

Abstract
Complement activation is tightly regulated to avoid excessive inflammatory and immune responses. Crry-/- is an embryonic lethal phenotype secondary to the maternal complement alternative pathway (AP) attacking a placenta deficient in this inhibitor. In this study, we demonstrate that Crry-/- mice could be rescued on a partial as well as on a complete factor B (fB)- or C3-deficient maternal background. The C3 and fB protein concentrations in Crry-/-C3+/- and Crry-/-fB+/- mice were substantially reduced for gene dosage secondary to enhanced AP turnover. Based on these observations, a breeding strategy featuring reduced maternal AP-activating capacity rescued the lethal phenotype. It led to a novel, stable line of Crry SKO mice carrying normal alleles for C3 and fB. Crry SKO mice also had accelerated C3 and fB turnover and therefore reduced AP-activating potential. These instructive results represent an example of a membrane regulatory protein being responsible for homeostasis of the complement system. They imply that there is constant turnover on cells of the AP pathway which functions as an immune surveillance system for pathogens and altered self.
Introduction
The complement system is an integral part of the innate immune and inflammatory response to microbes. The alternative pathway (AP)4 is an ancient activation mechanism (1). The complement cascade is tightly controlled via membrane-bound and fluid-phase regulatory proteins (2). Disassociation of the C3 and C5 convertases (decay-accelerating activity (DAA)) and cleavage of C4b and C3b by the plasma protease factor I (fI) in the presence of a cofactor protein (cofactor activity (CA)) are two means to inhibit the activation process (3, 4). Hemolytic uremic syndrome (HUS) and age-related macular degeneration are informative examples in which dysfunction of complement regulators plays a key role in mediating tissue injury via the AP (5– 8). In these diseases, mutations and polymorphisms of complement inhibitors allow for excessive activation for a given degree of injury, resulting in tissue damage in the renal microvasculature and retina (9).
Spontaneous, continuous, low-grade activation of complement occurs through a process termed “C3-tickover,” which involves cleavage (hydrolysis) of the unstable thioester bond in C3 (10). The activated C3 can now bind factor B (fB) to trigger the AP (11). Specifically, factor D cleaves the zymosan fB in the complex to generate the C3 convertase of the AP, which may be stabilized by properdin. Recent evidence points to the classical pathway as also having a tickover process (12). This complement turnover in blood may facilitate the system's response to foreign Ags. In this manner, the complement system functions as a sonar or radar system by continuously providing activated C3. Interactions in the fluid phase (no target) and with normal self (wrong target) are held in check by inhibitors while amplification takes place on foreign particles. Such a nontargeted turnover and sensing mechanism, coupled to an amplification loop, must be rigorously controlled to avoid undesirable tissue damage and to maintain physiologic levels of complement components for host defense.
A failure to adequately regulate complement activation is associated with several types of glomerulonephritis (13). Complete factor H (fH) deficiency allows the AP C3 convertase (C3bBb) to go unchecked, consuming in the fluid phase C3 and fB. In humans, pigs, and mice deficient in fH (14, 15), ∼5% of the normal concentration of C3 is found in blood due to this accelerated turnover of the AP (16). The complement fragments generated by the excessive consumption lead to membranoproliferative glomerulonephritis type II (MPGN II) with renal failure and death at an early age in pigs and man (before dialysis) and a similar, albeit milder pathologic phenotype in older mice. A similar degree of C3 consumption occurs in fI deficiency, although the renal pathologic changes are distinct from that observed in fH deficiency as MPGN II has not been reported (17, 18). Also, recent studies using fI deficient mice demonstrate that uncontrolled AP activation results in decreased C3, fB, and fH in the serum, but C3 deposition along the glomerular basement membrane or MPGN II was not observed in fI-deficient mice (19). Another example featuring glomerulonephritis involves C3 nephritic factor, an autoantibody that stabilizes the AP C3 convertase. Its presence often leads to gain of function and accelerated C3 and fB consumption (20). These data point to the necessity of control of the AP in plasma by cofactor activity.
In contrast to plasma proteins fH and fI, there is little evidence to suggest that membrane-bound complement regulatory proteins control C3 turnover. Normal C3 blood levels are present in DAF-/- mice (21, 22) and in humans with a deficiency of decay accelerating factor (DAF) on hematopoietic cells in paroxysmal nocturnal hemoglobinuria (23). Atypical HUS patients with CD46 mutations usually have normal C3 levels; however, most of these individuals are haploinsufficient (6, 24).
Crry (CR1-related gene/protein Y), a membrane-bound complement regulator (25), is only known to be present in rodents, namely, the mouse and rat. Mouse Crry is generally considered though to be the murine counterpart of human membrane cofactor protein (MCP; CD46). CD46 has wide tissue distribution in humans and other mammals, but its expression is restricted to male germ cells in the mouse and rat. CD46 and Crry are cofactors for the fI-mediated cleavage of C3b and C4b. In these rodents, Crry is the only ubiquitously expressed regulator with cofactor activity (26). As illustrated by this Crry and MCP comparison, complement regulation by CA is conserved across species. In contrast to MCP, Crry also possesses DAA for the classical pathway but weak DAA for the AP C3 convertase (27, 28). The relative importance of its DAA vs that of the two mouse DAF proteins (DAF-1 and DAF-2) in protecting host tissue is unclear (29, 30). DAF-1 encodes a protein with wide tissue expression while DAF-2's expression is limited to germ cells. These two forms of mouse DAF and human DAF possess DAA for both the classical and AP C3 convertases (31). These expression patterns, taken together with the lessons learned from deficiency states, indicate that CA and DAA are required to maintain homeostasis of the complement system.
The importance of Crry in controlling complement activation is highlighted by the embryonic lethal phenotype of Crry-/- (32). Although no developmental defect was observed, the null embryo died at ∼7.5 days due to placental destruction mediated by the maternal complement system. In addition, mouse DAF is absent in the early stage (as late as day 10.5) of the developing mouse placenta. Consequently, Crry is the only membrane-bound C3 regulator in the early embryonic stage (33). The lethal phenotype of Crry-/- mice is rescued by complete C3 or fB deficiency, but not by a deficiency of C4, indicating that embryonic death is triggered by the AP (33). This is an informative model system to study fetomaternal tolerance and immune-mediated embryo destruction; however, analysis of disease models featuring complement activation in other tissues has not been possible because of concomitant C3 or fB deficiency. A critical role for Crry is also supported by the rapid complement-dependent clearance of Crry-/- RBCs injected into wild-type (WT) mice and by complement activation fragments depositing on Crry null kidneys transplanted into WT mice leading to an inflammatory response, interstitial fibrosis, and, eventually, renal failure (34 –36).
To further study Crry function in vivo, we generated Crry+/- and Crry-/- on a C3 or fB heterozygous background. In these mice, there was enhanced turnover of the AP, leading to reduced C3 and fB concentrations. Using a breeding program featuring a partial deficiency of C3 or fB on the maternal side allowed for survival of Crry null embryos. This strategy led to the generation of a stable strain of mice lacking only the Crry gene, the Crry single knockout mice (Crry SKO). Accelerated AP-mediated C3 tickover is present in these Crry SKO mice. These results further establish a role for a membrane inhibitor in maintaining homeostasis of the AP of complement activation.
Materials and Methods
Mice
129Sv/J X C57BL/6 Crry+/- mice were generated by a standard genetargeting strategy (32). Mixed background Crry+/- mice were backcrossed into C57BL/6 WT mice for nine generations. C57BL/6 Crry+/- mice were used for the experiments described here. Crry-/-fB-/- and Crry-/-C3-/- mice were on the 129Sv/J X C57BL/6 background and were generated by mating Crry+/-fB-/- or Crry+/-C3-/-, respectively (33). To assess the role of AP in C3 turnover in Crry-/-C3+/- mice, Crry-/-fB-/- and Crry-/-C3-/- were crossed to generate F1 Crry-/-fB+/-C3+/- mice. These F1 mice were then intercrossed to produce mice with Crry-/-fB-/-C3+/- and Crry-/-fB+/-C3+/- genotypes. fB-/- and C3-/- mice were on the C57BL/6 background (37, 38).
The following primers were used to determine the genotypes of Crry, fB, and C3: for Crry; mCrry 16 TTGAGTTCAATGCACTGAGGAGG, EcoRI 16F CGCAGAATTCAATCTCTTTTCTTTGCC and S46Neo GCTACCCGTGATATTGCTGAAGAG; for fB, fB-F1 CCGAAGCATTCCTATCCTCC, fB-R1 GTAGTCTTGTCTGCTTTCTCC and fB-Neo CGAATGGGTGACCGCTTCC (39); and for C3; V787 C3 GATCCCCAGAGCTAATG, V789 C3 AGGGACCAGCCCAGGTTCAG, and V788 Neo TCGTCCTGCAGTTCATTCAG (40). Mice were housed and maintained in a pathogen free environment. All animal experiments were conducted under approved protocols of the Animal Studies Committee of Washington University School of Medicine (St. Louis, MO).
AP assay
Zymosan particles (ZP) were used to activate the AP (28). Briefly, a boiled ZP suspension was washed twice with gelatin-veronal-buffered saline (GVBS). EGTA (10 mM) along with MgCl2 (2 mM) was added to GVBS to block the classical and lectin pathways. Fresh sera at dilutions indicated and ZP (both in GVBS) were mixed and incubated at 37°C for 30 min. After three washes, ZP were stained with FITC-conjugated polyclonal anti-mouse C3 Ab (Valeant Pharmaceuticals) and analyzed by FACScan (BD Biosciences).
Cobra venom factor (CVF)-based C3 cleavage assay
For this assay, mouse serum (10 μl), GVBS, and CVF (final concentration of 33 μg/ml; Quidel) were incubated at 37°C for 30 min. Resulting cleaved products were analyzed by SDS-PAGE and Western blot.
Western blotting
Fresh serum or EDTA-treated plasma (diluted 1/100) was used to assess C3, fB, and fH by Western blot. Total liver lysates were used to characterize the C3 protein precursor by Western blot. Reduced (sera and plasma) or non reduced (liver lysates) samples were subjected to 10% SDS-PAGE and then blotted on nitrocellulose. The membranes were blocked overnight with 5% nonfat dried milk. Goat anti-mouse C3 (1/10,000 dilution; Valeant Pharmaceuticals), goat anti-human fB or fH Ab (both at a 1/5,000 dilution; Advanced Research Technologies) were incubated with the membranes for 1.5 h at room temperature. After three washes with TBS (Tris-NaCl) containing 0.05% Tween 20, secondary HRP-conjugated rabbit anti-goat IgG (Southern Biotechnology Associates) was added for 1 h at 37°C. After three washes with TBS-Tween 20, membranes were developed with a SuperSignal West Kit (Pierce).
RT-PCR
Liver tissues were homogenized by passage through a 40-μm cell strainer. A RNeasy Mini Kit (Qiagen) was used to extract total RNA. First-strand cDNA was synthesized using 1 μg of total RNA with oligo(dT) primers and SuperScript II reverse transcriptase (Invitrogen). C3 mRNA levels were detected using C3-specific primers (C3F301 GATTCCAGCCAGTAAGGAATTC and C3R708 CTCTGCGGAGAAGATCTGCTTC).
C3 ELISA
Goat anti-mouse C3 Ab was used to coat the ELISA plates (Immunol). After washing, they were blocked with PBS containing 1% BSA and 0.1% Tween 20 for 2 h at room temperature. Fresh sera (usually diluted 1/4000) were added for at least 2 h at room temperature. After washing, the plates were incubated with HRP-conjugated goat anti-mouse C3 Ab for 1.5 h at room temperature. Substrate reaction was developed by tetramethylbenzidine and the OD was assessed at 630 nm. WT and C3-/- sera served as controls.
Immunohistochemistry
Kidneys were fixed with formalin and routine H&E staining was performed (33). For monitoring C3 deposition in the kidneys, fixed tissue sections were incubated with 5% goat serum before using a primary FITC-conjugated anti-mouse C3 Ab (41).
In vivo delivery of RBC-targeted mouse Crry
HEK 293T cells (ATCC CRL-11268) were used to generate secreted Crry forms. They were maintained in 10% FCS/DMEM with supplements of L-glutamine, nonessential amino acids, and penicillin/streptomycin. Transfections were conducted with the TransIT-293 reagent according to the manufacturer's instructions (Mirus). For protein synthesis, the medium was replaced with fresh FCS-free DMEM 24 h posttransfection and the supernatants were harvested at 48–72 h. To obtain concentrated protein stocks, the FCS-free supernatants were applied to centrifugal filter devices with a 10-kDa cutoff (Millipore).
For the in vivo delivery of RBC-targeted mouse Crry, mice were injected i.v. with up to 300 μl of filtered (0.2 μM) concentrated culture supernatant. Blood samples (2 μl) from WT control and test animals were collected by tail clipping. FACS was performed on mouse RBCs to determine the expression of endogenous Crry, RBC-targeted recombinant Crry, and deposition of complement activation fragments.
Results
Accelerated complement turnover in Crry-/-C3+/- and Crry-/-fB+/- mice
Based on previous results showing that maternal C3 deficiency rescues Crry-/- embryo mortality (33), we bred Crry+/-C3+/+ males with Crry-/-C3-/- females to generate, as expected, ∼50% each of Crry+/-C3+/- (23 of 47) and Crry-/-C3+/- (24 of 47) mice. In this situation, Crry+/- and Crry-/- mice can be compared on the same C3 heterozygous background. Along this line, if we crossed Crry-/-C3-/- male mice to Crry+/-C3+/+ female mice, 92% of the offspring were Crry+/-C3+/-. Thus, most Crry-/-C3+/- mice died if the mother was C3+/+ (33). We desired to test how critical the activating capacity of the maternal AP is for survival of Crry null embryos.
Therefore, Crry+/-fB+/+ males were mated with Crry-/-fB-/- females, resulting in the predicted ∼50% Crry+/-fB+/- and Crry-/-fB+/- mice (80 of 156 were Crry+/-fB+/- vs 76 of 156 were Crry-/-fB+/-) (Table I, A). However, if Crry-/-fB-/- males were bred with Crr+/-fB+/+ females, only 1 (1.4%) of 73 mice was Crry-/-fB+/- and 72 (99%) of 73 were Crry+/-fB+/- (Table I, B). Thus, similar to the result with a C3-sufficient mother, most all Crry-/- mice died if fB was present in the mother. These results point out how the efficiency of maternal C3 activation via the AP accounts for embryonic lethality. They also provide an opportunity to compare Crry+/- and Crry-/- mice with the same gene dosage and “presumably” the same protein levels of C3 and fB.
Table 1. Genotypic analysis of Crry+/- and Crry-/- mice mating with fB or C3 deficient in paternal or maternal breeding partners.
| Male X Female | Crry+/- fB+/- | Crry-/- fB+/+ | Crry-/- fB+/- | Crry-/- fB-/- | Total |
|---|---|---|---|---|---|
| A. Crry+/- fB+/+ X Crry-/- fB-/- | 80 (51%, 50%) | NA | 76 (49%, 50%) | NA | 156 |
| B. Crry-/- fB-/- X Crry+/- fB+/+ | 72 (99%, 50%) | NA | 1 (1%, 50%) | NA | 73 |
| C. Crry+/- fB+/+ X Crry-/- fB+/- | 33 (25%, 25%) | 38 (29%, 25%) | 28 (21%, 25%) | NA | 131 |
| D. Crry-/- fB+/- X Crry+/- fB+/+ | 33 (58%, 25%) | 0 (0%, 25%) | 1 (1.7%, 25%) | NA | 57 |
| E. Crry-/- fB+/- X Crry-/- fB+/- | NA | 50 (26%, 25%) | 103 (53%, 50%) | 40 (21%, 25%) | 193 |
| F. Crry+/- fB+/- X Crry +/- fB+/- | 79 (32%, 25%) | 6 (2.4%, 6.25%) | 12 (4.9%, 12.5 %) | 3 (1.2%, 6.25%) | 243 |
|
| |||||
| Crry+/-C3+/- | Crry-/-C3+/+ | Crry-/-C3+/- | Crry-/-C3-/- | ||
|
| |||||
| G. Crry+/-C3+/+ X Crry-/-C3+/- | 44 (22%, 25%) | 54 (28%, 25%) | 47 (24%, 25%) | NA | 194 |
| H. Crry-/-C3+/- X Crry-/-C3+/- | NA | 10 (17%, 25%) | 36 (62%, 50%) | 12 (21%, 25%) | 58 |
| I. Crry+/-C3+/- X Crry+/-C3+/- | 50 (30%, 25%) | 0 (0%, 6.25%) | 1 (0.5%, 12.5%) | 0 (0%, 6.25%) | 169 |
|
| |||||
| Crry+/- C3+/+ fB+/+ | Crry-/-C3+/+ fB+/+ | ||||
|
| |||||
| J. Crry+/- fB+/+ C3+/+ X Crry-/- fB+/+ C3+/+ | 37 (43%, 50%) | 49 (57%, 50%) | 86 | ||
| K. Crry-/- fB+/+ C3+/+ X Crry+/- fB+/+ C3+/+ | 31 (100%, 50%) | 0 (0%, 50%) | 31 | ||
The first number in parentheses represents the experimentally obtained percent. The second number represents the expected ratio. NA indicates that this genotype is not possible from this breeding combination. Crry SKO mice labeled as Crry-/- fB+/+, Crry-/-C3+/+ or Crry-/-fB+/+ C3+/+ are highlighted by red.
To address this, AP activity was monitored using several assay systems. In a functional test, the Crry-/- mice with the same C3 or fB genotypes had reduced C3 deposition on the surfaces of zymosan particles compared with Crry+/- mice (Fig. 1). For example, there was ∼50% reduction of C3 fragments deposited on the surfaces of zymosan in mice lacking Crry in 10% serum and a reduction to <5% in 5% serum. These results raised the possibility of C3 or other components of the AP being different in the sera or a difference in C3 biosynthesis in the liver. We next measured serum C3 levels by Western blot (Fig. 2A) and ELISA (Fig. 2B). The concentration of C3 was reduced in Crry-deficient mice, even though the Crry+/- and Crry-/- mice carried the same C3 heterozygous genotype. This phenomenon was also observed on a fB heterozygous background (Fig. 2A, bottom panel). Since the coagulation pathway may activate complement during serum preparation, we also measured the concentration of C3 in EDTA-treated plasma samples. No differences were detected between measurements in serum and plasma (data not shown).
FIGURE 1.
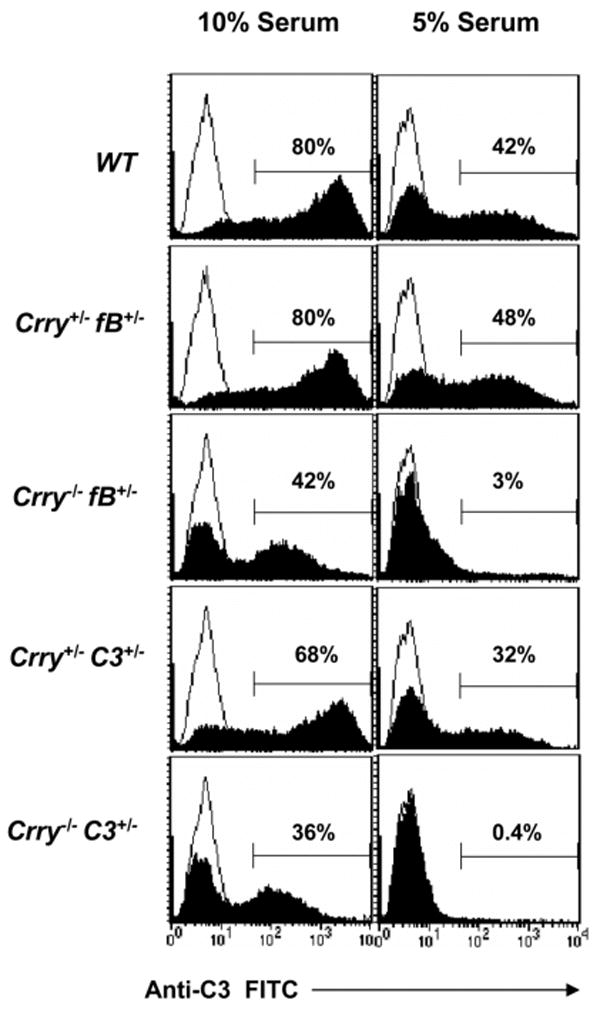
Impairment of AP in Crry+/- vs Crry-/- mice. ZP were used to activate the AP using 10 (left panel) and 5% mouse serum (right panel). C3 deposition was assessed by FACS using FITC-conjugated anti-C3 Ab. The line represents the EDTA-treated (negative) control sample. There was less AP activation in Crry-/-fB+/- and Crry-/-C3+/- mice. Sera from Crry+/-fB+/- or Crry+/-C3+/- were similar to the corresponding Crry+/+ strain (data not shown). This experiment was repeated twice with similar results.
FIGURE 2.
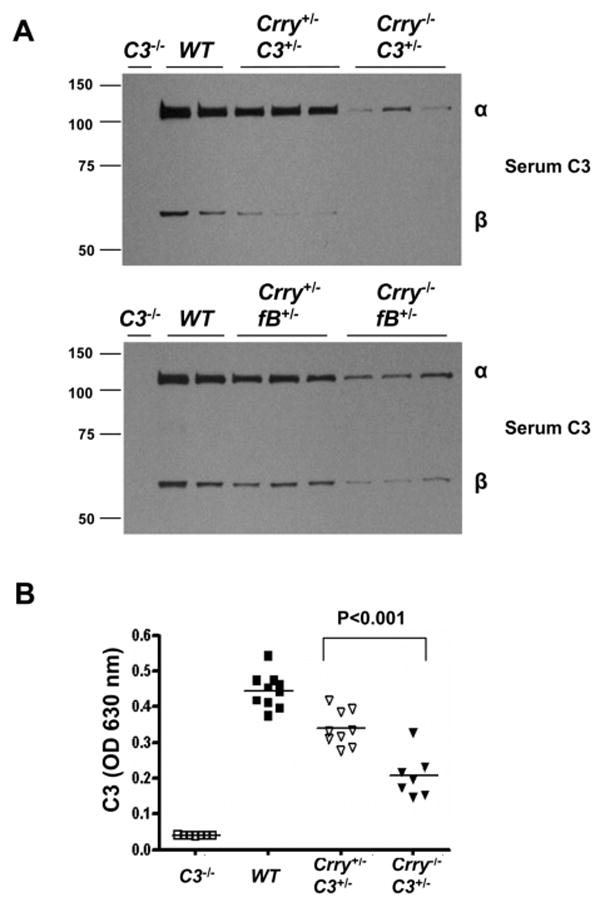
Critical role of membrane protein Crry in control of C3 levels. A, Western blot (reducing conditions) of serum C3 from Crry+/- and Crry-/- mice on a C3 or fB heterozygous background. C3-/- and WT mice served as controls. A reduced concentration of C3 or fB is present in mice completely deficient in Crry. B, Serum C3 in Crry+/-C3+/- and Crry-/-C3+/- mice were measured by ELISA. There was a 40–50% reduction of serum C3 in Crry-/-C3+/- mice compared with Crry+/-C3+/- mice. The asterisk indicates a statistically significant difference between the two groups (p <0.05; Student's t test).
Most of the C3 in fH-/- mice circulates as C3b (14). This failure to proteolytically inactivate C3b leads to renal pathology (14). However, we observed only an intact α-chain of C3 in the serum samples from Crry-/-fB+/- or Crry-/-C3+/- mice (Fig. 2A). To further characterize the status of C3, CVF was used to activate the AP of Crry+/-fB+/- and Crry-/-fB+/- mice. The quantity of the α2 fragment was proportional to the starting amount of the α-chain (data not shown). Thus, an increase in C3 activation products, including C3b, was not present in Crry-deficient mice. We propose that the presence of fH led to C3b cleavage and subsequent clearance from the circulation. Also, much of the C3 turnover could be occurring on cells and therefore the C3b is processed locally.
To further evaluate the ability of Crry to control C3 turnover in vivo, WT serum was transferred i.p. into Crry+/+C3-/- and Crry-/-C3-/- mice. Serum samples were collected from recipient mice and Western blots were performed (Fig. 3A). As early as 1–3 h after serum transfer, the C3 α-chain was reduced in Crry-/-C3-/- recipient mice, being comparable to approximately the 9-h data point in Crry+/+C3-/- recipients. The half-life (t1/2) of WT-derived C3 in Crry-/- hosts was ∼1 h and in Crry+/+ ∼8 h (Fig. 3B). Additional serum transfer experiments indicated that C3b cleavage fragments accumulated in the blood of Crry-/- mice (Fig. 3C). Also, 1 h following serum transfer, C3 deposition in liver but not in the kidney was detected in Crry-/- mice (Fig. 3D). This finding is consistent with the liver being a clearance site for C3c but could also reflect C3b deposition and processing. The accelerated turnover of C3 in a Crry-deficient host further demonstrates the essential role of Crry in the control of complement activation in vivo.
FIGURE 3.
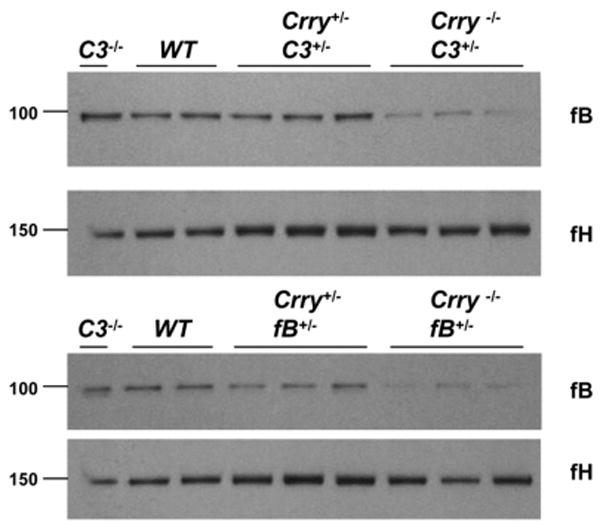
WT serum transfer experiments. A, WT serum was transferred by i.p. injection into Crry+/+C3-/- and Crry-/-C3-/- mice. Serum samples were collected from the recipient mice at the indicated time points following transfer. Western blots (reducing conditions) of the α-chain in the C3-deficient hosts (injected with WT serum) are shown. B, Densitometric scanning analysis of C3 survival in serum transfer experiments (WT to Crry+/+C3-/- or WT to Crry-/-C3-/-). The calculated t1/2 of C3 survival was 8 h in Crry+/+ mice and 1 h in Crry-/- mice. The experiment was performed three times with similar results. C, Intravenous injection of normal serum into Crry+/+C3-/- or Crry-/-C3-/- mice. Serum samples were collected at 10, 20, 30, 40, and 60 min. Western blot of C3 under reducing conditions. D, Increased C3 deposition was observed in the liver at 1 h after injection of WT serum in Crry-/-C3-/- mice. This experiment was performed twice with similar results.
RBC-targeted Crry does not prevent accelerated C3 turnover in vivo
Plasma is continuously in contact with RBCs. To address whether the RBC compartment is responsible for the enhanced turnover of C3 in Crry-/-C3+/- in vivo, this pool was coated with a recombinant fusion protein consisting of a single-chain Ab fragment (scFv) specific for mouse glycophorin A and Crry (Crry5-Ter) (42). This RBC-targeted Crry protects against classical or AP activation (34). Crry-/-C3+/- mice were injected with recombinant Crry5-Ter and the copy number of Crry was determined by FACS. There was a 3- to 5-fold excess of Crry compared with endogenous levels, sufficient to protect against C3 consumption on RBCs (34). However, Western blot analysis did not reveal an increase in C3 in the circulation of Crry-/-C3+/- mice (data not shown). Crry, like MCP and DAF, only protects against complement activation on the cell in which it is expressed (43, 44). An increased turnover is likely occurring on all cells exposed to blood and therefore coating only on one cell population with an inhibitor would have a modest effect on AP activation.
Crry maintains C3 homeostasis by regulating AP activation
Complement activation by the AP is the likely mechanism to explain the C3 consumption in Crry-deficient mice. Consistent with this proposal, there was reduced fB protein in the serum for the respective gene dosage in Crry-/-fB+/- and Crry-/-C3+/- mice (Fig. 4). A 52% decrease of serum fB was observed in Crry-/-C3+/- compared with that of Crry+/-C3+/- mice (n = 6 for Crry-/-C3+/-and n= 5 for Crry+/-C3+/-; p< 0.002). Also, using CVF in vitro to cleave fB into Ba and Bb, both fB and Bb were reduced in the sera of Crry-/-fB+/- mice (data not shown). fH levels of Crry-/-C3+/-and Crry-/-fB+/- mice were unchanged compared with Crry+/-C3+/- and Crry+/-fB+/- controls (Fig. 4). Thus, the concomitant reduction of C3 and fB further implicates excessive AP C3 convertase formation in Crry-deficient mice.
FIGURE 4.
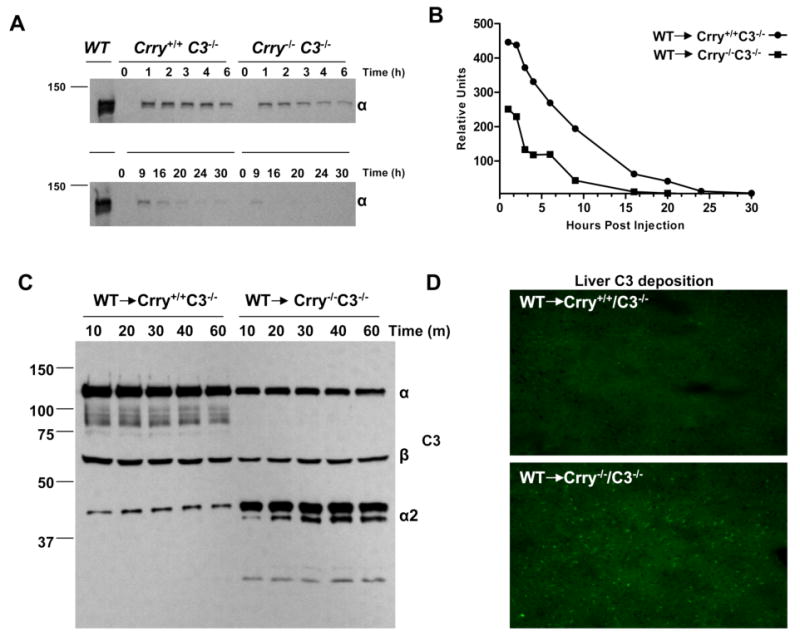
Excessive AP turnover in Crry-/-C3+/- and Crry-/-fB+/- mice. Western blot analysis of fB and fH from sera of Crry+/-C3+/- vs Crry-/-C3+/- and Crry+/-fB+/- vs Crry-/-fB+/- mice. Densitometric scanning analysis indicated that there was 52% reduction of fB in Crry-/-C3+/- compared with that of Crry+/-C3+/- mice (p < 0.002).
In contrast to the reduced C3 level in the sera of Crry-/-fB+/- mice (Fig. 5A, middle lanes), an equivalent quantity of C3 was observed in the Crry-/-fB-/-, Crry+/-fB+/-, and WT mice (Fig. 5A). Thus, complete fB deficiency or 50% of normal Crry levels prevented C3 turnover. To further prove that fB was necessary for the consumption of C3 in Crry-/-C3+/- mice, Crry-/-fB-/- and Crry-/-C3-/- mice were crossed to generate F1 Crry-/-fB+/-C3+/- mice. By intercrossing these F1 mice, we produced Crry-/-fB+/-C3+/- and Crry-/-fB-/-C3+/- mice. As shown in Fig. 5B, enhanced C3 turnover occurs in the Crry-/-fB+/-C3+/- mice compared with Crry+/-C3+/- mice; however, C3 levels in the setting of fB homozygosity (Crry-/-fB-/-C3+/- mice) are comparable to those in Crry+/-C3+/- mice. Thus, introducing fB deficiency into the Crry-/- mice normalized C3 levels for gene dosage.
FIGURE 5.
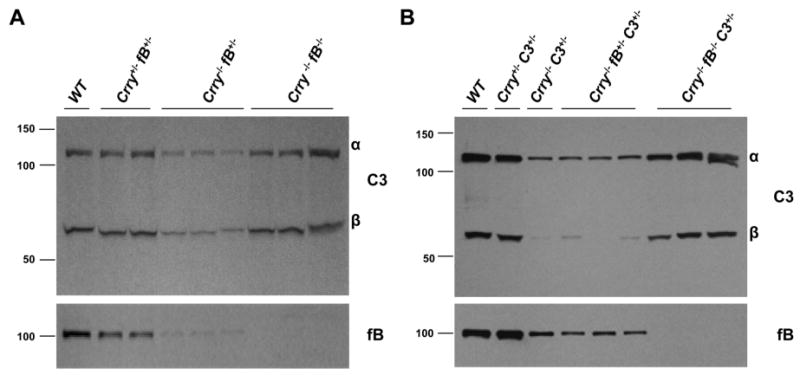
Accelerated C3 turnover in Crry-/-fB+/- and Crry-/-C3+/- mice is ameliorated by fB deficiency. A, Western blot (reducing conditions) of serum C3 and fB from Crry+/-fB+/-, Crry-/-fB+/-, and Crry-/-fB-/- mice. In the presence of fB deficiency, enhanced C3 turnover was prevented. B, Crry-/-C3-/- mice were crossed with Crry-/-fB-/- mice to generate F1 Crry-/-fB+/-C3+/- mice. These mice were crossed to generate Crry-/-fB+/-C3+/- and Crry-/-fB-/-C3+/- animals. C3 turnover was prevented in Crry-/-C3+/- mice by a deficiency of fB.
Although the experimental evidence is consistent with enhanced complement AP activation causing C3 turnover, a defect in C3 synthesis in the liver needed to be ruled out. We therefore measured hepatic C3 mRNA levels by semiquantitative RT-PCR and C3 precursor levels by Western blot. Liver C3 mRNA (Fig. 6A) and C3 protein precursor (Fig. 6B) were comparable between Crry+/-fB+/- and Crry-/-fB+/- mice. Thus, there was not a defect in hepatic C3 biosynthesis.
FIGURE 6.
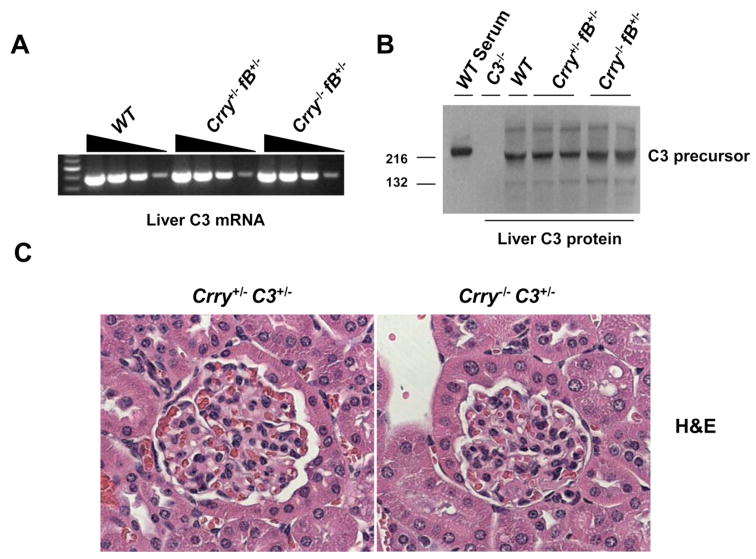
Hepatic liver C3 synthesis and renal kidney morphology in Crry+/- vs Crry-/- mice. A, Liver C3 mRNA levels as measured by RT-PCR are comparable in Crry+/-fB+/- and Crry-/-fB+/-mice. B, Hepatic C3 protein precursor, as detected by the Western blot, is comparable in Crry+/-fB+/- and Crry-/-fB+/- mice. C, Renal morphology in 9-mo-old Crry+/-C3+/- and Crry-/-C3+/- mice. Hypercellularity, mesangial expansion, and thickening of the glomerular basement membrane were not observed in Crry-/- mice. There was also no increase in C3 fragment deposition in Crry-/- mice (data not shown).
We monitored cohorts of Crry+/-C3+/- and Crry-/-C3+/- mice for up to 9 mo. The peripheral blood counts were in the normal range (data not shown). An increase in mortality was not observed in the Crry-/-C3+/- mice. There was also no increase of C3 deposition in the glomerulus of Crry-/-C3+/- mice (data not shown). At least up to 1-year of observation, Crry deficiency did not result in MPGN II (Fig. 6C).
Efficiency of maternal complement activation determines Crry-mediated embryonic mortality: the generation of the Crry single knockout mouse
One mouse with a genotype of Crry-/-fB+/+ was identified among 14 offspring when breeding pairs were fortuitously set up with two females (one Crry+/-fB+/- and one Crry-/-fB+/-) and 3 males (two Crry+/-fB+/- and one Crry-/-fB+/-). The other genotypes in these 14 offspring were 8 Crry+/-fB+/-, 1 Crry+/-fB-/-, 3 Crry-/-fB+/-, and 1 Crry-/-fB-/-. The possibility of a genotyping error was assessed because there had been no prior live births of Crry-/-fB+/+ mice; instead, Crry-/- mice only survived with either complete or partial fB deficiency (33). Nevertheless, Crry-/-fB+/+ remained a possible genotype when the males (Crry+/-fB+/- and Crry-/-fB+/-) and females (Crry+/-fB+/- and Crry-/-fB+/-) are used as breeding partners. The genotyping of Crry-/-fB+/+ was verified (Fig. 7A).
FIGURE 7.
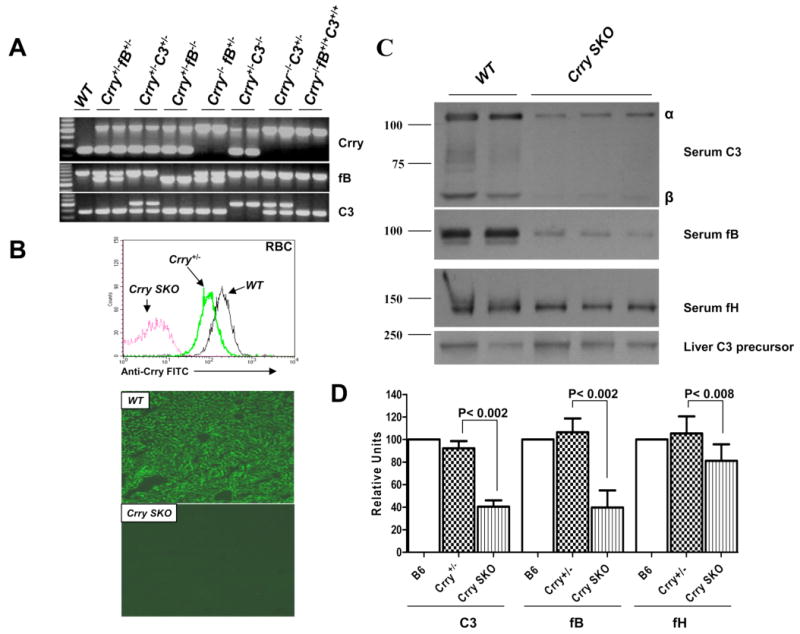
Development of Crry SKO mice. A, Genotyping. WT, Crry/C3, and Crry/fB double knockout mice served as controls. B, Protein expression. Crry is absent on the circulating RBCs (top) by FACS and in the liver by immunohistochemistry using an anti-mouse Crry Ab. C, Western blots demonstrate decreased concentrations of serum C3 and fB, a modest decrease of fH in Crry SKO mice, and normal levels of C3 precursor protein in the liver. C3 or fB split products were not detected. D, Densitometric scanning of C3, fB, and fH levels in WT, Crry+/-, and Crry SKO mice. There was an ∼60% reduction of C3 and fB in Crry SKO mice (p <0.001, n = 5 for C3 group and n = 8 for fB group) and a modest but significant decrease of fH in Crry SKO mice (p <0.008, n = 8).
We hypothesized that a fB heterozygous (fB+/-) background might allow for survival of Crry-/- embryos. By breeding Crry+/-fB+/+ males with Crry-/-fB+/- females (Table I, C), 131 live births were obtained with four genotypes: Crry+/-fB+/- (33 of 131, 25%), Crry-/-fB+/- (28 of 131, 21%), Crry-/-fB+/+ (38 of 131, 29%), and Crry+/-fB+/+ (32 of 131, 24%) (data not shown). However, breeding Crry-/-fB+/- males with Crry+/-fB+/+ females failed to generate Crry-/-fB+/+ mice (0 of 57, 0%) and resulted in only one Crry-/-fB+/- mouse (1 of 57, 1.7%; Table I, D).
The survival of Crry-/-fB+/+ mice was possible because of maternal AP insufficiency and represents the first example of a Crry single knockout (Crry SKO) mouse. Further evidence to support this conclusion was obtained using Crry-/-fB+/- breeding pairs. This cross produced 193 live births with three different genotypes: Crry-/-fB+/+ (50 of 193, 26%), Crry-/-fB+/- (103 of 193, 53%), and Crry-/-fB-/- (40 of 193, 21%) (Table I, E). Again, if the maternal genotype was heterozygous for fB, live births occurred in which the pups were knockouts for only Crry.
We next extended our examination by analyzing Crry-/- mice in the setting of a C3 heterozygous background. Crry+/-C3+/+ males were bred to Crry-/-C3+/- females to generate 194 live births. For 54 mice, the genotype was Crry-/-C3+/+ (Crry SKO) (54 of 194, 28%; Table I, G). In addition, breeding Crry-/-C3+/- to each other generated 10 of 58 live births as Crry-/-C3+/+ (10 of 58, 17%; Table I, H). This outcome is similar to that with Crry-/-fB+/- female mice and occurred with the expected Mendelian frequency. However, breeding Crry+/-C3+/- mice with each other failed to produce any Crry-/-C3+/+ (0 of 169, 0%), only one Crry-/-C3+/- (1 of 169, 0.5%), and no Crry-/-C3-/- (0 of 169, 0%) mice (Table I, I; the remainder of the genotypes from this breeding combination are not shown). Crry+/-fB+/- breedings generated a few less than the expected Mendelian distribution of live births: Crry-/-fB+/+ (6 of 243, 2.4%), Crry-/-fB+/- (12 of 243, 4.93%), and Crry-/-fB-/- genotypes (3 of 243, 1.23%) (Table I, F). Since there is no evidence for accelerated C3 turnover in mice on a Crry heterozygous background (Crry+/-, Crry+/-C3+/-, Crry+/-fB+/-) (Figs. 2 and 7D), these results point to a reduction in fB as being more crucial than a reduction in C3 relative to their contribution to complement-mediated embryo rejection.
The preceding data establish that C3 turnover, by reducing the activating capacity of the AP, allows for live births of Crry SKO mice. Breeding Crry+/-fB+/+C3+/+ males with Crry-/-fB+/+C3+/+ (Crry SKO) females produced Crry+/-fB+/+C3+/+ (37 of 86, 43%) and Crry-/-fB+/+C3+/+ (49 of 86, 57%) (Table I, J). However, breeding Crry-/-fB+/+C3+/+ males with Crry+/-fB+/+C3+/+ females produced no Crry-/-fB+/+C3+/+ mice, as 100% of the offspring were Crry+/-fB+/+C3+/+ mice (31 of 31, 100%) (Table I, K). In summary, a breeding strategy featuring reduced maternal AP-activating capacity generates a stable strain of Crry SKO.
Breeding Crry SKO mice generated a stable line lacking only the Crry gene (Fig. 7, A and B). In these mice C3 and fB are reduced by ∼50%, indicating accelerated AP-mediated C3 tickover (Fig. 7, C and D). Hepatic C3 precursor levels were comparable to those of WT and Crry+/- mice (Fig. 7C). Although fH was normal in Crry-/- mice carrying fB+/- or C3+/-, there was a modest but significant decrease in fH in the Crry SKO mice (Fig. 7D). This may reflect a larger quantity of C3 turnover in the latter that contributes to fH consumption (see Discussion). Taken together, these observations highlight the importance of a membrane protein, Crry, in controlling the complement system.
Discussion
Based on a series of breeding experiments involving crosses of Crry-, C3-, and fB-deficient mice, we conclude that the membrane complement regulator Crry helps to maintain homeostasis of the AP and demonstrates that a reduction in the efficiency of AP activation in the maternal circulation rescues the otherwise embryonically lethal Crry-/- phenotype. The former represents an example whereby a single membrane regulatory protein prevents excessive turnover of the complement system while the latter allows for the generation of Crry SKO mice (without a concomitant C3 or fB gene deficiency).
Turnover of the complement system in Crry deficiency
The complement system is continuously engaged. This was only recently shown for the classical pathway, based on the elevated C3 levels in C1q- and C2/fB-deficient mice (12), but was proposed 40 years ago for the AP (reviewed in Ref. 1). Evidence for this AP tickover in plasma (45) also comes from deficiencies of fH in humans, mouse, and pig and from fI deficiency in humans. In each species, C3 and fB are reduced to <10% of normal blood concentrations and reconstitution experiments demonstrate that low C3 and fB are secondary to accelerated AP turnover. This often leads to glomerulonephritis, featuring prominent C3 fragment deposition, as well as infections because of a secondary C3 deficiency (9).
In this study, reduction in the expected level of C3 was observed in three types of Crry-deficient mice, Crry-/-fB+/-, Crry-/-C3+/-, and Crry SKO. fB levels in blood were also lower than anticipated based on gene dosage, indicating that accelerated AP activation was the responsible mechanism. The normal C3 concentrations, based on gene dosage in Crry-/-fB-/- and Crry-/-fB-/-C3+/- mice also support this conclusion. In addition, using Crry-transgenic mice in which Crry expression was induced by zinc sulfate, C3 levels were increased (46). Also, administration of the function blocking anti-Crry Ab to WT mice led to an increase of C3 tickover as well as C3 deposition in the liver (47). Our in vivo rescue experiment in which a fB null allele was introduced into Crry-/-C3+/- mice is consistent with these data and suggests that the over-expressed Crry inhibited the AP. While this work was in progress, low C3 levels were reported in Crry-/-DAF-/-C3+/- mice; however, a double deficiency of Crry and DAF does not allow for dissection of the relative roles of Crry and DAF (48). Our results establish that a single membrane complement regulator contributes to the control of AP turnover and that CA, rather than DAA, is primarily responsible for holding the AP in check on cells.
We looked for but did not find renal pathology during the first year of life of the Crry-/-C3+/- mice. Several possibilities to explain this finding have been considered. The simplest is that the magnitude of C3 turnover is just not as great as in the complete fH or fI deficiency. This point coupled with reduced AP-activating capacity and relative normal levels of fH in plasma of Crry null mice appears to be sufficient to prevent the generated C3b from causing tissue damage. Also, the turnover in the case of the Crry deficient mice is likely to be primarily on cells which may be more tolerable because of repair mechanisms. However, we are following these mice into the second year and will be examining multiple tissues including the retina and renal glomeruli for the development of pathology.
Maintaining complement homeostasis through membrane and fluid-phase regulators
In fH-/- mice, <5% of normal C3 is detectable in blood and even fH heterozygous (fH+/-) mice have a lower concentration of C3 than WT mice (14). In mice deficient in Crry and heterozygous for C3 or fB, a reduction of C3 for gene dosage was also observed in both Crry-/-C3+/- and Crry-/-fB+/- mice. No reduction of C3 was observed if the mouse was heterozygous for Crry deficiency (Crry+/-C3+/-, Crry+/-fB+/-, and Crry+/-). These results shed light on the role played by fluid-phase and membrane regulators in maintaining homeostasis of the complement system. By inference and analogy to the fluid-phase turnover seen in a deficiency of fH or fI, these results imply that there is continuous turnover of the AP on cells and that CA is required both in the fluid phase (fH) and intrinsic to cells (Crry in rodents or MCP in humans and other mammals) to prevent excessive AP turnover. As pointed out in the Introduction, Crry and MCP are the only widely expressed membrane C3 regulators with CA in mice and humans, respectively.
Inactivation of the continuously generated C3b is an important mechanism to regulate complement activation. C3b represents not only a source of further activation products with a variety of biological functions but also initiates the amplification loop of the AP. This feedback loop has the potential to produce large amounts of C3b that may deposit on the microbial targets as well as on cells and matrices. The experimental evidence in this report emphasizes the critical role played by a membrane complement regulatory protein in regulating this loop. Although its effect on complement regulation is not as dramatic as the fluid phase regulators fI and fH, Crry deficiency leads to an ∼50% reduction of blood C3 (compared with >95% serum C3 consumption in fH-/- mice). Crry probably inactivates the continuously produced C3b which deposits on cell surfaces, primarily through its CA. Therefore, Crry, in combination with fI, is required to maintain homeostasis of the AP on cells. This conclusion is supported by the lack of C3 turnover in Crry-/-fB+/- and Crry-/-C3+/- mice if these mice are made null for fB.
Maternal complement activation and the generation of the Crry SKO mouse
In this report, we compared Crry+/- vs Crry-/- mice on a C3 or fB heterozygous background. As discussed above, substantial C3 turnover occurs in Crry-/-C3+/- or Crry-/-fB+/- mice secondary to decreased regulatory activity for the AP. Importantly, this Crry-dependent C3 turnover on the maternal side allowed for the generation of a stable mutant mouse strain lacking only the Crry gene because of decreased capacity of the AP in female mice.
The survival of Crry SKO mice in the setting of partial maternal C3 or fB deficiency further supports the crucial role of the AP in mediating embryonic death. Accelerated AP-mediated C3 turnover also occurs in Crry SKO mice. This leads to reduced levels of C3 in the circulation, which is essential for the establishment of the Crry SKO mouse line. However, since ∼50% of the normal fB and C3 levels remains in the circulation of Crry SKO mice, this strain can be used to study disease models in which the pathology depends upon complement activation.
The classical pathway does not play a role in embryo rejection since a deficiency of C4 did not allow for live births (33). In this report, we observed that C3 or fB deficiency on the maternal side rescues the Crry-deficient mouse. Of note, some Crry-/- mice survived from Crry+/-fB+/- breeding pairs while there were no Crry-/- live births from Crry+/-C3+/- breedings. This unexpected result suggests that fB plays a more critical role than C3 in embryo rejection; i.e., heterozygosity of fB allowed for live births of Crry-/- mice while loss of one allele of C3 did not.
The in vivo functions of Crry have also been explored by treatment with Crry-Ig and by using Crry-transgenic mice. For example, Crry plays a role in the pathogenesis of autoimmune disorders (such as MRL/lpr mice) (49, 50), ischemia/reperfusion injury (51), and Alzheimer's disease (52, 53). Using transplant models, Crry was shown to be essential to protect Crry deficient RBCs or kidneys from complement attack (35, 36). However, Crry deficiency could previously only be studied in Crry-/-C3-/- and Crry-/-fB-/- mice that completely lack AP activity. Accelerated AP-mediated C3 turnover occurs in the Crry SKO and this turnover has a direct impact on obtaining live births from Crry SKO breedings.
Biological significance of continuous complement turnover and its regulation
These data provide strong evidence for continuous turnover of the AP of complement activation on cells. We view this as a critical surveillance system to provide proinflammatory, opsonic and lytic activities to facilitate identification, clearance, and destruction of pathogens. Moreover, it likely serves a similar role relative to necrotic, apoptotic, and otherwise injured host cells and tissues where it would facilitate repair in some cases but foster elimination in others. A fundamental difference though between pathogen vs self-interactions in the innate immune response is the issue of instructing an adaptive immune response in the first but avoiding this response in the second. How this distinction is accomplished is unclear but it represents a fundamental problem in need of resolution.
The studies reported herein also illustrate the fine balance between regulation and activation of the AP. The lack of one regulator, Crry, on fetal-derived tissue allows for the “normal” maternal AP to attack the developing placenta. On the other hand, if a maternal AP-activating cascade is reduced by ∼50% (despite the total absence of a regulator), the embryo survives. In several examples in this study, a reduction in maternal complement activity capacity was secondary to accelerated complement consumption in the setting of deficient regulatory protein function. In particular, this report illustrates that a membrane inhibitor of complement is required to maintain physiologic levels of the activating pathways required for host defense. Implied in this conclusion is that there is a turnover of the complement-activating cascade on cells analogous to what has been previously shown in the fluid phase (plasma).
Acknowledgments
We greatly appreciate the assistance of Madonna Bogacki in the preparation of this manuscript.
Footnotes
This work was supported by National Institutes of Health Grants R01-AI037618 and R01-AI041592.
Abbreviations used in this paper: AP, alternative pathway; DAA, decay-accelerating activity; CA, cofactor activity; HUS, hemolytic uremic syndrome; fB, factor B; fH, factor H; fI, factor I; MCP, murine cofactor protein; WT, wild type; ZP, zymosan particle; GVBS, gelatin-veronal-buffered saline; CVF, cobra venom factor; DAF, decay-accelerating factor; MPGN II, membranoproliferative glomerulonephritis type II.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology” (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–1310. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 2.Liszewski MK, Farries TC, Lublin DM, Rooney IA, Atkinson JP. Control of the complement system. Adv Immunol. 1996;61:201–283. doi: 10.1016/s0065-2776(08)60868-8. [DOI] [PubMed] [Google Scholar]
- 3.Walport MJ. Complement: first of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 4.Walport MJ. Complement: second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 5.Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, Muslumanolu MH, Kavukcu S, Filler G, Pirson Y, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2003;100:12966–12971. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liszewski MK, Leung MK, Schraml B, Goodship TH, Atkinson JP. Modeling how CD46 deficiency predisposes to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44:1559–1568. doi: 10.1016/j.molimm.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Agerelated macular degeneration-emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zipfel PF, Heinen S, Jozsi M, Skerka C. Complement and diseases: Defective alternative pathway control results in kidney and eye diseases. Mol Immunol. 2006;43:97–106. doi: 10.1016/j.molimm.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Muller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57:321–347. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- 11.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway: acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. 1981;154:856–867. doi: 10.1084/jem.154.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manderson AP, Pickering MC, Botto M, Walport MJ, Parish CR. Continual low-level activation of the classical complement pathway. J Exp Med. 2001;194:747–756. doi: 10.1084/jem.194.6.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, Lambris JD, Lanning L, Lutz HU, Meri S, et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol. 2005;16:1392–1403. doi: 10.1681/ASN.2005010078. [DOI] [PubMed] [Google Scholar]
- 14.Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ, Botto M. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31:424–428. doi: 10.1038/ng912. [DOI] [PubMed] [Google Scholar]
- 15.Pickering MC, Warren J, Rose KL, Carlucci F, Wang Y, Walport MJ, Cook HT, Botto M. Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient mice. Proc Natl Acad Sci USA. 2006;103:9649–9654. doi: 10.1073/pnas.0601094103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogasen K, Jansen JH, Mollnes TE, Hovdenes J, Harboe M. Hereditary porcine membranoproliferative glomerulonephritis type II is caused by factor H deficiency. J Clin Invest. 1995;95:1054–1061. doi: 10.1172/JCI117751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vyse TJ, Spath PJ, Davies KA, Morley BJ, Philippe P, Athanassiou P, Giles CM, Walport MJ. Hereditary complement factor I deficiency. QJM. 1994;87:385–401. [PubMed] [Google Scholar]
- 18.Genel F, Sjoholm AG, Skattum L, Truedsson L. Complement factor I deficiency associated with recurrent infections, vasculitis and immune complex glomerulonephritis. Scand J Infect Dis. 2005;37:615–618. doi: 10.1080/00365540510034536. [DOI] [PubMed] [Google Scholar]
- 19.Rose KL, Paixao-Cavalcante D, Fish J, Manderson AP, Malik TH, Bygrave AE, Lin T, Sacks SH, Walport MJ, Cook HT, et al. Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice. J Clin Invest. 2008;118:608–618. doi: 10.1172/JCI32525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.West CD, McAdams AJ. Membranoproliferative glomerulonephritis type III: association of glomerular deposits with circulating nephritic factor-stabilized convertase. Am J Kidney Dis. 1998;32:56–63. doi: 10.1053/ajkd.1998.v32.pm9669425. [DOI] [PubMed] [Google Scholar]
- 21.Sun X, Funk CD, Deng C, Sahu A, Lambris JD, Song WC. Role of decay-accelerating factor in regulating complement activation on the erythrocyte surface as revealed by gene targeting. Proc Natl Acad Sci USA. 1999;96:628–633. doi: 10.1073/pnas.96.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin F, Emancipator SN, Salant DJ, Medof ME. Decay-accelerating factor confers protection against complement-mediated podocyte injury in acute nephrotoxic nephritis. Lab Invest. 2002;82:563–569. doi: 10.1038/labinvest.3780451. [DOI] [PubMed] [Google Scholar]
- 23.Parker CJ. The pathophysiology of paroxysmal nocturnal hemoglobinuria. Exp Hematol. 2007;35:523–533. doi: 10.1016/j.exphem.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 24.Fremeaux-Bacchi V, Moulton EA, Kavanagh D, Dragon-Durey MA, Blouin J, Caudy A, Arzouk N, Cleper R, Francois M, Guest G, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17:2017–2025. doi: 10.1681/ASN.2005101051. [DOI] [PubMed] [Google Scholar]
- 25.Wong WW, Fearon DT. p65: A C3b-binding protein on murine cells that shares antigenic determinants with the human C3b receptor (CR1) and is distinct from murine C3b receptor. J Immunol. 1985;134:4048–4056. [PubMed] [Google Scholar]
- 26.Li B, Sallee C, Dehoff M, Foley S, Molina H, Holers VM. Mouse Crry/p65: characterization of monoclonal antibodies and the tissue distribution of a functional homologue of human MCP and DAF. J Immunol. 1993;151:4295–4305. [PubMed] [Google Scholar]
- 27.Kim YU, Kinoshita T, Molina H, Hourcade D, Seya T, Wagner LM, Holers VM. Mouse complement regulatory protein Crry/p65 uses the specific mechanisms of both human decay accelerating factor and membrane cofactor protein. J Exp Med. 1995;181:151. doi: 10.1084/jem.181.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foley S, Li B, Dehoff M, Molina M, Holers VM. Mouse Crry/p65 is a regulator of the alternative pathway of complement activation. Eur J Immunol. 1993;23:1381–1384. doi: 10.1002/eji.1830230630. [DOI] [PubMed] [Google Scholar]
- 29.Kameyoshi Y, Matsushita M, Okada H. Murine membrane inhibitor of complement which accelerates decay of human C3 convertase. Immunology. 1989;68:439–444. [PMC free article] [PubMed] [Google Scholar]
- 30.Spicer AP, Seldin MF, Gendler SJ. Molecular cloning and chromosomal localization of the mouse decay-accelerating factor (DAF) genes: duplicated genes encode GPI-anchored and transmembrane forms. J Immunol. 1995;155:3079–3091. [PubMed] [Google Scholar]
- 31.Harris CL, Rushmere NK, Morgan BP. Molecular and functional analysis of mouse decay accelerating factor (CD55) Biochem J. 1999;341:821–829. [PMC free article] [PubMed] [Google Scholar]
- 32.Xu C, Mao D, Holers VM, Palanca B, Cheng AM, Molina H. A critical role for the murine complement regulator Crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- 33.Mao D, Wu X, Deppong C, Friend LD, Dolecki G, Nelson DM, Molina H. Negligible role of antibodies and C5 in pregnancy loss associated exclusively with C3-dependent mechanisms through complement alternative pathway. Immunity. 2003;19:813–822. doi: 10.1016/s1074-7613(03)00321-2. [DOI] [PubMed] [Google Scholar]
- 34.Spitzer D, Unsinger J, Mao D, Wu X, Molina H, Atkinson JP. In vivo correction of complement regulatory protein deficiency with an inhibitor targeting the RBC membrane. J Immunol. 2005;175:7763–7770. doi: 10.4049/jimmunol.175.11.7763. [DOI] [PubMed] [Google Scholar]
- 35.Miwa T, Zhou L, Hilliard B, Molina H, Song WC. Crry, but not CD59 and DAF, is indispensable for murine erythrocyte protection in vivo from spontaneous complement attack. Blood. 2002;99:3707–3716. doi: 10.1182/blood.v99.10.3707. [DOI] [PubMed] [Google Scholar]
- 36.Bao L, Wang Y, Chang A, Minto AW, Zhou J, Kang H, Haas M, Quigg RJ. Unrestricted C3 activation occurs in Crry-deficient kidneys and rapidly leads to chronic renal failure. J Am Soc Nephrol. 2007;18:811–822. doi: 10.1681/ASN.2006101176. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, Fugita S, Hidvegi T, Chaplin DD, Colten HR. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci USA. 1997;94:8720–8725. doi: 10.1073/pnas.94.16.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Circolo A, Garnier G, Fukuda W, Wang X, Hidvegi T, Szalai AJ, Briles DE, Volanakis JE, Wetsel RA, Colten HR. Genetic disruption of the murine complement C3 promoter region generates deficient mice with extrahepatic expression of C3 mRNA. Immunopharmacology. 1999;42:135–149. doi: 10.1016/s0162-3109(99)00021-1. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, Boackle SA, Colten HR, Gilkeson GS. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000;164:786–794. doi: 10.4049/jimmunol.164.2.786. [DOI] [PubMed] [Google Scholar]
- 40.Sekine H, Reilly CM, Molano ID, Garnier G, Circolo A, Ruiz P, Holers VM, Boackle SA, Gilkeson GS. Complement component C3 is not required for full expression of immune complex glomerulonephritis in MRL/lpr mice. J Immunol. 2001;166:6444–6451. doi: 10.4049/jimmunol.166.10.6444. [DOI] [PubMed] [Google Scholar]
- 41.Wu X, Peng SL. Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum. 2006;54:336–342. doi: 10.1002/art.21553. [DOI] [PubMed] [Google Scholar]
- 42.Spitzer D, Unsinger J, Bessler M, Atkinson JP. scFv-Mediated in vivo targeting of DAF to erythrocytes inhibits lysis by complement. Mol Immunol. 2004;40:911–919. doi: 10.1016/j.molimm.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 43.Oglesby TJ, Allen CJ, Liszewski MK, White DJG, Atkinson JP. Membrane cofactor protein (MCP;CD46) protects cells from complementmediated attack by an intrinsic mechanism. J Exp Med. 1992;175:1547–1551. doi: 10.1084/jem.175.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med. 1984;160:1558–1578. doi: 10.1084/jem.160.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lachmann PJ, Nicol P. Reaction mechanism of the alternative pathway of complement fixation. Lancet. 1973;1:465–467. doi: 10.1016/s0140-6736(73)91886-2. [DOI] [PubMed] [Google Scholar]
- 46.Kang HJ, Bao L, Xu Y, Quigg RJ, Giclas PC, Holers VM. Increased serum C3 levels in Crry transgenic mice partially abrogates its complement inhibitory effects. Clin Exp Immunol. 2004;136:194–199. doi: 10.1111/j.1365-2249.2004.02450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuo S, Ichida S, Takizawa H, Okada N, Baranyi L, Iguchi A, Morgan BP, Okada H. In vivo effects of monoclonal antibodies that functionally inhibit complement regulatory proteins in rats. J Exp Med. 1994;180:1619–1627. doi: 10.1084/jem.180.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miwa T, Zhou L, Tudoran R, Lambris JD, Madaio MP, Nangaku M, Molina H, Song WC. DAF/Crry double deficiency in mice exacerbates nephrotoxic serum-induced proteinuria despite markedly reduced systemic complement activity. Mol Immunol. 2007;44:139–146. doi: 10.1016/j.molimm.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 49.Alexander JJ, Jacob A, Bao L, Macdonald RL, Quigg RJ. Complement-dependent apoptosis and inflammatory gene changes in murine lupus cerebritis. J Immunol. 2005;175:8312–8319. doi: 10.4049/jimmunol.175.12.8312. [DOI] [PubMed] [Google Scholar]
- 50.Alexander JJ, Jacob A, Vezina P, Sekine H, Gilkeson GS, Quigg RJ. Absence of functional alternative complement pathway alleviates lupus cerebritis. Eur J Immunol. 2007;37:1691–1701. doi: 10.1002/eji.200636638. [DOI] [PubMed] [Google Scholar]
- 51.Thurman JM, Ljubanovic D, Royer PA, Kraus DM, Molina H, Barry NP, Proctor G, Levi M, Holers VM. Altered renal tubular expression of the complement inhibitor Crry permits complement activation after ischemia/reperfusion. J Clin Invest. 2006;116:357–368. doi: 10.1172/JCI24521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease: a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]