

Abstract
Titanium-mediated cross-coupling of allenic alcohols with alkynes has been investigated. Divergent reaction pathways were discovered that provide either stereodefined 1,4-dienes or substituted cross-conjugated trienes. In short, allene substitution plays a critical role in the determination of reaction pathway.
1. Introduction
The direct cross-coupling of unactivated π-systems could represent a powerful strategy for the convergent synthesis of complex molecules. Such processes provide an advantage over metal-catalyzed cross-coupling reactions1 that typically require the use of activated coupling partners (i.e. functionalized organometallic reagents and organic halides), as these “starting materials” often require numerous stoichiometric reactions for their preparation. While numerous examples of metal-mediated coupling reactions of unactivated π-systems have been described in intramolecular settings, few reports describe the success of such processes for intermolecular C–C bond formation.2 Where, in intramolecular C–C bond forming reactions, the control of reactivity and selectivity is controlled by the enforced proximity of the reacting π-systems, the analogous bimolecular processes are significantly more complex. To realize such intermolecular cross-coupling reactions, one must be able to control site selectivity (in the functionalization of each reacting π-component) and facial selectivity, while overcoming barriers associated with general reactivity.
Recently, we have described a variety of titanium-mediated cross-coupling reactions for the fusion of substituted and unactived π-systems (Scheme 1).3 While these coupling reactions have provided a convergent pathway to the synthesis of stereodefined tetrasubstituted 1,3-dienes (1), branched allylic systems (2) and 1,4-dienes (3), they represent only a small sample of the potential cross-coupling reactions possible based on the reactivity of metal–π complexes.
Scheme 1.
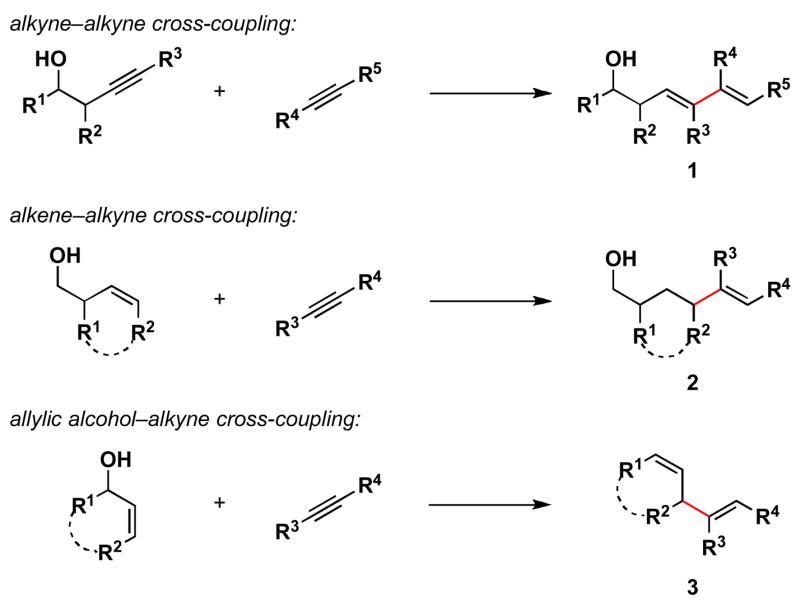
Directed cross-coupling reactions of unactivated π-systems.
Metal-mediated allene–alkyne cross-coupling is a relatively underdeveloped process.4 In the context of convergent synthesis of dienes, this mode of cross-coupling is known to be effective with only a limited subset of coupling partners, providing access to di- and trisubtituted products (Scheme 2). For example, allene hydrozirconation, followed by MAO-catalyzed allylzirconation of terminal alkynes provides a regio- and stereoselective route to 1,4-disubstituted 1,4-dienes (4+5→6).4d,e For more highly substituted products, cross-coupling between preformed titanium–alkyne complexes and monosubstituted allenes has been reported to provide trisubstituted 1,4-dienes, albeit with varying degrees of stereoselection (4+7→8).4a While cross-coupling of an alkyl substituted allene bearing a tethered benzyl ether (R1 = (CH2)2OBn) with a symmetrical alkyne (R2 and R3 = Bu) provides 1,4-dienes with a slight preference for the formation of the E-disubstituted olefin (E:Z = 64:36), selectivity is a complex function of the nature of the alkyne. For example, coupling of the same allene with a TMS substituted alkyne provides 1,4-diene products with a modest preference for the formation of the Z-olefin-containing 1,4-diene (E:Z = 25:75).
Scheme 2.
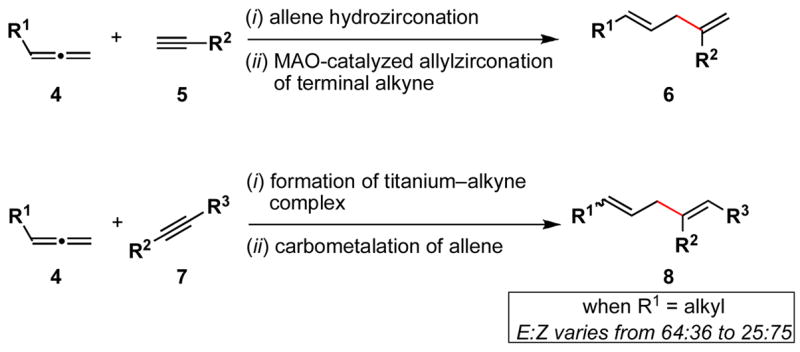
Known pathways to 1,4-dienes based on allene-alkyne cross-coupling.
Major challenges that exist in the development of a general allene–alkyne cross-coupling reaction include:
control in the functionalization of di- and tri-substituted allenes,
regioselection (for the functionalization of each π-component: allene and alkyne), and
stereoselection (in the formation of the substituted olefins resident in the products).
To address these issues, we initiated studies aimed at defining an alkoxide-directed allene–alkyne cross-coupling reaction.5 As depicted in Scheme 3, it was anticipated that formation of a titanium complex of 7, followed by exposure to a preformed allenic alkoxide (9) would afford a mixed titanate ester in a transient fashion. This reactive intermediate could then rearrange via directed carbometalation (A→10) or formal metallo-[3,3] rearrangment (B→11).6 Here, we describe titanium-mediated cross-coupling reactions between allenes and alkynes that provide either 1,4-dienes (10) or cross-conjugated trienes (11) in a stereoselective fashion.
Scheme 3.
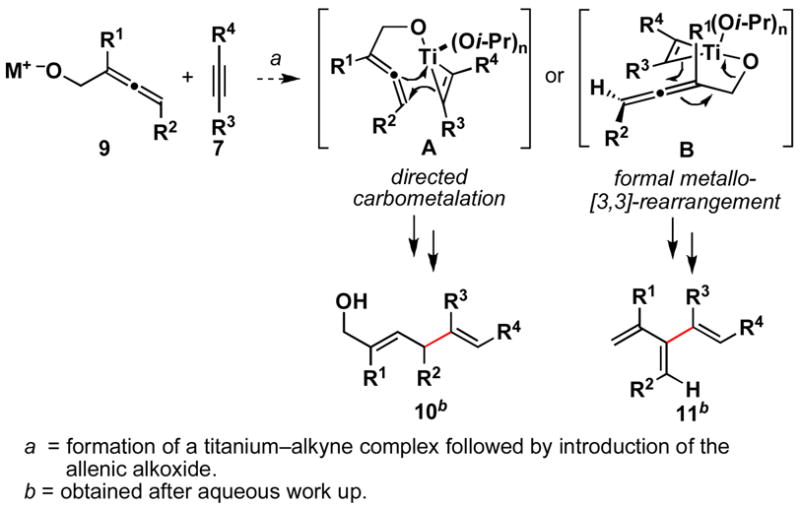
A titanium-mediated, alkoxide-directed allene–alkyne cross-coupling reaction.
2. Results
2.1. Synthesis of stereodefined 1,4-dienes
Our studies began with examination of the cross-coupling reaction between 2,3-butadiene-1-ol 12 and the symmetrical alkyne 13. As illustrated in Scheme 4, preformation of the metal–alkyne complex of 13, followed by addition of the lithium alkoxide of allene 12 and aqueous work up, provided a 3:1 mixture of coupled products in 53% yield. The major component of this mixture, identified as the stereodefined 1,4-diene 14, was accompanied by a small amount of a cross-conjugated triene (15). This initial result was viewed as quite promising, as no stereoisomeric 1,4-dienes were detected in the product mixture. Further, the stereoselection observed was consistent with a directed C–C bond forming process in preference to a non-directed process (the expected product from a non-directed cross-coupling is a 1,4-diene bearing a Z-allylic alcohol;4a not shown).
Scheme 4.
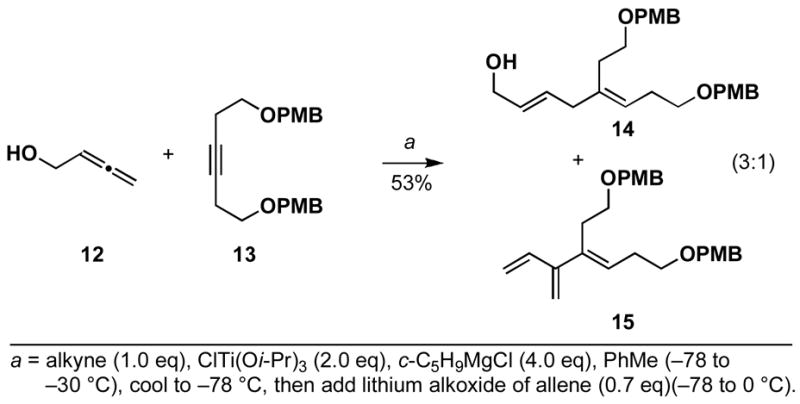
A titanium-mediated, alkoxide directed allene–alkyne cross-coupling reaction.
Cross-conjugated triene 15 was reasoned to be derived from a competition between the directed carbometalation pathway (A) and a formal metallo-[3,3] rearrangement process (B) (Scheme 3). In considering this competing pathway, it was reasoned that the mechanism of the reaction could derive from a pathway whereby C–C bond formation occurs in concert with C–O bond breaking (C; Figure 1),7 or by a pathway where directed carbometalation provides an intermediate bicyclo[3.2.0] metallacyclopentane, which then undergoes syn elimination (D→E→15).
Figure 1.
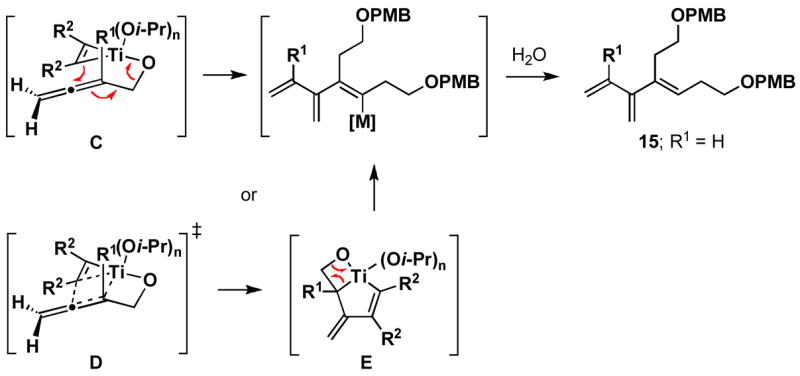
Possible origins of the cross-conjugated triene product 15.
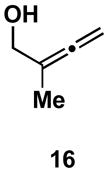
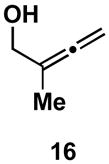
If production of the cross-conjugated triene proceeded by a pathway that involved full, or partial, C–Ti bond formation in the transition state (i.e. D), then allene substitution proximal to the alkoxide (R1 = alkyl) was anticipated to greatly impact the viability of this pathway. With R1 = alkyl, progression through D would require formation of a tertiary metal–carbon bond, a fact which was anticipated to greatly disfavor this pathway, and hence formation of a cross-conjugated triene. Alternatively, the reaction pathway for production of 1,4-dienes was not anticipated to be adversely affected by such substitution (see A; Scheme 3). In accord with these expectations, cross-coupling of 2-methyl-2,3-butadiene-1-ol 16 with alkyne 13 proceeds with good efficiency, and high selectivity for the formation of the E,E-1,4-diene 17 (Scheme 5). In this reaction, no evidence was found for the production of a cross-conjugated triene, or a stereiosomeric coupling product.
Scheme 5.
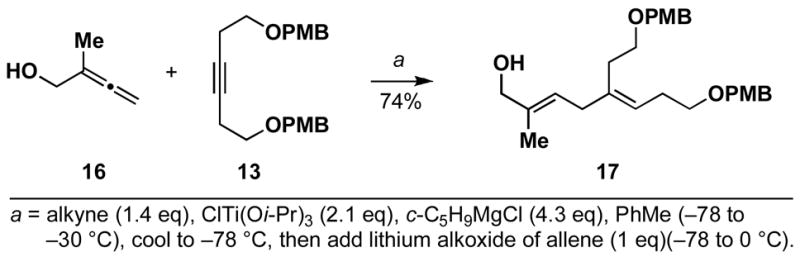
Cross-coupling of 1,1-disubstituted allenes with internal alkynes proceed with high levels of site-selectivity.
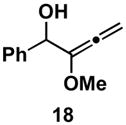
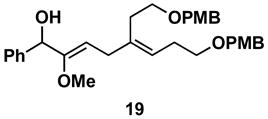
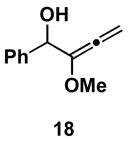
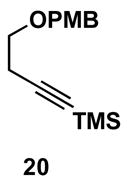
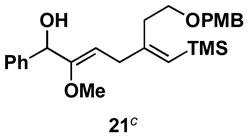
Table 1 illustrates that the 1,4-diene-selective cross-coupling of 1,1-disubstituted allenes with internal alkynes can be accomplished with a variety of functionalized substrates. For example, coupling of the methoxy-substituted allene 18 with symmetrical alkyne 13 affords the stereodefined enol ether-containing 1,4-diene 19 in 87% yield with ≥ 20:1 selectivity for the establishment of each stereodefined olefin (entry 1). In a more complex example, allene 18 can be coupled to the unsymmetrical alkyne 20 in a site- and stereoselective manner. In this case, the 1,4-diene 21, containing both a stereodefined enol ether and vinylsilane, is produced in 66% yield (entry 2). Although no evidence was found for the production of a stereoisomeric diene or cross-conjugated triene in this reaction, a minor isomer derived from regioisomeric functionalization of the TMS-alkyne was observed (rs = 4:1). This observation speaks to the complexity of the present cross-coupling reaction, where site- and stereoselective functionalization of each unsymmetric π-system is accomplished simultaneously.
Table 1.
Typical reaction conditions: alkyne (1.4 eq), ClTi(Oi-Pr)3 (2.1 eq), c-C5H9MgCl (4.2 eq),PhMe (−78 to −30 °C), cool to −78 °C then add allenyl alkoxide (1 eq), (−78 to 0 °C).
Selectivity refers to the olefin formed from the allene coupling partner.
rs = 4:1.
rr = 2:1.
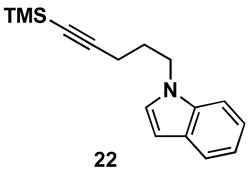
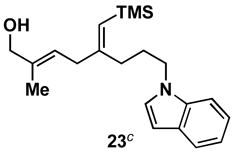
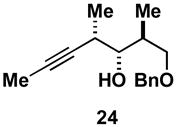
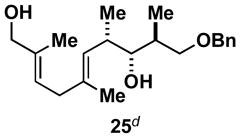
As depicted in entry 3, the heterocycle-containing TMS-alkyne 22 is also a suitable substrate for this cross-coupling process. Reaction with allene 16 provides the 1,4-diene product 23 as a single isomer (48% isolated yield). As observed previously, a minor isomer derived from regioisomeric functionalization of the unsymmetrical alkyne was generated (rs = 4:1). Finally, cross-coupling of allene 16 with the unsymmetrically substituted internal alkyne 24 provides 1,4-diene 25 in 62% yield (entry 4). This example demonstrates that site- and stereoselective C–C bond formation proceeds with free hydroxyl functionality tethered to each reacting π-system.
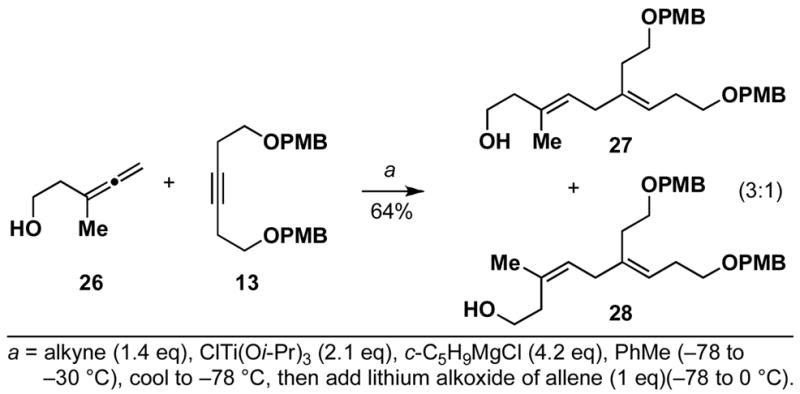
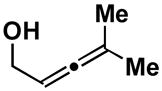
While allenic alkoxides uniformly provide 1,4-diene products with high levels of stereoselection in coupling reactions with internal alkynes (regio and diastereoselection is ≥ 20:1), the homoallenic alkoxide derived from 26 does not provide high levels of stereoselection in cross-coupling reactions with alkyne 13 (Scheme 6). Whereas the 1,4-diene 27 is the major product in this coupling reaction, the isomeric diene 28 is formed as a minor product (27:28 = 3:1). This erosion in selectivity, from allenic alcohol to homoallenic alcohol, is presumed to derive from a competition between directed and non-directed C–C bond forming processes (Figure 2).
Scheme 6.
Cross-coupling of homoallenic alcohols with alkynes leads to diminished stereoselection.
Figure 2.
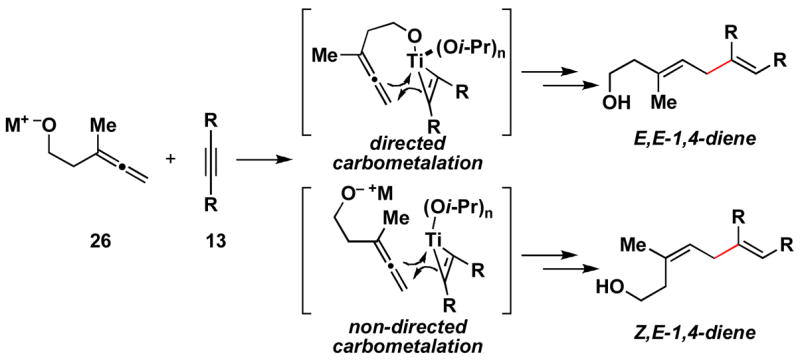
Competition between directed- and non-directed carbometalation.
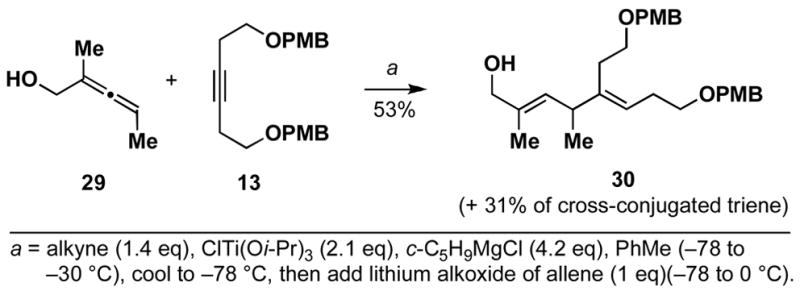
Trisubstituted allenes are also useful substrates in this stereoselective cross-coupling reaction. As depicted in Scheme 7, coupling of allene (+/−)-29 with the symmetrical alkyne 13 provides the highly substituted, stereodefined E,E-1,4-diene 30 in 53% yield. While the yield of the substituted diene is sufficient to render this reaction useful for the synthesis of highly substituted 1,4-dienes, unlike previously described cross-couplings, this reaction produces a significant amount of cross-conjugated triene (31%).
Scheme 7.
Cross-coupling of trisubstituted allenes with internal alkynes.
2.2. Convergent synthesis of stereodefined poly-substituted cross-conjugated trienes
2.2.1. Background to the utility of cross-conjugated trienes in complex molecule synthesis
Cross-conjugated trienes are a functional group with great potential in complex molecule synthesis.8 Since Blomquist and Verdol’s synthesis of 2-vinyl-1,3-butadiene (32), and subsequent demonstration of its diene transmissive Diels-Alder reaction with maleic anhydride,9 many have explored the potential of cross-conjugated trienes in carbocycle and heterocycle construction (Scheme 8).10
Scheme 8.
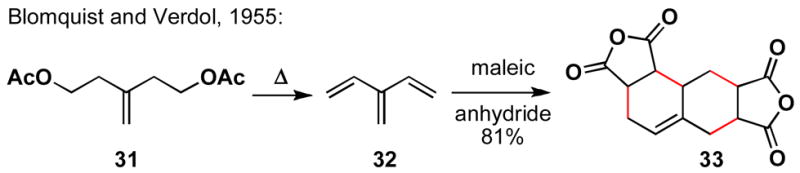
Synthesis of 2-vinyl-1,3-butadiene and its diene-transmissive Diels-Alder reaction with maleic anhydride.
Despite great interest in the use of this reactive structural motif, barriers still exist in the preparation of highly substituted and stereodefined cros-conjugated trienes. Whereas cyclic cross-conjugated trienes are accessible via metal-mediated cycloisomerization processes,11 the synthesis of acyclic cross-conjugated trienes is limited to the preparation of a small subset of sparsely substituted trienes.9,12 In such processes, issues concerning stereoselection in the generation of the substituted olefins of the cross-conjugated triene are pronounced.
Based on the potential utility of substituted cross-conjugated trienes in complex molecule synthesis, and the lack of general convergent pathways for their synthesis, we focused our attention on optimizing the allene–alkyne cross-coupling reaction for the synthesis of stereodefined cross-conjugated trienes.
2.2.2. Synthesis of cross-conjugated trienes
The decreased regioselection observed in cross-coupling of trisubstituted allene 29 with alkyne 13 could be derived from a destabilization of the transition state for directed carbometalation in pathway F (Figure 3). This destabilization would likely be based on the required build up of non-bonded 1,2-steric interactions in the development of the intermediate bicyclo-[3.3.0] metallacycle. Cross-coupling by C–C bond formation at the central carbon of the allene would avoid these destabilizing interactions, yet would require the formation (partial or full, depending on the mechanism) of a tertiary metal–carbon bond (G). Following this analysis, we reasoned that the reaction pathway for cross-coupling of allenes with alkynes could be diverted from the production of 1,4-dienes to stereodefined cross-conjugated trienes if the requirement of generating a tertiary metal–carbon bond was removed, while retaining the substitution at the terminus of the allene.
Figure 3.
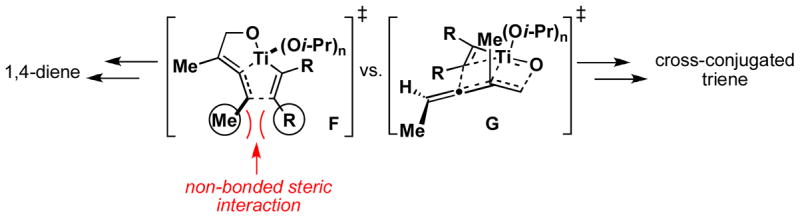
Competition between directed carbometalation and formal metallo-[3,3] rearrangement with trisubstituted allenes.
In line with this expectation, we were pleased to find that coupling of (+/−)-2,3-pentadiene-1-ol 34 with alkyne 13 affords the stereodefined cross-conjugated triene 35 (41% post HPLC; Scheme 9). Although this unoptimized coupling reaction did not proceed with high efficiency, no evidence was found for the production of a 1,4-diene.
Scheme 9.
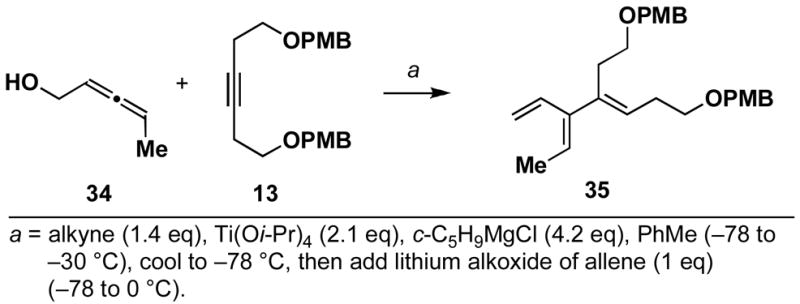
Coupling of (+/−)-2,3-pentadiene-1-ol 34 with alkyne 13.
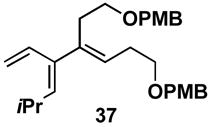
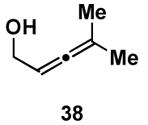
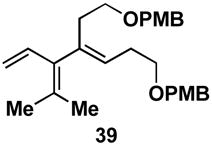
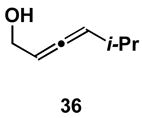
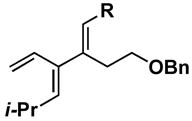
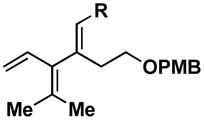
A variety of substituted allenes and alkynes can be employed in this site- and stereoselective cross-conjugated triene synthesis. As depicted in entry 1 of Table 2, coupling of the isopropyl-substituted allenic alcohol 36 with alkyne 13 provides the substituted triene 37 in 69% yield with ≥ 20:1 selectivity for the formation of each stereodefined trisubstituted olefin. Entry 2 demonstrates that this convergent coupling reaction can be employed to generate tetrasubstituted olefins. In this case, triene 39 can be prepared as a single isomer in 62% yield.
Table 2.
| entrya | allene | alkyne | yield (%) | (E):(Z)b | rsc | product |
|---|---|---|---|---|---|---|
| 1 |
|
13 | 69 | □20:1 | - |
|
| 2 |
|
13 | 62 | - | - |
|
| 3 |
|
|
57d | □20:1 | 3:1 |
|
|
|
|
||||
| 4 | 36 | 42; R=TMS | 75 | □20:1 | 4:1 | 43; R = TMS |
| 5 | 44; R= TIPS | 82 | □20:1 | 4:1 | 45; R = TIPS | |
|
|
|
||||
| 6 | 38 | 20; R = TMS | 71 | - | 4:1 | 46; R = TMS |
| 7 | 47; R = TIPS | 74 | - | 4:1 | 48; R = TIPS |
Typical reaction conditions: alkyne (1.4 eq),ClTi(OiPr)3 or Ti(Oi-Pr)4 (2.1 eq), c-C5H9MgCl (4.2 eq), PhMe (−78 to −30 °C), cool to −78 °C then add allenyl alkoxide (1 eq), (−78 to 0 °C).
Refers to the establishment of the substituted olefin derived from the allene (the other stereodefined olefin, derived from the alkyne is formed as a single isomer).
Refers to the site of alkyne functionalization.
Yield after deprotection with TBAF (See Supporting Information for details).
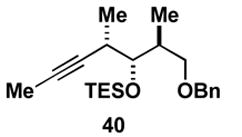
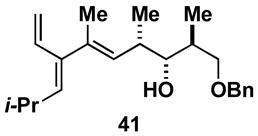
Aside from controlling olefin geometry and site-selectivity in allene-functionalization, one can also control site-selectivity in the functionalization of unsymmetrical alkynes. As illustrated in entry 3, the branched alkyne 40 can be selectively coupled to allene 36. In this reaction, C–C bond formation occurs preferentially distal to the branched architecture of alkyne 40, and affords cross-conjugated triene 41 in 57% yield (rs = 3:1, E:Z ≥ 20:1). The stereochemistry of these products was assigned on the basis of spectroscopic data, as depicted for 37 (Figure 4).
Figure 4.
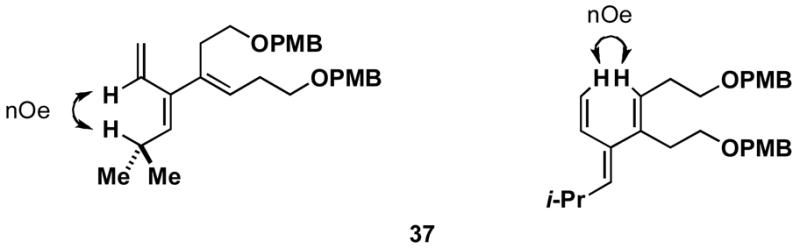
Stereochemical assignment of the cross-conjugated triene products was accomplished by analysis of nOe data as depicted for 37.
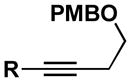
Similarly, entries 4–7 demonstrate that selective functionalization of silyl-substituted alkynes is possible.8 In these cases, the cross-conjugated triene products 43, 45, 46 and 48 can be generated in > 70% yield, with high levels of stereoselection (regioselection with respect to alkyne functionalization = 4:1).
The stereoselectivity observed in these reactions is consistent with C–C bond formation occuring anti to the distal alkyl substituent of the allene via geometry B (Scheme 3). To further explore the validity of this model, the cross-coupling reaction of a diastereomerically pure chiral allene, bearing a secondary hydroxyl directing group, with a symmetrical alkyne was investigated (Scheme 10). Coupling of allene 49 with alkyne 13 provides triene 50 in 50% yield. The observed stereoselection is consistent with reaction by way of a boat-like geometry H,3d where C–C bond formation occurs anti- to the terminal butyl substituent of the allene, with the allenic substituent (R1) occupying an equatorial position.
Scheme 10.
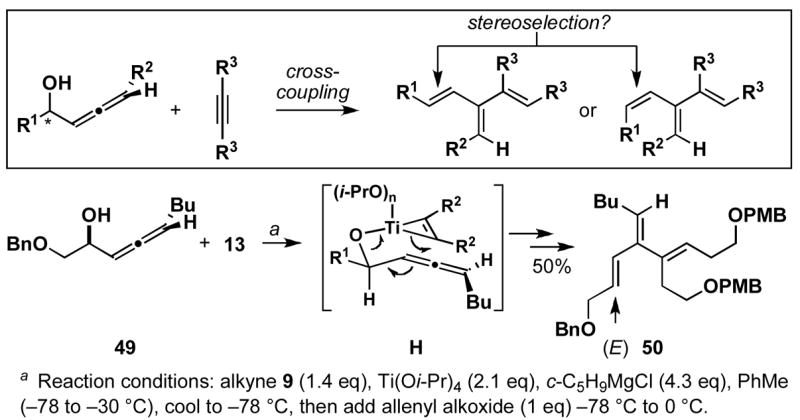
Allene-alkyne coupling for the synthesis of stereodefined tetrasubstituted cross-conjugated trienes.
The highly substituted cross-conjugated trienes produced from this cross-coupling reaction have great potential for facilitating the syntheses of complex carbocyclic and heterocyclic ring systems via site- and stereoselective cycloaddition processes. Whereas the reactivity of less-substituted acyclic cross-conjugated trienes are difficult to control in bimolecular [4+2] cycloadditions, as multiple cycloadditions often predominate,9,10,12 the substituted trienes generated here cleanly undergo mono-cycloaddition with activated dienophiles. For example, heating a toluene solution of 37 with maleimide 51 provides the cycloadduct 52 in 50% yield (endo:exo = 6:1; Scheme 11). This reactivity pattern is consistent with the decreased reactivity of (Z)-dienes in Diels-Alder reactions.13
Scheme 11.
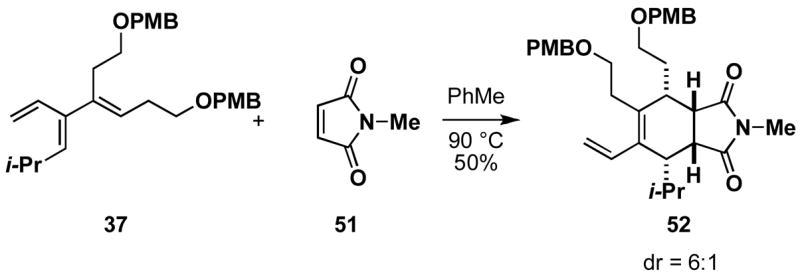
Thermal intermolecular site- and stereoselective Diels-Alder reaction of a trisubstituted cross-conjugated triene and an activated dienophile.
3. Conclusion
The cross-coupling of allenic alcohols with alkynes can be accomplished in a highly regio- and stereoselective manner. Applying an alkoxide-directed coupling reaction, tetrasubstituted 1,4-dienes are prepared by titanium-mediated coupling of 1,1-disubstituted allenes with internal alkynes. In these cases, stereoselection is consistent with the proposition that these processes proceed through the formation of bicyclo-[3.3.0] metallacyclopentenes, protonation of which affords 1,4-dienes 55 (Scheme 12). Alternatively, alkoxide-directed cross-coupling of 1,3-disubstituted allenes with internal alkynes provides a general strategy for the synthesis of substituted cross-conjugated trienes 57. These stereodefined substituted cross-conjugated trienes are useful intermediates in the synthesis of complex polycyclic systems, as they undergo site- and stereoselective mono [4+2] cycloaddition with activated dienophiles. Overall, these highly site-selective cross-coupling reactions provide stereoselective convergent access to structural motifs that are anticipated to be of great utility in the synthesis of complex molecules. Research directed at exploring these processes both mechanistically, and in the context of target-oriented syntehsis is underway.
Scheme 12.
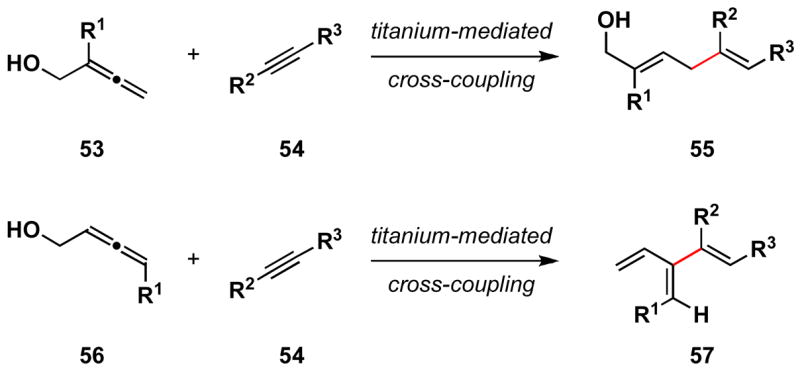
Allene-alkyne cross-coupling for the synthesis of 1,4-dienes or cross-conjugated trienes.
4. Experimental Section
4.1. General experimental information
All reactions were conducted in flame-dried glassware under nitrogen using anhydrous solvents. Toluene and tetrahydrofuran were distilled from sodium/benzophenone ketyl before use. Diethyl ether was used after passing through an activated alumina column. Titanium(IV) isopropoxide was used after distillation of the commercially available reagent. ClTi(Oi-Pr)3 was purchased as a 1M solution in hexanes from Aldrich and was used without further analysis or purification. All other commercially available reagents were used as received.
4.2. Representative procedure for the stereoselective synthesis of 1,4-dienes
Synthesis of (2E,5Z)-8-(4-methoxybenzyloxy)-5-(2-(4-methoxybenzyloxy)ethyl)-2-methylocta-2,5-dien-1-ol, 17
To a −78 °C solution of alkyne 13 (200 mg, 0.56 mmol) in 3.7 mL of PhMe was added 850 μL of ClTi(OiPr)3 (1.0M in hexanes, 0.85 mmol) and 860 μL of c-C5H9MgCl (1.96M in Et2O, 1.69 mmol) dropwise via a gas-tight syringe. The resulting clear, yellow solution turned dark reddish brown while warming slowly to −30 °C over 1 hr. The reaction mixture was stirred at −30 °C for 1 hr and then cooled to −78 °C. To a separate −78 °C solution of allene 16 (69 mg, 0.39 mmol) in 1.0 mL PhMe was added 160 μL of nBuLi (2.45M in hexanes, 0.39 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath and added to the −78 °C titanium solution dropwise via cannula. After warming slowly to 0 °C over 2 hr, the reaction was quenched with 5 mL of sat. NH4Cl solution. The mixture was warmed to room temperature before extracting with EtOAc (3 x 15 mL). The combined organic layer was washed with sat. NaHCO3 solution (1 x 30 mL), brine (1 x 30 mL) and dried over anhydrous Na2-SO4. Flash column chromatography of the crude material (20 % EtOAc/hexanes, then 50 % EtOAc/hexanes) provided 127 mg (74 %) of diene 17 as a clear, colorless oil. A small portion was further purified by HPLC [EtOAc/hexanes: gradient from 35 % to 55 % (0–20 min, 25 mL/min) on a Microsorb (Si 80-120-C5 H410119) column] to obtain a sample for analytical characterization. 1H NMR (400 MHz, CDCl3) δ 7.26-7.23 (m, 4H), 6.89-6.85 (m, 4H), 5.42-5.37 (m, 1H), 5.24 (t, J = 7.1 Hz, 1H), 4.42 (s, 2H), 4.41 (s, 2H), 4.00 (d, J = 5.8 Hz, 2H), 3.80 (s, 3H), 3.80 (s, 3H), 3.46-3.39 (m, 4H), 2.75 (d, J = 7.3 Hz, 2H), 2.34 (dt, J = 14.7, 7.6 Hz, 2H), 2.33 (t, J = 7.3 Hz, 2H), 1.64 (s, 3H), 1.34 (t, J = 6.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ159.1, 136.6, 136.0, 130.6, 130.5, 129.21, 129.16, 123.8, 123.1, 113.7, 77.2, 72.5, 69.8, 68.8, 68.6, 55.2, 35.7, 31.1, 28.6, 13.6; IR (thin film, NaCl) 3433, 2907, 2857, 1613, 1513, 1464, 1360, 1302, 1173, 1093, 1035, 802 cm−1; LRMS (EI, Na) calcd for C27H36O5Na, 463.26 m/z (M + Na); observed, 463.3 (M + Na)+ m/z.
4.3. Representative procedure for the stereoselective synthesis of cross-conjugated trienes
Synthesis of (3E,5E)-1-(4-methoxybenzyloxy)-4-(2-methoxybenzyloxy)ethyl)-5-vinyl-7-methyl-3,5-octadiene, 37
To a −78 °C solution of alkyne 13 (200 mg, 0.56 mmol) in 3.7 mL of PhMe was added 850 μL of ClTi(OiPr)3 (1.0M in hexanes, 0.85 mmol) and 860 μL of cC5H9MgCl (1.96M in Et2O, 1.68 mmol) dropwise via a gas-tight syringe. The resulting clear, yellow solution turned dark reddish brown while warming slowly to −30 °C over 1 hr. The reaction mixture was stirred at −30 °C for 1 hr and then cooled to −78 °C. To a separate −78 °C solution of allene 36 (44 mg, 0.39 mmol) in 1.0 mL PhMe was added 160 μL of nBuLi (2.45M in hexanes, 0.39 mmol) dropwise via gas-tight syringe. The resulting solution was stirred for 15 min, removed from the cold bath and added to the −78 °C titanium solution dropwise via cannula. After warming slowly to −30 °C over 1 hr, the reaction was quenched with 5 mL of sat. NH4Cl solution. The mixture was warmed to room temperature before extracting with EtOAc (3 x 15 mL). The combined organic layer was washed with sat. NaHCO3 solution (1 x 30 mL), brine (1 x 30 mL) and dried over anhydrous Na2SO4. Flash column chromatography of the crude material (5 % EtOAc/hexanes, then 7.5 % EtOAc/hexanes) provided 121 mg (69 %) of triene 37 as a clear, colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.26-7.21 (m, 4H), 6.88-6.83 (m, 4H), 6.53 (dd, J = 17.4, 10.6 Hz, 1H), 5.36 (t, J = 7.3 Hz, 1H), 5.15 (d, J = 10.1 Hz, 1H), 5.13-5.06 (m, 2H), 4.42 (s, 2H), 4.37 (s, 2H), 3.78 (s, 3H), 3.78 (s, 3H), 3.46 (t, J = 7.1 Hz, 2H), 3.36 (t, J = 7.3 Hz, 2H), 2.74-2.65 (m, 1H), 2.51 (t, J = 7.6 Hz, 2H), 2.42 (dt, J = 14.1, 7.3 Hz, 2H), 0.95 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 159.1, 159.0, 138.7, 138.5, 138.1, 132.6, 130.7, 130.6, 129.2, 129.1, 126.6, 116.4, 113.69, 113.65, 72.44, 72.39, 69.7, 68.7, 55.21, 55.20, 30.2, 28.8, 26.9, 23.1; IR (thin film, NaCl) 2957, 2864, 1613, 1513, 1464, 1360, 1302, 1248, 1173, 1097, 1037, 820 cm−1; LRMS (EI, Na) calcd for C29H38O4Na, 473.28 m/z (M + Na); observed, 473.5 (M + Na)+ m/z.
Supplementary Material
Supplementary Material
Complete experimental details for all preparative procedures along with spectral data for all products are provided.
Acknowledgments
We gratefully acknowledge financial support of this work by the American Cancer Society (RSG-06-117-01), the American Chemical Society (PRF-45334-G1), the Arnold and Mabel Beckman Foundation, Boehringer Ingelheim, Eli Lilly & Co., and the National Institutes of Health – NIGMS (GM80266). HLS acknowledges support from BMS in the form of a graduate student fellowship.
Footnotes
Dedicated to Professor John Hartwig on his receipt of the Tetrahedron Prize
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For a recent review of palladium-catalyzed cross-coupling in total synthesis, see: Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368.
- 2.For recent reviews of such metal-mediated coupling, see: Marek I, editor. Titanium and Zirconium in Organic Synthesis. Wiley-VCH: Weinheim; 2002. p. 512.Montgomery J. Angew Chem Int Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634.Trost BM, Toste FD, Pinkerson AB. Chem Rev. 2001;101:2067–2096. doi: 10.1021/cr000666b.Buchwald SL, Nielsen RB. Chem Rev. 1988;88:1047–1058.
- 3.For alkyne–alkyne cross-coupling, see: Ryan J, Micalizio GC. J Am Chem Soc. 2006;128:2764–2765. doi: 10.1021/ja057352w.Shimp HL, Micalizio GC. Org Lett. 2005;7:5111–5114. doi: 10.1021/ol052241n.For alkene–alkyne cross-coupling, see: Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440–1443. doi: 10.1002/anie.200603515.For allylic alcohol–alkyne cross-coupling, see: Kolundzic F, Micalizio GC. J Am Chem Soc. 2007;129:15112–15113. doi: 10.1021/ja075678u.For alkyne–imine cross-coupling, see: McLaughlin M, Takahashi M, Micalizio GC. Angew Chem Int Ed. 2007;46:3912–3914. doi: 10.1002/anie.200605060.For alkene–imine cross-coupling, see: Takahashi M, Micalizio GC. J Am Chem Soc. 2007;129:7514–7516. doi: 10.1021/ja071974v.
- 4.For examples of metal-mediated cross-coupling of allenes and alkynes, see: Hideura D, Urabe H, Sato F. Chem Commun. 1998:271–272.Tanaka R, Sasaki M, Sato F, Urabe H. Tetrahedron Lett. 2005;46:329–332.Suzuki K, Imai T, Yamanoi S, Chino M, Matsumoto T. Angew Chem Int Ed. 1997;36:2469–2471.Yamanoi S, Matsumoto T, Suzuki K. Tetrahedron Lett. 1998;39:9727–9730.Yamanoi S, Imai T, Matsumoto T, Suzuki K. Tetrahedron Lett. 1997;38:3031–3034.Trost BM, Kottirsch G. J Am Chem Soc. 1990;112:2816–2818.For examples of metal-mediated intramolecular coupling of allenes with alkynes, see: Kent JL, Wan H, Brummond KM. Tetrahedron Lett. 1995;36:2407–2410.Brummond KM, You L. Tetrahedron. 2005;61:6180–6185.Hicks FA, Kablaoui NM, Buchwald SL. J Am Chem Soc. 1996;118:9450–9451.Hicks FA, Kablaoui NM, Buchwald SL. J Am Chem Soc. 1999;121:5881–5898.Urabe H, Takeda T, Hideura D, Sato F. J Am Chem Soc. 1997;119:11295–11305.Urabe H, Sato F. Tetrahderon Lett. 1998:7329–7332.
- 5.For a preliminary account, see: Shimp HL, Micalizio GC. Chem Commun. 2007:4531–4533. doi: 10.1039/b708256h.
- 6. For an example of such a metal-mediated alkylation with allylic transposition, see ref 3d.
- 7.Vogel E, Ott KH, Gajek K. Liebigs Ann. 1961;644:172–188.Doering W, von E, Roth WR. Tetrahedron. 1963;19:715–737.For an example of a metallo-[3,3] rearrangement for isomerization of allylic alcohols, see: Chabardes P, Kuntz E, Varagnat J. Tetrahedron. 1977;33:1775–1783.
- 8.Winkler JD. Chem Rev. 1996;96:167–176. doi: 10.1021/cr950029z. [DOI] [PubMed] [Google Scholar]
- 9.Blomquist AT, Verdol JA. J Am Chem Soc. 1955;77:81–83.For related studies, see: Bailey WJ, Economy J. J Am Chem Soc. 1955;76:1133–1136.Bailey WJ, Economy J, Hermes ME. J Org Chem. 1962;27:3295–3299.
- 10.For recent examples, see: Woo S, Squires N, Fallis AG. Org Lett. 1999;1:573–575.Kwon O, Park SB, Schreiber SL. J Am Chem Soc. 2002;124:13402–13404. doi: 10.1021/ja028086e.Brummond KM, You L. Tetrahedron. 2005;61:6180–6185.
- 11.a) Llerena D, Aubert C, Malacria M. Tetrahedron Lett. 1996;37:7027–7030. [Google Scholar]; b) Yamazaki T, Urabe H, Sato F. Tetrahedron Lett. 1998;39:7333–7336. [Google Scholar]; c) Brummond KM, Chen H, Sill P, You L. J Am Chem Soc. 2002;124:15186–15187. doi: 10.1021/ja027588p. [DOI] [PubMed] [Google Scholar]
- 12.a) Tsuge O, Wada E, Kanemasa S. Chem Lett. 1983:239–242. [Google Scholar]; b) Arisawa M, Sugihara T, Yamaguchi M. Chem Commun. 1998:2615–2616. [Google Scholar]; c) Shi M, Shao LX. Synlett. 2004:807–810. [Google Scholar]; d) Bradford TA, Payne AD, Willis AC, Paddon-Row MN, Sherburn MS. Org Lett. 2007;9:4861–4864. doi: 10.1021/ol7021998. See also ref 10a. [DOI] [PubMed] [Google Scholar]
- 13.Roush WR, Barda DA. J Am Chem Soc. 1997;119:7402–7403. and references cited therein. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Complete experimental details for all preparative procedures along with spectral data for all products are provided.