

Summary
Methanosarcina acetivorans strain C2A is an acetate- and methanol-utilizing methane-producing organism for which the genome, the largest yet sequenced among the Archaea, reveals extensive physiological diversity. LC linear ion trap-FTICR mass spectrometry was employed to analyze acetate- vs. methanol-grown cells metabolically labeled with 14N vs. 15N, respectively, to obtain quantitative protein abundance ratios. DNA microarray analyses of acetate- vs. methanol-grown cells was also performed to determine gene expression ratios. The combined approaches were highly complementary, extending the physiological understanding of growth and methanogenesis. Of the 1081 proteins detected, 255 were ≥ 3-fold differentially abundant. DNA microarray analysis revealed 410 genes that were ≥ 2.5-fold differentially expressed of 1972 genes with detected expression. The ratios of differentially abundant proteins were in good agreement with expression ratios of the encoding genes. Taken together, the results suggest several novel roles for electron transport components specific to acetate-grown cells, including two flavodoxins each specific for growth on acetate or methanol. Protein abundance ratios indicated that duplicate CO dehydrogenase/acetyl-CoA complexes function in the conversion of acetate to methane. Surprisingly, the protein abundance and gene expression ratios indicated a general stress response in acetate- vs. methanol-grown cells that included enzymes specific for polyphosphate accumulation and oxidative stress. The microarray analysis identified transcripts of several genes encoding regulatory proteins with identity to the PhoU, MarR, GlnK, and TetR families commonly found in the Bacteria domain. An analysis of neighboring genes suggested roles in controlling phosphate metabolism (PhoU), ammonia assimilation (GlnK), and molybdopterin cofactor biosynthesis (TetR). Finally, the proteomic and microarray results suggested roles for two-component regulatory systems specific for each growth substrate.
Keywords: Quantitative proteomics, metabolic labeling, microarray, methanogenesis, acetate, methanol
Introduction
The microbiological conversion of organic matter to methane in diverse oxygen-free (anaerobic) environments is an important link in the global carbon cycle 1. The same process is employed for methane formation from biological waste and plant biomass (biomethanation), an alternative to fossil fuels 2. In the process, fermentative and acetogenic anaerobes convert the complex biomass to acetate, formate, CO2, and H2, such species being utilized for growth of methane-producing (methanogen) species by two distinct pathways 3. In the CO2-reduction pathway, either formate or H2 is oxidized which in turn provides electrons for the reduction of CO2 to methane. In the acetate fermentation pathway, acetate is cleaved, and the methyl group is reduced to methane with electrons derived from oxidation of the carbonyl group to CO2. Methanol, and other simple methyl-containing compounds, are also growth substrates for methylotrophic methanogens found in specialized anaerobic environments. The pathway for methane formation is a dismutation of the substrate in which methyl groups are either oxidized to CO2, generating reducing potential, or reduced to methane. Global proteomic and DNA microarray analyses have contributed to the understanding of diverse physiological aspects of species metabolizing methylotrophic substrates 4-13 or reducing CO214-21 to methane; however, few comprehensive global analyses of acetate metabolism are reported 10, 11.
Approximately two-thirds of the methane in nature derives from the fermentation of acetate by one of two genera, Methanosarcina and Methanosaeta. Thus, an understanding of growth and methane formation from acetate is essential to understanding the native ecology, and development of process parameters for control and optimization of the large-scale biomethanation of renewable plant biomass. Key to this understanding is the identification of proteins essential for acetate-dependent growth and methane formation. Several proteins have been discovered by either proteomic or DNA1 microarray approaches comparing acetate- vs. methanol-grown Methanosarcina species. DNA microarray analyses of the freshwater isolate Methanosarcina mazei11 has identified genes up regulated in response to growth with acetate vs. methanol, suggesting roles for the encoded proteins specific to each pathway.
Methanosarcina acetivorans is a marine isolate for which the genome, the largest yet among the Archaea, has been sequenced,22 suggesting extensive metabolic diversity. Several tools for genetic manipulation have been developed for M. acetivorans23-26 and the application of these tools exquisitely documented 27-29. Thus, M. acetivorans is particularly suited for investigation of the physiology of acetate-dependent growth and methanogenesis. A limited proteomic analysis of acetate- vs. methanol-grown M. acetivorans30 has revealed differences in the electron transport pathways of acetate-grown cells compared to M. mazei11.
A combined proteomic and DNA microarray approach to investigate any Methanosarcina species has not been reported. Here, we present a comprehensive quantitative analysis of the proteome, complemented by transcrptome analysis, of acetate- vs. methanol-grown M. acetivorans. In general, good agreement was observed between the proteomics and microarray data. The more advanced proteomic analyses combined with the microarray results provide a much larger and complementary in depth view of acetate and methanol metabolism that includes features of electron transport, regulation, and the general stress response.
Materials and Methods
Cell growth for proteomic studies
M. acetivorans acetate- and methanol-grown cells were cultured similar to that previously described, 31. The mineral medium contained in grams per liter: NaCl, 11.69 g; MgSO4·7H2O, 12.32 g; KCl, 0.76 g; CaCl2·2H2O, 0.14 g; NH4Cl, 0.5 g; Resazurin solution (1000 ×), 1ml; trace metal solution (100X) 10 ml 32; vitamin solution (100×) 10 ml 32; HCl (concentrated) 0.5ml; Na2HPO4·7H2O, 1.12 g; cysteine·HCl·H2O, 0.25 g; Na2CO3, 3.0 g. Methanol-grown cells were substituted with 15NH4Cl (98%) (Sigma, St. Louis, MO). An atmosphere (80:20) of nitrogen to carbon dioxide was used in the head-space. Cells from both cultures were harvested in mid-exponential growth at an OD600 of 0.8 and 0.6 for acetate and methanol cultures, respectively, as previously described 31.
Protein extraction, SDS/PAGE fractionation, and in-gel digestion
The cell pellet from about 40 ml of culture was re-suspended in 100 μl of 10 mM Tris-HCl containing 5 mM MgCl2 and 100 U DNase (Roche, Indianapolis, IN), and incubated on ice for 20 min. This treatment was followed by the addition of 900 μl of 8 M urea containing 0.05% SDS, and vortexing for 3 min. The whole cell lysate was cleared by centrifugation at 13,000 × g for 20 min at 4°C. The concentrations of whole-cell protein extracts, determined by the Bradford assay (Bio-Rad, Hercules, CA), from acetate- and methanol-grown cells were 5.8 and 3.5 mg/ml, respectively.
Whole-cell extracts of acetate- and methanol-grown cells were combined to generate a 1:1 (w/w) mixture of the 14N and 15N labeled proteins. An aliquot containing 40 μg of the mixture was diluted to 45 μl with SDS/PAGE sample buffer, consisting of 2% (w/v) SDS, 25% (v/v) glycerol, 100 mM DTT, 0.01% bromophenol blue, and 62.5 mM Tris-HCl (pH 7). The sample was resolved in a precast 12-well 10.5-14% linear gradient Criterion Tris-HCl gel (BioRad, Hercules, CA) developed at 160 V for 50 min. The gel was stained with silver as previously described 33. The lanes were cut into ten fractions, each of which contained roughly the same total density as estimated by visual inspection aided with a translucent illuminator. Each fraction was separately minced into ∼ 1 mm3 cubes and subjected to washing, in-gel digestion, and peptide extraction steps as described 34 except that the volume of solution added for each step was adjusted to accommodate the volume of gel pieces for each SDS/PAGE fraction. Sequencing grade trypsin (Promega, Madison, WI) was used as the digestion enzyme. The collected peptide extract solution for each fraction (∼1.2 ml) was concentrated to ∼30 μl final volume in a SPD1010 SpeedVac system (Thermo Savant, Holbrook, NY) at 45°C in a 1.5-ml microcentrifuge tube.
Protein identification and abundance ratio determination
Proteins were identified from the peptide extract of each 1D-SDS/PAGE gel fraction using a shotgun proteomics strategy similar to that previously described 35. Approximately 10 μl of peptide extract solution was loaded onto a 100 μm × 15 cm column packed with MagicC18 5 μm particles (Michrom BioResources, Auburn, CA), followed by a 75 min linear gradient of 2% to 35% ACN (v/v) in 0.1% formic acid using 300 nl/min flow rate. A hybrid linear ion trap - FTICR instrument (LTQ FT MS,ThermoFinnigan, San Jose, CA) was used for the analysis. In one data acquisition cycle, a single high resolution FT scan with accumulation of up to 2 × 106 ions was followed by acquisition of MS/MS spectra using the linear ion trap (accumulation of 3 × 104 ions) for up to the 7 most intense ions with the dynamic exclusion set to 1 min. A single data acquisition cycle was completed in approximately 3.5 sec. Each 1D-SDS/PAGE fraction was analyzed twice. The acquired data was searched against the NCBI database of M. acetivorans C2A in two separate Sequest searches, one corresponding to 14N and the other to 15N labeling. The precursor ion mass tolerance was set to ±1.4 Da, and trypsin was designated as the proteolytic enzyme with up to 2 missed cleavages. Peptides identified with Xcorr values greater than 1.5 (1+), 2.0 (2+), and 2.5(3+) in either 14N or 15N searches in two replicate LC-MS runs were initially selected, then only peptides with the precursor mass tolerance of ±10 ppm were accepted for quantitative analysis. In about 15% of peptide identifications, the second isotope was initially assigned as the precursor ion, resulting in 1 Da mass shift. In these cases, the shift was corrected for the monoisotopic peak before applying the 10 ppm precursor mass accuracy restriction. Relative abundance of identified peptides was then calculated using a lab-developed program 36 that determined ratios of chromatographic peak areas of the isotopically labeled peptide pairs. For successful quantitation, coeluting 14N, 15N pairs of peptides, with at least one peptide being identified using the database search criteria specified above and the mass difference between the peptides corresponding to the number of nitrogen atoms in the peptide sequence, had to be found. In the next step, the relative abundance ratios of proteins were calculated by averaging of identified peptide abundance ratios. Finally, only proteins quantitated with at least two peptides and protein probabilities greater than 0.95, estimated by Protein Prophet 37, were selected. Importantly, the combination of precursor mass tolerance (±10 ppm), Sequest Xcorr values the presence of a pair of co-eluting peaks, and ProteinProphet probabilities of 0.95 or higher provided highly confident protein identifications.
Cell growth for microarray experiments
M. acetivorans C2A was cultivated on a mineral medium identical to that described for proteomic studies. Following sterilization, the medium was supplemented with filter sterilized 0.1 ml 50 % methanol or 0.2 ml 5 M acetate per 10 ml medium.
For microarray experiments, eight independent cultures of M. acetivorans C2A cells were grown with either acetate (six) or methanol (two) with serial transfer (3 times to mid-exponential phase) to achieve steady state cell growth conditions. Cells were then harvested for RNA purification at mid-exponential growth (OD600 of 0.2 for acetate grown cultures and OD600 of 0.4 for the methanol grown cultures) as previously described 38. Duplicate samples of RNA from three individual acetate grown cultures were combined, since a lower cell yield was obtained relative to methanol cell growth.
Microarray probe selection and PCR primer design
Probes were designed, generated, and archived as previously described 39. The product sizes were generally 200 to 300 bp where a total of 3,981 ORFs were represented. Although M. acetivorans contains a total of 4,524 ORFs 22, unique PCR products could generated for 3,981 of the ORFs due to presence of multiple homologs of some ORFs; transposases, hypothetical proteins, and some uncharacterized proteins. These ORFs totaled 543 due to high DNA sequence similarity. The RT-PCR primers were created by a modified version of MyPROBES 38. The PCR product lengths were set to a range of 100-200 bp, melting temperature range 55-66°C, guanine plus cytosine content 55-65%, primer length 17-22 bases.
PCR amplification of microarray probes
PCR products were amplified from chromosomal DNA using the custom designed oligonucleotide pairs at an annealing temperature of 54°C for 30 s. The reactions were then extended for 30 s by rTaq Polymerase in 50μl reactions. Each original PCR product was then size verified by agrarose gel electrophoresis where only a singe product of the anticipated size was obtained in most cases. Where multiple bands were obtained, the correct sized fragment was selected by gel purification (QiaQuick Gel Purification Kit) for subsequent use. When no PCR product was obtained in the first round, temperature gradient PCR was performed in order to generate the desired DNA fragment. For the few remaining ORFs that failed using this approach, new oligonucleotides pairs were designed and used in a second attempt to generate the desired PCR product.
For the second round of DNA amplification, each of the initial amplified products (3933 ORFs) were used as templates and the gene specific primers to allow for more uniform product yield. Each PCR fragment was size verified and stocked for DNA cleanup. Prior to slide printing, each PCR generated DNA product was purified using Qiagen columns in a 96-well format (QiaQuick 96-well PCR Purification Kit). The purified DNA was quantified by UV spectrophotometry in 384-well format, and the concentration of each DNA level was determined. For slide printing, 2.4 μg of PCR product resuspended in 10μl 3xSSC with 0.0001% SDS solution was aliquoted into 384 well plates and dried. The remaining DNA was archived for future use in slide printing.
Microarray spotting
PCR products contained in 384 well plates were spotted onto GAPS II slides (Corning, Corning, New York) using a robotic arrayer Virtek ChipWriter Pro (Bio-Rad, Hercules, CA). The diameter of each spot was approximately 150 μm, and the distance between the centers of each spot was 220 μm. Slides were hydrated with steam for 2-3 s and snap dried on a 100°C heating block. The probes were cross-linked to the surface of the slide by UV light (Stratalinker, Stratagene, La Jolla, CA) at 250 mJ and the slides were then baked at 80°C for 3 hours. To minimize background, the slides were blocked by soaking for 15 min in 250 ml of 1-methyl-2-pyrrolidone with 4 g of succinic anhydride and 28 ml of 0.2 M sodium borate solution (pH 8.0). After blocking, the slides were washed with 95°C water for 2 min and transferred to 95% ethanol at room temperature for 1 min prior to drying by centrifugation.
Microarray experimental design
Cultures (10 ml) were incubated in anaerobic tubes at 37°C on a roller for 16-24 h to an OD600 of 0.4 for methanol and 70-80 h to an OD600 of 0.2 for acetate. Each tube was quick cooled in an ethanol/dry ice bath, cells were harvested by centrifugation with a Sorval ss-34 rotor (2000 rpm at 4°C), and resuspended in RNAlater (Ambion, Austin, TX) as previously described 38. Each cell growth condition was repeated two times. For microarray calibration, RNA samples from same culture were divided in half, labeled with Cy3 and Cy5 dyes (Amersham) and hybridized to 2 slides each. This calibration experiment was performed twice for each condition to obtain a reference distribution in the data analysis 40, 41.
RNA purification and labeling
For microarray experiments, total RNA was purified from 10 ml of cell samples using the RNAwiz (Ambion, Austin, TX) following the manufacturer's instructions. The purified RNA was treated with DNase I as described 42. Total RNA (60 μg) was labeled using indirect labeling with amino allyl-dUTP with either Cy3 or Cy5 monofunctional NHS-ester (Amersham Bioscience, Piscataway, NJ). The reverse transcription mixture, (40μl) including 600 units of Superscript II RNase H reverse transcriptase (Invitrogen), 4.5 μg random hexamers, 0.5 mM dATP, dCTP, and dGTP, 0.2 mM dTTP, and 0.3 mM aadUTP (Sigma, St. Louis, MO), was incubated at 42°C for 3 h. The Superscript enzyme was added in two steps, 300 units at time 0 h and 300 units after 1.5 h. After reverse transcription, the RNA was hydrolyzed by incubating at 65°C for 40 min after adding 10 μl of 0.5 M EDTA (pH 8.0) and 10 μl of 1 N NaOH. The cDNAs were neutralized with 25 μl 1M HEPES and purified with a Microcon-30 and dried. The dried cDNA was resuspended in 5 μl water plus 5 μl of 0.1M sodium bicarbonate, and transferred to a dry aliquot of the dye for the coupling reaction. The acetate samples were labeled with Cy3 and methanol were label with Cy5. The coupling reaction was quenched by adding 4.5 μl of 4 M hydroxylamine. The two dye-labeled cDNAs were combined, purified using a Qia-quick column (Qiagen, Valencia, CA), and concentrated to 1-2 μl using a Microcon-30 concentrator (Millipore).
Hybridization, scanning and data analysis
The concentrated Cy3 and Cy5 cDNA was hybridized and washed as described 42. The dried slides were analyzed using an Agilent DNA microarray scanner at 10 μm. The scanner creates a Tiff file for each channel, Cy3 and Cy5 where two images were simultaneously analyzed in an image analysis program Imagene5 (Biodiscovery, Marina Del Ray CA) to find spot intensities. Microarray spot intensity data were normalized and the 95% confidence level were estimated using the software lcDNA (http://receptor.seas.ucla.edu/lcDNA) 40, 41. Spot signals less than twice the mean background signal were removed. Outliers were then removed using a quality filtering routine, and the data were normalized using a rank-invariant method 41. The normalized data were then subjected to the Markov Chain Monte Carlo (MCMC) analysis method 40, 41.
Results and Discussion
Proteomic analysis
Previous 2-D gel MALDI TOF/TOF MS proteomic analyses of M. acetivorans identified 412 proteins 31 of which 34 were statistically differentially abundant between acetate- and methanol-grown cells 43. To obtain a deeper qualitative and quantitative analysis of the proteome, we first separated the lysate by SDS PAGE into 10 fractions and then utilized LC linear ion trap-FTICR MS to analyze acetate- vs. methanol-grown cells metabolically labeled with 14N vs.15N, respectively. Each gel fraction was analyzed twice by LC/MS. Using a conservative criterion (≥3-fold change) 44, 255 out of the 1081 quantitated proteins were found to be differentially abundant (≥3-fold change) between acetate- and methanol-grown cells as determined with two or more peptide pairs (Tables 1 and S1). These differentially abundant proteins were considered candidates for roles specific to growth with either methanol or acetate. Of the 255 proteins, 71 were more abundant in acetate- vs. methanol-grown cells, whereas 184 were more abundant in methanol- vs. acetate-grown M. acetivorans (Table 2).
Table 1.
Proteins with at least 3-fold differential abundance ratio, and genes with at least 2.5- fold differential expression ratio, in acetate- vs. methanol-grown M. acetivorans. See Table S1 for additional data.
| Loci | Annotationa | Proteome | Transcriptome (Me/Ac)b,e | |
|---|---|---|---|---|
| No. of peptides | (Me/Ac)b | |||
| MA0081 | polyphosphate kinase | ND | ND | 0.37 |
| MA0133 | small heat shock protein | ND | ND | 0.19 |
| MA0187 | molybdenum cofactor biosynthesis protein B (MoaB) | 8 | 5.8 ± 0.7 | ND |
| MA0304 | Molybdenum formylmethanofuran dehydrogenase (FmdE), subunit E | 4 | 10 ± 2 | ND |
| MA0305 | Molybdenum formylmethanofuran dehydrogenase (FmdF), subunit F | 4 | 13 ± 5 | ND |
| MA0306 | Molybdenum formylmethanofuran dehydrogenase (FmdA), subunit A | 8 | 30 ± 10 | ND |
| MA0307 | Molybdenum formylmethanofuran dehydrogenase (FmdC), subunit C | 2 | 30 ± 10 | 5.62 |
| MA0308 | Molybdenum formylmethanofuran dehydrogenase (FmdD), subunit D | 3 | 40 ± 20 | ND |
| MA0309 | Molybdenum formylmethanofuran dehydrogenase (FmdB), subunit B | 6 | 25 ± 9 | ND |
| MA0368 | transcriptional regulator, TetR family | 4 | 5.8 ± 0.4 | ND |
| MA0406 | iron-sulfur flavoprotein | ND | ND | 0.29 |
| MA0455 | methanol-5-hydroxybenzimidazolylcobamide co-methyltransferase (MtaC1), isozyme 1 | 5 | 500 ± 200 | ND |
| MA0456 | methanol-5-hydroxybenzimidazolylcobamide co-methyltransferase (MtaB1), isozyme 1 | 4 | 500 ± 200 | ND |
| MA0490 | sensory transduction histidine kinase | ND | ND | 0.23 |
| MA0619 | sensory transduction histidine kinase | ND | ND | 2.63 |
| MA0620 | sensory transduction histidine kinase | ND | ND | 0.28 |
| MA0630 | groES protein (Cpn10) | ND | ND | 5.33 |
| MA0659 | Na+-transporting NADH:ubiquinone oxidoreductase, subunit 1 (Ma-RnfC)c | 6 | 0.09 ± 0.04d | 0.42 |
| MA0660 | Na+-transporting NADH:ubiquinone oxidoreductase, subunit 2 (Ma-RnfD)c | ND | ND | 0.40 |
| MA0661 | Na+-transporting NADH:ubiquinone oxidoreductase, subunit 3 (Ma-RnfG)c | 3 | 0.08 ± 0.03d | ND |
| MA0662 | Na+-transporting NADH:ubiquinone oxidoreductase, subunit 4 (Ma-RnfE)c | ND | ND | 0.36 |
| MA0663 | Na+-transporting NADH:ubiquinone oxidoreductase, subunit 5 (Ma-RnfA)c | ND | ND | 0.26 |
| MA0664 | Na+-transporting NADH:ubiquinone oxidoreductase, subunit 6 (Ma-RnfB)c | 3 | 0.1 ± 0.03d | ND |
| MA0887 | phosphate ABC transporter, solute-binding protein | 3 | 0.01 ± 0.01 | ND |
| MA0889 | phosphate ABC transporter, permease protein | ND | ND | 0.08 |
| MA0891 | phosphate transport system regulatory protein | ND | ND | 0.33 |
| MA0924 | leucine responsive regulatory protein | ND | ND | 2.56 |
| MA1014 | carbon-monoxide dehydrogenase, (CdhC) gamma subunit | 5 | 0.04 ± 0.02d | 0.11 |
| MA1015 | carbon-monoxide dehydrogenase, (CdhB) beta subunit | 7 | 0.1 ± 0.03d | ND |
| MA1016 | carbon-monoxide dehydrogenase, (CdhA) alpha subunit | 6 | 0.04 ± 0.02d | 0.07 |
| MA1122 | transcriptional regulator, MarR family | ND | ND | 5.58 |
| MA1189 | Trp repressor binding protein | ND | ND | 0.26 |
| MA1469 | response regulator receiver | ND | ND | 3.51 |
| MA1477 | heat shock protein | 6 | 13 ± 6 | ND |
| MA1479 | heat shock protein 40 | ND | 7 ± 2 | ND |
| MA1487 | transcription regulation protein (TetR) | ND | ND | 2.57 |
| MA1616 | methanol-5-hydroxybenzimidazolylcobamide co-methyltransferase (MtaC3), isozyme 3 | 4 | 0.2 ± 0.1 | 0.29 |
| MA1617 | methanol-5-hydroxybenzimidazolylcobamide co-methyltransferase (MtaB3), isozyme 3 | 4 | 0.2 ± 0.1 | ND |
| MA1682 | Hsp60 | ND | ND | 0.13 |
| MA1763 | transcriptional regulator, CopG family | 3 | 15 ± 4 | ND |
| MA1799 | flavodoxin | ND | ND | 0.37 |
| MA1844 | sensory transduction histidine kinase | ND | ND | 4.68 |
| MA1991 | sensory transduction histidine kinase | ND | ND | 0.14 |
| MA2013 | sensory transduction histidine kinase | ND | ND | 0.40 |
| MA2082 | sensory transduction histidine kinase | ND | ND | 4.25 |
| MA2167 | iron-sulfur flavoprotein | ND | ND | 0.40 |
| MA2256 | sensory transduction histidine kinase | ND | ND | 0.33 |
| MA2351 | exopolyphosphatase | ND | ND | 0.34 |
| MA2699 | flavodoxin | ND | ND | 29.23 |
| MA2867 | polyferredoxin | 2 | 0.08 ± 0.04 | 0.35 |
| MA2914 | transcriptional regulator, Hth-3 family | 2 | 5 ± 1 | 2.72 |
| MA3212 | thioredoxin | ND | ND | 0.14 |
| MA3284 | universal stress protein | ND | ND | 0.31 |
| MA3743 | flavoprotein A | ND | ND | 0.37 |
| MA3860 | carbon-monoxide dehydrogenase, (CdhA) alpha subunit | 8 | 0.004 ± 0.002d | ND |
| MA3861 | carbon-monoxide dehydrogenase, (CdhB) beta subunit | 4 | 0.004 ± 0.002d | ND |
| MA3862 | carbon-monoxide dehydrogenase, (CdhC) gamma subunit | 5 | 0.006 ± 0.003d | ND |
| MA3863 | carbon-monoxide dehydrogenase, (CooC) accessory protein | ND | ND | 0.14 |
| MA3865 | carbon-monoxide dehydrogenase (CdhE), epsilon subunit | ND | ND | 0.35 |
| MA3916 | nitrogen regulatory protein P-II | ND | ND | 13.13 |
| MA3972 | conserved hypothetical protein | 8 | 0.1 ± 0.06 | 0.13 |
| MA4026 | sensory transduction histidine kinase | ND | ND | 0.16 |
| MA4103 | peroxiredoxin (alkyl hydroperoxide reductase) | ND | ND | 0.34 |
| MA4391 | methanol-5-hydroxybenzimidazolylcobamide co-methyltransferase (MtaC2), isozyme 2 | 2 | 90 ± 40 | ND |
| MA4392 | methanol-5-hydroxybenzimidazolylcobamide co-methyltransferase (MtaB2), isozyme 2 | 2 | 70 ± 40 | ND |
| MA4399 | carbon-monoxide dehydrogenase, subunit alpha | ND | ND | 0.27 |
| MA4413 | Hsp60 | 5 | 9 ± 4 | 6.03 |
| MA4542 | small heat shock protein | ND | ND | 0.26 |
| MA4566 | multiple resistance/pH regulation related protein G (Na+/H+) | ND | ND | 0.09 |
| MA4567 | multiple resistance/pH regulation related protein F (Na+/H+) | 2 | 0.025 ± 0.012d | 0.11 |
| MA4568 | multiple resistance/pH regulation related protein E (Na+/H+) | 3 | 0.03 ± 0.016d | 0.12 |
| MA4569 | multiple resistance/pH regulation related protein D (Na+/H+) | ND | ND | 0.13 |
| MA4570 | multiple resistance/pH regulation related protein C (Na+/H+) | ND | ND | 0.13 |
| MA4572 | multiple resistance/pH regulation related protein A (Na+/H+) | ND | ND | 0.19 |
| MA4671 | response regulator receiver | ND | ND | 0.36 |
Annotations are those listed at http://www.tigr.org.
Ac, acetate-grown cells; Me, methanol-grown cells.
Originally annotated as Na+-transporting NADH:ubiquinone oxidoreductase subunits 52, the deduced sequences were shown to have greater identity to subunits of Rnf complexes and proposed to be renamed Ma-RnfCDGEAB 30.
Previously reported 30.
P values were ≤0.04 (95% confidence).
ND, Not determined.
Table 2.
Summary of proteomic and microarray expression ratios.
| Proteomic analysis | ||||
|
|
||||
| Methanola | Acetateb | |||
|
|
||||
| Totals | 184 | 71 | ||
|
| ||||
| Microarray analysis | Methanola | 210 | 63c | 3c |
|
| ||||
| Acetateb | 200 | 5c | 17c | |
Genes up regulated, or proteins in greater abundance, in methanol- vs. acetate-grown cells.
Genes up regulated, or proteins in greater abundance, in acetate- vs. methanol-grown cells.
Genes identified by both proteomic and microarray analysis are included in the totals shown for both analyses.
DNA microarray analysis
Transcript profiling of acetate- vs. methanol-grown M. acetivorans was performed to complement the proteomic results. The cells used for microarray and proteomic analyses were cultured independently which provided biological replicates. Total RNA was isolated from cells following flash cooling (Materials and Methods), and cDNA was then prepared for each experimental condition and used to hybridize to each of two slides. The experimental protocol was repeated to give a total of four slides and a total of eight measurements for each ORF. The data were analyzed using lcDNA, a MCMC method estimating the confidence level of the data, and outliers were removed by a quality filtering routine (Materials and Methods). The normalized data were then subjected to the MCMC analysis method. Of the 3940 spotted genes, 1972 (ca. 50%) had measurable transcript levels under both growth conditions that were two-fold above the mean (data not shown). Of 410 genes showing 2.5-fold or greater differential expression, 200 were expressed greater in response to growth with acetate, and 210 greater in response to methanol (Tables 1 and S1). Due to a higher confidence level of ratios for microarray data, gene expression ratios with a cut-off of ≥2.5-fold were considered candidates for roles specific to growth with either substrate.
Comparison of proteomic and microarray results
A combined total of 577 gene transcripts or proteins were identified by the two approaches (Tables 1 and S1). A comparison of the approaches are shown in Table 2. Of the 255 proteins detected with at least 3-fold differential abundance, 88 of the encoding genes showed a differential expression of 2.5-fold or greater. Of these 88 genes, the expression ratios of 80 were consistent with the protein abundance ratios indicating good agreement between the two methods of analysis. Microarray analyses detected 322 differentially expressed genes for which the protein products were either not detected or less than 3-fold differentially abundant in the proteome. Conversely, proteomic analyses detected 167 differentially abundant proteins for which transcripts of the encoding genes were not detected or the gene expression ratios were less than 2.5-fold. Some of these differences undoubtedly arise from the thresholds arbitrarily set, while other differences are from the inherently different experimental and biological variables of the two methodologies. Nonetheless, the two methods validate and complement each other, and in a number of cases demonstrate similar trends. Indeed, the combined analysis of experimental data sets provides a superior view of the complex physiology of M. acetivorans relative to either approach alone.
As shown in Figure 1, the 577 genes for which both proteomic and genomic microarray data were obtained revealed a reasonable correlation coefficient (R = ∼0.61). Only the data with no error measurement associated with it, or where the calculated protein abundance ratio was smaller than its standard deviation (i.e., 30 ± 50), were removed prior to plotting. Finally, three outlier microarray data points were removed where prior RT-PCR data indicated probe errors due to high nucleotide identity of several gene probes. Since the majority of the genes revealed by the two experimental approaches were in reasonably good agreement (R = ∼0.61), this result suggests that modulation of gene expression is a major level of control for substrate-specific growth. It is interesting to speculate that divergence of the corresponding proteomic and microarray data identifies potential candidates for regulatory control at the post-transcriptional level. Further experiments are needed to test this notion.
Figure 1.
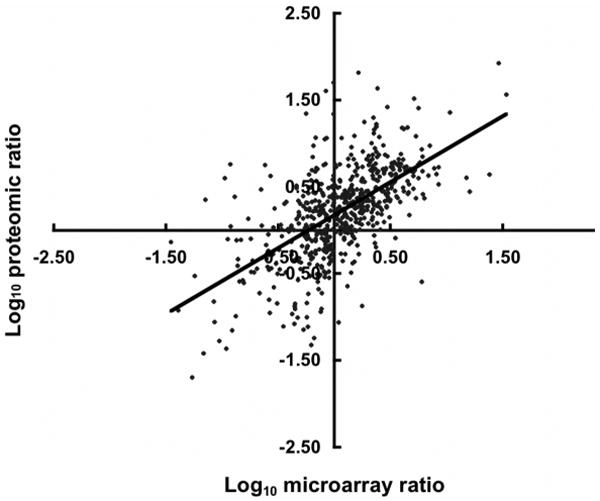
The differential expression of selected ORFs for proteomic and microarray data. For each ORF, the log change for proteomic data is plotted on the X axis and the corresponding microarray data on the Y axis. Data are available in Tables 1 and S1.
The genes presented in Tables 1 and S1 were classified using the COG functional classification (Table 3). For the genes identified by microarray analysis of acetate-grown cells, two functional categories that were statistically over or under represented were “energy production and conversion” and “replication, recombination and repair”, respectively. The former gene group suggests that a greater number of genes are needed to derive energy from acetate relative to methanol. The latter group is consistent with a reduced rate of cell replication with acetate relative to methanol. A similar COG group pattern was seen for the proteomic results except the “coenzyme transport and metabolism” group was additionally over represented. In methanol-grown cells, the three functional categories significantly elevated in expression relative to acetate-grown cells were “amino acid transport and metabolism”, “coenzyme transport and metabolism”, and “translation”. A similar COG group pattern was seen for the proteomic results except the “nucleotide transport and metabolism” and “replication, recombination and repair” groups were additionally over represented. Therefore, translation machinery demand appears elevated due to methanol dependent cell growth similar to that reported for M. mazei11 and consistent with the faster growth rate for these species on methanol compared to acetate 9-11.
Table 3.
Differentially expressed genes and differentially abundant proteins divided into functional groups.
| Functional Group | Total in groupa | Acetate-grown cells | Methanol-grown cells | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Microarray | Proteome | Microarray | Proteome | ||||||
| Sub-totalb | P-value | Sub-totalc | P-value | Sub-totalb | P-value | Sub-totalc | P-value | ||
| RNA processing and modification | 1 | 0 | 9.1E-01 | 0 | 9.57E-01 | 0 | 8.98E-01 | 0 | 8.98E-01 |
| Chromatin structure and dynamics | 1 | 0 | 9.1E-01 | 1 | 4.18E-02 | 0 | 8.98E-01 | 0 | 8.98E-01 |
| Energy production and conversion | 261 | 51d | 3.9E-07 | 35 | 2.37E-09 | 37 | 1.68E-02 | 33 | 4.67E-02 |
| Cell cycle control, mitosis and meiosis | 20 | 3 | 1.7E-01 | 4 | 9.93E-03 | 2 | 2.70E-01 | 3 | 1.94E-01 |
| Amino acid transport and metabolism | 253 | 27 | 6.2E-02 | 7 | 6.22E-02 | 57 | 8.27E-08 | 54 | 9.87E-07 |
| Nucleotide transport and metabolism | 62 | 6 | 1.6E-01 | 4 | 1.50E-01 | 13 | 1.01E-02 | 28 | 3.24E-10 |
| Carbohydrate transport and metabolism | 105 | 15 | 3.1E-02 | 6 | 1.33E-01 | 12 | 1.13E-01 | 7 | 5.69E-02 |
| Coenzyme transport and metabolism | 143 | 14 | 1.1E-01 | 18 | 4.52E-05 | 33 | 2.52E-05 | 32 | 6.03E-05 |
| Lipid transport and metabolism | 32 | 1 | 1.5E-01 | 0 | 2.46E-01 | 6 | 7.33E-02 | 8 | 1.55E-02 |
| Translation | 162 | 9 | 3.0E-02 | 4 | 8.74E-02 | 74 | 1.52E-25 | 83 | 7.17E-32 |
| Transcription | 180 | 14 | 8.3E-02 | 4 | 5.99E-02 | 17 | 8.44E-02 | 29 | 8.75E-03 |
| Replication, recombination and repair | 273 | 11e | 4.9E-04 | 3 | 1.55E-03 | 18 | 6.66E-03 | 12 | 1.08E-04 |
| Cell wall/membrane biogenesis | 102 | 8 | 1.2E-01 | 2 | 1.15E-01 | 16 | 3.53E-02 | 16 | 3.61E-02 |
| Cell motility | 40 | 3 | 2.1E-01 | 2 | 2.67E-01 | 1 | 5.82E-02 | 1 | 5.74E-02 |
| Posttranslational modification, protein turnover, chaperones | 116 | 13 | 9.1E-02 | 4 | 1.75E-01 | 14 | 9.77E-02 | 17 | 4.63E-02 |
| Inorganic ion transport and metabolism | 197 | 27 | 1.2E-02 | 9 | 1.34E-01 | 22 | 8.51E-02 | 15 | 3.61E-02 |
| Secondary metabolites biosynthesis, transport and catabolism | 98 | 11 | 1.0E-01 | 6 | 1.19E-01 | 8 | 1.01E-01 | 9 | 1.18E-01 |
| General function prediction only | 490 | 55 | 2.1E-02 | 18 | 7.18E-02 | 45 | 3.29E-02 | 65 | 1.20E-02 |
| Function unknown | 356 | 35 | 6.7E-02 | 19 | 6.45E-02 | 32 | 4.10E-02 | 29 | 1.99E-02 |
Total number of genome annotations for each functional group.
Number of differentially expressed genes detected for each functional group by microarray analyses.
Number of differentially abundant proteins detected for each functional group by proteomic analyses.
Genes shown in bold are statistically over represented.
Genes shown in bold and italics are statistically under represented.
Methanol conversion to methane
A total of 184 proteins were more abundant, and the expression of 210 genes up regulated, (Table 2) in methanol-grown M. acetivorans. Transcriptional profiling of M. mazei identified 212 genes at least 2.5-fold up regulated in response to growth on methanol 11, many of which encode proteins specific to the previously well-characterized pathway of methanol conversion to methane in Methanosarcina species 11, 45, 46. The proteomic and microarray analyses of methanol- vs. acetate-grown M. acetivorans (Tables 1 and S1) indicated that the pathway for conversion of methanol to methane is similar to all other Methanosarcina species investigated. Many of the transcripts and proteins found to be greater in methanol- vs. acetate-grown M. acetivorans are involved in translation and biosynthesis (Table S1), consistent with the transcriptional profile of M. mazei11. Both M. mazei11 and M. acetivorans31 have doubling times for growth on methanol that are 4-5 times greater than that for acetate, and it has been proposed 11 that the faster growth rate with methanol requires elevated levels of the translational apparatus and biosynthetic enzymes.
The genome of M. acetivorans is predicted to encode three two-subunit methyltransferase isozymes specific for methanogenesis from methanol, two of which (MA0455-0456 and MA4391-4392) were more abundant in methanol-grown cells, and the third (MA1616-1617) more abundant in acetate-grown cells (Table 1). Recent reporter gene studies of M. acetivorans are consistent with these results 47, and analogous to increased expression of genes in acetate- vs. methanol-grown M. mazei11 encoding one of three methanol-specific methyltransferase isozymes. It has been reported that individual disruption of genes encoding the three methyltransferase isozymes had no effect on acetate-dependent growth of M. acetivorans, indicating none of the isozymes are essential for growth on acetate 27. All of the above results are in agreement with a report where it was proposed that one of three methyltransferase isozymes synthesized in acetate-grown Methanosarcina thermophila facilitates transition from growth on acetate to growth on methanol 13.
Several group I chaperonins (MA1477, MA1479 and MA4413), and transcripts of genes encoding group I chaperonins (MA0630 and MA4413), were elevated in methanol-grown M. acetivorans (Table 1). MA0630 encodes GroEL that is clustered with MA0631 encoding GroES (Fig. 2) consistent with co-transcription and the coordinated roles for folding proteins described for species from the Bacteria domain. MA1477 and MA1479 encode GrpE and DnaJ, and are clustered with MA1478 encoding DnaK (Fig. 2) consistent with co-transcription and function in the Hsp70 system shown to assist protein folding in species from the Bacteria domain. The results confirm a previous report that MA1477 is up regulated in methanol-grown M. acetivorans, consistent with the reported up regulation of genes encoding the Hsp70 system in heat-shocked M. mazei48-50 and Methanococcoides burtonii grown at high temperature 4. A role for the group I chaperonins in M. acetivorans grown on methanol vs. acetate is less obvious, although it was previously proposed that the higher growth rate on methanol requires chaperonins to meet the increased demand for protein synthesis 10.
Figure 2.

Organization of differentially expressed genes and genes encoding differentially abundant proteins. Arrows indicate the length and orientation of genes. Hatched arrows indicate genes not represented in the proteome or transcriptome. The numbers above the arrows correspond to the gene loci. See Table 1 for gene annotations.
Thus, it appears there are few differences between growth on methanol between M. acetivorans and M. mazei, with one notable exception. A gene annotated to encode a flavodoxin (MA2699) was expressed 29-fold greater in methanol- vs. acetate-grown M. acetivorans (Table 1), consistent with an electron transport function during growth on methanol. The only gene (MM0261) in the genome of M. mazei with sequence identity to MA2699 encodes a putative flavoprotein with only 26% identity to the flavodoxin encoded by MA2699 11. These results suggest that the M. acetivorans flavodoxin (MA2699) does not have a functional equivalent in M. mazei and may reflect differences in electron transport between these marine and freshwater species.
Conversion of acetate to methane
Proteomic analyses identified 71 proteins that were more abundant in acetate-grown M. acetivorans, and microarray analyses identified a greater expression of 200 genes in acetate-grown cells (Tables 1 and S1). These results are similar to the number of genes reported to be up regulated in acetate- vs. methanol-grown M. mazei11.
The CO dehydrogenase/acetyl-CoA synthase (Cdh) is central to the pathway for conversion of acetate to methane in freshwater Methanosarcina species and M. acetivorans30, 51 where it cleaves acetyl-CoA releasing the methyl group for eventual reduction to methane and oxidizing the carbonyl group to carbon dioxide with transfer of electrons to ferredoxin. The genome of M. acetivorans is annotated with duplicate gene clusters (MA1011-1016 and MA3860-3865), each encoding the five subunits of the Cdh complex (CdhABCDE). The amino acid sequences of each cluster share greater than 90% identity 52. A remarkable feature of the genomic sequences of M. mazei and M. acetivorans is the extensive gene redundancy which has raised questions regarding the expression and physiological significance of duplicated genes 22, 53. The genome of M. mazei also contains >90% identical duplicate gene clusters encoding the Cdh complex; however, transcriptional profiling was unable to distinguish the level of expression between clusters with strict confidence due to the high sequence identity and potential cross hybridization 11. This same limitation applies to the DNA microarray results for M. acetivorans reported here, although data for MA1014, MA1016, MA3863, and MA3865 were in agreement with the proteomic results (Table 1) indicating up regulation of both operons in acetate- vs. methanol-grown cells. The three subunits encoded by MA3860-3862 and MA1014-1016 (Table 1) were in substantially greater abundance in acetate-grown vs. methanol grown cells (Table 1). Each of the six proteins was identified by high confidence MS/MS peptide sequencing of at least three unique peptide pairs. Co-transcription of all the genes in each cluster from M. acetivorans is supported by arrangement in the genome 52 (Fig. 2) which is identical to the operon characterized for M. thermophila54. These results strongly indicate that both Cdh complexes function in acetate-grown cells. The microarray results show MA4399, encoding the CdhA subunit of the Cdh complex, is up regulated in acetate-grown cells suggesting a role for this additional CdhA subunit in the conversion of acetate to methane. However, MA4399 is remote from the two complete cdh operons and not clustered with genes encoding the other four subunits of the complex; thus, the role for this additional CdhA subunit is not immediately apparent. Finally, these results further demonstrate how the combination of proteomic and microarray approaches extends the information beyond that achievable by either approach alone.
A previous report 30 described a co-transcribed gene cluster (MA0658-0665) in M. acetivorans proposed to encode an eight-subunit membrane-bound complex (Ma-Rnf). The report also showed three of the subunits are in greater abundance for acetate- vs. methanol-grown M. acetivorans30. The microarray analysis reported here showed four of the genes were up regulated in acetate-grown cells (Table 1) consistent with the previously reported proteomic analyses 30. The proposed function for Ma-Rnf is to transfer electrons from reduced ferredoxin to methanophenazine generating a Na+ gradient (high outside) (Fig. 3A). This electron transport chain is distinct from that proposed for freshwater Methanosarcina species 55-57 (Fig. 3B) for which the Ech hydrogenase oxidizes ferredoxin producing H2 that is oxidized by the Vho hydrogenase generating a H+ gradient (high outside). Transcriptional profiling (Table 1) also showed greater expression of genes (MA4566-4570 and MA4572) in acetate vs. methanol-grown M. acetivorans previously shown to be co-transcribed with genes (MA4566-4572) annotated to encode a seven-subunit Na+/H+ antiporter (Ma-Mrp). The relative levels of the transcripts were consistent with the previously reported proteomic analyses 30 for which two of the gene products (MA4567 and MA4568) were in greater abundance in acetate-grown cells. The antiporter is proposed to function during growth on acetate by exchanging the Ma-Rnf-generated Na+ gradient (Fig. 3A) for a H+ gradient that drives ATP synthesis 30.
Figure 3.
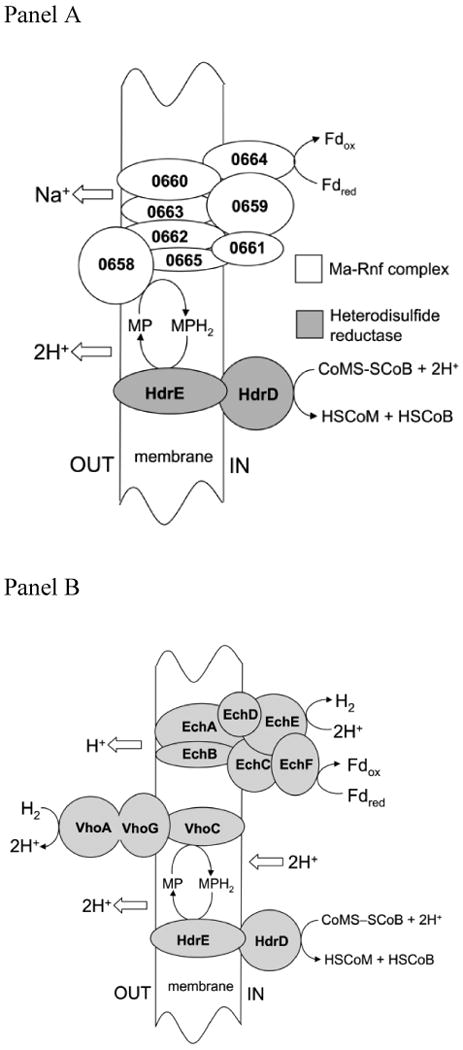
Panel A. Electron transport proposed for acetate-grown Methanosarcina acetivorans. The numbers represent the loci of genes encoding subunits of Ma-Rnf. Fd, ferredoxin; MP, methanophenazine; CoM; coenzyme M, CoB; coenzyme B; HdrDE; heterodisulfide reductase. Panel B. Electron transport proposed for acetate-grown freshwater Methanosarcina species. EchA-F, subunits of the Ech hydrogenase; Fd, ferredoxin; VhoACG, subunits of the Vho hydrogenase; MP, methanophenazine; CoM; coenzyme M, CoB; coenzyme B; HdrDE; heterodisulfide reductase.
A role for flavodoxins in the pathway of acetate conversion to methane has not been reported for any methanogenic species. Microarray analysis (Table 1) identified a flavodoxin gene (MA1799) up regulated 3-fold in acetate-grown M. acetivorans. This flavodoxin has only 26% identity to the flavodoxin encoded by MA2699 that is expressed 29-fold greater in methanol-grown cells (Table 1), consistent with different electron transport functions for each flavodoxin that are specific for growth with the respective substrates. The only flavodoxin encoded in the M. mazei genome (MM0261) with identity to MA1799 (27%) was not reported among differentially expressed genes 11. The only other flavodoxin gene (MM0637) reported up regulated in acetate- vs. methanol-grown M. mazei has no homologs in the genome of M. acetivorans52. These results suggest different flavodoxins function in acetate-grown M. acetivorans and M. mazei that further distinguishes electron transport in marine vs. freshwater Methanosarcina species.
A polyferredoxin, and expression of the encoding gene (MA2867), was greater in acetate- vs. methanol-grown cells (Table 1) consistent with a role in electron transport during growth with acetate. MA2867 is clustered with MA2868 (Fig. 2), the product of which was in greater abundance in acetate-grown cells. Interestingly, MA2868 is a fusion of hdrA and mvhD. HdrA is a subunit of the HdrABC-type of heterodisulfide reductase that functions to reduce CoM-S-S-CoB in the pathway for reduction of CO2 to methane with H2 as the electron donor. HdrA is the entry point for transfer of electrons to the HdrBC subunits harboring the site for reduction of the heterodisulfide 58, 59. MvhD is a subunit of the Mvh hydrogenase that functions in H2-utilizing CO2-reducing species other than Methanosarcina52. It is proposed that the MvhD subunit mediates electron transfer to the HdrA subunit of the HdrABC-type of heterodisulfide reductase 59. These results are consistent with an electron transport role for the putative HdrA∷MvhD fusion protein during growth on acetate. However, M. acetivorans is unable to reduce CO2 to methane with H2, and the HdrDE-type of heterodisulfide reductase (Fig. 3A) is postulated to function during growth on acetate 30. Thus, a role for the HdrABC-type during growth on acetate is unlikely and raises the question of a role for the putative HdrA∷MvhD fusion protein during growth on acetate.
Iron-sulfur flavoprotein (Isf) was previously hypothesized to function in the electron transport scheme of acetate-grown M. thermophila60, and more recently for M. mazei11. Remarkably, the genome of M. acetivorans is annotated with nineteen isf homologs 52 for which microarray analysis showed two (MA2167 and MA0406) were up regulated in acetate- vs. methanol-grown cells (Table 1). This result is consistent with M. mazei based on the up regulation in acetate-grown cells of an isf homolog 11, among eleven isf homologs, in the genome of M. mazei52. The recent crystal structures of Isf from M. thermophila and Archaeoglobus fulgidus shows similarity to rubredoxin:oxygen oxidoreductase, an enzyme proposed to function in oxidative stress by reducing O2 to water 61. Indeed, the prototypic Isf from M. thermophila reduces O2 and H2O2 to water 62. Thus, it is possible Isf paralogs function in the oxidative stress response rather than an electron transport function in the acetate pathway.
The protein product of MA3972 was in greater abundance, and expression of the gene up regulated in acetate-grown cells (Table 1). Although MA3972 is annotated as a conserved hypothetical protein with a molecular mass of 59 kDa 52, the sequence is 90% identical to MM0940 of M. mazei encoding a 59-kDa putative flavoprotein of unknown function that is also up regulated in acetate-grown cells 11. The results suggest that the product of MM0940 and MA3972 is a core flavoprotein with an unknown function in the acetate pathways of freshwater and marine Methanosarcina species.
Stress
The previously reported 2-D gel proteomic analysis of M. acetivorans identified a peroxiredoxin (MA4103) that was in greater amounts in acetate- vs. methanol-grown cells 10. Peroxiredoxins reduce H2O2 to water in response to oxidative stress. The microarray results reported here identified seven additional genes, with potential to respond to oxidative stress, that were up regulated in acetate-grown cells (Table 1 and Fig. 4). MA4103 encodes a peroxiredoxin, and MA0406 and MA2167 encode paralogs of the Isf family for which the prototype reduces O2 and H2O2 to water 62. MA3743 is annotated to encode flavoprotein A which reduces O2 to water in Methanobrevibacter arboriphilus63. MA3212 is annotated as thioredoxin, a family of proteins acting as general disulfide reductases in the oxidative stress response. In contrast, methanol-grown cells showed no elevated expression of genes or elevated abundance of proteins involved in the oxidative stress response. Further, MA1189 is annotated to encode a tryptophan repressor binding protein (WrbA) previously proposed to be a regulatory protein in Escherichia coli; however, it was recently shown that WrbA from E. coli is the prototype of a new family of NAD(P)H:quinone oxidoreductases implicated in the oxidative stress response of procaryotes from both the Bacteria and Archaea domains 64. Nonetheless, a regulatory role in the stress response cannot be ruled out at this juncture. Overall, these results indicate that an oxidative stress response is induced in acetate-grown vs. methanol-grown M. acetivorans. Cells cultured with either substrate were grown under strict anaerobic conditions with the same amounts of reducing agent. One possible explanation for the oxidative stress response in acetate-grown cells is the long doubling time (84 h) compared to methanol-grown cells (42 h) which induced a general stress response that includes oxidative stress. Genes encoding other stress-related proteins (MA0133, MA1682, MA3284, MA4542) were also expressed greater in acetate- vs. methanol-grown cells (Table 1).
Figure 4.
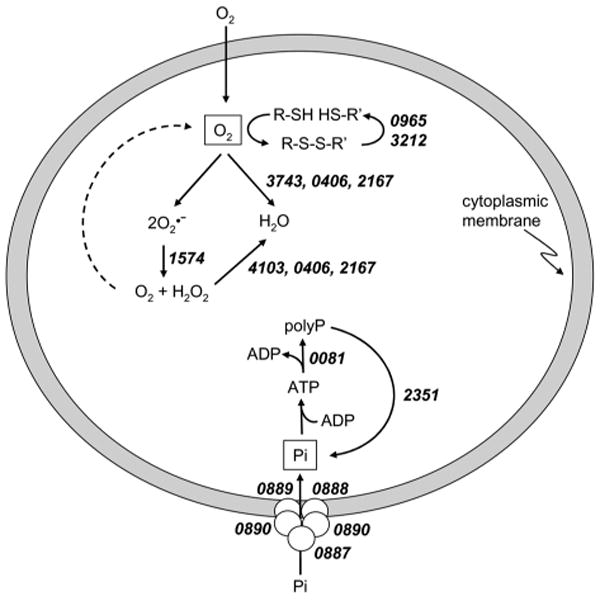
Genes implicated in the stress response of acetate-grown Methanosarcina acetivorans. See Table 1 for gene annotations.
Proteomic and microarray approaches also identified a suite of genes that function in polyphosphate (polyP) formation for which the protein products were more abundant or expression of the genes were greater in acetate-grown cells (Table 1, Fig. 4). The gene cluster MA0081-0083 (Fig. 2) encodes for polyphosphate kinase (MA0081) and exopolyphosphatase (MA0083), both of which have been characterized from the Bacteria domain 65. The former catalyzes the synthesis of polyP from the gamma phosphate of ATP, and the latter cleaves inorganic phosphate from polyP in a processive manner to maintain homeostasis of polyP levels. Both enzymes in Escherichia coli are encoded on an operon 66 reminiscent of the MA0081-0083 gene cluster in M. acetivorans except for an intervening gene (MA0082) of unknown function (Fig. 2). Microarray analysis showed expression of a gene encoding a second exopolyphosphatase (MA2351) was also up regulated in acetate-grown cells (Table 1). Also, either the protein product or gene expression increased in response to acetate for three genes (MA0887, MA0889 and MA0891) of a five-gene cluster (MA0887-0891) (Fig. 2) encoding proteins of the Pst phosphate transport system described in species from the Bacteria domain 67. Overall, these results are consistent with greater expression of genes in acetate-grown cells of M. acetivorans that function in polyP formation. The organization of genes encoding the Pst system (Fig. 2) is identical to that in E. coli which includes a gene encoding the regulatory protein PhoU (MA0891). In the Bacteria domain, PhoU is part of the phosphate regulon which functions to assimilate phosphate wherein PhoU is a negative effector of the response regulator in a two-component regulatory system 67. The results are consistent with a role for the product of MA0891 in the regulation of phosphate uptake by one of several uncharacterized two-component systems in M. acetivorans discussed in the next section.
A role has been proposed for polyP in response to a variety of stressors, including oxidative stress, for members of the Bacteria domain 65, 68, 69. In the Bacteria, ppk- mutants lacking polyP fail to express rpoS encoding a regulatory sigma factor that governs the expression of genes essential to resist a variety of stress conditions during stationary phase 70. Thus, enzymes required for polyP synthesis in acetate-grown cells of M. acetivorans is consistent with a role in the general stress response. Although a specific function has yet to be determined, it is interesting to note that polyphosphate kinase is necessary to protect Salmonella enterica from stress imposed by growth in the presence of acetic acid 71. It is proposed that polyphosphate acts as a chemical chaperonin, helping to refold proteins denatured by acetic acid and oxidative stress.
Regulation
The genome of M. acetivorans is annotated with an unusually large number (fifty) of sensory transduction histidine kinase proteins and a considerably smaller number (eighteen) of cognate proteins with response regulator receiver domains 22. Further, only one of the proteins with a receiver domain is of sufficient length to also harbor an effector domain. These anomalies compared to the two-component regulatory systems in the Bacteria domain raise questions regarding the expression and roles for the putative sensory and response proteins of M. acetivorans. The results indicate that fifteen genes encoding sensory proteins were expressed in M. acetivorans (not shown) of which nine were differentially expressed between methanol- and acetate-grown cells (Table 1), a result suggesting possible roles for these proteins during growth on acetate or methanol. Furthermore, genes encoding five proteins with receiver domains were expressed (not shown) for which two were differentially expressed (Table 1).
More energy is available when converting methanol (ΔG°′ = -106.5 kJ) vs. acetate (ΔG°′ = -36 kJ) to methane; thus, Methanosarcina species regulate genes specific for each substrate with a preference for methanol 45. However, little is understood regarding the mechanism of gene regulation in response to the growth substrate. Several regulatory proteins were in greater abundance (MA0368, MA1763, and MA2914), and expression of genes encoding regulatory proteins up regulated (MA0924, MA1122, MA1487, MA2914, and MA3916) in methanol-grown M. acetivorans (Table 1). The results suggest roles for these proteins in regulating the expression of genes specific to the growth substrate. MA0368 and MA1487 encode TetR family regulatory proteins, and the genome of M. acetivorans is annotated with an additional eleven genes encoding TetR family proteins 52 suggesting a role in regulation. MA1487 is clustered with MA1486, annotated as a MoaA/NifB family protein, that is also expressed greater in methanol- vs. acetate-grown cells. MoaA functions in the synthesis of molybdopterin, a cofactor of the formyl-methanofuran dehydrogenase (MA0304-0309) which functions in the pathway of methanol conversion to methane and is in greater abundance in methanol-grown cells (Table 1). MoaB (MA0187), also involved in molybdopterin biosynthesis, was also in greater abundance in methanol-grown cells. The MarR family regulatory protein encoded by MA1122 has 76% identity to MarR encoded by MM3195 in M. mazei that is also expressed greater in methanol- vs. acetate-grown cells of M. mazei11. All other genes encoding regulatory proteins expressed in M. acetivorans were not among the genes reported to be differentially expressed in M. mazei11.
Microarray analysis showed MA3916 encoding the GlnK regulatory protein was up regulated in methanol-grown cells (Table 1) and previously shown to be a regulator of ammonium assimilation in M. mazei72. MA3916 is clustered with MA3917 and MA3918 (Fig. 2), both encoding ammonium transporters, also up regulated in methanol-grown cells (Table 1), consistent with the regulatory function of GlnK. The apparent requirement for increased ammonium in methanol- vs. acetate-grown cells could reflect the greater growth rate in methanol-grown cells and an increased need for nutrients.
Conclusions
A combination of advanced proteomics and DNA microarray analyses of M. acetivorans, the first for any methanosarcina species, has extended the physiological understanding for growth and methanogenesis with either acetate or methanol as substrates. The two experimental approaches were in good agreement, and the different capabilities were highly complementary leading to discovery not possible by each method alone. The proteomic results indicated that duplicate CO dehydrogenase/acetyl-CoA complexes function in the conversion of acetate to methane, a question not resolvable by microarray analysis alone. The gene expression ratios for stress-related proteins were greater in acetate- versus methanol-grown cells supporting an earlier proteomic study suggesting that a general stress response is elicited by growth on acetate. Novel roles were suggested for electron transport proteins specific for either growth substrate. The results also identified several regulatory proteins, commonly found in the Bacteria domain, suggested to function during growth of M. acetivorans with acetate or methanol. Finally, the results also suggested roles for two-component regulatory systems specific for each growth substrate.
Supplementary Material
Table S1. Proteins with at least 3-fold, and transcripts with at least 2.5-fold, difference, in acetate- versus methanol-grown M. acetivorans.
Acknowledgments
This work was supported by NSF grant MCB-0110762 to J.G.F., NIH grant GM15847 to B.L.K, and a Department of Energy Biosciences Division grant award DE-FG03-86ER13498 to R.P.G. Contribution number 886 from the Barnett Institute.
Footnotes
The abbreviations used are: DNA, deoxyribonucleic acid; NCBI, National Center for Biotechnology Information; RT, reverse transcriptase; DNase, deoxyribonuclease; dATP, deoxyadenosine triphosphate; dCTP, deoxycytidine triphosphate; dGTP, deoxyguanosine triphosphate; dTTP, deoxythimadine triphosphate; dUTP, deoxyuridine triphosphate; RNase, ribonuclease; cDNA, complementary deoxyribonucleic acid; MCMC, Markov Chain Monte Carlo; COG, clusters of orthologous groups; THMPT, tetrahydromethanopterin.
References
- 1.Zinder S. Physiological ecology of methanogens. In: Ferry JG, editor. Methanogenesis. Chapman and Hall; New York, NY: 1993. pp. 128–206. [Google Scholar]
- 2.McCarty PL. The development of anaerobic treatment and its future. Water Sci Technol. 2001;44(8):149–56. [PubMed] [Google Scholar]
- 3.Ferry JG. One-carbon metabolism in methanogenic anaerobes. In: Ljungdahl LG, Adams MW, Barton LL, Ferry JG, Johnson MK, editors. Biochemistry and Physiology of Anaerobic Bacteria. Springer-Verlag; New York: 2003. pp. 143–156. [Google Scholar]
- 4.Goodchild A, Saunders NFW, Ertan H, Raftery M, Guilhaus M, Curmi PMG, Cavicchioli R. A proteomic determination of cold adaptation in the antarctic archaeon, Methanococcoides burtonii. Mol Microbiol. 2004;53:309–321. doi: 10.1111/j.1365-2958.2004.04130.x. [DOI] [PubMed] [Google Scholar]
- 5.Goodchild A, Raferty M, Saunders NFW, Guilhaus M, Cavicchioli R. Biology of the cold-adapted archaeon, Methanococcoides burtonii, determined by proteomics using liquid chromatography-tandem mass spectrometry. J Proteome Res. 2004;3:1164–1176. doi: 10.1021/pr0498988. [DOI] [PubMed] [Google Scholar]
- 6.Saunders NFW, Goodchild A, Raftery M, Guilhaus M, Curmi PMG, Cavicchioli R. Predicted roles for hypothetical proteins in the low-temperature expressed proteome of the antarctic archaeon Methanococcoides burtonii. J Proteome Res. 2005;4:464–472. doi: 10.1021/pr049797+. [DOI] [PubMed] [Google Scholar]
- 7.Goodchild A, Raftery M, Saunders NFW, Guilhaus M, Cavicchioli R. Cold adaptation of the antarctic archaeon, Methanococcoides burtonii, assessed by proteomics using ICAT. J Proteome Res. 2005;4:473–480. doi: 10.1021/pr049760p. [DOI] [PubMed] [Google Scholar]
- 8.Nichols DS, Miller MR, Davies NW, Goodchild A, Raftery M, Cavicchioli R. Cold adaptation in the antarctic archaeon Methanococcoides burtonii involves membrane lipid unsaturation. J Bacteriol. 2004;186(24):8508–8515. doi: 10.1128/JB.186.24.8508-8515.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Q, Li L, Rejtar T, Karger BL, Ferry JG. The proteome of Methanosarcina acetivorans. Part I, an expanded view of the biology of the cell. J Proteome Res. 2005;4:112–128. doi: 10.1021/pr049832c. [DOI] [PubMed] [Google Scholar]
- 10.Li Q, Li L, Rejtar T, Karger BL, Ferry JG. The proteome of Methanosarcina acetivorans. Part II, comparison of protein levels in acetate- and methanol-grown cells. J Proteome Res. 2005;4:129–136. doi: 10.1021/pr049831k. [DOI] [PubMed] [Google Scholar]
- 11.Hovey R, Lentes S, Ehrenreich A, Salmon K, Saba K, Gottschalk G, Gunsalus RP, Deppenmeier U. DNA microarray analysis of Methanosarcina mazei Go1 reveals adaptation to different methanogenic substrates. Mol Genet Genomics. 2005;273:225–239. doi: 10.1007/s00438-005-1126-9. [DOI] [PubMed] [Google Scholar]
- 12.Veit K, Ehlers C, Ehrenreich A, Salmon K, Hovey R, Gunsalus RP, Deppenmeier U, Schmitz RA. Global transcriptional analysis of Methanosarcina mazei strain Go1 under different nitrogen availabilities. Mol Genet Genomics. 2006 doi: 10.1007/s00438-006-0117-9. [DOI] [PubMed] [Google Scholar]
- 13.Ding YH, Zhang SP, Tomb JF, Ferry JG. Genomic and proteomic analyses reveal multiple homologs of genes encoding enzymes of the methanol:coenzyme M methyltransferase system that are differentially expressed in methanol- and acetate-grown Methanosarcina thermophila. FEMS Microbiol Lett. 2002;215(1):127–132. doi: 10.1111/j.1574-6968.2002.tb11381.x. [DOI] [PubMed] [Google Scholar]
- 14.Farhoud MH, Wessels HJ, Steenbakkers PJ, Mattijssen S, Wevers RA, van Engelen BG, Jetten MS, Smeitink JA, van den Heuvel LP, Keltjens JT. Protein complexes in the archaeon Methanothermobacter thermautotrophicus analyzed by Blue Native/SDS-PAGE and mass spectrometry. Mol Cell Proteomics. 2005;4(11):1653–63. doi: 10.1074/mcp.M500171-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Zhu W, Reich CI, Olsen GJ, Giometti CS, Yates JR., 3rd Shotgun proteomics of Methanococcus jannaschii and insights into methanogenesis. J Proteome Res. 2004;3(3):538–48. doi: 10.1021/pr034109s. [DOI] [PubMed] [Google Scholar]
- 16.Giometti CS, Reich C, Tollaksen S, Babnigg G, Lim H, Zhu W, Yates J, Olsen G. Global analysis of a “simple” proteome: Methanococcus jannaschii. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;782(12):227–43. doi: 10.1016/s1570-0232(02)00568-8. [DOI] [PubMed] [Google Scholar]
- 17.Porat I, Kim W, Hendrickson EL, Xia Q, Zhang Y, Wang T, Taub F, Moore BC, Anderson IJ, Hackett M, Leigh JA, Whitman WB. Disruption of the operon encoding Ehb hydrogenase limits anabolic CO2 assimilation in the archaeon Methanococcus maripaludis. J Bacteriol. 2006;188(4):1373–1380. doi: 10.1128/JB.188.4.1373-1380.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia Q, Hendrickson EL, Zhang Y, Wang T, Taub F, Moore BC, Porat I, Whitman WB, Hackett M, Leigh JA. Quantitative proteomics of the archaeon Methanococcus maripaludis validated by microarray analysis and real time PCR. Mol Cell Proteomics. 2006;5(5):868–881. doi: 10.1074/mcp.M500369-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forbes AJ, Patrie SM, Taylor GK, Kim YB, Jiang L, Kelleher NL. Targeted analysis and discovery of posttranslational modifications in proteins from methanogenic Archaea by top-down MS. Proc Natl Acad Sci U S A. 2004;101:2678–2683. doi: 10.1073/pnas.0306575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukhopadhyay B, Johnson EF, Wolfe RS. A novel pH2 control on the expression of flagella in the hyperthermophilic strictly hydrogenotrophic methanarchaeaon Methanococcus jannaschii. Proc Natl Acad Sci U S A. 2000;97(21):11522–11527. doi: 10.1073/pnas.97.21.11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giometti CS, Reich CI, Tollaksen SL, Babnigg G, Lim H, Yates JR, Olsen GJ. Structural modifications of Methanococcus jannaschii flagellin proteins revealed by proteome analysis. Proteomics. 2001;1:1033–1042. doi: 10.1002/1615-9861(200108)1:8<1033::AID-PROT1033>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 22.Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, FitzHugh W, Calvo S, Engels R, Smirnov S, Atnoor D, Brown A, Allen N, Naylor J, Stange-Thomann N, DeArellano K, Johnson R, Linton L, McEwan P, McKernan K, Talamas J, Tirrell A, Ye W, Zimmer A, Barber RD, Cann I, Graham DE, Grahame DA, Guss AM, Hedderich R, Ingram-Smith C, Kuettner HC, Krzycki JA, Leigh JA, Li W, Liu J, Mukhopadhyay B, Reeve JN, Smith K, Springer TA, Umayam LA, White O, White RH, de Macario EC, Ferry JG, Jarrell KF, Jing H, Macario AJ, Paulsen I, Pritchett M, Sowers KR, Swanson RV, Zinder SH, Lander E, Metcalf WW, Birren B. The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 2002;12(4):532–42. doi: 10.1101/gr.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metcalf WW, Zhang JK, Apolinario E, Sowers KR, Wolfe RS. A genetic system for Archaea of the genus Methanosarcina. Liposome-mediated transformation and construction of shuttle vectors. Proc Natl Acad Sci USA. 1997;94:2626–2631. doi: 10.1073/pnas.94.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang JK, Pritchett MA, Lampe DJ, Roberton HM, Metcalf WW. In vivo transposon mutagenesis of the methanogenic archaeon Methanosarcina acetivorans C2A using a modified version of the insect mariner-family transposable element Himar1. Proc Natl Acad Sci. 2000;97(17):9665–9670. doi: 10.1073/pnas.160272597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang JK, White AK, Kuettner HC, Boccazzi P, Metcalf WW. Directed mutagenesis and plasmid-based complementation in the methanogenic archaeon Methanosarcina acetivorans C2A demonstrated by genetic analysis of proline biosynthesis. J Bacteriol. 2002;184(5):1449–1454. doi: 10.1128/JB.184.5.1449-1454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pritchett MA, Zhang JK, Metcalf WW. Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl Environ Microbiol. 2004;70(3):1425–1433. doi: 10.1128/AEM.70.3.1425-1433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pritchett MA, Metcalf WW. Genetic, physiological and biochemical characterization of multiple methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Mol Microbiol. 2005;56(5):1183–94. doi: 10.1111/j.1365-2958.2005.04616.x. [DOI] [PubMed] [Google Scholar]
- 28.Rother M, Boccazzi P, Bose A, Pritchett MA, Metcalf WW. Methanol-dependent gene expression demonstrates that methyl-coenzyme M reductase is essential in Methanosarcina acetivorans C2A and allows isolation of mutants with defects in regulation of the methanol utilization pathway. J Bacteriol. 2005;187(16):5552–5559. doi: 10.1128/JB.187.16.5552-5559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahapatra A, Patel A, Soares JA, Larue RC, Zhang JK, Metcalf WW, Krzycki JA. Characterization of a Methanosarcina acetivorans mutant unable to translate UAG as pyrrolysine. Mol Microbiol. 2006;59(1):56–66. doi: 10.1111/j.1365-2958.2005.04927.x. [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Li L, Rejtar T, Lessner DJ, Karger BL, Ferry JG. Electron transport in the pathway of acetate conversion to methane in the marine archaeon Methanosarcina acetivorans. J Bacteriol. 2006;188:702–710. doi: 10.1128/JB.188.2.702-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Q, Li l, Rejtar T, Karger BL, Ferry JG. The proteome of Methanosarcina acetivorans. Part I, an expanded view of the biology of the cell. J Proteome Res. 2004;4:112–128. doi: 10.1021/pr049832c. [DOI] [PubMed] [Google Scholar]
- 32.Wolfe RS, Higgins IJ. Microbial biochemistry of methane: a study in contrasts. University Park Press; Baltimore: 1979. [Google Scholar]
- 33.Nesterenko MV, Tilley M, Upton SJ. A simple modification of Blum's silver stain method allows for 30 minute detection of proteins in polyacrylamide gels. J Biochem Biophys Methods. 1994;28:239–242. doi: 10.1016/0165-022x(94)90020-5. [DOI] [PubMed] [Google Scholar]
- 34.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 35.Washburn MP, Ulaszek R, Deciu C, Schieltz DM, Yates JR., 3rd Analysis of quantitative proteomic data generated via multidimensional protein identification technology. Anal Chem. 2002;74:1650–1657. doi: 10.1021/ac015704l. [DOI] [PubMed] [Google Scholar]
- 36.Andreev VP, Li L, Rejtar T, Li Q, Ferry JG, Karger BL. New algorithm for 15N/14N quantitation with LC-ESI-MS using an LTQ-FT mass spectrometer. J Proteome Res. 2006;5:2039–2045. doi: 10.1021/pr060105m. [DOI] [PubMed] [Google Scholar]
- 37.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 38.Rohlin L, Trent JD, Salmon K, Kim U, Gunsalus RP, Liao JC. Heat shock response of Archaeoglobus fulgidus. J Bacteriol. 2005;187(17):6046–6057. doi: 10.1128/JB.187.17.6046-6057.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pruss BM, Campbell JW, Van Dyk TK, Zhu C, Kogan Y, Matsumura P. FlhD/FlhC is a regulator of anaerobic respiration and the Entner-Doudoroff pathway through induction of the methyl-accepting chemotaxis protein Aer. J Bacteriol. 2003;185(2):534–43. doi: 10.1128/JB.185.2.534-543.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hyduke DR, Rohlin L, Kao KC, Liao JC. A software package for cDNA microarray data normalization and assessing confidence intervals. Omics. 2003;7(3):227–34. doi: 10.1089/153623103322452369. [DOI] [PubMed] [Google Scholar]
- 41.Tseng GC, Oh MK, Rohlin L, Liao JC, Wong WH. Issues in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res. 2001;29(12):2549–57. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oh MK, Rohlin L, Kao KC, Liao JC. Global expression profiling of acetate-grown Escherichia coli. J Biol Chem. 2002;277(15):13175–13183. doi: 10.1074/jbc.M110809200. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Li L, Rejtar T, Karger BL, Ferry JG. The proteome of Methanosarcina acetivorans. Part II, comparison of protein levels in acetate- and methanol-grown cells. J Proteome Res. 2004;4:129–136. doi: 10.1021/pr049831k. [DOI] [PubMed] [Google Scholar]
- 44.Julka S, Regnier F. Quantification in proteomics through stable isotope coding: a review. J Proteome Res. 2004;3(3):350–63. doi: 10.1021/pr0340734. [DOI] [PubMed] [Google Scholar]
- 45.Jablonski PE, DiMarco AA, Bobik TA, Cabell MC, Ferry JG. Protein content and enzyme activities in methanol- and acetate-grown Methanosarcina thermophila. J Bacteriol. 1990;172:1271–1275. doi: 10.1128/jb.172.3.1271-1275.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deppenmeier U. The unique biochemistry of methanogenesis. Prog Nucleic Acid Res Mol Biol. 2002;71:223–283. doi: 10.1016/s0079-6603(02)71045-3. [DOI] [PubMed] [Google Scholar]
- 47.Bose A, Pritchett MA, Rother M, Metcalf WW. Differential regulation of the three methanol methyltransferase Isozymes in Methanosarcina acetivorans C2A. J Bacteriol. 2006;188(20):7274–7283. doi: 10.1128/JB.00535-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lange M, Tolker-Nielsen T, Molin S, Ahring BK. In situ reverse transcription-PCR for monitoring gene expression in individual Methanosarcina mazei S-6 cells. Appl Environ Microbiol. 2000;66(5):1796–800. doi: 10.1128/aem.66.5.1796-1800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Biase A, Macario AJL, de Macario EC. Effect of heat stress on promoter cytosol of the archae binding by transcription factors in the on Methanosarcina mazeii. Gene. 2002;282(12):189–197. doi: 10.1016/s0378-1119(01)00832-0. [DOI] [PubMed] [Google Scholar]
- 50.Conway De Macario E, Clarens M, Macario AJ. Archaeal grpE: transcription in two different morphologic stages of Methanosarcina mazei and comparison with dnaK and dnaJ. J Bacteriol. 1995;177(3):544–550. doi: 10.1128/jb.177.3.544-550.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferry JG. Enzymology of one-carbon metabolism in methanogenic pathways. FEMS Microbiol Rev. 1999;23(1):13–38. doi: 10.1111/j.1574-6976.1999.tb00390.x. [DOI] [PubMed] [Google Scholar]
- 52.http://www.tigr.org.
- 53.Deppenmeier U, Johann A, Hartsch T, Merkl R, Schmitz RA, Martinez-Arias R, Henne A, Wiezer A, Baumer S, Jacobi C, Bruggemann H, Lienard T, Christmann A, Bomeke M, Steckel S, Bhattacharyya A, Lykidis A, Overbeek R, Klenk HP, Gunsalus RP, Fritz HJ, Gottschalk G. The genome of Methanosarcina mazei: evidence for lateral gene transfer between Bacteria and Archaea. J Mol Microbiol Biotechnol. 2002;4(4):453–61. [PubMed] [Google Scholar]
- 54.Maupin-Furlow JA, Ferry JG. Analysis of the CO dehydrogenase/acetyl-coenzyme A synthase operon of Methanosarcina thermophila. J Bacteriol. 1996;178:6849–6856. doi: 10.1128/jb.178.23.6849-6856.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hedderich R. Energy-converting [NiFe] hydrogenases from Archaea and extremophiles: ancestors of complex I. J Bioenerg Biomembr. 2004;36:65–75. doi: 10.1023/b:jobb.0000019599.43969.33. [DOI] [PubMed] [Google Scholar]
- 56.Meuer J, Bartoschek S, Koch J, Kunkel A, Hedderich R. Purification and catalytic properties of Ech hydrogenase from Methanosarcina barkeri. Eur J Biochem. 1999;265(1):325–335. doi: 10.1046/j.1432-1327.1999.00738.x. [DOI] [PubMed] [Google Scholar]
- 57.Meuer J, Kuettner HC, Zhang JK, Hedderich R, Metcalf WW. Genetic analysis of the archaeon Methanosarcina barkeri Fusaro reveals a central role for Ech hydrogenase and ferredoxin in methanogenesis and carbon fixation. Proc Natl Acad Sci U S A. 2002;99(8):5632–5637. doi: 10.1073/pnas.072615499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Setzke E, Hedderich R, Heiden S, Thauer RK. H2:heterodisulfide oxidoreductase complex from Methanobacterium thermoautotrophicum: composition and properties. Eur J Biochem. 1994;220:139–148. doi: 10.1111/j.1432-1033.1994.tb18608.x. [DOI] [PubMed] [Google Scholar]
- 59.Stojanowic A, Mander GJ, Duin EC, Hedderich R. Physiological role of the F420-non-reducing hydrogenase (Mvh) from Methanothermobacter marburgensis. Arch Microbiol. 2003;180(3):194–203. doi: 10.1007/s00203-003-0577-9. [DOI] [PubMed] [Google Scholar]
- 60.Latimer MT, Painter MH, Ferry JG. Characterization of an iron-sulfur flavoprotein from Methanosarcina thermophila. J Biol Chem. 1996;271:24023–24028. doi: 10.1074/jbc.271.39.24023. [DOI] [PubMed] [Google Scholar]
- 61.Andrade SLA, Cruz F, Drennan CL, Ramakrishnan V, Rees DC, Ferry JG, Einsle O. Structures of the iron-sulfur flavoproteins from Methanosarcina thermophila and Archaeoglobus fulgidus. J Bacteriol. 2005;187:3848–3854. doi: 10.1128/JB.187.11.3848-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cruz FC, Ferry JG. Interaction of iron-sulfur flavoprotein with oxygen and hydrogen peroxide. Biochim Biophys Acta. 2006;1760:858–864. doi: 10.1016/j.bbagen.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 63.Seedorf H, Dreisbach A, Hedderich R, Shima S, Thauer R. K, F420H2 oxidase (FprA) from Methanobrevibacter arboriphilus, a novel coenzyme F420 dependent enzyme involved in O2 detoxification. Arch Microbiol. 2004;182:126–137. doi: 10.1007/s00203-004-0675-3. [DOI] [PubMed] [Google Scholar]
- 64.Patridge EV, Ferry JG. WrbA from Escherichia coli and Archaeoglobus fulgidus is an NAD(P)H:quinone oxidoreductase. J Bacteriol. 2006;188(10):3498–3506. doi: 10.1128/JB.188.10.3498-3506.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kornberg A, Rao NN, Ault-Riche D. Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem. 1999;68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- 66.Akiyama M, Crooke E, Kornberg A. An exopolyphosphatase of Escherichia coli. The enzyme and its ppx gene in a polyphosphate operon. J Biol Chem. 1993;268(1):633–9. [PubMed] [Google Scholar]
- 67.van Veen HW. Phosphate transport in prokaryotes: molecules, mediators and mechanisms. Antonie Van Leeuwenhoek. 1997;72(4):299–315. doi: 10.1023/a:1000530927928. [DOI] [PubMed] [Google Scholar]
- 68.Crooke E, Akiyama M, Rao NN, Kornberg A. Genetically altered levels of inorganic polyphosphate in Escherichia coli. J Biol Chem. 1994;269(9):6290–5. [PubMed] [Google Scholar]
- 69.Rao NN, Kornberg A. Inorganic polyphosphate supports resistance and survival of stationary-phase Escherichia coli. J Bacteriol. 1996;178(5):1394–400. doi: 10.1128/jb.178.5.1394-1400.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shiba T, Tsutsumi K, Yano H, Ihara Y, Kameda A, Tanaka K, Takahashi H, Munekata M, Rao NN, Kornberg A. Inorganic polyphosphate and the induction of rpoS expression. Proc Natl Acad Sci U S A. 1997;94(21):11210–5. doi: 10.1073/pnas.94.21.11210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Price-Carter M, Fazzio TG, Vallbona EI, Roth JR. Polyphosphate kinase protects Salmonella enterica from weak organic acid stress. J Bacteriol. 2005;187(9):3088–99. doi: 10.1128/JB.187.9.3088-3099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ehlers C, Grabbe R, Veit K, Schmitz RA. Characterization of G1nK(1) from Methanosarcina mazei strain Go1: Complementation of an Escherichia coli glnK mutant strain by GlnK(1) J Bacteriol. 2002;184(4):1028–1040. doi: 10.1128/jb.184.4.1028-1040.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Proteins with at least 3-fold, and transcripts with at least 2.5-fold, difference, in acetate- versus methanol-grown M. acetivorans.