

Summary
Background
Cell growth arrest and autophagy are required for autophagic cell death in Drosophila. Maintenance of growth by expression of either activated Ras, Dp110, or Akt is sufficient to inhibit autophagy and cell death in fly salivary glands, but the mechanism that controls growth arrest is unknown. While the Warts (Wts) tumor-suppressor has emerged as a critical regulator of tissue growth in animals, it is not clear how this signaling pathway controls cell growth.
Results
Here we show that genes in the Wts pathway are required for salivary gland degradation, and that wts mutants have defects in cell growth arrest, caspase activity, and autophagy. Expression of Atg1, a regulator of autophagy, in salivary glands is sufficient to rescue wts mutant salivary gland destruction. Surprisingly, expression of Yorkie (Yki) and Scalloped (Sd) in salivary glands fails to phenocopy wts mutants. By contrast, mis-expression of the Yki target microRNA bantam was able to inhibit salivary gland cell death, even though mutations in bantam fail to suppress the wts mutant salivary gland persistence phenotype. Significantly, wts mutant salivary glands possess altered phosphoinositide signaling and phospho-Akt localization, and decreased function of the class I PI3K pathway genes chico and TOR suppressed wts defects in autophagic cell death.
Conclusions
These data provide the first evidence that the Wts tumor-suppressor pathway regulates autophagy and autophagic cell death. Our data suggest that the previously described Wts pathway involving Yki, Sd, and bantam fails to function in salivary glands, and that Wts regulates salivary gland cell death in a PI3K-dependent manner involving Chico and TOR.
Keywords: autophagy, cell death, apoptosis, growth, Drosophila, development
Introduction
Cell growth, division, and death are important determinants of tissue and animal size [1], and disruption in their balance can lead to physiological disorders including cancer [2]. While the mechanisms that integrate cell division checkpoints with cell death are relatively well studied [3], less is known about the relationship between cell growth and death.
Apoptosis and autophagic cell death are the two most prominent morphological forms of cell death that occur during animal development [4, 5]. The mechanisms that regulate apoptosis have been extensively studied, but far less is known about autophagic cell death. Drosophila larval salivary glands are an excellent system for investigating autophagic cell death during development. A rise in the steroid hormone 20-hydroxyecdysone (ecdysone) 12 hours after puparium formation triggers future adult head eversion, salivary gland cell death, and the synchronized degradation of salivary gland cells is completed by 16 hours after puparium formation [6]. Both caspases and autophagy are induced following the rise in ecdysone that triggers cell death [7, 8]. Caspases and autophagy function in an additive manner in dying salivary glands, as evidenced by the finding that the combined inhibition of both caspases and autophagy results in a stronger salivary gland persistence phenotype than inhibition of either caspases or autophagy alone [9]. Cell growth stops prior to salivary gland cell death, and maintenance of growth is sufficient to suppress both autophagy and degradation of this tissue [9], but the mechanism that regulates this growth arrest is not clear.
The insulin-triggered class I phosphoinositide 3-kinase (PI3K) pathway is highly conserved and regulates cell and tissue growth [10]. Binding of insulin to the insulin receptor leads to the phosphorylation of the receptor and the insulin receptor substrate protein Chico [11]. This phosphorylation cascade activates the catalytic subunit Dp110 of the class I PI3K pathway [10, 12]. Activated Dp110 converts phosphatidylinositol-4, 5-P (2) to the second messenger phosphatidylinositol-3, 4, 5-P (3) (PIP3). The pleckstrin homology (PH) domain of Akt interacts with PIP3 on the cell membrane and activates the downstream effector target-of-rapamycin (TOR), an evolutionarily conserved kinase [13]. TOR influences a wide range of cellular processes such as protein translation, cell cycle progression, growth and autophagy. Although activation of the class I PI3K pathway by expression of either activated Ras, Dp110, or Akt is sufficient to inhibit autophagy and degradation of salivary glands [9], the mechanism that is responsible for the regulation of PI3K-dependent growth arrest in this tissue is not fully understood.
Recent studies in Drosophila have identified the evolutionarily conserved Warts (Wts) signaling pathway as an important regulator of tissue growth. Wts, Fat, Merlin (Mer), Expanded (Ex), Hippo (Hpo), Salvador (Sav), and Mats are members of a kinase cascade that negatively regulates tissue growth [14–27]. Mutations in any of these recessive genes causes increased cell division, and several of these mutants exhibit decreased cell death. These Wts pathway defects in cell division and cell death are caused by altered levels of the cell cycle regulator Cyclin E and the inhibitor of apoptosis DIAP1 [20]. Wts, also known as the large tumor suppressor Lats, encodes a NDR family kinase that phosphorylates the transcriptional coactivator Yorkie (Yki) [28], and inactivates Yki by exclusion from the nucleus [29]. Yki, the orthologue of mammalian Yes associated protein Yap, is a positive regulator of growth, and over-expression of Yki results in overgrowth phenotypes in tissues that resemble loss-of-function mutations in members of the Wts pathway [28]. Yki functions with the TEAD/TEF DNA binding family member protein Scalloped (Sd) to regulate transcription of the inhibitor of apoptosis diap1 [30, 31], and presumably other Wts signaling targets, including the microRNA bantam and the cell cycle regulator cyclin E [14, 15, 18, 32, 33]. While mutations in Wts pathway genes, and over-expression of Yki, cause tissue overgrowth, it is not clear how this pathway influences cell growth, and if the PI3K pathway is influenced by this tumor suppressor pathway.
Here we show that wts mutant salivary glands fail to arrest growth, exhibit decreased caspase activity, have attenuated autophagy, and are not degraded. Although previous studies indicate that expression of Yki phenocopies wts mutants, this was not the case in salivary glands. By contrast, expression of the Yki target bantam was sufficient to induce cell growth and inhibit salivary gland cell death. However, bantam loss-of-function mutations failed to suppress the wts mutant salivary gland persistence phenotype. These data suggest that Wts is capable of regulating growth and autophagy independent of Yki. wts mutants had altered PI3K markers and required the function of TOR and chico to inhibit salivary gland degradation. These data suggest that Wts influences the PI3K signaling pathway, growth, and autophagy in a manner that is distinct from its regulation of Yki.
Results
Wts, Sav and Mats are required for salivary gland degradation
Wts was identified as a protein that is expressed during autophagic cell death of Drosophila larval salivary glands using a high throughput proteomics approach [34]. This was surprising, as we failed to detect wts RNA using DNA microarrays [35]. Therefore, we investigated whether Wts is present in salivary glands, and determined that it is constitutively expressed at stages preceding and following the rise in ecdysone that triggers autophagic cell death (Figure 1A). Significantly, 2 forms of Hpo are expressed during stages preceding salivary gland cell death, suggesting that phosphorylated Hpo is present in these cells and that this signaling pathway is activated (Figure 1A).
Figure 1. Wts signaling pathway is active and required for salivary gland cell death.
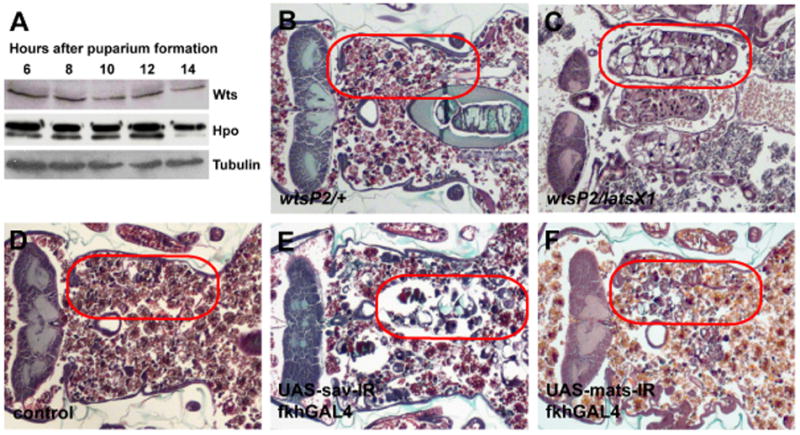
(A) Immunoblot showing that Wts and Hpo are present in salivary glands at 6, 8, 10, 12 and 14 hours after puparium formation. (B–F) Paraffin sections of pupae 12 hours after head eversion. (B and D) Salivary glands are completely degraded in control animals. (C) Loss of wts in wtsP2/latsX1 inhibits salivary gland degradation. Expression of sav-IR (E) and mats-IR (F) in a salivary specific manner causes incomplete degradation of salivary glands. Red circles outline the persistent salivary gland tissue in the pupae.
We had previously shown that animals that are homozygous for the hypomorphic wtsP2 allele, that is caused by a P element insertion [22], are defective in salivary gland cell death [34]. Since strong loss-of-function wts mutants are homozygous lethal prior to the stage of salivary gland cell death, we tested if the wtsP2 allele in combination with the stronger X-ray-induced latsx1 allele [17] caused a defect in salivary gland degradation. In control animals, ecdysone-triggered head eversion occurs 12 hours after puparium formation and salivary glands are absent 24 hours after puparium formation (12 hours after head eversion) (Figures 1B). By contrast, wtsP2/latsX1 mutants fail to degrade salivary glands by 12 hours after head eversion (Figure 1C). Previous studies indicated that hpo is required to complete degradation of salivary glands [34]. Therefore, we tested if other Wts pathway components are required for salivary gland degradation. While control animals had no salivary gland remnants 24 hours after puparium formation (Figure 1D), knock-down of sav by tissue-specific expression of RNAi (sav-IR) inhibited the degradation with 58% of the animals having incompletely degraded vacuolated cell fragments (Figure 1E). Similarly, knock-down of mats by tissue-specific expression of RNAi (mats-IR) inhibited the degradation with 62% of the animals possessing incompletely degraded salivary gland cell fragments (Figure 1F). These data indicate that wts, sav and mats are required for degradation of salivary glands in Drosophila.
wts influences caspase activity, autophagy and cell growth
Previous studies have shown that both caspases and autophagy are induced prior to and function in salivary gland autophagic cell death [7–9]. Therefore, we investigated whether caspases are altered in wts mutant salivary glands. Caspase-dependent DNA fragmentation was detected by TUNEL assay in both control wtsP2/wild-type and wtsP2/wtsP2 mutant salivary glands 1.5 hours after head eversion even though wts mutant salivary glands fail to degrade (Figures 2A and 2B). Although wts mutant salivary glands appeared to possess approximately half as many TUNEL-positive nuclei compared to controls, the qualitative nature of this assay limits our ability to make strong conclusions about caspase activity based on this approach. Loss of nuclear lamins and increased levels of cleaved caspase-3 were also observed in both control and mutant salivary glands (data not shown). In addition, we detected caspase-3-like activity by cleavage of the caspase substrate DEVD-AMC in both wtsP2/wild-type control and wtsP2/wtsP2 mutant animals 4 hours after puparium formation, although caspase-3-like activity was reduced in homozygous mutant animals (Figure 2C). Together, these data indicate that caspases are present but reduced in homozygous wtsP2mutant animals compared to controls.
Figure 2. Caspases are reduced in wts mutants, and DIAP1-induced inhibition of salivary gland degradation is enhanced by reduced Atg1 function.
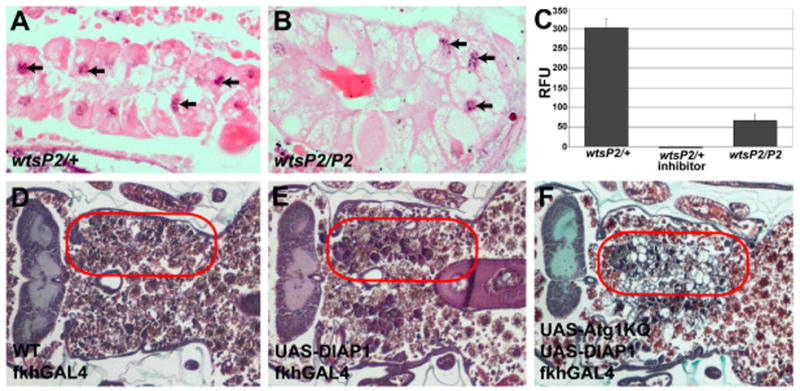
(A–B) Visualization of DNA fragmentation by TUNEL assay. TUNEL-positive nuclei are indicated by black arrowheads in both control wtsP2/+ (A) and wts homozygous mutant wtsP2/P2 (B) salivary glands. (C) Cleavage of the caspase-3-like substrate Z-DEVD-AMC was measured in whole pupae staged 4 hours after puparium formation in control (wtsP2/+), control plus Ac-DEVD-CHO inhibitor, and experimental wtsP2/P2 mutant pupae. Data are presented as the mean ± SE, n = 3/treatment. (D–F) Paraffin sections 24 hours after puparium formation. (D) Salivary glands are completely degraded in control animals. (E) Partial degradation occurs in salivary glands expressing DIAP1. (F) Coexpression of dominant negative Atg1KQ and DIAP1 increases the persistence of salivary glands. Red circles outline the persistent salivary gland tissue in the pupae.
The inhibitor of apoptosis DIAP1 suppresses caspases, functions downstream of wts, and DIAP1 was previously shown to be elevated in wts mutant cells [14, 15, 20, 28]. Therefore, we tested if expression of DIAP1 is sufficient to inhibit salivary gland autophagic cell death. In control animals, salivary glands are completely degraded 24 hours after puparium formation (Figure 2D). By contrast, salivary glands are partly degraded in DIAP1-expressing salivary glands (Figure 2E), which is consistent with previous studies indicating that salivary glands are partially degraded when caspases are blocked by either expression of p35, or caspase loss-of-function mutants [7–9]. Significantly, combined inhibition of caspases and autophagy by co-expression of DIAP1 and a dominant negative form of Atg1, Atg1KQ, resulted in an almost complete inhibition of salivary gland cell death (Figure 2F). These results support the conclusion that both caspases and autophagy function in an additive manner during cell death of salivary glands.
The number of autophagosomes increases following the rise in steroid that triggers autophagic cell death of salivary glands, and autophagy is required for degradation of this tissue [7, 9]. Therefore, we analyzed the number of autophagosomes in dying salivary glands of control and wts mutants using the autophagy reporter GFP-LC3. Interestingly, the number of GFP-LC3 puncta were reduced in homozygous wtsP2 mutant salivary glands compared to control wtsP2/wild-type salivary glands (Figure 3A – 3C). Previous studies have shown that expression of Atg1 is sufficient to induce autophagy [9, 36], so we tested if expression of Atg1 in wts mutant salivary glands is sufficient to suppress the wts mutant salivary gland degradation phenotype. While control wtsP2/wtsP2 mutant animals all posses salivary glands 24 hours after puparium formation (Figures 3D an 3F), expression of Atg1 in salivary glands leads to almost complete degradation of this tissue in wtsP2/wtsP2 animals (Figures 3E and 3F) even though they had salivary glands during early pupal stages (data not shown). Together these results indicate that decreased autophagy contributes to the cell death defect in wts mutant animals.
Figure 3. Autophagy is decreased in wts mutants, and expression of Atg1 rescues wts salivary gland persistence.
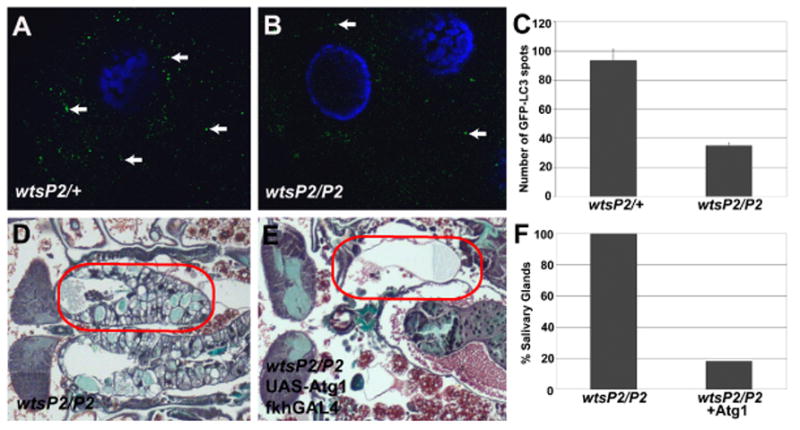
(A–C) Autophagy is indicated by the presence of GFP-LC3 puncta (autophagosomes) in salivary glands. (A and C) Abundant GFP-LC3 spots are detected in control salivary glands 1.5 hours after head eversion. (B and C) wts mutant salivary glands possess attenuated autophagy based on the detection of fewer GFP-LC3 spots than in control salivary glands. (D and E) Histological sections 12 hours after head eversion. (D and F) Loss of wts in wtsP2/P2 animals prevent salivary gland degradation. (E and F) Expression of Atg1 in wts mutant salivary glands suppresses the persistence phenotype. (F) Quantification of the percent of pupae with persistent salivary glands (n > 20 pupae/genotype). Red circles outline the persistent salivary gland tissue in the pupae.
Growth arrest is required for induction of autophagy and salivary gland degradation [9]. Therefore, we investigated whether decreased autophagy in wts mutants is associated with altered salivary gland cell growth by measuring the cell area in control wtsP2/wild-type and experimental wtsP2/wtsP2 salivary glands. While the cell area of control and homozygous wtsP2 mutant salivary gland cells were similar at the onset of puparium formation (Figures 4A and 4B), wtsP2mutant salivary gland cells were 2.5-fold larger than control salivary glands 6 hours after puparium formation (Figures 4C, 4D and 4E). We observed similar numbers of salivary gland nuclei in control wtsP2/wild-type (mean = 117.7, n = 15) and homozygous wtsP2 mutants (mean = 118.9, n = 15), and larval developmental period was similar in control and homozygous wtsP2 mutants (Figure 4F). These data indicate that a failure in cell growth arrest at the onset of puparium formation is responsible for the larger size of wtsP2mutant salivary gland cells.
Figure 4. wts mutant salivary gland cells fail to arrest growth at pupariation.

(A–D) Paraffin sections of salivary glands. (A, B and E) Cells of control wtsP2/+ and mutant wtsP2/P2 salivary glands are similar in size at puparium formation. (C, D and E) While control wtsP2/+ salivary gland cells have not significantly increased in size by 6 hours after puparium formation, mutant wtsP2/P2 salivary gland cells doubled in size during this period. (E) Cell area measurements of control and wts mutant salivary glands are presented as fold increase in cell area compared to control salivary gland cells at puparium formation, and are presented as mean ± SE. (F) Timing of pupariation in control (solid line) and wts mutant (dotted line) animals show that they have comparable developmental period.
Expression of Bantam, but not Yki and Sd, phenocopies wts in salivary glands
The transcriptional coactivator Yorkie (Yki) is phosphorylated by Wts inactivating its influence on cell cycle and cell death effectors [28]. Yki, the DNA binding protein Sd, and their target the microRNA bantam are positive regulators of tissue growth [28, 30–33]. Over-expression of either Yki or bantam in developing adult eyes phenocopies wts loss-of-function mutants in this tissue [28, 32, 33], and expression of Sd enhances Yki-induced growth and target gene activities [31]. Therefore, we tested if expression of Yki in salivary glands prevents the death of this tissue. Surprisingly, expression of Yki fails to inhibit salivary gland degradation (Figures 5A and 5B) even though expression of this transgene causes overgrowth in developing adult eyes (data not shown). In addition, expression of either Sd alone, or co-expression of Sd and Yki, induces premature degradation of salivary glands by 6 hours after puparium (Figures 5C and S1). Expression of Yki alone failed to induce premature salivary gland cell death in animals 6 hours after puparium formation (Figure 5B). These data indicate that expression of neither Yki, Sd, nor Yki and Sd together phenocopies the loss-of-function phenotype of wts in salivary glands. Significantly, our data also indicate that the Wts pathway that has been described in developing adult Drosophila tissues is different in salivary glands. To test this possibility, we determined if DIAP1 levels are altered in wts mutant salivary glands. Indeed, DIAP1 protein levels are not altered in homozygous wtsP2 mutants (Figure 5D), further supporting our data that Wts alters salivary gland growth and cell death in a Yki- and Sd-independent manner.
Figure 5. Expression of Yki and Sd fail to phenocopy the wts salivary gland phenotype, and DIAP1 levels are not altered in wts mutants.
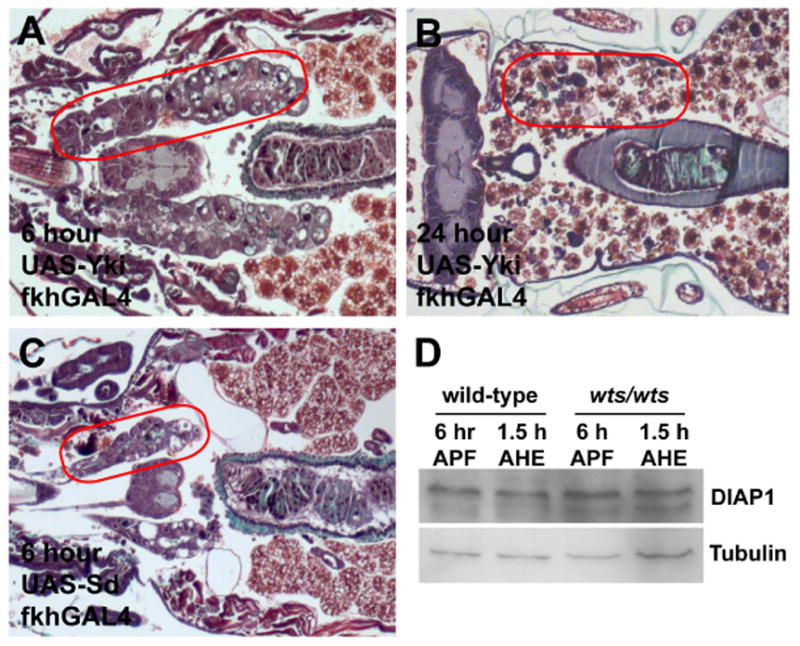
(A–C) Paraffin sections of animals 6 and 24 hours after puparium formation. (A) Paraffin sections of animals mis-expressing Yki in salivary glands at 6 hours after puparium formation have normal salivary glands. (B) Paraffin sections of animals mis-expressing Yki in salivary glands completely lack this tissue 24 hours after puparium formation. (C) Mis-expression of Sd in salivary glands causes premature degradation such that almost no tissue is present 6 hours after puparium formation. (D) Immunoblot showing the DIAP-1 levels are similar in control (wtsP2/+) and wts mutant (wtsP2/P2) salivary glands. The lanes contain salivary gland extracts isolated from control and mutant animals staged at 6 hours after puparium formation (APF) and 1.5 hours after head eversion (AHE). Red circles outline the persistent salivary gland tissue in the pupae.
Our data suggests that salivary gland cells are not sensitive to Yki- and Sd-induced growth, but it is not clear if this is because of differences in the response of Yki and Sd target genes, or if Yki and Sd targets are not sufficient to induce the growth and inhibit degradation of these cells. Expression of DIAP1 is sufficient to inhibit complete salivary gland degradation (Figure 2E), but DIAP1 does not influence salivary gland cell growth (data not shown). By contrast, mis-expression of bantam in salivary glands was sufficient to inhibit salivary gland degradation and induce significant cell growth (Figure 6A and and data not shown). Co-expression of dominant negative TOR (Torted) with bantam failed to suppress the bantam-induced salivary gland persistence phenotype compared to control animals 24 hours after puparium formation (Figures 5B and 5C). In addition, loss-of-function mutations in the insulin receptor substrate othologue chico also failed to suppress the bantam-induced defect in salivary gland degradation 24 hours after puparium formation (Figure 5D). Consistent with these genetic findings, mis-expression of bantam is not sufficient to maintain cortical localization of the reporter of Class I PI3K activity sensor tGPH sensor (data not shown). These data indicate that expression of bantam, but not Yki, phenocopies the loss-of-function phenotype of wts, but does so in a manner that is independent of two conserved genes in the PI3K pathway. These data support the conclusion that Yki and Sd target gene promoters, such as bantam, may not be active in salivary glands. Consistent with this conclusion, we failed to detect evidence of bantam RNA in late larval and prepupal salivary glands using a bantam sensor (data not shown). In addition, bantam loss-of-function mutants failed to suppress the homozygous wts mutant salivary gland cell death phenotype (Figures 6E and 6F), even though bantam mutants animals are smaller as previously described [37]. These data support the conclusion that Wts regulates growth and death of salivary glands using a mechanism that is different from the pathway that has been described in developing adult tissues of Drosophila.
Figure 6. bantam mis-expression inhibits salivary gland degradation in a PI3K-independent manner.
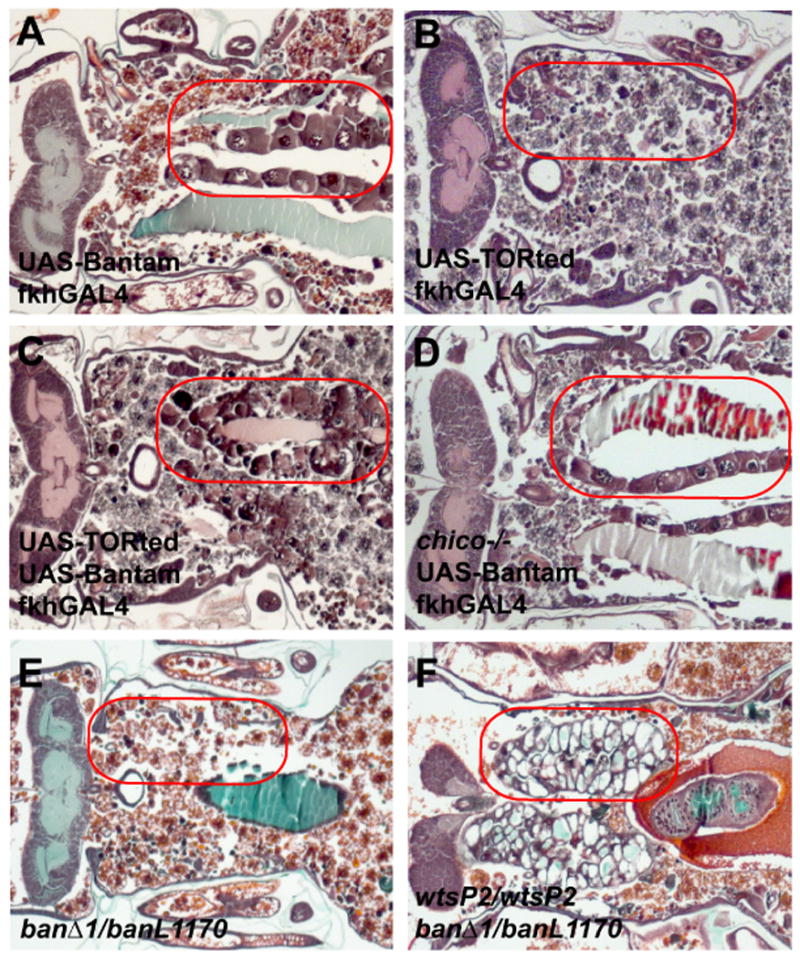
(A–F) Paraffin sections of animals 24 hours after puparium formation. (A) Animals that mis-express bantam in salivary glands fail to degrade this tissue 24 hours after puparium formation. (B) Salivary glands expressing TORted are degraded by 24 hours after puparium formation. (C) Coexpression of TORted does not overcome inhibition of salivary gland removal caused by mis-expression of bantam. (D) chico mutants fail to suppress salivary gland persistence that is induced by mis-expression of bantam. (E) Salivary glands are degraded in bantam mutants (banΔ1/banL1170). (F) Salivary glands fail to degrade in wtsP2 and bantam double-mutant animals. Red circles outline the persistent salivary gland tissue in the pupae.
PI3K signaling is required for wts to inhibit cell death
Maintenance of growth by expression of positive regulators of the class I PI3K pathway inhibits autophagy and salivary gland degradation [9]. Since Yki and Sd expression failed to phenocopy the wts mutant salivary gland phenotype, and bantam appears to regulate growth in a PI3K-independent manner, we investigated if wts may influence PI3K signaling. We monitored PI3K activity in salivary glands using the tGPH (tubulin-GFP-Pleckstrin Homology) reporter [38]. During the larval feeding stage when animals are growing, tGPH is cortically localized in salivary gland cells of both control and homozygous wts mutant animals (data not shown). The cortical localization of tGPH is lost in salivary glands of control wtsP2/wild-type animals when they stop feeding and growth arrest occurs (Figure 7A). By contrast, cortical localization of tGPH was maintained in wtsP2/wtsP2 and wtsP2/latsX1 mutant animals even after the onset of puparium formation (data not shown) and continued during prepupal development (Figure 7B). Consistent with the larger salivary gland cell size of wtsP2mutants (Figures 4C, 4D, and 4E), these data indicate that wts mutant salivary gland cells fail to arrest growth at puparium formation.
Figure 7. Wts regulates growth in a PI3K pathway-dependent manner.
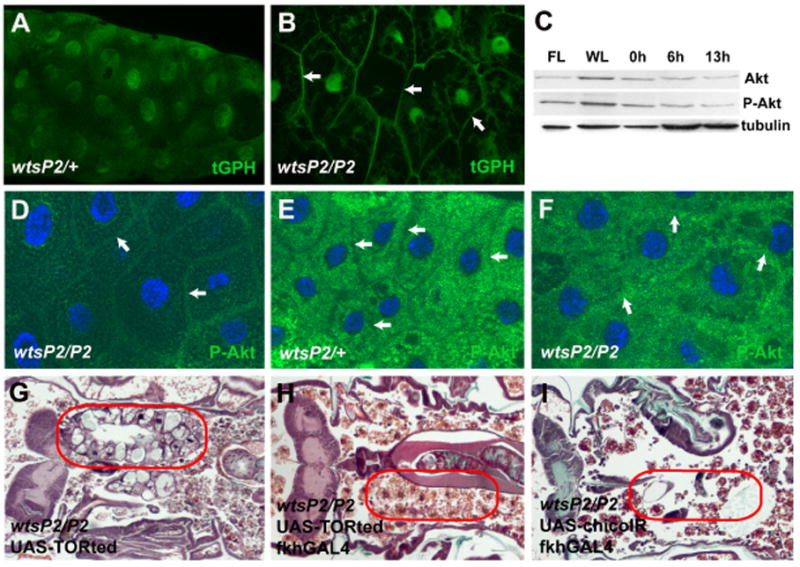
(A) Salivary glands of control wtsP2/+ animals 6 hours after puparium formation lack cortical tGPH indicating that growth has arrested. (B) wts mutant (wtsP2/P2) salivary glands isolated 6 hours after puparium formation possess cortically localized tGPH indicating that they have not arrested growth. (C) Immunoblot showing the levels of Akt and its activated form phosphorylated Akt (P-Akt) in wild-type salivary glands during larval and pupal stages. The lanes contain salivary gland extracts isolated from feeding larvae (FL), wandering larvae (WL), and stages 0, 6, and 13 hours after puparium formation. (D–F) Salivary glands stained with anti-phospho-Akt (P-Akt). (D) Salivary glands from both control (not shown) and wts mutant feeding larvae contain P-Akt that is associated with the cell cortex (white arrows). (E) P-Akt changes localization to the cytosol and is excluded from the cell cortex (white arrows) in control wtsP2/+ 6 hours after puparium formation. (F) Although some P-Akt is relocalized in the cytosol, it remains associated with the cell cortex (white arrows) in wts mutant salivary gland cells 6 hours after puparium formation. (G–I) Paraffin sections of pupae 24 hours after puparium formation. (G) The presence of dominant negative TORted without any GAL4 driver in wts mutant animals does not prevent salivary gland degradation. (H) Expression of TOR ted in salivary glands of wts mutant animals results in complete degradation of salivary glands. (I) Decreased function of chico by expression of RNAi (chico-IR) in salivary glands suppresses the wts mutant persistent salivary gland phenotype. Red circles outline the persistent salivary gland tissue in the pupae.
The maintenance of growth and presence of tGPH at the cell cortex in wts mutant salivary glands suggests that the class I PI3K pathway remains activated following puparium formation in this tissue. Activation of Class I PI3K pathway causes recruitment of Akt to the plasma membrane where phosphorylated Akt initiates downstream signaling via TOR to regulate growth [13]. To our surprise, the levels Akt and phosphorylated Akt were very similar in wild-type and wts mutant animals following growth arrest of salivary glands at the onset of puparium formation (Figures 7C and S2). However, immuno-localization of phosphorylated Akt changed from cortical (Figure 7D) to cytoplasmic in control salivary glands following puparium formation (Figure 7E), while much of the phosphorylated Akt remained associated with the cell cortex in homozygous wts mutant salivary glands (Figure 7F).
tGPH and phosphorylated Akt localization indicated that the class I PI3K pathway is altered in wts mutant salivary glands. Therefore, we investigated whether wts mutant salivary gland degradation is dependent on the PI3K pathway and TOR. Salivary glands are present in control wtsP2/wtsP2 animals that possess a dominant negative TOR (UAS-Torted) transgene that is not expressed because they lack the fkhGAL4 activator (Figures 7G). By contrast, expression of Torted in salivary glands suppressed the wts mutant degradation defect in this tissue (Figure 7H). Similarly, decreased function of the insulin receptor substrate encoding gene chico by expression of chico-RNAi in salivary glands attenuated the wts mutant salivary gland persistence phenotype (Figure 7I). Together these data indicate that Wts is regulating salivary gland degradation in a PI3K-dependent manner.
Discussion
Our studies indicate that Wts and other core components of this tumor suppressor pathway are required for autophagic cell death of Drosophila salivary glands. wts is required for cell growth arrest, and proper regulation of caspases and autophagy that contribute to the destruction of salivary glands. Although it is well known that cell division, cell growth and cell death are important regulators of tissue and tumor size [1], it has been unclear if a mechanistic relationship exists between cell growth and control of cell death.
It is possibile that wts, and associated downstream growth regulatory mechanisms, could suppress cell death in other animals and cell types. Autophagic cell death morphology has been reported in diverse taxa [5, 39], but we know little about the mechanisms that control this form of cell death, and this is likely related to the limited investigation of physiologically relevant of models of this process. Here we have used steroid-activated autophagic cell death of salivary glands as a system to study the relationship between cell growth and cell death. It is logical that cell growth influences death in salivary glands, as autophagy is known to be regulated by class I PI3K signaling that contributes to the death of these cells [9]. It is unclear if growth arrest is a determinant of autophagic cell death in other cell types and animals, and this is important to resolve because of the importance of growth and autophagy in multiple disorders including cancer [40]. wts mutant salivary gland cells fail to arrest growth at the onset of puparium formation, and this suppresses the induction of autophagy. As previously reported, the inhibitor of apoptosis DIAP1 influences salivary gland cell death [41] and is one of the best characterized target genes of the Wts signaling pathway [14–16, 18], but DIAP1 levels are not altered in wts mutant salivary glands. Significantly, our data provide the first evidence that Wts regulates autophagy, and support previous studies indicating that caspases and autophagy function in an additive manner during autophagic cell death [7, 9]. Given the importance of both the Wts pathway and autophagy in human health [29, 40, 42], it is critical to determine if this relationship exists in other cells.
Cell growth and division are often considered to be synonymous even though they are controlled by independent mechanisms. The Wts signaling pathway must influence cell growth, but most studies have emphasized the influence of this pathway on cell division and death. bantam is the only previously studied gene that is regulated by the Wts pathway that is known to regulate cell growth [32, 33]. However, the mechanism of bantam action remains obscure. Our studies suggest the possibility that Wts may regulate growth via different mechanisms, and that this may depend on cell context. It is premature to conclude that bantam regulates a completely novel cell growth program, but the fact that mis-expression of bantam stimulates cell growth in the absence of changes in a phosphoinositide marker, and that chico and TOR fail to suppress the bantam-induced salivary gland persistence phenotype, minimally suggests that this microRNA regulates genes downstream of TOR. Significant progress has been made in the identification of microRNA targets [43–45], and future studies should resolve mechanism underlying bantam regulation of cell growth.
Recent studies of Wts signaling in Drosophila have identified a linear pathway that terminates with Yki and Sd regulation of effector genes that influence cell growth, cell division and cell death [29]. Our studies indicate that the Wts pathway may not always regulate downstream effector genes via Yki and Sd, as Yki expression was not able to phenocopy the wts mutant salivary gland destruction, and expression of Sd induced premature degradation of salivary glands. Although bantam expression was sufficient to induce growth and inhibit cell death in salivary glands, bantam function was not required for the wts mutant phenotype. wts mutant salivary glands possess altered markers of PI3K signaling, and their defect in cell death is suppressed by chico and TOR. Combined, these results indicate that Wts regulates cell growth and cell death via a PI3K-dependent, and Yki- and Sd-independent, mechanism. Future studies will determine if Wts regulates cell growth in a PI3K-dependent manner in other cells and animals.
Experimental Procedures
Drosophila Strains
For loss of function studies, wtsP2 [22], latsX1 [17], chico1 [11], and banΔ1 [37] strains were analyzed. sav-IR, mats-IR and chico-IR stocks were obtained from the VDRC stock center. For ectopic expression studies, UAS-DIAP1 [46], UAS-Atg1KQ [36], UAS-Atg1 [36], UAS-Yki [28], UAS-Sd [47], UAS-Bantam A [48], UAS-TORted [49] were used. UAS-GFP-LC3 [50] was used as a marker of autophagy and tGPH served as a PI3K activity sensor [38]. Canton-S wild-type was used as a control.
Protein Extracts and Western Blotting
Salivary glands were dissected from wild-type Canton S and wtsP2 homozygous animals staged as feeding larvae, wandering larvae, and 0, 6, 8, 10, 12, and 14 hours following puparium formation at 25°C. Salivary glands were homogenized in Laemmli buffer (0.1% glycerol, 2%SDS, 0.125% 1M Tris (pH6.8), 0.05% β-mercaptoethanol, and 0.05% Bromo-phenol blue) and boiled for 5 minutes at 100°C. Equal amounts of proteins were separated on 10–12% SDS polyacrylamide gels. Proteins were transferred to 0.45μm Immobilon-P membranes (Millipore) following standard procedures. Blots were stripped using low pH stripping buffer (25mM glycine-HCl, pH 2, 15(w/v) SDS) between antibodies. Primary antibodies used were rabbit anti-Wts (1:5000)(Cho et al., 2006), guineapig anti-Hpo (1:2000) [21], mouse anti-DIAP1 (1:100) [51], rabbit anti-Akt (1:1000) (Cell Signaling), rabbit anti phospho-Drosophila Akt (Ser505)(1:1000)(Cell Signaling) and mouse anti-beta-tubulin (1:50)(Developmental Studies Hybridoma Bank).
Caspase Substrate Assays
The EnzChek Caspase-3 Assay (Molecular Probes) was used for caspase substrate assays. Whole wtsP2/Canton-S (control) and wtsP2/wtsP2 (experimental) pupae were staged at 4 hours after puparium formation, homogenized in lysis buffer and reaction buffer was added to the lysates. After centrifugation, clear lysates were assayed with Z-DEVD-AMC to detect caspase-3-like activity. To confirm the specificity of this assay to detect caspase-3-like activity, Ac-DEVD-CHO was added to the control (wtsP2/Canton-S) lysate as a competitive inhibitor. All the genotypes for caspase substrate assays were analyzed in triplicate.
Salivary Gland Histology
Animals of the indicated genotypes were aged to 6, 13.5 and 24 hours after puparium formation at 25°C, fixed in FAAG, dehydrated, embedded in paraffin, sectioned and stained with Weigert’s Hematoxylin and Pollack Trichrome as previously described [52]. TUNEL assay was performed using the Apoptag kit (Chemicon) as previously described [7] and examined using Zeiss Axio Imager.Z1 microscope. For TUNEL assays, a minimum of 10 pupae were examined for each genotype. For all other experiments, a minimum of 20 pupae were analyzed for each genotype. Cell area measurements were done using ImageJ software [53]. Area measurements represent an average of at least 6 cells per salivary gland and a minimum of 7 animals for each genotype.
Fluorescence Microscopy
For autophagy assays, salivary glands of fkhGAL4; UAS-GFP-LC3; wtsP2/wtsP2 experimental animals and fkhGAL4; UAS-GFP-LC3; wtsP2/wild-type controls were isolated 1.5 hr after head eversion, stained with Hoechst 33342, and imaged immediately as unfixed tissue using Zeiss Axio Imager.Z1 with apotome. The numbers of GFP-LC3 punctate spots were counted using Zeiss image counting software. To detect PI3K activity using the tGPH sensor, tGPH; wtsP2/wtsP2and fkhGAL4; tGPH/wild-type; UAS-Bantam A experimental, and tGPH/wild-type salivary glands were dissected from third instar larvae, staged 6 hours after puparium formation, and immediately imaged with a Zeiss Axio Imager.Z1 microscope. To count salivary gland nuclei, salivary glands were dissected from wtsP2/wildtype control and wtsP2/wtsP2 experimental animals staged 6 hours after puparium formation. Dissected glands were then mounted with vectashield with DAPI (Vector Laboratories) and imaged using a Zeiss Axio Imager.Z1 with apotome. The numbers of nuclei per salivary gland was determined using Zeiss image counting software.
Immunohistochemistry
For immunohistochemistry, wtsP2/Canton-S control and wtsP2/wtsP2 experimental salivary glands were dissected from feeding larvae and animals staged 6 hours after puparium formation, fixed with 4% paraformaldehyde, and processed according to standard procedures [8]. Rabbit anti-phospho-Akt (1:500)(Cell Signaling) and Toto-3 (Molecular Probes) were used to stain salivary glands, and they were imaged using Zeiss Axiovert confocal microscope.
Supplementary Material
Acknowledgments
We thank A. Bergmann, S. Cohen, B. Edgar, G. Halder, B. Hay, K. Irvine, T. Neufeld, D. Pan, the Bloomington Stock Center, and the Developmental Studies Hybridoma Bank for fly stocks and antibodies, and L. Pick, D. Berry, C. McPhee and members of the Baehrecke lab for helpful discussions and comments on the manuscript. This work was supported NIH grants GM59136 and GM079431 to EHB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Conlon I, Raff M. Size control in animal development. Cell. 1999;96:235–244. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 2.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 3.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 4.Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- 5.Clarke PGH. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 6.Jiang C, Baehrecke EH, Thummel CS. Steroid regulated programmed cell death during Drosophila metamorphosis. Development. 1997;124:4673–4683. doi: 10.1242/dev.124.22.4673. [DOI] [PubMed] [Google Scholar]
- 7.Lee CY, Baehrecke EH. Steroid regulation of autophagic programmed cell death during development. Development. 2001;128:1443–1455. doi: 10.1242/dev.128.8.1443. [DOI] [PubMed] [Google Scholar]
- 8.Martin DN, Baehrecke EH. Caspases function in autophagic cell death in Drosophila. Development. 2004;131:275–284. doi: 10.1242/dev.00933. [DOI] [PubMed] [Google Scholar]
- 9.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozma SC, Thomas G. Regulation of cell size in growth, development and human disease: PI3K, PKB and S6K. Bioessays. 2002;24:65–71. doi: 10.1002/bies.10031. [DOI] [PubMed] [Google Scholar]
- 11.Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell. 1999;97:865–875. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- 12.Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 1996;15:6584–6594. [PMC free article] [PubMed] [Google Scholar]
- 13.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457–467. doi: 10.1016/s0092-8674(03)00557-9. [DOI] [PubMed] [Google Scholar]
- 15.Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol. 2003;5:921–927. doi: 10.1038/ncb1051. [DOI] [PubMed] [Google Scholar]
- 16.Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol. 2003;5:914–920. doi: 10.1038/ncb1050. [DOI] [PubMed] [Google Scholar]
- 17.Xu T, Wang W, Zhang S, Stewart RA, Yu W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development. 1995;121:1053–1063. doi: 10.1242/dev.121.4.1053. [DOI] [PubMed] [Google Scholar]
- 18.Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- 19.Lai ZC, Wei X, Shimizu T, Ramos E, Rohrbaugh M, Nikolaidis N, Ho LL, Li Y. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell. 2005;120:675–685. doi: 10.1016/j.cell.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 20.Tapon N, Harvey KF, Bell DW, Wahrer DC, Schiripo TA, Haber DA, Hariharan IK. salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell. 2002;110:467–478. doi: 10.1016/s0092-8674(02)00824-3. [DOI] [PubMed] [Google Scholar]
- 21.Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8:27–36. doi: 10.1038/ncb1339. [DOI] [PubMed] [Google Scholar]
- 22.Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9:534–546. doi: 10.1101/gad.9.5.534. [DOI] [PubMed] [Google Scholar]
- 23.Kango-Singh M, Nolo R, Tao C, Verstreken P, Hiesinger PR, Bellen HJ, Halder G. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development. 2002;129:5719–5730. doi: 10.1242/dev.00168. [DOI] [PubMed] [Google Scholar]
- 24.Silva E, Tsatskis Y, Gardano L, Tapon N, McNeill H. The tumor-suppressor gene fat controls tissue growth upstream of expanded in the hippo signaling pathway. Curr Biol. 2006;16:2081–2089. doi: 10.1016/j.cub.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Bennett FC, Harvey KF. Fat cadherin modulates organ size in Drosophila via the Salvador/Warts/Hippo signaling pathway. Curr Biol. 2006;16:2101–2110. doi: 10.1016/j.cub.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 26.Willecke M, Hamaratoglu F, Kango-Singh M, Udan R, Chen CL, Tao C, Zhang X, Halder G. The fat cadherin acts through the hippo tumor-suppressor pathway to regulate tissue size. Curr Biol. 2006;16:2090–2100. doi: 10.1016/j.cub.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Cho E, Feng Y, Rauskolb C, Maitra S, Fehon R, Irvine KD. Delineation of a Fat tumor suppressor pathway. Nat Genet. 2006;38:1142–1150. doi: 10.1038/ng1887. [DOI] [PubMed] [Google Scholar]
- 28.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14:388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Ren F, Zhang Q, Chen Y, Wang B, Jiang J. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev Cell. 2008;14:377–387. doi: 10.1016/j.devcel.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nolo R, Morrison CM, Tao C, Zhang X, Halder G. The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr Biol. 2006;16:1895–1904. doi: 10.1016/j.cub.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 33.Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126:767–774. doi: 10.1016/j.cell.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 34.Martin DN, Balgley B, Dutta S, Chen J, Rudnick P, Cranford J, Kantartzis S, DeVoe DL, Lee C, Baehrecke EH. Proteomic analysis of steroid-triggered autophagic programmed cell death during Drosophila development. Cell Death Differ. 2007;14:916–923. doi: 10.1038/sj.cdd.4402098. [DOI] [PubMed] [Google Scholar]
- 35.Lee CY, Clough EA, Yellon P, Teslovich TM, Stephan DA, Baehrecke EH. Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol. 2003;13:350–357. doi: 10.1016/s0960-9822(03)00085-x. [DOI] [PubMed] [Google Scholar]
- 36.Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hipfner DR, Weigmann K, Cohen SM. The bantam gene regulates Drosophila growth. Genetics. 2002;161:1527–1537. doi: 10.1093/genetics/161.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Britton JS, Lockwood WK, Li L, Cohen SM, Edgar BA. Drosophila’s insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell. 2002;2:239–249. doi: 10.1016/s1534-5807(02)00117-x. [DOI] [PubMed] [Google Scholar]
- 39.Baehrecke EH. How death shapes life during development. Nature Reviews Mol Cell Biol. 2002;3:779–787. doi: 10.1038/nrm931. [DOI] [PubMed] [Google Scholar]
- 40.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin VP, Thummel CS, Bashirullah A. Down-regulation of inhibitor of apoptosis levels provides competence for steroid-triggered cell death. J Cell Biol. 2007;178:85–92. doi: 10.1083/jcb.200703206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell. 2008;13:188–192. doi: 10.1016/j.ccr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 43.Stark A, Brennecke J, Russell RB, Cohen SM. Identification of Drosophila MicroRNA targets. PLoS Biol. 2003;123:1133–1146. doi: 10.1371/journal.pbio.0000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burgler C, Macdonald PM. Prediction and verification of microRNA targets by MovingTargets, a highly adaptable prediction method. BMC Genomics. 2005;6:88. doi: 10.1186/1471-2164-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–1864. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hay BA, Wassarman DA, Rubin GM. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- 47.Simmonds AJ, Liu X, Soanes KH, Krause HM, Irvine KD, Bell JB. Molecular interactions between Vestigial and Scalloped promote wing formation in Drosophila. Genes Dev. 1998;12:3815–3820. doi: 10.1101/gad.12.24.3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113 doi: 10.1016/s0092-8674(03)00231-9. in press. [DOI] [PubMed] [Google Scholar]
- 49.Hennig KM, Neufeld TP. Inhibition of cellular growth and proliferation by dTOR overexpression in Drosophila. Genesis. 2002;34:107–110. doi: 10.1002/gene.10139. [DOI] [PubMed] [Google Scholar]
- 50.Rusten TE, Lindmo K, Juhasz G, Sass M, Seglen PO, Brech A, Stenmark H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7:179–192. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 51.Yoo SJ, Huh JR, Muro I, Yu H, Wang L, Wang SL, Feldman RM, Clem RJ, Muller HA, Hay BA. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat Cell Biol. 2002;4:416–424. doi: 10.1038/ncb793. [DOI] [PubMed] [Google Scholar]
- 52.Muro I, Berry DL, Huh JR, Chen CH, Huang H, Yoo SJ, Guo M, Baehrecke EH, Hay BA. The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development. 2006;133:3305–3315. doi: 10.1242/dev.02495. [DOI] [PubMed] [Google Scholar]
- 53.Abràmoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.