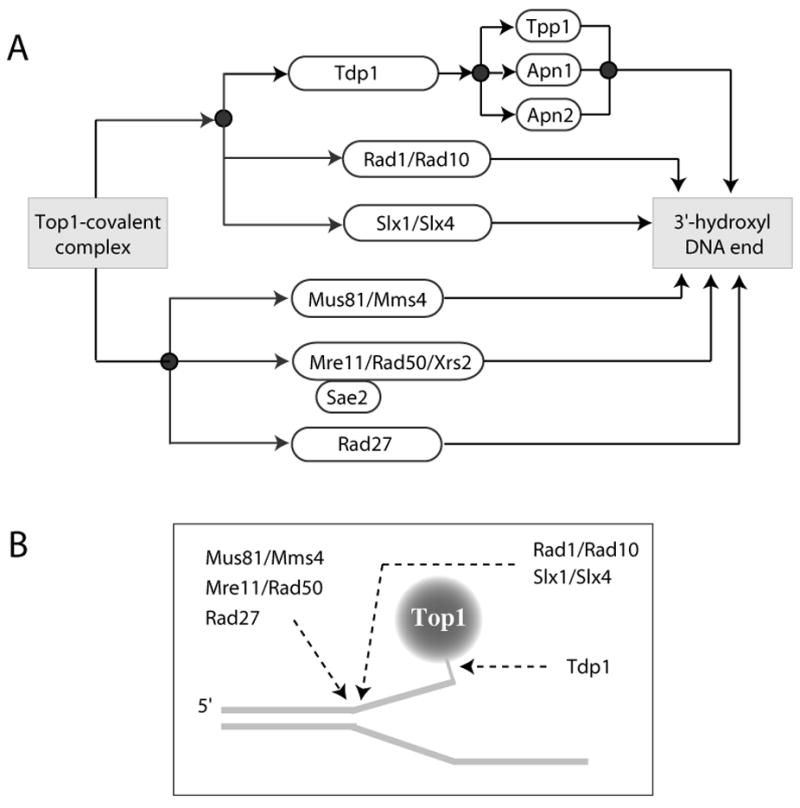
A. Schematic representation of the genetic pathways implicated in the removal of the Top1-DNA covalent complexes. Tdp1 appear to function in alternative pathways with Rad1/Rad10 and Slx1/Slx4. The other endonucleases (Mus81/Mms4; Mre11/Rad50/Xrs2 and Rad27) appear to function in parallel. Sae2 is a cofactor for the endonuclease activity of Mre11. Tpp1, Apn1 and Apn2 are 3′-phosphatases that remove the 3′-phosphate left after Tdp1 hydrolyzes the tyrosyl-DNA adduct (see Figs. 5B and 6C). B. Shematic representation of the sites of attack for Tdp1 and the endonucleases described in panel A. Srs2 helicase is also shown.
In humans, Tdp1 and PNKP form a multiprotein complex with XRCC1, poly(ADP)ribose-polymerase (PARP), β-polymerase and ligase III [116, 117] (Fig. 5B, bottom). This complex contains the critical elements for base excision repair. Mammalian cells deficient for XRCC1 or PARP are also hypersensitive to camptothecin (Fig. 7) (Table 1) [78, 116, 118]. The hypersensitivity of PARP−/− cells to camptothecin (Fig. 7A) can be related to a functional defect in Tdp1 (Fig. 7B–C). It is not explained by abnormal levels of Tdp1 or Top1 proteins (Barcelo and Pommier, unpublished). Recently, a novel protein, aprataxin has been found to associate with the XRCC1 complex (Fig. 5B) [119]. Aprataxin is a 342 amino acid protein encoded by the APTX gene whose homozygote mutation produces AOA1 (Ataxia-oculomotor Apraxia; the most common autosomal recessive ataxia in Japan) [120, 121]. AOA1 cells are hypersensitive to camptothecin [122] (Table 1).
Fig. 7. Hypersensitivity of PARP-1−/− cells to camptothecin and functional Tdp1 deficiency in PARP-1 −/− cells.
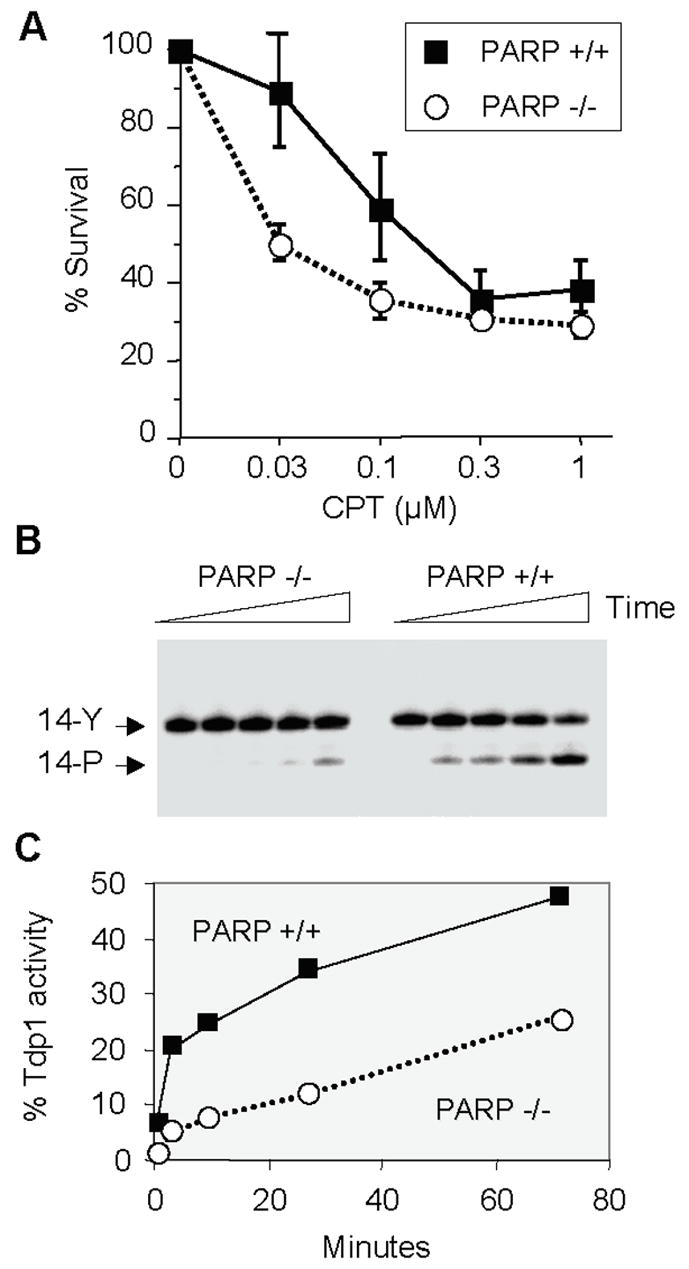
A. Mouse fibroblasts were exposed to camptothecin (concentrations indicated on X axis) for 1 hour, washed, kept in culture for 4 days and counted. The curves represent the averages and standard deviations from five independent experiments. B. A 14-mer single-stranded oligonucleotide with a phosphotyrosine at the 3′-end (see Fig. 6) (14-Y) [106] was incubated with nuclear extracts obtained from PARP-1−/− and PARP-1 +/+ cells [195] in the presence of 50 mM EDTA and absence of MgCl2 to eliminate PNKP activity, which requires MgCl2. A representative experiment is shown. TDP1 activity was determined as a shift in band position (from 14-Y to 14-P). C. Quantitation of the results shown in panel B using ImageQuant (Molecular Dynamics, Sunnyvale, CA).
There is no pharmacological inhibitor of Tdp1 reported to date. Vanadate and tungstate act as phosphate mimetic in co-crystal structures and block Tdp1 activity at millimolar concentrations [102]. It would, however, be rational to develop Tdp1 inhibitors for cancer treatment in combination with camptothecins. The anticancer activity of Tdp1 inhibitors may prove dependent on the presence of cancer-related genetic abnormalities, since camptothecin hypersensitivity in Tdp1-defective yeast is conditional for deficiencies in the checkpoint (Rad9) [88, 97, 98]. A Rad9 defect in a Tdp1-deficient background confers marked sensitization to camptothecin [88], and it is tempting to speculate that Tdp1 is primarily required when the checkpoints are deficient as in the case of the yeast RAD9 mutant. A second group of conditional genes (with respect to Tdp1 deficiencies) includes three sets of genes from the 3′-flap endonuclease pathway: Rad1/Rad10, Mre11/Rad50, and Mus81/Eme1 [88, 97, 98]. Mutation in each of these genes renders Tdp1-deficient cells highly sensitive to camptothecin (see Table 3). Hence, colon cancers, which are commonly mutated for Mre11 might be selectively sensitive to the combination of camptothecin with a Tdp1 inhibitor. The potent activity of Tdp1 against a number of artificial substrates (Fig. 6E) [105] can be used to design high throughput screens (our unpublished observations) [123, 124], and it is likely that Tdp1 inhibitors will be reported in the near future.
III. Top1 excision by Endonucleases (Fig. 5C)
Studies in yeast demonstrate the existence of alternative pathways besides Tdp1 for removing the Top1 covalent complexes. Figure 8 summarizes the multiple endonuclease genetic pathways implicated in the repair of Top1 cleavage complexes [97–99].
Rad1/Rad10 and Slx1/Slx4 appear to function in parallel and redundant pathways with Tdp1 [97–99] (Fig. 8A). Rad1/Rad10 is the ortholog of the human endonuclease XPF/ERCC1, which cleaves DNA 3′ from bulky adduct during nucleotide excision repair [125]. Like Tdp1, Rad1/Rad10 requires a single-stranded gap between the 3′-end to be processed and the 5′-end of the DNA (Fig. 8B) [126], suggesting that Tdp1 and Rad1/Rad10 share common substrates. SLX1 and SLX4 were originally identified as genes synthetically lethal with mutations in SGS1 and TOP3 [127]. The dimeric complex has strong endonuclease activity with a wide range of substrates [128], and Slx4/Slx1 appears to function as an alternative pathway in the absence of Tdp1 [99] (Fig. 8).
Mus81/Mms4 (the ortholog of budding yeast Mms4 is Eme1 in humans and fission yeast –see Tables 3 & 4) preferentially cleaves broken replication forks and requires the presence of duplex DNA near the 3′-end to be processed [see Fig. 8B and Fig. 4B in [78]] [126, 129, 130]. Mus81- and Rad50-deficient yeasts are highly sensitive to camptothecin (Tables 3 & 4) [98, 99, 126, 129, 131].
The Mre11/Rad50/Xrs2 complex (MRX) (the human orthologs are Mre11/Rad50/Nbs1 [MRN]) preferentially cleaves gapped substrates (Fig. 8B) and hairpin structures [132]. MRX appears to function independently from the Tdp1 pathway, which is also the case for Mus81/Mms4 and Rad27 [97–99] (Fig. 8A). Sae1 is required for the endonuclease activity of MRX, and sae2 deletion strains are among the strains most sensitive to camptothecin [99]. The MRN complex also possesses checkpoint functions, which probably contribute to cell survival in response to camptothecin [97, 98].
Surprisingly, the 5′-flap endonuclease, Rad27 (the human ortholog is FEN-1) also contributes to the repair of Top1 covalent complexes (Fig. 8). Deletion of RAD27 causes mild sensitivity to camptothecin (Table 3) [98, 99, 133]. The apparent discrepancy between the established 5′-flap endonuclease activity of Rad27 (FEN-1) and its role in Top1 repair can be explained by a recent study demonstrating that Rad27 in coordination with the Werner syndrome protein and replication protein A (RPA) possesses gap 5′-endonuclease activity (GEN activity) and can process substrates that mimic stalled replication forks. Human FEN-1 is able to rescue the defect in resistance to camptothecin and UV in a yeast FEN-1 null mutant [133].
The recent yeast genome-wide screen to detect novel genes that are important for protection against growth inhibition and killing produced by camptothecin identified a large number of genes for further exploration [99]. SRS2 was among the most critical genes [98, 99]. SRS2 is a DNA helicase that might contribute to the local unwinding (Fig. 8B) that would provide access to the endonucleases described above. A number of other genes involved in transcription, replication, ubiquitination and protein degradation await further studies (Table 3) [99, 134–136]. However, extrapolating the results obtained in budding yeast to human cells is not straightforward. Cells from Mus81 knockout mice have recently been tested and failed to show significant hypersensitivity to camptothecin in spite of their hypersensitivity to DNA crosslinking agents (cisplatin and nitrogen mustards) [137]. Similarly, XPF cells do not demonstrate hypersensitivity to camptothecin (Pourquier and Pommier, unpublished). These apparent discrepancies may be related to the presence of additional repair pathways and different checkpoint controls in humans.
E. Checkpoint response to Top1-associated DNA damage
Cellular responses to Top1 poisons determine both tumor response and host toxicity. Efficient repair is probably coupled with checkpoint activation. Cell cycle arrest following checkpoint activation would have two beneficial consequences: 1/ it would give time for the repair of DNA damage; and 2/ it would prevent further replication-dependent DNA damage. Both the S-phase and the G2 checkpoints, as well as the p53/p21 pathways are activated by Top1-mediated DNA damage [57, 138]. Because cell cycle checkpoints are also connected to the apoptosis machinery, it is likely that extensive DNA damage activates apoptosis by involving the same DNA damage sensors and checkpoints [42]. Thus, an exciting challenge is to elucidate the relationships between sensor proteins, checkpoints, DNA repair and apoptosis. Integration of these pathways should explain the cellular determinants of response to Top1 poisons. We will focus on the Chk1 and Chk2 pathways/responses elicited by Top1 poisons, and how defects in these pathways can sensitize tumors to Top1-mediated DNA damage. We will not review the roles of p53, c-Abl, and the stress kinase (JNK/SAPK) pathways, which have been detailed elsewhere [42, 78, 139].
I. Chk1 activation by camptothecin
Chk1 is an evolutionarily conserved kinase and essential member of the DNA damage checkpoint [140–142]. Deletion of Chk1 is embryonic lethal with massive apoptosis in stem cells [143, 144]. Chk1 deletion is not lethal in budding and fission yeast or chicken somatic cells (DT40). However, these cells display an inability to recover from replication blockade and are hypersensitive to ionizing radiation [145–147]. In budding yeast, the upstream kinase Tel1 activates Chk1 in concert with the “9-1-1 PCNA –like” clamp proteins: Rad17, Mec3 and Rad24. Strains defective for the Tel1 or the 9-1-1 clamp proteins are hypersensitive to camptothecin (Table 3). In xenopus and humans, ATR (the Tel1 ortholog) with the Rad9-Rad1-Hus1 (“9-1-1”) complex regulates Chk1 via phosphorylation. The DNA-binding protein claspin also associates with Chk1 and is required for Chk1 activation [143, 148–150]. Other kinases, including ATM [151, 152], might also activate Chk1 by phosphorylation [152–154].
The first reports of Chk1 activation by camptothecin were obtained in the fission yeast [155]. Blocking DNA synthesis with a hydroxyurea pre-treatment blocked camptothecin-induced Chk1 phosphorylation/activation, and cells lacking functional Chk1 were hypersensitive to camptothecin [155–157]. Phosphorylation of Chk1 is rapid following exposure to camptothecin and reaches a plateau between 60 minutes and 3 hours [155]. Chk1 activation requires phosphorylation by Rad3 and is required for cell cycle arrest and interaction with Rad24 and Rad25 (14-3-3 orthologs), which sequester (and functionally inactivate) Cdc25 [158–164]. In mammalian cells, camptothecin also activates Chk1, and Chk1 anti-sense oligonucleotides abolish the checkpoint response to camptothecin [165]. Chk1 activation requires phosphorylation by ATR on S345. G2/M arrest is mediated by phosphorylation of Cdc25C by Chk1 and subsequent inactivation of Cdc25C [161–164], and S-phase arrest is mediated by Chk1-mediated phosphorylation of Cdc25A and degradation of Cdc25A [166]. Chk1 can also phosphorylate Rad51 on T309. Rad51 is involved in homologous recombination and provides a link between Chk1 activation, DNA repair and survival after replicative stress [167].
Camptothecin also induces Chk1 degradation by the ubiquitin-proteosome pathway [168] via the E4 ligase complex containing cullins (Cul1 and Cul4A). Phosphorylation at S345 marks Chk1 for proteolytic degradation and may be indicative of a negative feedback mechanism that may promote the checkpoint termination. Chk1 degradation had been previously suggested to occur via a proteosomal pathway after exposure to geldanamycin, an Hsp90 binding agent that stimulate proteasome-mediated degradation of several other proteins [169]. Downregulation of Chk1 by siRNA potentiates the cytotoxicity of camptothecin in cancer cell lines [162, 170]. Consistently, Chk1 inhibition by UCN-01 (7-hydroxystaurosporine) markedly potentiates the cytoxicity of camptothecin via abrogation of the S-phase checkpoint [57]. Preincubation with UCN-01 prevents camptothecin-induced degradation of Cdc25A and cyclin E in a p53-dependent manner, which is consistent with an abrogation of S phase arrest [171]. In another study, concurrent treatment with camptothecins and UCN-01 resulted in S-phase checkpoint abrogation, increased phosphorylation of γ-H2AX (a marker of DNA double strand breaks) and cell killing independently of p53 [172].
II. Chk2 activation by camptothecin
Chk2 shares no sequence homology with Chk1 [for recent review see [173]]. Chk2 knockout mice are viable and are defective in p53 stabilization following DNA double-strand breaks [174, 175]. Chk2 functions as a checkpoint and apoptotic kinase after being activated via phosphorylation primarily by ATM [141, 176, 177], and also by ATR [178], DNA-PK (via interactions with Ku70/80) [179], Polo-like kinases, Plk-1 and Plk-3, [180–184] and TTK/hMPS1 kinases [185]. Following initial phosphorylation on T68, Chk2 undergoes dimerization via the FHA domain of a second Chk2 molecule [186]. This interaction is followed by a cascade of autophosphorylation steps on T387/T383 and S516, which are required for full activation of Chk2 [reviewed in [187] and (http://discover.nci.nih.gov/mim/)].
Human kidney embryonic cells expressing anti-sense Chk2 display defective S-phase delay and enhanced cell killing in response to replication-mediated DNA damage induced camptothecin [188]. Chk2 siRNA experiments also demonstrate a role for Chk2 for cell survival after camptothecin [162]. However, the phenotype of Chk2-deficient cells may depend on the cell type. In cortical neurons, ATM deficiency, but not Chk2 deficiency, attenuates cell death and significantly inhibits the induction of p53 phosphorylation on S15 and p53 levels induced by camptothecin [189]. Further analyses in cells with fully functional replication machinery are warranted to determine the potential role of Chk2 in Top1-induced DNA replication stress. More specifically, the relative roles of ATR and ATM remain to be elucidated. Chk2 is rapidly phosphorylated by ATM in camptothecin-treated cells [190], and phosphorylated Chk2 forms nuclear foci that are associated with sites of DNA damage marked by 53BP1, γ-H2AX and NBS1 [191]. Camptothecin also reduces the amount of chromatin-associated Chk2, especially the active T68-phosphorylated forms of Chk2 [179]. Soluble Chk2 might be implicated in the transmission of signals to remote chromatin sites from the DNA damage.
Chk2 has a dual role on cell cycle checkpoints and apoptosis via phosphorylation of its downstream substrates including Cdc25A, Cdc25C, p53, BRCA1, E2F1, and PML [reviewed in [173]] (http://discover.nci.nih.gov/mim/). Chk2 provides an unexplored therapeutic target in cancer cells with inherent defects in G1 checkpoint function. By virtue of Chk2’s role in both cell cycle checkpoint regulation and apoptosis, selective inhibition of Chk2 could improve the therapeutic index of DNA-damaging agents such as camptothecin [Reviewed in [173]]. This may be especially true in p53-deficient tumors where the p53-dependent apoptotic response is deficient. In normal tissues Chk2 may act as a pro-apoptotic effector, thus Chk2 inhibitors may protect normal tissues. However, no clinical agent currently presents selective Chk2 inhibition.
III. Chk1 vs. Chk2 activation by camptothecin and Top1-mediated DNA damage
As discussed above, Top1-induced DNA damage activates both the Chk1 and Chk2 kinases. However, the relative timing of such activations has not been described in detail. Understanding the kinetics of activation of each has the potential for presenting a better target for inhibition in combination with camptothecins. A recent study [192] reported that both Chk1 and Chk2 are rapidly activated following low dose exposure to camptothecin, suggesting concomitant activation of both the ATR-Chk1 and ATM-Chk2 pathways. Additionally, a deficiency of ATM kinase prevented the activation of Chk2, with no effects on Chk1 activity or the degradation of Cdc25A [192]. The authors proposed that the ATR-Chk1 pathway is sufficient, though not the only pathway, to induce checkpoint-mediated degradation of Cdc25A. One explanation for the ATM-independent analysis might be the cautionary finding of a highly activated ATR-Chk1 pathway in AT cells treated with ionizing radiation [193]. Recent reports identifying multiple phosphorylation sites on Chk1 and Chk2 and their unraveling functions also provide a rationale for further studies that are needed to detail the kinetics of Chk1 and Chk2 activation by camptothecin. Overall, the redundancy in activation of both Chk1 and Chk2 by camptothecin may be indicative of distinct functions for each kinase in collecting, sustaining and deploying the DNA damage signal to its various substrates for favorable use by the cell. Abrogation of these kinases, and consequently the cell cycle arrest checkpoint, thus presents novels opportunities for enhancing drug efficacy.
F. Conclusion and perspective
Studies performed in yeast have provided major insights in the DNA damage and repair of Top1 cleavage complexes. These insights include the demonstration that Top1 is the selective target of camptothecin, the discovery of Tdp1, the identification of genetic defects (in recombination pathways and endonucleases) that sensitize yeast to Top1-mediated DNA damage. It is clear that more lessons remain to be learned from yeast including the roles of chromatin structure, ubiquitin and protein modification pathways. However, not all yeast pathways can be translated to human cells even when the orthologs are present in both. For instance, the Tdp1 mutant in budding yeast is only sensitive to camptothecin if a checkpoint pathway (Rad9) or an endonuclease pathway (Rad1/Rad10) is also defective. By contrast, human cells deficient for Tdp1 (cells from SCAN1 patients) appear to be sensitive with this single alteration. Mus81-deficient yeast is highly sensitive to camptothecin whereas murine Mus81-knockout cells are not. These differences must be related to the “genetic background” of the cells.
The divergent phenotypes (and genotypes) of mammalian cells in culture provide therapeutic opportunities for cancer treatment. Programmed genetic and pharmacological deficiencies should have different consequences with respect to cellular response to Top1-mediated DNA damage depending on the genomic context of the cell. This challenge brings the opportunity to find out which genetic contexts provide selective sensitivity or resistance to Top1 inhibitors, and the rationale for developing therapies tailored to the genetic deficiencies selective to particular tumors. It is in this context that one could foresee the use of inhibitors of Tdp1 and Chk1/2 in combination with Top1 inhibitors once it is established which tumor-specific deficiencies provide the greatest sensitization to Top1 inhibitors.
Acknowledgments
This research is supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
We wish to thank Dr. Mark Smulson for providing us with the PARP−/− and PARP+/+ cells. We wish to thank the past and current member of the Laboratory of Molecular Pharmacology (Drs. Smitha Antony, Laurent Debethune, Takahisa Furuta, Linghua Meng, Philippe Pourquier, Haruyuki Takemura, and Qiang Yu), and more particularly Dr. Kurt W. Kohn for outstanding contribution to the topoisomerase inhibitor studies and for sustained input and encouragements.
References
- 1.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 2.Zhang H, Barcelo JM, Lee B, Kohlhagen G, Zimonjic DB, Popescu NC, Pommier Y. Human mitochondrial topoisomerase I. Proc Natl Acad Sci U S A. 2001;98:10608–10613. doi: 10.1073/pnas.191321998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang H, Meng LH, Zimonjic DB, Popescu NC, Pommier Y. Thirteen-exon-motif signature for vertebrate nuclear and mitochondrial type IB topoisomerases. Nucleic Acids Res. 2004;32:2087–2092. doi: 10.1093/nar/gkh525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 5.Harmon FG, Brockman JP, Kowalczykowski SC. RecQ helicase stimulates both DNA catenation and changes in DNA topology by topoisomerase III. J Biol Chem. 2003;278:42668–42678. doi: 10.1074/jbc.M302994200. [DOI] [PubMed] [Google Scholar]
- 6.Morham S, Kluckman KD, Voulomanos N, Smithies O. Targeted disruption of the mouse topoisomerase I gene by camptothecin selection. Mol Cell Biol. 1996;16:6804–6809. doi: 10.1128/mcb.16.12.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang CX, Chen AD, Gettel NJ, Hsieh TS. Essential functions of DNA topoisomerase I in Drosophila melanogaster. Dev Biol. 2000;222:27–40. doi: 10.1006/dbio.2000.9704. [DOI] [PubMed] [Google Scholar]
- 8.Eng WK, Faucette L, Johnson RK, Sternglanz R. Evidence that DNA topoisomerase I is necessary for the cytotoxic effects of camptothecin. Mol Pharmacol. 1988;34:755–760. [PubMed] [Google Scholar]
- 9.Baker SD, Wadkins RM, Stewart CF, Beck WT, Danks MK. Cell cycle analysis of amount and distribution of nuclear DNA topoisomerase I as determined by fluorescence digital imaging microscopy. Cytometry. 1995;19:134–145. doi: 10.1002/cyto.990190208. [DOI] [PubMed] [Google Scholar]
- 10.Muller MT, Pfund WP, Mehta VB, Trask DK. Eukaryotic type I topoisomerase is enriched in the nucleolus and catalytically active on ribosomal DNA. Embo J. 1985;4:1237–1243. doi: 10.1002/j.1460-2075.1985.tb03766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Wang JC, Liu LF. Involvement of DNA topoisomerase I in transcription of human ribosomal RNA genes. Proc Natl Acad Sci USA. 1988;85:1060–1064. doi: 10.1073/pnas.85.4.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pommier Y, Kohlhagen G, Wu C, Simmons DT. Mammalian DNA topoisomerase I activity and poisoning by camptothecin are inhibited by simian virus 40 large T antigen. Biochemistry. 1998;37:3818–3823. doi: 10.1021/bi972067d. [DOI] [PubMed] [Google Scholar]
- 13.Champoux JJ. DNA TOPOISOMERASES: Structure, Function, and Mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 14.Leppard JB, Champoux JJ. Human DNA topoisomerase I: relaxation, roles, and damage control. Chromosoma. 2005;114:75–85. doi: 10.1007/s00412-005-0345-5. [DOI] [PubMed] [Google Scholar]
- 15.Xu CJ, Grainge I, Lee J, Harshey RM, Jayaram M. Unveiling two distinct ribonuclease activities and a topoisomerase activity in a site-specific DNA recombinase. Molecular Cell. 1998;1:729–739. doi: 10.1016/s1097-2765(00)80072-6. [DOI] [PubMed] [Google Scholar]
- 16.Pourquier P, Jensen AD, Gong SS, Pommier Y, Rogler CE. Human DNA topoisomerase I-mediated cleavage and recombination of duck hepatitis B virus DNA in vitro. Nucleic Acids Res. 1999;27:1919–1923. doi: 10.1093/nar/27.8.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng C, Shuman S. Recombinogenic flap ligation pathway for intrinsic repair of topoisomerase IB-induced double-strand breaks. Mol Cell Biol. 2000;20:8059–8068. doi: 10.1128/mcb.20.21.8059-8068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soret J, Gabut M, Dupon C, Kohlhagen G, Stevenin J, Pommier Y, Tazi J. Altered serine/arginine-rich protein phosphorylation and exonic enhancer-dependent splicing in Mammalian cells lacking topoisomerase I. Cancer Res. 2003;63:8203–8211. [PubMed] [Google Scholar]
- 19.Rossi F, Labourier E, Forne T, Divita G, Derancourt J, Riou JF, Antoine E, Cathala G, Brunel C, Tazi J. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature. 1996;381:80–82. doi: 10.1038/381080a0. [DOI] [PubMed] [Google Scholar]
- 20.Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A model for the mechanism of human topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 21.Lesher DT, Pommier Y, Stewart L, Redinbo MR. 8-Oxoguanine rearranges the active site of human topoisomerase I. Proc Natl Acad Sci U S A. 2002;99:12102–12107. doi: 10.1073/pnas.192282699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carey JF, Schultz SJ, Sisson L, Fazzio TG, Champoux JJ. DNA relaxation by human topoisomerase I occurs in the closed clamp conformation of the protein. Proc Natl Acad Sci U S A. 2003;100:5640–5645. doi: 10.1073/pnas.1031537100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo MH, Losasso C, Guo H, Pattarello L, Benedetti P, Bjornsti MA. Locking the DNA topoisomerase I protein clamp inhibits DNA rotation and induces cell lethality. Proc Natl Acad Sci U S A. 2003;100:13767–13772. doi: 10.1073/pnas.2235886100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koster DA, Croquette V, Dekker C, Shuman S, Dekker NH. Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature. 2005;434:671–674. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- 26.Pommier Y, Cherfils J. Interfacial protein inhibition: a nature’s paradigm for drug discovery. Trends Pharmacol Sci. 2005;28:136–145. doi: 10.1016/j.tips.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Pommier Y, Marchand C. Interfacial inhibitors of protein-nucleic acid interactions. Curr Med Chem Anti-Canc Agents. 2005;5:421–429. doi: 10.2174/1568011054222337. [DOI] [PubMed] [Google Scholar]
- 28.Chrencik JE, Staker BL, Burgin AB, Pourquier P, Pommier Y, Stewart L, Redinbo MR. Mechanisms of camptothecin resistance by human topoisomerase I mutations. J Mol Biol. 2004;339:773–784. doi: 10.1016/j.jmb.2004.03.077. [DOI] [PubMed] [Google Scholar]
- 29.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 30.Ioanoviciu A, Antony S, Pommier Y, Staker BL, Stewart L, Cushman M. Synthesis and Mechanism of Action Studies of a Series of Norindenoisoquinoline Topoisomerase I Poisons Reveal an Inhibitor with a Flipped Orientation in the Ternary DNA-Enzyme-Inhibitor Complex As Determined by X-ray Crystallographic Analysis. J Med Chem. 2005;48:4803–4814. doi: 10.1021/jm050076b. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8:641–661. [PubMed] [Google Scholar]
- 32.Meng LH, Liao ZY, Pommier Y. Non-camptothecin DNA topoisomerase I inhibitors in cancer chemotherapy. Curr Topics Med Chem. 2003;3:305–320. doi: 10.2174/1568026033452546. [DOI] [PubMed] [Google Scholar]
- 33.Covey JM, Jaxel C, Kohn KW, Pommier Y. Protein-linked DNA strand breaks induced in Mammalian cells by camptothecin, an inhibitor of topoisomerase I. Cancer Res. 1989;49:5016–5022. [PubMed] [Google Scholar]
- 34.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential induction of topoisomerase I-DNA cleavage complexes by the indenoisoquinoline MJ-III-65 (NSC 706744) and camptothecin: base sequence analysis and activity against camptothecin-resistant topoisomerases I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- 35.Jaxel C, Capranico G, Kerrigan D, Kohn KW, Pommier Y. Effect of local DNA sequence on topoisomerase I cleavage in the presence or absence of camptothecin. J Biol Chem. 1991;266:20418–20423. [PubMed] [Google Scholar]
- 36.Tanizawa A, Kohn KW, Kohlhagen G, Leteurtre F, Pommier Y. Differential stabilization of eukaryotic DNA topoisomerase I cleavable complexes by camptothecin derivatives. Biochemistry. 1995;34:7200–7206. doi: 10.1021/bi00021a035. [DOI] [PubMed] [Google Scholar]
- 37.Pourquier P, Pommier Y. Topoisomerase I-mediated DNA damage. Adv Cancer Res. 2001;80:189–216. doi: 10.1016/s0065-230x(01)80016-6. [DOI] [PubMed] [Google Scholar]
- 38.Sokhansanj BA, Wilson DM., 3rd Oxidative DNA damage background estimated by a system model of base excision repair. Free Radic Biol Med. 2004;37:422–427. doi: 10.1016/j.freeradbiomed.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Pourquier P, Ueng LM, Fertala J, Wang D, Park HJ, Essigman JM, Bjornsti MA, Pommier Y. Induction of reversible complexes between eukaryotic DNA topoisomerase I and DNA-containing oxidative base damages. J Biol Chem. 1999;274:8516–8523. doi: 10.1074/jbc.274.13.8516. [DOI] [PubMed] [Google Scholar]
- 40.Pourquier P, Ueng LM, Kohlhagen G, Mazumder A, Gupta M, Kohn KW, Pommier Y. Effects of uracil incorporation, DNA mismatches, and abasic sites on cleavage and religation activities of mammalian topoisomerase I. J Biol Chem. 1997;272:7792–7796. doi: 10.1074/jbc.272.12.7792. [DOI] [PubMed] [Google Scholar]
- 41.Pourquier P, Pilon A, Kohlhagen G, Mazumder A, Sharma A, Pommier Y. Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps: importance of DNA end phosphorylation and camptothecin effects. J Biol Chem. 1997;272:26441–26447. doi: 10.1074/jbc.272.42.26441. [DOI] [PubMed] [Google Scholar]
- 42.Sordet O, Khan Q, Kohn KW, Pommier Y. Apoptosis induced by topoisomerase inhibitors. Curr Med Chem Anticancer Agents. 2003;3:271–290. doi: 10.2174/1568011033482378. [DOI] [PubMed] [Google Scholar]
- 43.Sordet O, Khan QA, Plo I, Pourquier P, Urasaki Y, Yoshida A, Antony S, Kohlhagen G, Solary E, Saparbaev M, Laval J, Pommier Y. Apoptotic Topoisomerase I-DNA Complexes Induced by Staurosporine-mediated Oxygen Radicals. J Biol Chem. 2004;279:50499–50504. doi: 10.1074/jbc.M410277200. [DOI] [PubMed] [Google Scholar]
- 44.Sordet O, Khan QA, Pommier Y. Apoptotic Topoisomerase I-DNA Complexes Induced by Oxygen Radicals and Mitochondrial Dysfunction. Cell Cycle. 2004;3:1095–1097. [PubMed] [Google Scholar]
- 45.Sordet O, Liao Z, Liu H, Antony S, Stevens EV, Kohlhagen G, Fu H, Pommier Y. Topoisomerase I-DNA complexes contribute to arsenic trioxide-induced apoptosis. J Biol Chem. 2004;279:33968–33975. doi: 10.1074/jbc.M404620200. [DOI] [PubMed] [Google Scholar]
- 46.Burgin AB, Huizenga BN, Nash HA. A novel suicide substrate for DNA topoisomerases and site-specific recombinases. Nucleic Acids Res. 1995;23:2973–2979. doi: 10.1093/nar/23.15.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shuman S. Vaccinia DNA topoisomerase I promotes illegitimate recombination in Eschrichia coli. Proceedings of the National Academy of Sciences, USA. 1989;86:3489–3493. doi: 10.1073/pnas.86.10.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holm C, Covey JM, Kerrigan D, Pommier Y. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res. 1989;49:6365–6368. [PubMed] [Google Scholar]
- 49.Hsiang YH, Lihou MG, Liu LF. Arrest of DNA replication by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 50.Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol. 1996;134:757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol. 2000;20:3977–3987. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, Nussenzweig A, Aladjem MI, Bonner WM, Pommier Y. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA-double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem. 2003;278:20303–20312. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- 54.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 55.Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 56.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shao RG, Cao CX, Shimizu T, O’Connor P, Kohn KW, Pommier Y. Abrogation of an S-phase checkpoint and potentiation of camptothecin cytotoxicity by 7-hydroxystaurosporine (UCN-01) in human cancer cell lines, possibly influenced by p53. Cancer Res. 1997;57:4029–4035. [PubMed] [Google Scholar]
- 58.Kohn EA, Ruth ND, Brown MK, Livingstone M, Eastman A. Abrogation of the S Phase DNA Damage Checkpoint Results in S Phase Progression or Premature Mitosis Depending on the Concentration of 7- Hydroxystaurosporine and the Kinetics of Cdc25C Activation. J Biol Chem. 2002;277:26553–26564. doi: 10.1074/jbc.M202040200. [DOI] [PubMed] [Google Scholar]
- 59.Huberman JA. New views of the biochemistry of eucaryotic DNA replication revealed by aphidicolin, an unusual inhibitor of DNA polymerase alpha. Cell. 1981;23:647–648. doi: 10.1016/0092-8674(81)90426-8. [DOI] [PubMed] [Google Scholar]
- 60.Horwitz SB, Chang CK, Grollman AP. Studies on camptothecin. I. Effects of nucleic acid and protein synthesis. Mol Pharmacol. 1971;7:632–644. [PubMed] [Google Scholar]
- 61.Kessel D. Effects of camptothecin on RNA synthesis in leukemia cells. Biochim Biophys Acta. 1971;246:225–232. doi: 10.1016/0005-2787(71)90131-6. [DOI] [PubMed] [Google Scholar]
- 62.Kann HE, Jr, Kohn KW. Effects of deoxyribonucleic acid-reactive drugs on ribonucleic acid synthesis in leukemia L1210 cells. Mol Pharmacol. 1972;8:551–560. [PubMed] [Google Scholar]
- 63.Collins I, Weber A, Levens D. Transcriptional consequences of topoisomerase inhibition. Mol Cell Biol. 2001;21:8437–8451. doi: 10.1128/MCB.21.24.8437-8451.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stewart AF, Herrera RE, Nordheim A. Rapid induction of c-fos transcription reveals quantitive linkage of RNA polymerase II and DNA topoisomerase I enzyme activities. Cell. 1990;60:141–146. doi: 10.1016/0092-8674(90)90724-s. [DOI] [PubMed] [Google Scholar]
- 65.Zhou Y, Gwadry FG, Reinhold WC, Miller LD, Smith LH, Scherf U, Liu ET, Kohn KW, Pommier Y, Weinstein JN. Transcriptional regulation of mitotic genes by camptothecin-induced DNA damage: microarray analysis of dose- and time-dependent effects. Cancer Res. 2002;62:1688–1695. [PubMed] [Google Scholar]
- 66.Daoud SS, Munson PJ, Reinhold W, Young L, Prabhu V, Yu Q, LaRose J, Kohn KW, Weinstein JN, Pommier Y. Impact of p53 knockout and topotecan treatment on gene expression profiles in Human colon carcinoma cells: a pharmacogenomic study. Cancer Res. 2003;63:2782–2793. [PubMed] [Google Scholar]
- 67.Ljungman M, Hanawalt PC. The anti-cancer drug camptothecin inhibits elongation but stimulates initiation of RNA polymerase II transcription. Carcinogenesis. 1996;17:31–35. doi: 10.1093/carcin/17.1.31. [DOI] [PubMed] [Google Scholar]
- 68.Bendixen C, Thomsen B, Alsner J, Westergaard O. Camptothecin-stabilized topoisomerase I-DNA adducts cause premature termination of transcription. Biochemistry. 1990;29:5613–5619. doi: 10.1021/bi00475a028. [DOI] [PubMed] [Google Scholar]
- 69.Wu J, Liu LF. Processing of topoisomerase I cleavable complexes into DNA damage by transcription. Nucleic Acids Res. 1997;25:4181–4186. doi: 10.1093/nar/25.21.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mondal N, Parvin JD. DNA topoisomerase IIalpha is required for RNA polymerase II transcription on chromatin templates. Nature. 2001;413:435–438. doi: 10.1038/35096590. [DOI] [PubMed] [Google Scholar]
- 71.Duann P, Sun M, Zhang LC-TH, Liu LF. Plasmid linking number change induced by topoisomerase I-mediated DNA damage. Nucleic Acids Res. 1999;27:2905–2911. doi: 10.1093/nar/27.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun M, Duann P, Lin CT, Zhang H, Liu LF. Rapid chromatin reorganization induced by topoisomerase I-mediated DNA damage. Ann N Y Acad Sci. 2000;922:340–342. doi: 10.1111/j.1749-6632.2000.tb07059.x. [DOI] [PubMed] [Google Scholar]
- 73.Merino A, Madden KR, Lane WS, Champoux JJ, Reinberg D. DNA topoisomerase I is involved in both repression and activation of transcription. Nature. 1993;365:227–232. doi: 10.1038/365227a0. [DOI] [PubMed] [Google Scholar]
- 74.Shykind BM, Kim J, Stewart L, Champoux JJ, Sharp PA. Topoisomerase I enhances TFIID-TFIIA complex assembly during activation of transcription. Genes & Development. 1997;11:397–407. doi: 10.1101/gad.11.3.397. [DOI] [PubMed] [Google Scholar]
- 75.Desai SD, Zhang H, Rodriguez-Bauman A, Yang JM, Wu X, Gounder MK, Rubin EH, Liu LF. Transcription-dependent degradation of topoisomerase I-DNA covalent complexes. Mol Cell Biol. 2003;23:2341–2350. doi: 10.1128/MCB.23.7.2341-2350.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Squires S, Ryan AJ, Strutt HL, Johnson RT. Hypersensitivity of Cockayne’s syndrome cells to camptothecins is associated with the generation of abnormally high levels of double strand breaks in nascent DNA. Cancer Res. 1993;53:2012–2019. [PubMed] [Google Scholar]
- 77.Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF. Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res. 2001;61:5926–5932. [PubMed] [Google Scholar]
- 78.Pommier Y, Redon C, Rao A, Seiler JA, Sordet O, Takemura H, Antony S, Meng LH, Liao ZY, Kohlhagen G, Zhang H, Kohn KW. Repair of and Checkpoint Response to Topoisomerase I-Mediated DNA Damage. Mutat Res. 2003;532:173–203. doi: 10.1016/j.mrfmmm.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 79.Rao A, Fan AM, Meng L, Doe CL, North PS, Hickson I, Pommier Y. Phosphorylation of BLM, dissociation for topoisomerase IIIα and colocalization with γ-H2AX after topoisomerase I-induced replication damage. Mol Cell Biol. 2005;25:8925–8937. doi: 10.1128/MCB.25.20.8925-8937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 81.Bjergbaek L, Cobb JA, Tsai-Pflugfelder M, Gasser SM. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. Embo J. 2005;24:405–417. doi: 10.1038/sj.emboj.7600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Imamura O, Fujita K, Itoh C, Takeda S, Furuichi Y, Matsumoto T. Werner and Bloom helicases are involved in DNA repair in a complementary fashion. Oncogene. 2002;21:954–963. doi: 10.1038/sj.onc.1205143. [DOI] [PubMed] [Google Scholar]
- 83.Van Den Boom V, Jaspers NG, Vermeulen W. When machines get stuck-obstructed RNA polymerase II: displacement, degradation or suicide. Bioessays. 2002;24:780–784. doi: 10.1002/bies.10150. [DOI] [PubMed] [Google Scholar]
- 84.Pommier Y, Jenkins J, Kohlhagen G, Leteurtre F. DNA recombinase activity of eukaryotic DNA topoisomerase I; Effects of camptothecin and other inhibitors. Mutat Res. 1995;337:135–145. doi: 10.1016/0921-8777(95)00019-g. [DOI] [PubMed] [Google Scholar]
- 85.Huang CC, Han CS, Yue XF, Shen CM, Wang SW, Wu FG, Xu B. Cytotoxicity and sister chromatid exchanges induced in vitro by six anticancer drugs developed in the People’s Republic of China. J Natl Cancer Inst. 1983;71:841–847. [PubMed] [Google Scholar]
- 86.Pinero J, Lopez Baena M, Ortiz T, Cortes F. Sister chromatid exchange induced by DNA topoisomerases poisons in late replicating heterochromatin: influence of inhibition of replication and transcription. Mutat Res. 1996;354:195–201. doi: 10.1016/0027-5107(96)00050-4. [DOI] [PubMed] [Google Scholar]
- 87.Fasullo M, Zeng L, Giallanza P. Enhanced stimulation of chromosomal translocations by radiomimetic DNA damaging agents and camptothecin in Saccharomyces cerevisiae rad9 checkpoint mutants. Mutat Res. 2004;547:123–132. doi: 10.1016/j.mrfmmm.2003.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topo I covalent complexes. Science. 1999;286:552–555. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 89.Yang SW, Burgin AB, Huizenga BN, Robertson CA, Yao KC, Nash HA. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci USA. 1996;93:11534–11539. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Interthal H, Pouliot JJ, Champoux JJ. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc Natl Acad Sci U S A. 2001;98:12009–12014. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32:267–272. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 92.Barthelmes HU, Grue P, Feineis S, Straub T, Boege F. Active DNA topoisomerase IIalpha is a component of the salt-stable centrosome core. J Biol Chem. 2000;275:38823–38830. doi: 10.1074/jbc.M007044200. [DOI] [PubMed] [Google Scholar]
- 93.Nivens MC, Felder T, Galloway AH, Pena MM, Pouliot JJ, Spencer HT. Engineered resistance to camptothecin and antifolates by retroviral coexpression of tyrosyl DNA phosphodiesterase-I and thymidylate synthase. Cancer Chemother Pharmacol. 2004;53:107–115. doi: 10.1007/s00280-003-0717-6. [DOI] [PubMed] [Google Scholar]
- 94.Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ. SCAN1 mutant Tdp1 accumulates the enzyme--DNA intermediate and causes camptothecin hypersensitivity. Embo J. 2005;24:2224–2233. doi: 10.1038/sj.emboj.7600694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou T, Lee JW, Tatavarthi H, Lupski JR, Valerie K, Povirk LF. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1) Nucleic Acids Res. 2005;33:289–297. doi: 10.1093/nar/gki170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pouliot JJ, Robertson CA, Nash HA. Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells. 2001;6:677–687. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 97.Liu C, Pouliot JJ, Nash HA. Repair of topoisomerase I covalent complexes in the absence of the tyrosyl-DNA phosphodiesterase Tdp1. Proc Natl Acad Sci U S A. 2002;99:14970–14975. doi: 10.1073/pnas.182557199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vance JR, Wilson TE. Yeast Tdp1 and Rad1-Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc Natl Acad Sci U S A. 2002;99:13669–13674. doi: 10.1073/pnas.202242599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deng C, Brown JA, You D, Brown JM. Multiple Endonucleases Function to Repair Covalent Topoisomerase I Complexes in Saccharomyces cerevisiae. Genetics. 2005 doi: 10.1534/genetics.104.028795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Inamdar KV, Pouliot JJ, Zhou T, Lees-Miller SP, Rasouli-Nia A, Povirk LF. Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double-strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J Biol Chem. 2002;277:27162–27168. doi: 10.1074/jbc.M204688200. [DOI] [PubMed] [Google Scholar]
- 101.Davies DR, Interthal H, Champoux JJ, Hol WG. The crystal structure of human tyrosyl-DNA phosphodiesterase, Tdp1. Structure (Camb) 2002;10:237–248. doi: 10.1016/s0969-2126(02)00707-4. [DOI] [PubMed] [Google Scholar]
- 102.Davies DR, Interthal H, Champoux JJ, Hol WG. Insights into substrate binding and catalytic mechanism of human tyrosyl-DNA phosphodiesterase (Tdp1) from vanadate and tungstate-inhibited structures. J Mol Biol. 2002;324:917–932. doi: 10.1016/s0022-2836(02)01154-3. [DOI] [PubMed] [Google Scholar]
- 103.Davies DR, Interthal H, Champoux JJ, Hol WG. Crystal structure of a transition state mimic for tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived Peptide. Chem Biol. 2003;10:139–147. doi: 10.1016/s1074-5521(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 104.Stuckey JA, Dixon JE. Crystal structure of a phospholipase D family member. Nat Struct Biol. 1999;6:278–284. doi: 10.1038/6716. [DOI] [PubMed] [Google Scholar]
- 105.Interthal H, Chen HJ, Champoux JJ. Human Tdp1 cleaves a broad spectrum of substrates including phosphoamide linkages. J Biol Chem. 2005;280:36518–36528. doi: 10.1074/jbc.M508898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Debethune L, Kohlhagen G, Grandas A, Pommier Y. Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2002;30:1198–1204. doi: 10.1093/nar/30.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Beidler DR, Chang JY, Zhou BS, Cheng YC. Camptothecin resistance involving steps subsequent to the formation of protein-linked DNA breaks in human camptothecin-resistant KB cell lines. Cancer Res. 1995;56:345. [PubMed] [Google Scholar]
- 108.Desai SD, Liu LF, Vazquez-Abad D, D’Arpa P. Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J Biol Chem. 1997;272:24159–24164. doi: 10.1074/jbc.272.39.24159. [DOI] [PubMed] [Google Scholar]
- 109.Raymond AC, Staker BL, Burgin AB., Jr Substrate specificity of tyrosyl-DNA phosphodiesterase I (Tdp1) J Biol Chem. 2005;280:22029–22035. doi: 10.1074/jbc.M502148200. [DOI] [PubMed] [Google Scholar]
- 110.Vance JR, Wilson TE. Uncoupling of 3′-phosphatase and 5′-kinase functions in budding yeast. Characterization of Saccharomyces cerevisiae DNA 3′-phosphatase (TPP1) J Biol Chem. 2001;276:15073–15081. doi: 10.1074/jbc.M011075200. [DOI] [PubMed] [Google Scholar]
- 111.Vance JR, Wilson TE. Repair of DNA strand breaks by the overlapping functions of lesion-specific and non-lesion-specific DNA 3′ phosphatases. Mol Cell Biol. 2001;21:7191–7198. doi: 10.1128/MCB.21.21.7191-7198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Meijer M, Karimi-Busheri F, Huang TY, Weinfeld M, Young D. Pnk1, a DNA kinase/phosphatase required for normal response to DNA damage by gamma-radiation or camptothecin in Schizosaccharomyces pombe. J Biol Chem. 2002;277:4050–4055. doi: 10.1074/jbc.M109383200. [DOI] [PubMed] [Google Scholar]
- 113.Karimi-Busheri F, Daly G, Robins P, Canas B, Pappin DJC, Sgouros J, Miller GG, Fakhrai H, Davis EM, Le Beau MM, Weinfeld M. Molecular Characterization of a Human DNA Kinase. J Biol Chem. 1999;274:24187–24194. doi: 10.1074/jbc.274.34.24187. [DOI] [PubMed] [Google Scholar]
- 114.Jilani A, Ramotar D, Slack C, Ong C, Yang XM, Scherer SW, Lasko DD. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J Biol Chem. 1999;274:24176–24186. doi: 10.1074/jbc.274.34.24176. [DOI] [PubMed] [Google Scholar]
- 115.Rasouli-Nia A, Karimi-Busheri F, Weinfeld M. Stable down-regulation of human polynucleotide kinase enhances spontaneous mutation frequency and sensitizes cells to genotoxic agents. Proc Natl Acad Sci U S A. 2004;101:6905–6910. doi: 10.1073/pnas.0400099101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 117.El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 118.Chatterjee S, Cheng MF, Trivedi D, Petzold SJ, Berger NA. Camptothecin hypersensitivity in poly(adenosine diphosphate-ribose) polymerase-deficient cell lines. Cancer Commun. 1989;1:389–394. doi: 10.3727/095535489820875129. [DOI] [PubMed] [Google Scholar]
- 119.Clements PM, Breslin C, Deeks ED, Byrd PJ, Ju L, Bieganowski P, Brenner C, Moreira MC, Taylor AM, Caldecott KW. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst) 2004;3:1493–1502. doi: 10.1016/j.dnarep.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 120.Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29:189–193. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- 121.Paulson HL, Miller VM. Breaks in coordination: DNA repair in inherited ataxia. Neuron. 2005;46:845–848. doi: 10.1016/j.neuron.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 122.Mosesso P, Piane M, Palitti F, Pepe G, Penna S, Chessa L. The novel human gene aprataxin is directly involved in DNA single-strand-break repair. Cell Mol Life Sci. 2005;62:485–491. doi: 10.1007/s00018-004-4441-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cheng TJ, Rey PG, Poon T, Kan CC. Kinetic studies of human tyrosyl-DNA phosphodiesterase, an enzyme in the topoisomerase I DNA repair pathway. Eur J Biochem. 2002;269:3697–3704. doi: 10.1046/j.1432-1033.2002.03059.x. [DOI] [PubMed] [Google Scholar]
- 124.Rideout MC, Raymond AC, Burgin AB., Jr Design and synthesis of fluorescent substrates for human tyrosyl-DNA phosphodiesterase I. Nucleic Acids Res. 2004;32:4657–4664. doi: 10.1093/nar/gkh796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 126.Bastin-Shanower SA, Fricke WM, Mullen JR, Brill SJ. The mechanism of mus81-mms4 cleavage site selection distinguishes it from the homologous endonuclease rad1-rad10. Mol Cell Biol. 2003;23:3487–3496. doi: 10.1128/MCB.23.10.3487-3496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. Requirement for Three Novel Protein Complexes in the Absence of the Sgs1 DNA Helicase in Saccharomyces cerevisiae. Genetics. 2001;157:103–118. doi: 10.1093/genetics/157.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fricke WM, Brill SJ. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Doe CL, Ahn JS, Dixon J, Whitby MC. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem. 2002;277:32753–32759. doi: 10.1074/jbc.M202120200. [DOI] [PubMed] [Google Scholar]
- 130.Ciccia A, Constantinou A, West SC. Identification and characterization of the human Mus81/Eme1 endonuclease. J Biol Chem. 2003 doi: 10.1074/jbc.M302882200. [DOI] [PubMed] [Google Scholar]
- 131.Bennett CB, Lewis LK, Karthikeyan G, Lobachev KS, Jin YH, Sterling JF, Snipe JR, Resnick MA. Genes required for ionizing radiation resistance in yeast. Nat Genet. 2001;29:426–434. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- 132.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 133.Zheng L, Zhou M, Chai Q, Parrish J, Xue D, Patrick SM, Turchi JJ, Yannone SM, Chen D, Shen B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005;6:83–89. doi: 10.1038/sj.embor.7400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Reid RJ, Fiorani P, Sugawara M, Bjornsti MA. CDC45 and DPB11 are required for processive DNA replication and resistance to DNA topoisomerase I-mediated DNA damage. Proc Natl Acad Sci U S A. 1999;96:11440–11445. doi: 10.1073/pnas.96.20.11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fiorani P, Reid RJ, Schepis A, Jacquiau HR, Guo H, Thimmaiah P, Benedetti P, Bjornsti MA. The deubiquitinating enzyme Doa4p protects cells from DNA topoisomerase I poisons. J Biol Chem. 2004;279:21271–21281. doi: 10.1074/jbc.M312338200. [DOI] [PubMed] [Google Scholar]
- 136.Jacquiau HR, van Waardenburg RC, Reid RJ, Woo MH, Guo H, Johnson ES, Bjornsti MA. Defects in SUMO (Small Ubiquitin-related Modifier) Conjugation and Deconjugation Alter Cell Sensitivity to DNA Topoisomerase I-induced DNA Damage. J Biol Chem. 2005;280:23566–23575. doi: 10.1074/jbc.M500947200. [DOI] [PubMed] [Google Scholar]
- 137.Dendouga N, Gao H, Moechars D, Janicot M, Vialard J, McGowan CH. Disruption of murine Mus81 increases genomic instability and DNA damage sensitivity but does not promote tumorigenesis. Mol Cell Biol. 2005;25:7569–7579. doi: 10.1128/MCB.25.17.7569-7579.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nelson WG, Kastan MB. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol. 1994;14:1815–1823. doi: 10.1128/mcb.14.3.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kohn KW, Pommier Y. Molecular interaction map of the p53 and Mdm2 logic elements, which control the Off-On switch of p53 in response to DNA damage. Biochem Biophys Res Commun. 2005;331:816–827. doi: 10.1016/j.bbrc.2005.03.186. [DOI] [PubMed] [Google Scholar]
- 140.Chen Y, Sanchez Y. Chk1 in the DNA damage response: conserved roles from yeasts to mammals. DNA Repair (Amst) 2004;3:1025–1032. doi: 10.1016/j.dnarep.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 141.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:893–897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 142.Ljungman M. Activation of DNA damage signaling. Mutat Res. 2005;577:203–216. doi: 10.1016/j.mrfmmm.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 143.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 144.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 145.Zachos G, Rainey MD, Gillespie DA. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. Embo J. 2003;22:713–723. doi: 10.1093/emboj/cdg060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- 148.Chini CC, Chen J. Claspin, a regulator of Chk1 in DNA replication stress pathway. DNA Repair (Amst) 2004;3:1033–1037. doi: 10.1016/j.dnarep.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 149.Sorensen CS, Syljuasen RG, Lukas J, Bartek J. ATR Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle. 2004;3:941–945. [PubMed] [Google Scholar]
- 150.Gottifredi V, Prives C. The S phase checkpoint: when the crowd meets at the fork. Semin Cell Dev Biol. 2005;16:355–368. doi: 10.1016/j.semcdb.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 151.Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J, Khanna KK. ATM and NBS1 dependent phosphorylation of CHK1 on S317 in response to IR. J Biol Chem. 2003;278:14806–14811. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- 152.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 153.Guerra B, Issinger OG, Wang JY. Modulation of human checkpoint kinase Chk1 by the regulatory beta-subunit of protein kinase CK2. Oncogene. 2003;22:4933–4942. doi: 10.1038/sj.onc.1206721. [DOI] [PubMed] [Google Scholar]
- 154.Shtivelman E, Sussman J, Stokoe D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr Biol. 2002;12:919–924. doi: 10.1016/s0960-9822(02)00843-6. [DOI] [PubMed] [Google Scholar]
- 155.Wan S, Capasso H, Walworth NC. The topoisomerase I poison camptothecin generates a Chk1-dependent DNA damage checkpoint signal in fission yeast. Yeast. 1999;15:821–828. doi: 10.1002/(SICI)1097-0061(199907)15:10A<821::AID-YEA422>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 156.Tsao YP, D’Arpa P, Liu LF. The involvement of active DNA synthesis in camptothecin-induced G2 arrest: altered regulation of p34cdc2/cyclin B. Cancer Res. 1992;52:1823–1829. [PubMed] [Google Scholar]
- 157.Wan S, Walworth NC. A novel genetic screen identifies checkpoint-defective alleles of Schizosaccharomyces pombe chk1. Curr Genet. 2001;38:299–306. doi: 10.1007/s002940000172. [DOI] [PubMed] [Google Scholar]
- 158.Dunaway S, Liu HY, Walworth NC. Interaction of 14-3-3 protein with Chk1 affects localization and checkpoint function. J Cell Sci. 2005;118:39–50. doi: 10.1242/jcs.01570. [DOI] [PubMed] [Google Scholar]
- 159.Chen L, Liu TH, Walworth NC. Association of Chk1 with 14.3.3 proteins is stimulated by DNA damage. Genes Develop. 1999;13:675–685. doi: 10.1101/gad.13.6.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Capasso H, Palermo C, Wan S, Rao H, John UP, O’Connell MJ, Walworth NC. Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J Cell Sci. 2002;115:4555–4564. doi: 10.1242/jcs.00133. [DOI] [PubMed] [Google Scholar]
- 161.Yin MB, Hapke G, Wu J, Azrak RG, Frank C, Wrzosek C, Rustum YM. Chk1 signaling pathways that mediated G(2)M checkpoint in relation to the cellular resistance to the novel topoisomerase I poison BNP1350. Biochem Biophys Res Commun. 2002;295:435–444. doi: 10.1016/s0006-291x(02)00683-6. [DOI] [PubMed] [Google Scholar]
- 162.Flatten K, Dai NT, Vroman BT, Loegering D, Erlichman C, Karnitz LM, Kaufmann SH. The role of checkpoint kinase 1 in sensitivity to topoisomerase I poisons. J Biol Chem. 2005;280:14349–14355. doi: 10.1074/jbc.M411890200. [DOI] [PubMed] [Google Scholar]
- 163.Cliby WA, Lewis KA, Lilly KK, Kaufmann SH. S Phase and G2 Arrests Induced by Topoisomerase I Poisons Are Dependent on ATR Kinase Function. J Biol Chem. 2002;277:1599–1606. doi: 10.1074/jbc.M106287200. [DOI] [PubMed] [Google Scholar]
- 164.Hapke G, Yin MB, Wu J, Frank C, Rustum YM. Phosphorylation of chk1 at serine-345 affected by topoisomerase I poison SN-38. Int J Oncol. 2002;21:1059–1066. [PubMed] [Google Scholar]
- 165.Wang JL, Wang X, Wang H, Iliakis G, Wang Y. CHK1-regulated S-phase checkpoint response reduces camptothecin cytotoxicity. Cell Cycle. 2002;1:267–272. [PubMed] [Google Scholar]
- 166.Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, Zhang H. Chk1 Mediates S and G2 Arrests through Cdc25A Degradation in Response to DNA-damaging Agents. J Biol Chem. 2003;278:21767–21773. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- 167.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 168.Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 169.Nomura M, Nomura N, Yamashita J. Geldanamycin-induced degradation of Chk1 is mediated by proteasome. Biochem Biophys Res Commun. 2005;335:900–905. doi: 10.1016/j.bbrc.2005.07.160. [DOI] [PubMed] [Google Scholar]
- 170.Xiao Z, Xue J, Sowin TJ, Rosenberg SH, Zhang H. A novel mechanism of checkpoint abrogation conferred by Chk1 downregulation. Oncogene. 2005;24:1403–1411. doi: 10.1038/sj.onc.1208309. [DOI] [PubMed] [Google Scholar]
- 171.Levesque AA, Kohn EA, Bresnick E, Eastman A. Distinct roles for p53 transactivation and repression in preventing UCN-01-mediated abrogation of DNA damage-induced arrest at S and G2 cell cycle checkpoints. Oncogene. 2005;24:3786–3796. doi: 10.1038/sj.onc.1208451. [DOI] [PubMed] [Google Scholar]
- 172.Tse AN, Schwartz GK. Potentiation of cytotoxicity of topoisomerase i poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res. 2004;64:6635–6644. doi: 10.1158/0008-5472.CAN-04-0841. [DOI] [PubMed] [Google Scholar]
- 173.Pommier Y, Sordet O, Rao A, Zhang H, Kohn KW. Targeting Chk2 kinase: molecular interaction maps and therapeutic rationale. Curr Pharm Des. 2005;11:2855–2872. doi: 10.2174/1381612054546716. [DOI] [PubMed] [Google Scholar]
- 174.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA Damage-Induced Activation of p53 by the Checkpoint Kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 175.Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, Nagashima K, Sawa H, Ikeda K, Motoyama N. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. Embo J. 2002;21:5195–5205. doi: 10.1093/emboj/cdf506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ahn J, Urist M, Prives C. The Chk2 protein kinase. DNA Repair (Amst) 2004;3:1039–1047. doi: 10.1016/j.dnarep.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 177.Ahn JY, Schwartz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–5936. [PubMed] [Google Scholar]
- 178.Helt CE, Cliby WA, Keng PC, Bambara RA, O’Reilly MA. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem. 2005;280:1186–1192. doi: 10.1074/jbc.M410873200. [DOI] [PubMed] [Google Scholar]
- 179.Li J, Stern DF. Regulation of CHK2 by DNA-dependent protein kinase. J Biol Chem. 2005;280:12041–12050. doi: 10.1074/jbc.M412445200. [DOI] [PubMed] [Google Scholar]
- 180.Bahassiel M, Conn CW, Myer DL, Hennigan RF, McGowan CH, Sanchez Y, Stambrook PJ. Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene. 2002;21:6633–6640. doi: 10.1038/sj.onc.1205850. [DOI] [PubMed] [Google Scholar]
- 181.Myer DL, Bahassiel M, Stambrook PJ. The Plk3-Cdc25 circuit. Oncogene. 2005;24:299–305. doi: 10.1038/sj.onc.1208278. [DOI] [PubMed] [Google Scholar]
- 182.Tsvetkov L, Stern DF. Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage. Cell Cycle. 2005;4:166–171. doi: 10.4161/cc.4.1.1348. [DOI] [PubMed] [Google Scholar]
- 183.Tsvetkov L, Xu X, Li J, Stern DF. Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. J Biol Chem. 2003;278:8468–8475. doi: 10.1074/jbc.M211202200. [DOI] [PubMed] [Google Scholar]
- 184.Tsvetkov LM, Tsekova RT, Xu X, Stern DF. The Plk1 Polo box domain mediates a cell cycle and DNA damage regulated interaction with Chk2. Cell Cycle. 2005;4:609–617. [PubMed] [Google Scholar]
- 185.Wei JH, Chou YF, Ou YH, Yeh YH, Tyan SW, Sun TP, Shen CY, Shieh SY. TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. J Biol Chem. 2005;280:7748–7757. doi: 10.1074/jbc.M410152200. [DOI] [PubMed] [Google Scholar]
- 186.Ahn JY, Li X, Davis HL, Canman CE. Phosphorylation of threonine 68 promotes oligomerization and autophosphorylation of the Chk2 protein kinase via the forkhead-associated domain. J Biol Chem. 2002;277:19389–19395. doi: 10.1074/jbc.M200822200. [DOI] [PubMed] [Google Scholar]
- 187.Schwarz JK, Lovly CM, Piwnica-Worms H. Regulation of the Chk2 Protein Kinase by Oligomerization-Mediated cis- and trans-Phosphorylation. Mol Cancer Res. 2003;1:598–609. [PubMed] [Google Scholar]
- 188.Yu Q, La Rose JH, Zhang H, Pommier Y. Inhibition of Chk2 activity and radiation-induced p53 elevation by the cell cycle checkpoint abrogator 7-hydroxystaurosporine (UCN-01) Proc Am Assoc Cancer Res. 2001;42:800. [Google Scholar]
- 189.Keramaris E, Hirao A, Slack RS, Mak TW, Park DS. Ataxia telangiectasia-mutated protein can regulate p53 and neuronal death independent of Chk2 in response to DNA damage. J Biol Chem. 2003;278:37782–37789. doi: 10.1074/jbc.M304049200. [DOI] [PubMed] [Google Scholar]
- 190.Chaturvedi P, Eng WK, Zhu Y, Mattern MR, Mishra R, Hurle MR, Zhang X, Annan RS, Lu Q, Faucette LF, Scott GF, Li X, Carr SA, Johnson RK, Winkler JD, Zhou BB. Mammalian Chk2 is a downstream effector of the ATM-dependent DNA damage checkpoint pathway. Oncogene. 1999;18:4047–4054. doi: 10.1038/sj.onc.1202925. [DOI] [PubMed] [Google Scholar]
- 191.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 192.Agner J, Falck J, Lukas J, Bartek J. Differential impact of diverse anticancer chemotherapeutics on the Cdc25A-degradation checkpoint pathway. Exp Cell Res. 2005;302:162–169. doi: 10.1016/j.yexcr.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 193.Wang X, Khadpe J, Hu B, Iliakis G, Wang Y. An overactivated ATR/CHK1 pathway is responsible for the prolonged G2 accumulation in irradiated AT cells. J Biol Chem. 2003;278:30869–30874. doi: 10.1074/jbc.M301876200. [DOI] [PubMed] [Google Scholar]
- 194.Goulaouic H, Roulon T, Flamand O, Grondard L, Lavelle F, Riou JF. Purification and characterization of human DNA topoisomerase IIIα. Nucleic Acids Res. 1999;27:2443–2450. doi: 10.1093/nar/27.12.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rosenthal DS, Simbulan-Rosenthal CM, Liu WF, Velena A, Anderson D, Benton B, Wang ZQ, Smith W, Ray R, Smulson ME. PARP determines the mode of cell death in skin fibroblasts, but not keratinocytes, exposed to sulfur mustard. J Invest Dermatol. 2001;117:1566–1573. doi: 10.1046/j.0022-202x.2001.01578.x. [DOI] [PubMed] [Google Scholar]
- 196.Pommier Y, Pourquier P, Fan Y, Strumberg D. Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim Biophys Acta. 1998;1400:83–105. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- 197.Pourquier P, Takebayashi Y, Urasaki Y, Gioffre C, Kohlhagen G, Pommier Y. Induction of topoisomerase I cleavage complexes by 1-β-D-arabinofuranosylcytosine (Ara-C) in vitro and in ara-C-treated cells. Proc Natl Acad Sci USA. 2000;97:1885–1890. doi: 10.1073/pnas.97.4.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chrencik JE, Burgin AB, Pommier Y, Stewart L, Redinbo MR. Structural impact of the leukemia drug Ara-C on the covalent human topoisomerase I DNA complex. J Biol Chem. 2003;278:12461–12466. doi: 10.1074/jbc.M212930200. [DOI] [PubMed] [Google Scholar]
- 199.Pourquier P, Gioffre C, Kohlhagen G, Urasaki Y, Goldwasser F, Hertel LW, Yu S, Pon RT, Gmeiner WH, Pommier Y. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin Cancer Res. 2002;8:2499–2504. [PubMed] [Google Scholar]
- 200.Leteurtre F, Kohlhagen G, Fesen MR, Tanizawa A, Kohn KW, Pommier Y. Effects of DNA methylation on topoisomerase I and II cleavage activities. J Biol Chem. 1994;269:7893–7900. [PubMed] [Google Scholar]
- 201.Antony S, Arimondo PB, Sun JS, Pommier Y. Position- and orientation-specific enhancement of topoisomerase I cleavage complexes by triplex DNA structures. Nucleic Acids Res. 2004;32:5163–5173. doi: 10.1093/nar/gkh847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lanza A, Tornatelli S, Rodolfo C, Scanavini MC, Pedrini AM. Human DNA topoisomerase I-mediated cleavages stimulated by ultraviolet light-induced DNA damage. J Biol Chem. 1996;271:6978–6986. doi: 10.1074/jbc.271.12.6978. [DOI] [PubMed] [Google Scholar]
- 203.Subramanian D, Rosenstein BS, Muller MT. Ultraviolet-induced DNA damage stimulates topoisomerase I-DNA complex formation in vivo: possible relationship with DNA repair. Cancer Res. 1998;58:976–984. [PubMed] [Google Scholar]
- 204.Pourquier P, Waltman JL, Urasaki Y, Loktionova NA, Pegg AE, Nitiss JL, Pommier Y. Topoisomerase I-mediated cytotoxicity of N-methyl-N′-nitro-N- nitrosoguanidine: trapping of topoisomerase I by the O6-methylguanine. Cancer Res. 2001;61:53–58. [PubMed] [Google Scholar]
- 205.Pommier Y, Laco GS, Kohlhagen G, Sayer JM, Kroth H, Jerina DM. Position-specific trapping of topoisomerase I-DNA cleavage complexes by intercalated benzo[a]-pyrene diol epoxide adducts at the 6-amino group of adenine. Proc Natl Acad Sci U S A. 2000;97:10739–10744. doi: 10.1073/pnas.190312697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pommier Y, Kohlhagen G, Pourquier P, Sayer JM, Kroth H, Jerina DM. Benzo[a]pyrene epoxide adducts in DNA are potent inhibitors of a normal topoisomerase I cleavage site and powerful inducers of other topoisomerase I cleavages. Proc Natl Acad Sci USA. 2000;97:2040–2045. doi: 10.1073/pnas.040397497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pommier Y, Kohlhagen G, Laco GS, Kroth H, Sayer JM, Jerina DM. Different effects on human topoisomerase I by minor groove and intercalated deoxyguanosine adducts derived from two polycyclic aromatic hydrocarbon diol epoxides at or near a normal cleavage site. J Biol Chem. 2002;277:13666–13672. doi: 10.1074/jbc.M200209200. [DOI] [PubMed] [Google Scholar]
- 208.Pourquier P, Bjornsti MA, Pommier Y. Induction of topoisomerase I cleavage complexes by the vinyl chloride adduct, 1,N6-ethenoadenine. J Biol Chem. 1998;273:27245–27249. doi: 10.1074/jbc.273.42.27245. [DOI] [PubMed] [Google Scholar]
- 209.Antony S, Theruvathu JA, Brooks PJ, Lesher DT, Redinbo M, Pommier Y. Enhancement of camptothecin-induced topoisomerase I cleavage complexes by the acetaldehyde adduct N2-ethyl-2′-deoxyguanosine. Nucleic Acids Res. 2004;32:5685–5692. doi: 10.1093/nar/gkh902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Moser BA, Brondello JM, Baber-Furnari B, Russell P. Mechanism of caffeine-induced checkpoint override in fission yeast. Mol Cell Biol. 2000;20:4288–4294. doi: 10.1128/mcb.20.12.4288-4294.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Smith PJ, Makinson TA, Watson JV. Enhanced sensitivity to camptothecin in ataxia telangiectasia cells and its relationship with the expression of DNA topoisomerase I. Int J Radiat Biol. 1989;55:217–231. doi: 10.1080/09553008914550271. [DOI] [PubMed] [Google Scholar]
- 212.Johnson MA, Jones NJ. The isolation and genetic analysis of V79-derived etoposide sensitive Chinese hamster cell mutants: two new complementation groups of etoposide sensitive mutants. Mutation Res. 1999;435:271–282. doi: 10.1016/s0921-8777(99)00055-5. [DOI] [PubMed] [Google Scholar]
- 213.Jones NJ, Ellard S, Waters R, Parry EM. Cellular and chromosomal hypersensitivity to DNA crosslinking agents and topoisomerase inhibitors in the radiosensitive Chinese hamster irs mutants: phenotypic similarities to ataxia telangiectasia and Fanconi’s anaemia cells. Carcinogenesis. 1993;14:2487–2494. doi: 10.1093/carcin/14.12.2487. [DOI] [PubMed] [Google Scholar]
- 214.Johnson MA, Bryant PE, Jones NJ. Isolation of camptothecin-sensitive Chinese hamster cell mutants: phenotypic heterogeneity within the ataxia telangiectasia-like XRCC8 (irs2) complementation group. Mutagenesis. 2000;15:367–374. doi: 10.1093/mutage/15.4.367. [DOI] [PubMed] [Google Scholar]
- 215.Walton MI, Whysong D, O’Connor PM, Hockenbery D, Korsmeyer SJ, Kohn KW. Constitutive expression of human Bcl-2 modulates nitrogen mustard and camptothecin induced apoptosis. Cancer Res. 1993;53:1853–1861. [PubMed] [Google Scholar]
- 216.Fedier A, Steiner RA, Schwarz VA, Lenherr L, Haller U, Fink D. The effect of loss of Brca1 on the sensitivity to anticancer agents in p53-deficient cells. Int J Oncol. 2003;22:1169–1173. [PubMed] [Google Scholar]
- 217.Rahden-Staron II, Szumilo M, Grosicka E, Kraakman Van Der Zwet M, Zdzienicka MZ. Defective Brca2 influences topoisomerase I activity in mammalian cells. Acta Biochim Pol. 2003;50:139–144. [PubMed] [Google Scholar]
- 218.Yu Q, Rose JH, Zhang H, Pommier Y. Antisense inhibition of Chk2/hCds1 expression attenuates DNA damage- induced S and G2 checkpoints and enhances apoptotic activity in HEK-293 cells. FEBS Lett. 2001;505:7–12. doi: 10.1016/s0014-5793(01)02756-9. [DOI] [PubMed] [Google Scholar]
- 219.Hinz JM, Helleday T, Meuth M. Reduced apoptotic response to camptothecin in CHO cells deficient in XRCC3. Carcinogenesis. 2003;24:249–253. doi: 10.1093/carcin/24.2.249. [DOI] [PubMed] [Google Scholar]
- 220.Culmsee C, Bondada S, Mattson MP. Hippocampal neurons of mice deficient in DNA-dependent protein kinase exhibit increased vulnerability to DNA damage, oxidative stress and excitotoxicity. Brain Res Mol Brain Res. 2001;87:257–262. doi: 10.1016/s0169-328x(01)00008-0. [DOI] [PubMed] [Google Scholar]
- 221.Kraakman-van der Zwet M, Overkamp WJ, Friedl AA, Klein B, Verhaegh GW, Jaspers NG, Midro AT, Eckardt-Schupp F, Lohman PH, Zdzienicka MZ. Immortalization and characterization of Nijmegen Breakage syndrome fibroblasts. Mutat Res. 1999;434:17–27. doi: 10.1016/s0921-8777(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 222.Chatterjee S, Cheng MF, Berger NA. Hypersensitivity to clinically useful alkylating agents and radiations in poly(ADP-ribose) polymerase-deficient cell lines. Cancer Communication. 1990;2:401–407. doi: 10.3727/095535490820873958. [DOI] [PubMed] [Google Scholar]
- 223.Godthelp BC, Wiegant WW, van Duijn-Goedhart A, Scharer OD, van Buul PP, Kanaar R, Zdzienicka MZ. Mammalian Rad51C contributes to DNA cross-link resistance, sister chromatid cohesion and genomic stability. Nucleic Acids Res. 2002;30:2172–2182. doi: 10.1093/nar/30.10.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J Biol Chem. 2004;279:55618–55625. doi: 10.1074/jbc.M405042200. [DOI] [PubMed] [Google Scholar]
- 225.Gupta M, Fan S, Zhan Q, Kohn KW, O’Connor PM, Pommier Y. Inactivation of p53 increases the cytotoxicity of camptothecin in human colon HCT116 and breast MCF-7 cancer cells. Clin Cancer Res. 1997;3:1653–1660. [PubMed] [Google Scholar]
- 226.Han Z, Wei W, Dunaway S, Darnowski JW, Calabresi P, Sedivy J, Hendrickson EA, Balan KV, Pantazis P, Wyche JH. Role of p21 in Apoptosis and Senescence of Human Colon Cancer Cells Treated with Camptothecin. J Biol Chem. 2002;277:17154–17160. doi: 10.1074/jbc.M112401200. [DOI] [PubMed] [Google Scholar]
- 227.Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of proliferative capacity. Proc Natl Acad Sci USA. 1998;95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Poot M, Gollahon KA, Rabinovitch PS. Werner syndrome lymphoblastoid cells are sensitive to camptothecin- induced apoptosis in S-phase. Hum Genet. 1999;104:10–14. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- 229.Pichierri P, Franchitto A, Mosesso P, Palitti F. Werner’s syndrome cell lines are hypersensitive to camptothecin-induced chromosomal damage. Mutat Res. 2000;456:45–57. doi: 10.1016/s0027-5107(00)00109-3. [DOI] [PubMed] [Google Scholar]
- 230.Caldecott K, Jeggo P. Cross-sensitivity of gamma-ray-sensitive hamster mutants to cross- linking agents. Mutat Res. 1991;255:111–121. doi: 10.1016/0921-8777(91)90046-r. [DOI] [PubMed] [Google Scholar]
- 231.Barrows LR, Holden JA, Anderson M, D’Arpa P. The CHO XRCC1 mutant, EM9, deficient in DNA ligase III activity, exhibits hypersensitivity to camptothecin independent of DNA replication. Mutat Res. 1998;408:103–110. doi: 10.1016/s0921-8777(98)00022-6. [DOI] [PubMed] [Google Scholar]
- 232.Park SY, Lam W, Cheng YC. X-ray repair cross-complementing gene I protein plays an important role in camptothecin resistance. Cancer Res. 2002;62:459–465. [PubMed] [Google Scholar]
- 233.Gueven N, Becherel OJ, Kijas AW, Chen P, Howe O, Rudolph JH, Gatti R, Date H, Onodera O, Taucher-Scholz G, Lavin MF. Aprataxin, a novel protein that protects against genotoxic stress. Hum Mol Genet. 2004;13:1081–1093. doi: 10.1093/hmg/ddh122. [DOI] [PubMed] [Google Scholar]
- 234.Thacker J, Ganesh AN. DNA-break repair, radioresistance of DNA synthesis, and camptothecin sensitivity in the radiation-sensitive irs mutants: comparisons to ataxia-telangiectasia cells. Mutat Res. 1990;235:49–58. doi: 10.1016/0921-8777(90)90057-c. [DOI] [PubMed] [Google Scholar]
- 235.Nitiss J, Wang JC. DNA topoisomerase-targeting antitumor drugs can be studied in yeast. Proc Natl Acad Sci USA. 1988;85:7501–7505. doi: 10.1073/pnas.85.20.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Simon JA, Szankasi P, Nguyen DK, Ludlow C, Dunstan HM, Roberts CJ, Jensen EL, Hartwell LH, Friend SH. Differential toxicities of anticancer agents among DNA repair and checkpoint mutants of Saccharomyces cerevisiae. Cancer Res. 2000;60:328–333. [PubMed] [Google Scholar]
- 237.Redon C, Pilch D, Rogakou E, Orr AH, Lowndes NF, Bonner WM. Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep. 2003;4:1–7. doi: 10.1038/sj.embor.embor871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hryciw T, Tang M, Fontanie T, Xiao W. MMS1 protects against replication-dependent DNA damage in Saccharomyces cerevisiae. Mol Genet Genomics. 2002;266:848–857. doi: 10.1007/s00438-001-0605-x. [DOI] [PubMed] [Google Scholar]
- 239.Zhang H, Siede W. Validation of a novel assay for checkpoint responses: characterization of camptothecin derivatives in Saccharomyces cerevisiae. Mutat Res. 2003;527:37–48. doi: 10.1016/s0027-5107(03)00074-5. [DOI] [PubMed] [Google Scholar]
- 240.Walowsky C, Fitzhugh DJ, Castaño IB, Ju JY, Levin NA, Christman MF. The Topoisomerase-related Function Gene TRF4 Affects Cellular Sensitivity to the Antitumor Agent Camptothecin. J Biol Chem. 1999;274:7302–7308. doi: 10.1074/jbc.274.11.7302. [DOI] [PubMed] [Google Scholar]
- 241.Scheller J, Schurer A, Rudolph C, Hettwer S, Kramer W. MPH1, a yeast gene encoding a DEAH protein, plays a role in protection of the genome from spontaneous and chemically induced damage. Genetics. 2000;155:1069–1081. doi: 10.1093/genetics/155.3.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Redon C, Pilch DR, Bonner WM. Genetic analysis of Saccharomyces cerevisiae H2A serine 129 mutant suggests a functional relationship between H2A and the sister chromatid cohesion partners Csm3-Tof1 for the repair of Topoisomerase I-induced DNA damage. Genetics. 2005 doi: 10.1534/genetics.105.046128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–415. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 244.Choy JS, Kron SJ. NuA4 subunit Yng2 function in intra-S-phase DNA damage response. Mol Cell Biol. 2002;22:8215–8225. doi: 10.1128/MCB.22.23.8215-8225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Lewis LK, Karthikeyan G, Cassiano J, Resnick MA. Reduction of nucleosome assembly during new DNA synthesis impairs both major pathways of double-strand break repair. Nucleic Acids Res. 2005;33:4928–4939. doi: 10.1093/nar/gki806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Fiorani P, Bjornsti MA. Mechanisms of DNA topoisomerase I-induced cell killing in the yeast Saccharomyces cerevisiae. Ann N Y Acad Sci. 2000;922:65–75. doi: 10.1111/j.1749-6632.2000.tb07026.x. [DOI] [PubMed] [Google Scholar]
- 247.Doe CL, Whitby MC. The involvement of Srs2 in post-replication repair and homologous recombination in fission yeast. Nucleic Acids Res. 2004;32:1480–1491. doi: 10.1093/nar/gkh317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Doe CL, Osman F, Dixon J, Whitby MC. DNA repair by a Rad22-Mus81-dependent pathway that is independent of Rhp51. Nucleic Acids Res. 2004;32:5570–5581. doi: 10.1093/nar/gkh853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Collura A, Blaisonneau J, Baldacci G, Francesconi S. The fission yeast Crb2/Chk1 pathway coordinates the DNA damage and spindle checkpoint in response to replication stress induced by topoisomerase I inhibitor. Mol Cell Biol. 2005;25:7889–7899. doi: 10.1128/MCB.25.17.7889-7899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Francesconi S, Smeets M, Grenon M, Tillit J, Blaisonneau J, Baldacci G. Fission yeast chk1 mutants show distinct responses to different types of DNA damaging treatments. Genes Cells. 2002;7:663–673. doi: 10.1046/j.1365-2443.2002.00552.x. [DOI] [PubMed] [Google Scholar]
- 251.Dalgaard JZ, Klar AJ. swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell. 2000;102:745–751. doi: 10.1016/s0092-8674(00)00063-5. [DOI] [PubMed] [Google Scholar]