

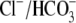
Abstract
To analyze the cardiac functions of AE3, we disrupted its gene
(Slc4a3) in mice.
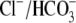 exchange coupled with Na+-dependent acid extrusion can mediate
pH-neutral Na+ uptake, potentially affecting Ca2+
handling via effects on Na+/Ca2+ exchange. AE3 null mice
appeared normal, however, and AE3 ablation had no effect on
ischemia-reperfusion injury in isolated hearts or cardiac performance in
vivo. The NKCC1 Na+-K+-2Cl–
cotransporter also mediates Na+ uptake, and loss of NKCC1 alone
does not impair contractility. To further stress the AE3-deficient myocardium,
we combined the AE3 and NKCC1 knock-outs. Double knock-outs had impaired
contraction and relaxation both in vivo and in isolated ventricular
myocytes. Ca2+ transients revealed an apparent increase in
Ca2+ clearance in double null cells. This was unlikely to result
from increased Ca2+ sequestration, since the ratio of
phosphorylated phospholamban to total phospholamban was sharply reduced in all
three mutant hearts. Instead, Na+/Ca2+ exchanger
activity was found to be enhanced in double null cells. Systolic
Ca2+ was unaltered, however, suggesting more direct effects on the
contractile apparatus of double null myocytes. Expression of the catalytic
subunit of protein phosphatase 1 was increased in all mutant hearts. There was
also a dramatic reversal, between single null and double null hearts, in the
carboxymethylation and localization to the myofibrillar fraction, of the
catalytic subunit of protein phosphatase 2A, which corresponded to the loss of
normal contractility in double null hearts. These data show that AE3 and NKCC1
affect Ca2+ handling, PLN regulation, and expression and
localization of major cardiac phosphatases and that their combined loss
impairs cardiac function.
exchange coupled with Na+-dependent acid extrusion can mediate
pH-neutral Na+ uptake, potentially affecting Ca2+
handling via effects on Na+/Ca2+ exchange. AE3 null mice
appeared normal, however, and AE3 ablation had no effect on
ischemia-reperfusion injury in isolated hearts or cardiac performance in
vivo. The NKCC1 Na+-K+-2Cl–
cotransporter also mediates Na+ uptake, and loss of NKCC1 alone
does not impair contractility. To further stress the AE3-deficient myocardium,
we combined the AE3 and NKCC1 knock-outs. Double knock-outs had impaired
contraction and relaxation both in vivo and in isolated ventricular
myocytes. Ca2+ transients revealed an apparent increase in
Ca2+ clearance in double null cells. This was unlikely to result
from increased Ca2+ sequestration, since the ratio of
phosphorylated phospholamban to total phospholamban was sharply reduced in all
three mutant hearts. Instead, Na+/Ca2+ exchanger
activity was found to be enhanced in double null cells. Systolic
Ca2+ was unaltered, however, suggesting more direct effects on the
contractile apparatus of double null myocytes. Expression of the catalytic
subunit of protein phosphatase 1 was increased in all mutant hearts. There was
also a dramatic reversal, between single null and double null hearts, in the
carboxymethylation and localization to the myofibrillar fraction, of the
catalytic subunit of protein phosphatase 2A, which corresponded to the loss of
normal contractility in double null hearts. These data show that AE3 and NKCC1
affect Ca2+ handling, PLN regulation, and expression and
localization of major cardiac phosphatases and that their combined loss
impairs cardiac function.
At least four electroneutral
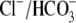 exchangers, termed AE1, AE2, AE3 (Slc4a1–3), and PAT1 or CFEX
(Slc26a6), are expressed in cardiac muscle
(1–6).
Despite their abundance in the heart, which exceeds that of most other tissues
(1,
6,
7), and the wealth of
information about their epithelial functions, the physiological roles of
sarcolemmal
exchangers, termed AE1, AE2, AE3 (Slc4a1–3), and PAT1 or CFEX
(Slc26a6), are expressed in cardiac muscle
(1–6).
Despite their abundance in the heart, which exceeds that of most other tissues
(1,
6,
7), and the wealth of
information about their epithelial functions, the physiological roles of
sarcolemmal
 exchangers in the heart remain unclear. In epithelial tissues,
exchangers in the heart remain unclear. In epithelial tissues,
 exchange coupled with Na+/H+ exchange or
exchange coupled with Na+/H+ exchange or
 cotransport mediates NaCl absorption across apical membranes, serves as a NaCl
uptake system on basolateral membranes to facilitate anion secretion, and
contributes to increases in cell volume
(8–13).
Because their activities are balanced, the resulting fluxes of acid-base
equivalents and electrolytes take place with little or no change in
pHi.3
Similarly, in cardiac myocytes,
cotransport mediates NaCl absorption across apical membranes, serves as a NaCl
uptake system on basolateral membranes to facilitate anion secretion, and
contributes to increases in cell volume
(8–13).
Because their activities are balanced, the resulting fluxes of acid-base
equivalents and electrolytes take place with little or no change in
pHi.3
Similarly, in cardiac myocytes,
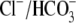 exchange acting alone would acidify the cell, impairing contractility;
instead, there is evidence that such
exchange acting alone would acidify the cell, impairing contractility;
instead, there is evidence that such
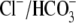 exchange operates in concert with Na+-coupled acid extrusion
(14). Cardiac myocytes contain
an abundance of Na+ transport mechanisms mediating substantial
levels of inward Na+ flux with major physiological effects
(15,
16). We hypothesized that
major ion fluxes resulting from these coupled transport mechanisms involving
exchange operates in concert with Na+-coupled acid extrusion
(14). Cardiac myocytes contain
an abundance of Na+ transport mechanisms mediating substantial
levels of inward Na+ flux with major physiological effects
(15,
16). We hypothesized that
major ion fluxes resulting from these coupled transport mechanisms involving
 exchange would significantly impact Ca2+ handling and cardiac
contractility.
exchange would significantly impact Ca2+ handling and cardiac
contractility.
Ischemia causes pHi to decline and Cl–
levels to rise in cardiac myocytes or hearts
(17). Nonspecific stilbene
derivatives, known to inhibit
 exchange and other
exchange and other  transporters,
reduce these changes in pHi and Cl–
levels (18), suggesting that
transporters,
reduce these changes in pHi and Cl–
levels (18), suggesting that
 exchange contributes to the observed perturbations caused by ischemia.
Stilbene derivatives also limited Ca2+ loading, which leads to
hypercontracture injury during reoxygenation
(17), indicating that
Na+ loading coupled to
exchange contributes to the observed perturbations caused by ischemia.
Stilbene derivatives also limited Ca2+ loading, which leads to
hypercontracture injury during reoxygenation
(17), indicating that
Na+ loading coupled to
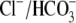 exchange may increase [Na+]i and therefore
Ca+ loading beyond what would occur as a result of
Na+/H+ exchange alone. However, it is unclear whether
the beneficial effects of stilbene derivatives are due to inhibition of one or
more of the
exchange may increase [Na+]i and therefore
Ca+ loading beyond what would occur as a result of
Na+/H+ exchange alone. However, it is unclear whether
the beneficial effects of stilbene derivatives are due to inhibition of one or
more of the
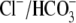 exchangers or to inhibition of
exchangers or to inhibition of
 cotransport in the heart, since stilbene derivatives also inhibit
cotransport in the heart, since stilbene derivatives also inhibit
 cotransport, which is functionally equivalent to Na+/H+
exchange and appears to be involved in ischemia-reperfusion injury
(19).
cotransport, which is functionally equivalent to Na+/H+
exchange and appears to be involved in ischemia-reperfusion injury
(19).
In order to gain a better understanding of the functions of the
 exchangers in cardiac muscle, we developed a mouse model in which the AE3 gene
was disrupted and analyzed both the response to ischemia-reperfusion injury in
isolated hearts and cardiac performance in vivo. The absence of an
overt cardiac phenotype suggested the possibility that the effects of AE3
deficiency were being masked by the activities of the three remaining
exchangers in cardiac muscle, we developed a mouse model in which the AE3 gene
was disrupted and analyzed both the response to ischemia-reperfusion injury in
isolated hearts and cardiac performance in vivo. The absence of an
overt cardiac phenotype suggested the possibility that the effects of AE3
deficiency were being masked by the activities of the three remaining
 exchangers, coupled with one or more of the Na+-dependent acid
extrusion systems in the heart, and the NKCC1
Na+-K+-2Cl– cotransporter
(Slc12a2). Although loss of NKCC1 alone has shown no effect on
cardiac contractility (20),
NKCC1 ablation reduced Ca2+ levels in astrocytes
(21) via effects on
Na+/Ca2+ exchange. Thus, to further stress the
Na+ loading capacity of the AE3-deficient myocardium, we combined
the AE3 and NKCC1 knock-outs.
exchangers, coupled with one or more of the Na+-dependent acid
extrusion systems in the heart, and the NKCC1
Na+-K+-2Cl– cotransporter
(Slc12a2). Although loss of NKCC1 alone has shown no effect on
cardiac contractility (20),
NKCC1 ablation reduced Ca2+ levels in astrocytes
(21) via effects on
Na+/Ca2+ exchange. Thus, to further stress the
Na+ loading capacity of the AE3-deficient myocardium, we combined
the AE3 and NKCC1 knock-outs.
We hypothesized that cardiac function would be compromised in double null
hearts because of effects on Ca2+ handling. Our data showed that
loss of AE3 and NKCC1 together impaired cardiac contractility and altered
Ca2+ handling via modulation of both NCX
(Na+/Ca2+ exchanger)-mediated Ca2+ transport
and PLN expression and phosphorylation. However, since these alterations did
not significantly impact SR Ca2+ stores, it seemed likely that the
impaired contractility in double null hearts was the result of effects on the
contractile apparatus. Biochemical studies revealed that ablation of either
AE3 or NKCC1 had similar effects on the expression and/or localization of the
two principal phosphatases PP1 and PP2A (protein phosphatase 1 and 2A), both
of which are known to regulate myofibrillar proteins and cardiac
contractility. These results demonstrate, for the first time, that
AE3-mediated
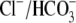 exchange and NKCC1-mediated Na+-K+-2Cl–
cotransport are intimately involved in the regulation of cardiac contractility
and suggest that they serve very similar functions.
exchange and NKCC1-mediated Na+-K+-2Cl–
cotransport are intimately involved in the regulation of cardiac contractility
and suggest that they serve very similar functions.
EXPERIMENTAL PROCEDURES
Generation of Null Mutant Mice and Genotype Analysis—A 1.9-kb SmaI fragment extending from exon 2 to exon 6 of the mouse AE3 gene (22) and a 3.3-kb BamHI fragment extending from intron 7 to intron 14 were inserted into the pMJKO targeting vector (23). Electroporation, selection, and analysis of 129SvJ embryonic stem cells and blastocyst-mediated transgenesis were performed as described previously (23). The probe used for Southern blot analysis was a 0.54-kb BamHI-SmaI fragment extending from exon 1 to exon 2, which allowed analysis of homologous recombination at the 5′-end of the gene. To confirm homologous recombination at the 3′-end of the gene, the 3′-arm of the targeting construct was used as a probe. Chimeric male mice were bred with Black Swiss mice, and mutant offspring were identified by Southern blot analysis.
For the I/R injury studies, the AE3 null and wild-type mice were of the original mixed 129SvJ and Black Swiss background. For studies of cardiovascular performance in vivo, AE3 null and wild-type mice of both the mixed background and an inbred FVB/N background were analyzed. For in vivo cardiovascular studies in which the NKCC1 knock-out (24) was crossed with the AE3 knock-out, mice of the mixed 129SvJ and Black Swiss lines were used. AE3 null mutant and wild-type mice used for in vivo studies were generated by breeding AE3 heterozygous mice. AE3/NKCC1 double null mutant and wild-type mice were generated by breeding double heterozygous mutants. Mutant and wild-type mice for each experiment were derived from the same group of breeding pairs, and sibling pairs were used as often as possible. The AE3/NKCC1 double null and wild-type mice used for in vivo studies were obtained from the initial double knock-out breeders; the in vitro Ca2+ imaging and myocyte mechanics studies used double KOs and WTs derived from double heterozygous breeders from later generations of this colony.
Genotype analysis was performed by PCR using DNA from tail biopsies and sets of three primers that simultaneously amplify fragments from the wild-type and mutant alleles. For AE3, the PCR contained a primer (5′-GACAATAGCAGGCATGCTGG-3′) derived from the 3′-end of the neoR gene, a forward primer (5′-GATGAAGATGACAGCCCAGG-3′) from exon 5 of the AE3 gene, and a reverse primer (5′-GACTAGGTGGGGACACCCTC-3′) from the region of intron 6 preceding the heart-specific exon. These primers amplify 785-base pair and 605-base pair products from the wild-type and mutant alleles, respectively. For NKCC1, PCR genotyping of wild-type and mutant alleles was performed as described previously (24).
Northern Blot Analysis—Total RNA was isolated from the heart and brain of adult wild-type, AE3 heterozygous, and AE3 homozygous mutant mice using the Tri-Reagent kit (Molecular Research Center, Cincinnati, OH), and Northern blots were prepared and hybridized as described previously (23) using a 1.2-kilobase AE3 probe spanning codons 124–528, which are part of the common region present in both AE3 mRNAs.
Langendorff Heart Studies—Susceptibility of wild-type and AE3 null hearts (n = 6 hearts/genotype) to impaired cardiac function resulting from I/R injury was analyzed using a Langendorff apparatus as described for the Na+/H+ exchanger 1 knock-out (25), except that a 30-min period of no-flow ischemia was used.
Analysis of in Vivo Cardiac Performance—Left ventricular function and mean arterial blood pressure of anesthetized adult male and female mice were analyzed using the intact closed chest model, as described previously (26, 27). Left ventricular pressure was recorded via a high fidelity transducer (Millar Instruments, Houston, TX) inserted into the left ventricle, and arterial blood pressure was recorded from a fluid-filled catheter in the femoral artery. A femoral vein catheter was used for infusion of dobutamine, a β-adrenergic agonist.
Ca2+ Transient Analysis and Ventricular Myocyte Mechanics—Cardiomyocytes were enzymatically dissociated from ventricles of 3–5-month-old mutant and wild-type hearts mounted in a Langendorff perfusion apparatus (28), and Ca2+ imaging was carried out as described before (29). Analysis of Ca2+ transients elicited by application of caffeine was carried out as previously described (30). Briefly, adult ventricular myocytes were initially stimulated at a constant frequency of 0.5 Hz. 10–15 s after the last stimulation, 10 mm caffeine was added to the bath solution, and the rate and amplitude of fluorescent changes were measured using a dual excitation fluorescence photomultiplier system. Ventricular myocyte mechanics were determined using an edge detection camera that allowed simultaneous recordings of cell length changes.
Immunoblot Analysis—Mice were anesthetized with avertin, and hearts were excised, rinsed in ice-cold phosphate-buffered saline, frozen in liquid N2, and stored at –80 °C until further use. Each heart was manually pulverized in liquid N2 and then homogenized in Buffer H (10 mm Tris (pH 7.4), 1 mm EDTA, 0.25% Nonidet P-40, 2 mm dithiothreitol, plus protease and phosphatase inhibitors) using a Polytron 3000 mechanical homogenizer. Protein concentration was determined using the Coomassie Protein Assay Reagent (Pierce). Total homogenate was separated into soluble and myofibrillar fractions, as previously described (31), with minor variations. Briefly, 2 mg of total homogenate was resuspended in 1 ml of 20 mm Tris (pH 7.4 plus protease and phosphatase inhibitors), homogenized using the Polytron 3000 homogenizer, and fractionated by centrifugation at 3000 × g for 10 min at 4 °C. The pellet, identified as the myofibril-enriched fraction, was resuspended in the fractionation buffer. Proteins were resolved on a discontinuous, reducing SDS-polyacrylamide gel, transferred to nitrocellulose or polyvinylidene difluoride membranes, and probed for specific proteins using the following antibodies: monoclonal anti-SERCA2a (sarco(endo)plasmic reticulum isoform 2a; clone 2A7-A1; Abcam Inc., Cambridge, MA), monoclonal anti-NCX (catalog number R3F1; Swant, Bellinzona, Switzerland), anti-PLN (catalog number MA3-922; Affinity Bioreagents, Golden, CO), anti-phosphoserine 16-phospholamban and anti-phosphothreonine 17-phospholamban (catalog numbers A010–12 and A010–13; Badrilla Ltd., Leeds, UK), anti-nonmethylated PP2A-C (catalytic subunit of PP2A; catalog number 4957 clone 4B7; Cell Signaling Technology, Danvers, MA), anti-PP2A-C (catalog number 05-421, clone 1D6; Millipore, Temecula, CA), anti-PP1-C (catalytic subunit of PP1) antibody raised against recombinant human PP1α and expected to cross-react with PP1β and PP1γ in addition to detecting PP1α (catalog number MAB3000, clone 319319; R&D Systems, Minneapolis, MN), and horseradish peroxidase-conjugated secondary antibodies (KPL Inc., Gaithersburg, MD). Chemiluminescence was developed using the LumiGlo chemiluminescent substrate kit (KPL), and autoradiograms were developed using BioMax Films ML and MR (Eastman Kodak Co.). Anti-PP2A-C antibody (clone 1D6) is methylation-sensitive (32) and binds demethylated PP2A-C with greater affinity than the methylated PP2A-C. Demethylation of PP2A-C was carried out by cold base treatment of the membrane as previously described (33).
Statistics—For group comparisons, a mixed factor analysis of variance with repeated measures on the second factor was used. Individual comparisons were performed using Student's t test.
RESULTS
AE3–/– Mice Were Indistinguishable from Wild-type Littermates and Were Fertile—The targeting strategy (Fig. 1A) was designed to eliminate both the 1227-amino acid AE3 variant in brain and the 1030-amino acid variant in heart. Any mRNA transcribed from the promoter for the long form would lack codons 209–319 and would either be disrupted by the neomycin resistance gene or contain a frameshift if it were deleted. The absence of the transcription start site and first two exons of the heart-specific transcript ensured the loss of this mRNA. Targeted embryonic stem cells were identified by Southern blot analysis (Fig. 1B), and breeding of heterozygous mutants yielded offspring of all three genotypes (Fig. 1C) in a normal Mendelian ratio. Northern blot analysis (Fig. 1D) revealed abundant mRNA for the long and short forms of AE3 in wild-type brain and heart, respectively, but not in brain or heart of AE3–/– mice.
FIGURE 1.

AE3 gene targeting, genotyping, and mRNA analysis. A, targeting strategy. Top, intron-exon organization of the wild-type allele, with filled boxes indicating exons. Middle, targeting construct, with the neomycin resistance gene (NEO) replacing exon 6, the first exon of the heart-specific variant (exon C1), and exon 7. The herpes simplex virus thymidine kinase gene (TK) was included for negative selection. Bottom, structure of the targeted allele. The locations of the wild-type 4.4- and 3.3-kb BamHI fragments and the mutant 7.4-kb BamHI fragment are shown above the corresponding allele. The locations of the diagnostic probes are indicated below the targeted allele. SmaI (S) and BamHI (B) restriction sites used for preparation of the construct and probes are shown in each allele. B, Southern blot analysis of BamHI-digested wild-type (+/+) and targeted (+/–) embryonic stem cell DNA using the outside probe (left) and tail DNA using the inside probe (right). C, PCR genotyping revealed the presence of wild-type, heterozygous, and null mutant offspring (785 bp, wild-type allele; 605 bp, mutant allele). D, RNA (10 μg/lane) from brain and heart of wild-type (+/+) and null mutant (–/–) mice was hybridized with an AE3 cDNA probe corresponding to sequences from exons 8–14.
AE3–/– mice grew normally and were indistinguishable from wild-type mice in both appearance and behavior, and both male and female mutants were fertile. Heart weight/body weight ratios for AE3–/– and wild-type mice of the original 129SvJ and Black Swiss mixed background were 5.97 ± 0.17 mg/g and 6.98 ± 0.39 mg/g, respectively (p = 0.034, n = 7 for each genotype). As discussed below, however, loss of AE3 by itself had no apparent effect on I/R injury or cardiac performance in vivo, and heart weight/body weight ratios for AE3/NKCC1 double knock-out mice, where a defect in cardiac performance was observed, were not significantly different from wild-type controls.
Loss of AE3 Did Not Affect Ischemia-Reperfusion Injury—Loss
of Na+/H+ exchange or
 cotransport activity protects against I/R injury
(19,
25,
34,
35) by reducing the
Na+ loading that leads to excess Ca2+ loading. Because
cotransport activity protects against I/R injury
(19,
25,
34,
35) by reducing the
Na+ loading that leads to excess Ca2+ loading. Because
 exchange can operate in concert with these acid extrusion mechanisms, thereby
enhancing their activities, and has an opposite effect on
pHi when operating alone, we analyzed the effect of the
AE3 null mutation on I/R injury (Fig.
2). Using the Langendorff heart preparation, basal values of left
ventricular developed pressure (107.0 ± 2.8 and 103.4 ± 2.8 mm
Hg in AE3–/– and wild-type hearts, respectively) or
left ventricular end diastolic pressure (7.3 ± 0.5 and 6.0 ± 0.5
mm Hg in AE3–/– and wild-type hearts, respectively) did
not differ significantly between the two genotypes (n = 6 for each
genotype). Following 30 min of ischemia, no significant differences were
observed in either of these parameters between wild-type and
AE3–/– hearts (Fig.
2). After 20–30 min of reperfusion, left ventricular
developed pressure (69.6 ± 2.8 and 64.6 ± 4.6 mm Hg in
AE3–/– and wild-type hearts, respectively) and LVEDP
(25.3 ± 2.9 and 27.4 ± 3.8 mm Hg in
AE3–/– and wild-type hearts, respectively) were similar
in both genotypes.
exchange can operate in concert with these acid extrusion mechanisms, thereby
enhancing their activities, and has an opposite effect on
pHi when operating alone, we analyzed the effect of the
AE3 null mutation on I/R injury (Fig.
2). Using the Langendorff heart preparation, basal values of left
ventricular developed pressure (107.0 ± 2.8 and 103.4 ± 2.8 mm
Hg in AE3–/– and wild-type hearts, respectively) or
left ventricular end diastolic pressure (7.3 ± 0.5 and 6.0 ± 0.5
mm Hg in AE3–/– and wild-type hearts, respectively) did
not differ significantly between the two genotypes (n = 6 for each
genotype). Following 30 min of ischemia, no significant differences were
observed in either of these parameters between wild-type and
AE3–/– hearts (Fig.
2). After 20–30 min of reperfusion, left ventricular
developed pressure (69.6 ± 2.8 and 64.6 ± 4.6 mm Hg in
AE3–/– and wild-type hearts, respectively) and LVEDP
(25.3 ± 2.9 and 27.4 ± 3.8 mm Hg in
AE3–/– and wild-type hearts, respectively) were similar
in both genotypes.
FIGURE 2.

Effects of AE3 ablation on cardiac function during ischemia and reperfusion. Hearts from WT and AE3–/– mice of the mixed 129SvJ and Black Swiss background, retrogradely perfused in a Langendorff apparatus, were subjected to a 25-min equilibration period (Eq), followed by a 30-min period of no-flow ischemia (shaded boxes) and a 30-min period of reperfusion. The hearts were paced at 400 beats/min during the initial equilibration; pacing was terminated during ischemia and then reinitiated 3 min after the start of the reperfusion period. Loss of AE3 did not elicit cardioprotective effects during ischemia and reperfusion. No significant differences were observed between wild-type and AE3 null hearts (n = 6 of each genotype), with respect to left ventricular developed pressure (LVDP)(A) or LVEDP (B). Values are means ± S.E.
Cardiovascular Performance Was Normal in AE3–/– Mice—Cardiovascular performance was measured in vivo using mice of both the original 129SvJ and Black Swiss mixed background and an FVB/N inbred background (Fig. 3). In response to β-adrenergic stimulation, the chronotropic effect was slightly higher in AE3–/– mice than in wild-type mice (Fig. 3A); however, the differences were not significant, and there were no significant differences between the two genotypes with respect to mean arterial pressure, left ventricular systolic pressure, left ventricular end diastolic pressure, and maximum dP/dt (Fig. 3, B–E). There were possible background differences in MAP, since the knock-out exhibited slightly elevated basal MAP on the mixed background (78.5 ± 6.3 and 71.6 ± 4.8 mm Hg in AE3–/– and wild-type mice, respectively) and reduced MAP on the FVB/N background (72.6 ± 4.1 and 79.2 ± 3.5 mm Hg in AE3–/– and wild-type mice, respectively). Corresponding differences were observed in basal systolic LVP, which is consistent with reduced afterload (lower basal MAP) in the FVB/N mice. Thus, dobutamine dose-response relationships for dP/dt40 were calculated for wild-type and AE3–/– mice on each background separately and combined (Fig. 3F). Because dP/dt40 is determined at a specific pressure and occurs before opening of the aortic valve, it serves as an index of contractility that provides a correction for possible differences in afterload (26). Although there was a difference in basal dP/dt40 between the two mouse strains, with higher values in the FVB/N background than in the mixed 129SvJ and Black Swiss background (data not shown), there were no significant differences between wild-type and AE3–/– mice on either background, indicating that the loss of AE3 does not impair cardiac contractility.
FIGURE 3.

Cardiovascular performance of wild-type and AE3 null mutant mice. Pressure measurements from the left ventricle and right femoral artery were obtained from anesthetized adult mice of both the mixed and FVB/N backgrounds under control conditions and in response to increasing levels of β-adrenergic stimulation (intravenous infusion of dobutamine). Results shown include heart rate (A), mean arterial pressure (B), systolic left ventricular pressure (C), left ventricular end-diastolic pressure (D), maximum dP/dt (E), and dP/dt40 (F)(dP/dt calculated at an intraventricular pressure of 40 mm Hg during the contractile phase). For each genotype, n = 6 mice of the mixed background and 7 mice of the FVB/N background. Values shown are means ± S.E. No significant differences were observed between WT and AE3–/– mice. Although there was a difference in the dP/dt40 values between the two mouse strains (data not shown), there was no significant difference between WT and AE3–/– mice in either strain. HR, heart rate.
Impaired Cardiovascular Performance in AE3/NKCC1 Double Null Mutant
Mice—When no major differences in cardiac function were observed
between wild-type and AE3–/– mice, we considered the
possibility that robust mechanisms for electrolyte homeostasis in the cardiac
myocyte might mask the phenotypic consequences of AE3 ablation. NKCC1,
operating alone, also provides a mechanism for Na+ and
Cl– entry into the cardiac myocyte
(36,
37) and frequently contributes
to the same physiological processes as
 exchange coupled with Na+-dependent acid extrusion (see
“Discussion”). The loss of NKCC1 was shown previously to cause a
reduction in blood pressure due to a vascular defect
(20); however, a deficit in
cardiac function was not observed. Therefore, to impose a further deficit in
Na+ and Cl– uptake that might exceed the
compensatory mechanisms of the myocardium, we crossed the AE3 mutants with
mice carrying a null mutation in NKCC1. The appearance and behavior of double
null mice seemed identical to that of NKCC1-deficient mice
(24); the additional loss of
AE3 caused no outwardly apparent changes in the NKCC1 phenotype. Heart
weight/body weight ratios for AE3/NKCC1 null mice (5.22 ± 0.22 mg/g,
n = 13) and wild-type mice (5.11 ± 0.26 mg/g, n = 11)
were essentially the same (p = 0.74).
exchange coupled with Na+-dependent acid extrusion (see
“Discussion”). The loss of NKCC1 was shown previously to cause a
reduction in blood pressure due to a vascular defect
(20); however, a deficit in
cardiac function was not observed. Therefore, to impose a further deficit in
Na+ and Cl– uptake that might exceed the
compensatory mechanisms of the myocardium, we crossed the AE3 mutants with
mice carrying a null mutation in NKCC1. The appearance and behavior of double
null mice seemed identical to that of NKCC1-deficient mice
(24); the additional loss of
AE3 caused no outwardly apparent changes in the NKCC1 phenotype. Heart
weight/body weight ratios for AE3/NKCC1 null mice (5.22 ± 0.22 mg/g,
n = 13) and wild-type mice (5.11 ± 0.26 mg/g, n = 11)
were essentially the same (p = 0.74).
In the closed chest model, there were no significant differences between wild-type and AE3/NKCC1 double null mice with respect to chronotropic effects or LVEDP (Fig. 4, A and D), but MAP and systolic LVP were significantly reduced in double knock-out mice at all levels of β-adrenergic stimulation (Fig. 4, B and C), as observed previously for the NKCC1 knock-out (20). The absolute values of both maximum and minimum dP/dt were significantly lower in the double knock-outs under basal conditions and at all levels of β-adrenergic stimulation, compared with wild-type controls (Fig. 5, A and B). The dobutamine dose-response relationship for dP/dt40 was also significantly reduced in the double knock-outs (Fig. 5C). This was in contrast to the results with the single AE3 (Fig. 3) and single NKCC1 (20) knock-outs, in which cardiac contractility was unaffected by the loss of each transporter alone. These data show that loss of both transporters together impairs cardiac contractility.
FIGURE 4.

Cardiovascular performance of wild-type and AE3/NKCC1 double null mutant mice. Experiments were performed as in Fig. 3, using mice of the mixed 129SvJ and Black Swiss background. Heart rate (A), mean arterial pressure (B), left ventricular systolic pressure (C), and left ventricular end diastolic pressure (D), were determined for wild-type and double null mice (n = 7 for each genotype) under basal conditions and in response to increasing β-adrenergic stimulation. Values are means ± S.E. *, significant group effect between wild-type and double null mice for MAP (p < 0.007) and systolic LVP (p < 0.01). No significant differences were observed for heart rate (HR)(p = 0.53) or LVEDP (p = 0.85).
FIGURE 5.

Dobutamine dose-response relationships for maximum dP/dt, minimum dP/dt, and dP/dt40 in wild-type and AE3/NKCC1 double null mutant mice. Data from the same experiments shown in Fig. 4 were used to calculate maximum dP/dt (A), minimum dP/dt (B), and dP/dt at 40 mm Hg (C) for wild-type and double null mice (n = 7 for each genotype) under basal conditions and in response to increasing β-adrenergic stimulation. Values are means ± S.E. *, significant group effect between wild-type and double null mice for maximum dP/dt (p < 0.004), minimum dP/dt (p < 0.005), and dP/dt40 (p < 0.03) was observed.
Reduced Basal Contraction of Adult Ventricular Myocytes Isolated from AE3/NKCC1 Double Null Hearts—In order to further confirm the finding of impaired contractility, we analyzed myocyte mechanics and cell shortening of adult ventricular myocytes isolated from double null mutant hearts and wild-type controls. When paced at 0.5 Hz, as shown in Fig. 6A, fractional shortening (8.6 ± 0.7% in the double null myocytes compared with 11.2 ± 0.6% in wild-type controls), maximal rates of myocyte shortening (+dL/dt; 89.6 ± 7.8 μm/s for double null myocytes versus 151.0 ± 10.4 μm/s for controls) and relengthening (-dL/dt; 68.7 ± 7.6 μm/s for the double null myocytes versus 122.2 ± 10.6 μm/s for controls) were significantly decreased in cells lacking both AE3 and NKCC1, suggesting reduced basal contraction. Furthermore, time to 80% relengthening was slightly, although not significantly, increased in these cells (data not shown). In contrast, no significant differences were observed between wild-type and single null (AE3–/– or NKCC1–/–) cells (Fig. 6B).
FIGURE 6.

Contractile function and mechanics in adult cardiomyocytes isolated from AE3/NKCC1 double null, AE3 null, and NKCC1 null hearts. Ventricular myocytes isolated from adult hearts were paced at 0.5 Hz, and cell shortening was measured. A, significant reductions were observed in fractional shortening relative to diastolic length (p = 0.004), the maximal rates of myocyte shortening (+dL/dt, p < 0.0001), and myocyte relengthening (–dL/dt, p < 0.001) in AE3/NKCC1 double null myocytes when compared with wild-type controls, consistent with impaired contractility in vivo (Fig. 5). B, cardiomyocytes from single AE3 and NKCC1 null mutants did not exhibit impaired mechanics, consistent with normal contractile parameters in AE3 null (Fig. 3) and NKCC1 null (20) mice.
Ca2+ Transient Analysis Revealed Increased Rates of Ca2+ Clearance and Normal Amplitude of Transient in Double Null Myocytes—A possible reason for the reduction in contractility was impaired Ca2+ handling in the double null cells. We therefore performed analysis of Ca2+ transients in isolated adult ventricular myocytes. No significant reduction in the amplitude of the Ca2+ transient was observed in double null myocytes (Table 1). In contrast, time for 70% decay of the Ca2+ transient was significantly reduced in the double null mutants, as was the time constant (τ) of recovery to base line. These findings indicated that the rate of Ca2+ clearance from the cytosol was increased in AE3/NKCC1 double null cells, although a reduction in NCX-mediated Ca2+ entry in the later portion of the Ca2+ transient could contribute to a more rapid decline in the Ca2+ transient (see “Discussion”).
TABLE 1.
Ca2+ transients in ventricular myocytes from wild-type and AE3/NKCC1 double knock-out mice
Ca2+ transients were measured in isolated myocytes loaded with Fura-2/AM and paced at 0.5 Hz. Although the amplitude of the transients was not significantly altered between WT and AE3/NKCC1 double null myocytes, the time for 70% decay of transient (TRC 70%), and the time constant (τ) of recovery to base line were significantly reduced in the double null myocytes, indicating an increased Ca2+ removal mechanism in these cells compared with WT controls.
| No. of cells/No. of mice | Diastolic Fura-2 ratio (base line) | Amplitude, 340/380 nm (systolic-diastolic) | TRC 70% | Time constant of recovery to base line | |
|---|---|---|---|---|---|
| s | s | ||||
| Wild type | 89/5 | 1.59 ± 0.02 | 0.53 ± 0.02 | 0.71 ± 0.01 | 0.52 ± 0.02 |
| AE3-/-NKCC1-/- | 81/5 | 1.55 ± 0.02a | 0.58 ± 0.02 | 0.62 ± 0.02b | 0.42 ± 0.02c |
p = 0.029.
p = 2.68E—5.
p = 0.000108.
PLN Expression and Phosphorylation Were Altered in Mutant Hearts—Immunoblot analysis was performed to determine if alterations in the SR Ca2+ sequestration complex might account for the increased removal of Ca2+ from the cytosol in double null myocytes. Expression of SERCA2a, the SR Ca2+-sequestering pump, was unaltered in AE3/NKCC1 double null (Fig. 7C) and in AE3 and NKCC1 single null hearts (Fig. 8). In contrast, PLN levels were increased 1.6-fold in double null hearts (Fig. 7, A and D) and 1.25-fold in AE3 and NKCC1 single null hearts (Fig. 8). Although the absolute levels of phosphorylated PLN were not reduced in any of the mutant hearts (Figs. 7B and 8) when compared with wild-type controls, relative levels of phosphorylated PLN (PS16 levels normalized to total PLN) were reduced in AE3 null (to 70 ± 10% of wild type), NKCC1 null (to 57 ± 15% of wild type), and double null (to 51 ± 14% of wild type) hearts (Fig. 9).
FIGURE 7.

Expression of sarcoplasmic reticulum-associated Ca2+-handling proteins in AE3/NKCC1 double null hearts. Proteins from whole tissue homogenates of hearts from WT and AE3/NKCC1 DKO mice were resolved by polyacrylamide gel electrophoresis, and immunoblot (A–C) and densitometric (D) analyses were performed using antibodies specific for PLN (A), PLN phosphorylated on Ser16 (PS16) and Thr17 (PT17)(B), and SERCA2a (C). When compared with WT levels, PLN (#, p = 0.014) expression was significantly increased in double null hearts. SERCA2a levels and the absolute levels of PS16 and PT17 were not significantly different from wild-type controls. Statistical analysis was carried out using the paired t test.
FIGURE 8.

Expression of sarcoplasmic reticulum-associated Ca2+-handling proteins in AE3 and NKCC1 single null hearts. Proteins from whole tissue homogenates of AE3 KO and WT hearts (A) and from NKCC1 KO and WT hearts (B) were resolved by polyacrylamide gel electrophoresis, and immunoblot analyses were performed using antibodies specific for SERCA2a (top of A and B), PLN (middle of A and B), and PLN phosphorylated on Ser16 (PS16)(bottom of A and B). Compared with respective WT levels, PLN was increased in AE3 (†, p < 0.03) and NKCC1 (p = 0.1) null hearts. Statistical analysis was carried out using the paired t test.
FIGURE 9.

Phosphorylation of phospholamban in AE3 null, NKCC1 null, and AE3/NKCC1 double null hearts. PLN protein expression and levels of PLN phosphorylated on Ser16 (PS16) were determined using specific antibodies. PS16 levels, when normalized to PLN expression, were significantly reduced in AE3 (*, p = 0.01), NKCC1 (*, p = 0.03), and AE3/NKCC1 (*, p < 0.001) null hearts.
NCX-mediated Ca2+ Efflux Is Increased with No Corresponding Increase in NCX Protein Levels in AE3/NKCC1 Double Null Myocytes—In light of the reduction in relative PLN phosphorylation levels in double null hearts, it was unlikely that SR-mediated Ca2+ sequestration could account for the observed increase in the rate of decay of the Ca2+ transient. We therefore explored the possibility that NCX-mediated Ca2+ efflux was increased in double null myocytes. NCX protein levels were not significantly changed (Fig. 10A). Analysis of Ca2+ transients in response to the rapid and sustained application of caffeine was carried out. Under these conditions, SR Ca2+-stores are emptied and decay of the transient occurs entirely via non-SR-related Ca2+ removal mechanisms. Although the amplitude of the Ca2+ transient was not significantly altered (Fig. 10B), the time to 70% recovery was significantly reduced (Fig. 10C), demonstrating that the rate of decay of the Ca2+ transient and, therefore, NCX-mediated Ca2+ efflux were increased in double null cells.
FIGURE 10.

Expression of NCX in double null hearts and determination of NCX-mediated Ca2+ efflux in AE3/NKCC1 double null myocytes. A, proteins from whole heart homogenates from WT and DKO mice were resolved by polyacrylamide gel electrophoresis. Immunoblot analysis was performed using a monoclonal antibody against NCX (R3F1). Densitometric analysis revealed no significant alteration in NCX levels (p = 0.2). SR Ca2+ content in WT (n = 19 cells/4 mice) and double null (DKO; n = 22 cells/4 mice) ventricular myocytes was measured by the rapid and sustained application of caffeine (10 mm). B, fluorescent changes measured using a dual excitation fluorescence photomultiplier system revealed no significant reduction in total SR Ca2+ store levels (amplitude (340/380 nm) was 1.12 ± 0.09 for WT and 0.90 ± 0.07 for DKO). In the same experiment, the rate of decay of the caffeine-induced transient was determined. C, time for 70% recovery of transient (TRC 70%) was significantly reduced in DKO myocytes compared with WT controls (5.6 ± 0.5 in WT versus 4.2 ± 0.27 in DKO; p < 0.5).
PP1-C Expression Is Up-regulated in Mutant Hearts—PP1 accounts for much of the SR-associated phosphatase activity (38, 39) and has been identified as the principal negative regulator of PLN phosphorylation. Increased expression of PP1-C (gene symbols Ppp1ca/b/c), the catalytic subunit of PP1, has been shown to reduce PLN phosphorylation on Ser16 (40). Given the reduction in relative levels of phosphorylated PLN (on Ser16) in hearts lacking AE3, NKCC1, or both, it was important to determine if expression of PP1 was altered in these mutant hearts. Immunoblot analysis revealed that expression of PP1-C was increased 1.7-fold in AE3 null hearts, 1.6-fold in NKCC1 null hearts, and 2.2-fold in AE3/NKCC1 double null hearts (Fig. 11).
FIGURE 11.

Expression of PP1-C in AE3 null, NKCC1 null, and AE3/NKCC1 double null hearts. Proteins from whole heart homogenates from WT and mutant mice were resolved by polyacrylamide gel electrophoresis. Expression of PP1-C was determined in AE3 null (A), NKCC1 null (B), and AE3/NKCC1 double null (C) hearts. Densitometric analyses (D) revealed that PP1-C expression was significantly increased in AE3 null (*, p < 0.02), NKCC1 null (*, p < 0.05), and double null hearts (*, p < 0.02).
Carboxymethylation and Localization of PP2A-C Is Altered in Mutant Hearts—Localization of the PP2A holoenzyme is determined by the association of PP2A-C (gene symbols: Ppp2ca/b) with various B subunits and is regulated by reversible carboxymethylation of PP2A-C (33, 41). Given the impaired cardiac contractility in the double null mice and increased expression of PP1-C (Fig. 11) in all three mutants, we wanted to determine whether PP2A was also affected. Immunoblot analysis, using methylation-sensitive antibodies that preferentially bind nonmethylated PP2A-C, revealed a significant decrease in nonmethylated PP2A-C in AE3 (50 ± 9.5% of wild type) and NKCC1 (39 ± 11% of wild type) null hearts (Fig. 12, A, B, and D). These data indicated that carboxymethylation of PP2A-C was increased in AE3 and NKCC1 single null hearts. In contrast, levels of nonmethylated PP2A-C were increased ∼15% in AE3/NKCC1 double null hearts (Fig. 12, C and D), indicating that carboxymethylation of PP2A-C was reduced.
FIGURE 12.

Carboxymethylation of PP2A-C in AE3 null, NKCC1 null, and AE3/NKCC1 double null hearts. Proteins from whole heart homogenates from wild-type and mutant mice were resolved by polyacrylamide gel electrophoresis. Immunoblot analyses were performed using monoclonal antibodies specific for unmethylated PP2A-C (clone 1D6 and clone 4B7). The PP2A-C subunits migrate either as single or double bands, varying for the same sample between immunoblots, a pattern that has been previously reported (33). Levels of unmethylated PP2A-C were significantly reduced in AE3 null hearts (A and D,*, p < 0.001) and NKCC1 null hearts (B and D,*, p < 0.01). In contrast, levels of unmethylated PP2A-C were significantly increased in AE3/NKCC1 double null hearts (C and D,*, p < 0.05). Total PP2A-C levels were determined upon cold base treatment of nitrocellulose membranes, which results in complete demethylation of PP2A-C (32). Total PP2A-C levels were slightly reduced in AE3 single null hearts (to 86 ± 1.6% of wild type; bottom of A; p < 0.001) and was unaltered in NKCC1 single null and double null hearts (B and C).
Altered carboxymethylation of PP2A-C was found to be associated with a dramatic shift in the localization of total PP2A-C in mutant hearts. PP2A-C levels associated with the myofibrillar fraction were significantly reduced in both the AE3 (Fig. 13, A and D, 17 ± 6.2% of wild-type) and NKCC1 (Fig. 13, B and D, 16 ± 6.9% of wild type) single null hearts. In contrast, PP2A-C was found to be relocalized to the myofibrillar fraction of AE3/NKCC1 double null hearts (Fig. 13, C and D, 79 ± 11.3% of wild type).
FIGURE 13.

Localization of PP2A-C in the myofibrillar fraction of AE3 null, NKCC1 null, and AE3/NKCC1 double null hearts. The myofibrillar fraction (3000 × g pellet) was generated as described under “Experimental Procedures.” Proteins were resolved by polyacrylamide gel electrophoresis, and levels of total PP2A-C were determined as described in Fig. 12, upon cold base treatment of nitrocellulose membranes. Localization of PP2A-C to the myofibrillar fraction was dramatically reduced in AE3 (A and D; p < 0.0001) and NKCC1 (B and D; p < 0.0001) single null hearts. In contrast, PP2A-C levels in the myofibrillar fraction of AE3/NKCC1 double null hearts were comparable with wild-type levels (C and D; p = 0.16).
DISCUSSION
AE3 null mutants were born in a normal Mendelian ratio, were fertile, and appeared healthy at all ages, consistent with studies of another AE3-deficient mouse model, in which null mutants exhibited no apparent defects other than increased susceptibility to epileptic seizures (42). In our own studies, we analyzed the cardiac functions of AE3.
Role of Acid-Base and Electrolyte Transporters in the
Heart—Cardiac myocytes contain at least four
 exchangers (Fig. 14). Two of
these, AE3 (1) and PAT1
(6,
7), are expressed at higher
levels in the heart than in any other tissue, and both AE3
(3) and AE1
(1) have heart-specific
variants. The remarkable abundance and diversity of
exchangers (Fig. 14). Two of
these, AE3 (1) and PAT1
(6,
7), are expressed at higher
levels in the heart than in any other tissue, and both AE3
(3) and AE1
(1) have heart-specific
variants. The remarkable abundance and diversity of
 exchangers in cardiac tissue suggest that they serve important functions in
the heart. In addition to acid-loading
exchangers in cardiac tissue suggest that they serve important functions in
the heart. In addition to acid-loading
 exchangers, cardiac muscle contains NKCC1
(36,
37) and a number of
Na+-dependent acid extrusion proteins
(Fig. 14) that serve as major
Na+-loading mechanisms
(15,
36,
43–46).
The functional coupling of acid-loading and acid-extruding transporters is
well established in epithelial tissues, where they contribute to
transepithelial NaCl and fluid transport
(6–12,
47).
Na+-K+-2Cl– cotransport serves some of
the same functions as those served by the concerted activities of
exchangers, cardiac muscle contains NKCC1
(36,
37) and a number of
Na+-dependent acid extrusion proteins
(Fig. 14) that serve as major
Na+-loading mechanisms
(15,
36,
43–46).
The functional coupling of acid-loading and acid-extruding transporters is
well established in epithelial tissues, where they contribute to
transepithelial NaCl and fluid transport
(6–12,
47).
Na+-K+-2Cl– cotransport serves some of
the same functions as those served by the concerted activities of
 exchangers and Na+-dependent acid extruders
(48), and there is evidence
that NKCC1 activity is rapidly increased when
exchangers and Na+-dependent acid extruders
(48), and there is evidence
that NKCC1 activity is rapidly increased when
 exchange is deficient
(49).
exchange is deficient
(49).
FIGURE 14.

Acid-base and electrolyte transporter activities have the potential to
alter intracellular Ca2+ handling. The diagram shows the
 exchangers (AE1, AE2, AE3, and PAT1), Na+-dependent transporters
that extrude H+ (Na+/H+ exchanger) or take up
exchangers (AE1, AE2, AE3, and PAT1), Na+-dependent transporters
that extrude H+ (Na+/H+ exchanger) or take up
 (NBCn1, NBC1, and NBC4
(NBCn1, NBC1, and NBC4
 cotransporters), the NKCC1 Na+-K+-2Cl–
cotransporter, and the NCX that have been identified in heart. It should be
noted that NBC4 has been identified in the human heart
(45) but may not be present in
the rodent heart (46). The
grouping of transporters is according to general function and is not meant to
indicate a polarized distribution or specific location in the cell. Both NKCC1
by itself and the Na+-dependent acid-base transporters, when
coupled with
cotransporters), the NKCC1 Na+-K+-2Cl–
cotransporter, and the NCX that have been identified in heart. It should be
noted that NBC4 has been identified in the human heart
(45) but may not be present in
the rodent heart (46). The
grouping of transporters is according to general function and is not meant to
indicate a polarized distribution or specific location in the cell. Both NKCC1
by itself and the Na+-dependent acid-base transporters, when
coupled with
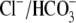 exchangers to prevent an increase in pHi from inhibiting
their activities, have the potential to mediate sustained Na+
loading in cardiac myocytes. Na+ loading can reduce the rate of
Ca2+ extrusion via inhibition or reversal of the NCX.
exchangers to prevent an increase in pHi from inhibiting
their activities, have the potential to mediate sustained Na+
loading in cardiac myocytes. Na+ loading can reduce the rate of
Ca2+ extrusion via inhibition or reversal of the NCX.
The hypothesis that
 exchangers and Na+-dependent acid extrusion transporters function
as coupled systems in cardiac muscle is supported by the observation that
stimulation of Na+/H+ exchange by angiotensin II in the
cardiac myocyte, which causes a positive inotropic response, is not
accompanied by an increase in pHi
(50). This indicated that
H+ extrusion via Na+/H+ exchange was balanced
by an equivalent
exchangers and Na+-dependent acid extrusion transporters function
as coupled systems in cardiac muscle is supported by the observation that
stimulation of Na+/H+ exchange by angiotensin II in the
cardiac myocyte, which causes a positive inotropic response, is not
accompanied by an increase in pHi
(50). This indicated that
H+ extrusion via Na+/H+ exchange was balanced
by an equivalent  efflux mechanism.
This conclusion was supported by the finding that
efflux mechanism.
This conclusion was supported by the finding that
 exchange was increased by angiotensin II
(14,
51), thus countering the
alkalinizing effects of Na+/H+ exchange
(14) and contributing to
pHi-neutral Na+ and Cl–
uptake. These and other studies
(52–57)
suggest that Na+ loading can, via modulation of NCX activity,
affect Ca2+ handling, cardiac contractility, and heart disease.
exchange was increased by angiotensin II
(14,
51), thus countering the
alkalinizing effects of Na+/H+ exchange
(14) and contributing to
pHi-neutral Na+ and Cl–
uptake. These and other studies
(52–57)
suggest that Na+ loading can, via modulation of NCX activity,
affect Ca2+ handling, cardiac contractility, and heart disease.
AE3 Ablation Does Not Reduce IR Injury—The first issue we
examined was whether loss of AE3 affected cardiac I/R injury. Ischemia is
known to cause acidification of the cardiac myocyte, and, during reperfusion,
NHE1-mediated Na+/H+ exchange leads to increased
intracellular Na+, which in turn causes inhibition or reversal of
Na+/Ca2+ exchange
(34). The resulting
Ca2+ overload, along with recovery of pHi,
leads to hypercontracture injury. Inhibition
(34) or genetic ablation
(25) of NHE1, which serves as
a major mechanism of Na+ loading, has been shown to protect against
I/R injury. Previous studies indicated that inhibition of
 exchange might also protect against I/R injury
(17,
18). Loss of AE3, however, had
no apparent effect on left ventricular developed pressure or end diastolic
pressure in response to ischemia and reperfusion using Langendorff heart
preparations. With at least three other
exchange might also protect against I/R injury
(17,
18). Loss of AE3, however, had
no apparent effect on left ventricular developed pressure or end diastolic
pressure in response to ischemia and reperfusion using Langendorff heart
preparations. With at least three other
 exchangers in the heart, the loss of AE3 alone may not have caused a
sufficient reduction in total
exchangers in the heart, the loss of AE3 alone may not have caused a
sufficient reduction in total
 exchange to noticeably protect cardiac function from I/R injury. The stilbene
disulfonates used in studies suggesting that inhibition of
exchange to noticeably protect cardiac function from I/R injury. The stilbene
disulfonates used in studies suggesting that inhibition of
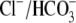 exchange protects against I/R injury
(17,
18) may act primarily on one
of the other isoforms, or, by acting on multiple isoforms, they may reduce
total
exchange protects against I/R injury
(17,
18) may act primarily on one
of the other isoforms, or, by acting on multiple isoforms, they may reduce
total 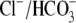 exchange well beyond that caused by the loss of AE3 alone. Alternatively,
these stilbene derivatives could provide protection via inhibition of
exchange well beyond that caused by the loss of AE3 alone. Alternatively,
these stilbene derivatives could provide protection via inhibition of
 cotransport (35). In fact,
inhibition of the NBC1
cotransport (35). In fact,
inhibition of the NBC1
 cotransporter, which is sensitive to stilbene disulfonates, has been reported
to protect against I/R injury
(19).
cotransporter, which is sensitive to stilbene disulfonates, has been reported
to protect against I/R injury
(19).
AE3 or NKCC1 Activities Are Necessary for Normal Cardiac Performance—We anticipated that the loss of AE3 would impair cardiac performance, secondary to effects on Ca2+ handling. However, loss of AE3 by itself caused no reduction in basal contractility, and the increase in contractile parameters in response to β-adrenergic stimulation was normal. Given the considerations discussed above, it seemed possible that the additional loss of NKCC1 on the AE3 null background would further stress the myocardium and impair contractility. MAP and systolic LVP were significantly reduced in the double knock-outs, as observed previously in the NKCC1 mutant (20). Unlike the NKCC1 knock-out however, the double knock-outs exhibited impaired contraction and relaxation, with absolute values of both maximum and minimum dP/dt being significantly lower in the mutant mice. The use of dP/dt40 as an index of contractility provided a correction for the effects of reduced afterload (26). Furthermore, analysis of isolated myocytes showed that the contractile defect was intrinsic to the AE3/NKCC1 double null cells rather than being secondary to systemic effects. In contrast, no reduction in contractile parameters was observed in AE3 or NKCC1 single null myocytes, consistent with the normal cardiac performance in AE3 (Fig. 3) and NKCC1 (20) single knock-outs in vivo. Importantly, these results also provided a direct comparison between AE3/NKCC1 double null and NKCC1 single null myocytes.
Loss of AE3 and NKCC1 Affects Ca2+ Handling and PLN Regulation—We explored the possibility that Ca2+ handling may be impaired in myocytes lacking both AE3 and NKCC1, accounting for the reduced contractility observed in double null hearts. Although the amplitude of the Ca2+ transient elicited in response to field stimulation at 0.5 Hz was unaltered, decay of the transient occurred significantly faster in double null myocytes, indicating that the rate of Ca2+ clearance was enhanced in these cells. Removal of cytosolic Ca2+ is known to occur predominantly by sequestration of Ca2+ into the SR and NCX-mediated Ca2+ efflux across the sarcolemma. Enhanced Ca2+ sequestration into the SR was unlikely to account for the observed increase in Ca2+ clearance, since expression of PLN, the principal negative regulator of SERCA2a activity (58), was increased 1.65-fold. Relative PLN phosphorylation was reduced by almost 50%, indicating a sharp increase in the ratio of inhibitory PLN to SERCA2a. Increased inhibitory function of PLN has been shown to depress rates of Ca2+ sequestration, prolong decay parameters of Ca2+ transients, and lower Ca2+ stores and Ca2+ transients in myocytes (58–60).
NKCC1 is known to provide a substantial capacity for Na+ influx in cardiac myocytes (36), and its activity has been shown to affect NCX-mediated Na+/Ca2+ exchange in astrocytes (21). The combined loss of AE3 and NKCC1 is therefore likely to reduce subsarcolemmal [Na+], providing an increased driving force for NCX activity in the forward mode. Indeed, the rate of decay of caffeine-induced Ca2+ transients, resulting from non-SR-related Ca2+ efflux mechanisms and predominantly mediated by NCX in mouse cardiac myocytes (61), was significantly increased in double null myocytes. There was no corresponding increase in NCX protein levels, strongly indicating that the enhanced NCX-mediated Ca2+ efflux was the result of an increased driving force in myocytes lacking both AE3 and NKCC1.
Studies on transgenic mice overexpressing NCX in the heart (61–63) have shown that increased NCX-mediated Ca2+ efflux does not reduce SR Ca2+ stores. On the contrary, there is evidence that, in mouse cardiac myocytes, NCX facilitates Ca2+ entry (reverse mode of activity) both at rest and in the later relaxation phase of the Ca2+ transient (61). Conditions leading to reduced subsarcolemmal [Na+], which would increase NCX-mediated Ca2+-efflux in double null myocytes, would also reduce NCX-mediated Ca2+ entry. This might explain the significant increase in the decay of the Ca2+ transient and the more rapid recovery to base line observed in double null myocytes paced at 0.5 Hz (Table 1).
Ablation of AE3 and/or NKCC1 Affects PP1 Expression—Despite changes in Ca2+ handling and PLN regulation, the absence of a reduction in Ca2+ transient amplitude discounted the likelihood that impaired contractility in double null hearts was a result of reduced cytosolic Ca2+ fluxes. Furthermore, the enhanced decay of the Ca2+ transient suggested that the impaired relaxation was not due to alterations in Ca2+ handling, but more likely due to changes in the myofibrillar apparatus of the mutant myocytes. Myofibrillar proteins are key determinants of cardiac function. Their functions are, in turn, regulated by phosphorylation and dephosphorylation events controlled by the expression, localization, and activity of various kinases and phosphatases (64). In mammalian hearts, PP1 and PP2A have been identified as the two principal phosphatases.
PP1 serves as a negative regulator of cardiac function and is the major phosphatase acting on PLN. Heart-specific overexpression of inhibitor-1 or inhibitor-2, both of which block PP1 activity, improves cardiac function (65, 66), and inhibition of PP1 restores cardiac performance in a heart failure model (65). Overexpression of PP1-C or ablation of a PP1-specific inhibitor causes a reduction in PLN phosphorylation on Ser16 (40). Thus, it is likely that the increase in PP1-C levels in AE3, NKCC1, and AE3/NKCC1 null hearts contributes to the reduction in relative levels of phosphorylated PLN in these hearts. It may also contribute more directly to impaired contractile function in double null hearts, although a major caveat is that the specific effects of PP1 overexpression, other than those on PLN, remain largely undefined. The increased levels of PP1-C in AE3 and NKCC1 single null hearts were not accompanied by alterations in cardiac function. This suggested that additional changes had occurred that countered the negative regulatory effects of increased PP1-C expression in single null hearts. These could include changes in the expression and localization of the second major phosphatase, PP2A.
Ablation of AE3 and/or NKCC1 Affects PP2A-C Methylation and Localization—PP2A regulates many key determinants of cardiac contractility (67). Total PP2A-C levels were slightly reduced in AE3 single null hearts but not in NKCC1 or AE3/NKCC1 null hearts. PP2A activity and substrate specificity are also regulated by the subcellular distribution of the holoenzyme, which is determined by the choice of the regulatory B subunit that associates with the core enzyme. Association between the core enzyme, comprising the catalytic C and the scaffold A subunits, and regulatory B subunits is regulated by methylation of PP2A-C (33, 41, 68, 69). There is also a second subset of B subunits whose association with the core enzyme is independent of PP2A-C methylation (33). The reversible methylation of PP2A-C may therefore allow the formation of distinct groups of PP2A holoenzymes, with different localization and substrate specificities (70).
Levels of unmethylated PP2A-C were significantly reduced in hearts lacking either AE3 or NKCC1, indicating that methylation of PP2A-C was increased in single null hearts. In contrast, levels of unmethylated PP2A-C were increased in AE3/NKCC1 double null hearts. Given the probable effects of altered PP2A-C methylation on PP2A holoenzyme assembly and localization, it was likely that subcellular localization of PP2A-C was different between the single and double null hearts. PP2A-C was found to be dramatically excluded from the myofibrillar fraction of single null hearts, a finding that was strikingly reversed in the double null hearts, where PP2A-C relocalized to the myofibrillar fraction. Localization of PP2A-C to the myofibrillar fraction in cardiac myocytes has been associated with reduced contractility and the anti-adrenergic effects mediated by adenosine A1 receptors (67, 71–73). The impaired contractility in double null hearts, associated with the relocalization of PP2A-C to the myofibrillar fraction, suggested that the exclusion of PP2A-C from the myofibrillar fraction of single null hearts served a compensatory function and was necessary for sustaining normal contractility. Furthermore, the expression of either AE3 or NKCC1 appeared to be essential for the translocation of PP2A-C away from the myofibrillar fraction.
Conclusions—Our studies demonstrate that the loss of AE3 and
NKCC1 together impairs cardiac contractility, with effects on 1) PLN
expression and phosphorylation; 2) Ca2+ handling, as indicated by
increased NCX-mediated Ca2+-efflux; and 3) PP1-C expression and
PP2A-C methylation and localization. The effects on PP1 and PP2A-C show that
AE3 and NKCC1 activities have a major impact on these pleiotropic signaling
mechanisms. Despite the absence of impaired cardiac function in the single
mutants, a number of significant changes, including the effects on PLN
phosphorylation, PP1 expression, and PP2A methylation and localization,
occurred in both single null mutants, and the changes were either additive
(PLN and PP1) or completely reversed (PP2A) in the double mutants. These
results provide the first direct demonstration that AE3-mediated
 exchange and NKCC1-mediated Na+-K+-2Cl–
cotransport serve similar and important functions in the regulation of cardiac
contractility and suggest that each transport activity can compensate to some
degree for loss of the other activity.
exchange and NKCC1-mediated Na+-K+-2Cl–
cotransport serve similar and important functions in the regulation of cardiac
contractility and suggest that each transport activity can compensate to some
degree for loss of the other activity.
Acknowledgments
We thank Dr. David C. Pallas (Emory University School of Medicine) for extremely helpful discussions on PP2A-C.
This work was supported, in whole or in part, by National Institutes of Health Grants HL61974, DK50594, DK57552, HL081859, and HL080686. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: pHi, intracellular pH; I/R, ischemia-reperfusion; AE3–/– mice/cells, AE3 homozygous null mutant mice/cells; LVEDP, left ventricular end diastolic pressure; MAP, mean arterial pressure; LVP, left ventricular pressure; NKCC1–/– mice/cells, Na+-K+-2Cl– cotransporter null mutant mice/cells; KO, knock-out; DKO, double knock-out.
References
- 1.Kudrycki, K. E., Newman, P. R., and Shull, G. E. (1990) J. Biol. Chem. 265 462–471 [PubMed] [Google Scholar]
- 2.Kopito, R. R., Lee, B. S., Simmons, D. M., Lindsey, A. E., Morgans, C. W., and Schneider, K. (1989) Cell 59 927–937 [DOI] [PubMed] [Google Scholar]
- 3.Linn, S. C., Kudrycki, K. E., and Shull, G. E. (1992) J. Biol. Chem. 267 7927–7935 [PubMed] [Google Scholar]
- 4.Richards, S. M., Jaconi, M. E., Vassort, G., and Puceat, M. (1999) J. Cell Sci. 112 1519–1528 [DOI] [PubMed] [Google Scholar]
- 5.Alvarez, B. V., Kieller, D. M., Quon, A. L., Markovich, D., and Casey, J. R. (2004) J. Physiol. 561 721–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knauf, F., Yang, C. L., Thomson, R. B., Mentone, S. A., Giebisch, G., and Aronson, P. S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 9425–9430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang, Z., Petrovic, S., Mann, E., and Soleimani, M. (2002) Am. J. Physiol. 282 G573–G579 [DOI] [PubMed] [Google Scholar]
- 8.Knickelbein, R., Aronson, P. S., Schron, C. M., Seifter, J., and Dobbins, J. W. (1985) Am. J. Physiol. 249 G236–G245 [DOI] [PubMed] [Google Scholar]
- 9.Melvin, J. E., Yule, D., Shuttleworth, T., and Begenisich, T. (2005) Annu. Rev. Physiol. 67 445–469 [DOI] [PubMed] [Google Scholar]
- 10.Gawenis, L. R., Greeb, J. M., Prasad, V., Grisham, C., Sanford, L. P., Doetschman, T., Andringa, A., Miller, M. L., and Shull, G. E. (2005) J. Biol. Chem. 280 12781–12789 [DOI] [PubMed] [Google Scholar]
- 11.Grubb, B. R., Lee, E., Pace, A. J., Koller, B. H., and Boucher, R. C. (2000) Am. J. Physiol. 279 G707–G718 [DOI] [PubMed] [Google Scholar]
- 12.Walker, N. M., Flagella, M., Gawenis, L. R., Shull, G. E., and Clarke, L. L. (2002) Gastroenterology 123 531–541 [DOI] [PubMed] [Google Scholar]
- 13.Jiang, L. Chernova, M. N., and Alper, S. L. (1997) Am. J. Physiol. 272 C191–C202 [DOI] [PubMed] [Google Scholar]
- 14.Camilion de Hurtado, M. C., Alvarez, B. V., Perez, N. G., Ennis, I. L., and Cingolani, H. E. (1998) Circ. Res. 82 473–481 [DOI] [PubMed] [Google Scholar]
- 15.Bers, D. M., Barry, W. H., and Despa, S. (2003) Cardiovasc. Res. 57 897–912 [DOI] [PubMed] [Google Scholar]
- 16.Pieske, B., Houser, S. R., Hasenfuss, G., and Bers, D. M. (2003) Cardiovasc. Res. 57 871–872 [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki, H., Otani, H., Mishima, K., Imamura, H., and Inagaki, C. (2001) Eur. J. Pharmacol. 411 35–43 [DOI] [PubMed] [Google Scholar]
- 18.Lai, Z. F., and Nishi, K. (1998) Am. J. Physiol. 275 H1613–H1619 [DOI] [PubMed] [Google Scholar]
- 19.Khandoudi, N., Albadine, J., Robert, P., Krief, S., Berrebi-Bertrand, I., Martin, X., Bevensee, M. O., Boron, W. F., and Bril, A. (2001) Cardiovasc. Res. 52 387–396 [DOI] [PubMed] [Google Scholar]
- 20.Meyer, J. W., Flagella, M., Sutliff, R. L., Lorenz, J. N., Nieman, M. L., Weber, C. S., Paul, R. J., and Shull, G. E. (2002) Am. J. Physiol. 283 H1846–H1855 [DOI] [PubMed] [Google Scholar]
- 21.Lenart, B., Kintner, D. B., Shull, G. E., and Sun, D. (2004) J. Neurosci. 24 9585–9597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linn, S. C., Askew, G. R., Menon, A. G., and Shull, G. E. (1995) Circ. Res. 76 584–591 [DOI] [PubMed] [Google Scholar]
- 23.Gawenis, L. R., Ledoussal, C., Judd, L. M., Prasad, V., Alper, S. L., Stuart-Tilley, A., Woo, A. L., Grisham, C., Sanford, L. P., Doetschman, T., Miller, M. L., and Shull, G. E. (2004) J. Biol. Chem. 279 30531–30539 [DOI] [PubMed] [Google Scholar]
- 24.Flagella, M., Clarke, L. L., Miller, M. L., Erway, L. C., Gianella, R. A., Andringa, A., Gawenis, L. R., Kramer, J., Duffy, J. J., Doetschman, T., Lorenz, J. N., Yamoah, E. N., Cardell, E. L., and Shull, G. E. (1999) J. Biol. Chem. 274 26946–26955 [DOI] [PubMed] [Google Scholar]
- 25.Wang, Y., Meyer, J. W., Ashraf, M., and Shull, G. E. (2003) Circ. Res. 93 776–782 [DOI] [PubMed] [Google Scholar]
- 26.Lorenz, J. N., and Robbins, J. (1997) Am. J. Physiol. 272 H1137–H1146 [DOI] [PubMed] [Google Scholar]
- 27.Periasamy, M., Reed, T. D., Liu, L. H., Ji, Y., Loukianov, E., Paul, R. J., Nieman, M. L., Riddle, T., Duffy, J. J., Doetschman, T., Lorenz, J. N., and Shull, G. E. (1999) J. Biol. Chem. 274 2556–2562 [DOI] [PubMed] [Google Scholar]
- 28.Zhou, Y. Y., Wang, S. Q., Zhu, W. Z., Chruscinski, A., Kobilka, B. K., Ziman, B., Wang, S., Lakatta, E. G., Cheng, H., and Xiao, R. P. (2000) Am. J. Physiol. 279 H429–H436 [DOI] [PubMed] [Google Scholar]
- 29.Braz, J. C., Gregory, K., Pathak, A., Zhao, W., Sahin, B., Klevitsky, R., imball, T. F., Lorenz, J. N., Nairn, A. C., Liggett, S. B., Bodi, I., Wang, S., Schwartz, A., Lakatta, E. G., Depaoli-Roach, A. A., Robbins, J., Hewett, T. E., Bibb, J. A., Westfall, M. V., Kranias, E. G., and Molkentin, J. D. (2004) Nat. Med. 10 248–254 [DOI] [PubMed] [Google Scholar]
- 30.Ji, Y., Zhao, W., Li, B., Desantiago, J., Picht, E., Kaetzel, M. A., Schultz, J el, J., Kranias, E. G. Bers, D. M., and Dedman, J. R. (2006) Am. J. Physiol. 290 H599–H606 [DOI] [PubMed] [Google Scholar]
- 31.Golenhofen, N., Ness, W., Koob, R., Htun, P., Schaper, W., and Drenckhahn, D. (1998) Am. J. Physiol. 274 H1457–H1464 [DOI] [PubMed] [Google Scholar]
- 32.Wei, H., Ashby, D. G., Moreno, C. S., Ogris, E., Yeong, F. M., Corbett, A. H., and Pallas, D. C. (2001) J. Biol. Chem. 276 1570–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu, X. X., Du, X., Moreno, C. S., Green, R. E., Ogris, E., Feng, Q., Chou, L., McQuoid, M. J., and Pallas, D. C. (2001) Mol. Biol. Cell 12 185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karmazyn, M., Gan, X. T., Humphreys, R. A., Yoshida, H., and Kusumoto, K. (1999) Circ. Res. 85 777–786 [DOI] [PubMed] [Google Scholar]
- 35.Schafer, C., Ladilov, Y. V., Siegmund, B., and Piper, H. M. (2000) Am. J. Physiol. 278 H1457–H1463 [DOI] [PubMed] [Google Scholar]
- 36.Frelin, C., Chassande, O., and Lazdunski, M. (1986) Biochem. Biophys. Res. Commun. 134 326–331 [DOI] [PubMed] [Google Scholar]
- 37.Mihailidou, A. S., Buhagiar, K. A., and Rasmussen, H. H. (1998) Am. J. Physiol. 274 C175–C181 [DOI] [PubMed] [Google Scholar]
- 38.MacDougall, L. K., Jones, L. R., and Cohen, P. (1991) Eur. J. Biochem. 196 725–734 [DOI] [PubMed] [Google Scholar]
- 39.Steenaart, N. A., Ganim, J. R., Di Salvo, J., and Kranias, E. G. (1992) Arch. Biochem. Biophys. 293 17–24 [DOI] [PubMed] [Google Scholar]
- 40.Carr, A. N., Schmidt, A. G., Suzuki, Y., del Monte, F., Sato, Y., Lanner, C., Breeden. K., Jing, S.-L., Allen, P. B., Greengard, P., Yatani, A., Hoit, B. D., Grupp, I. L., Hajjar, R. J., Depaoli-Roach, A. A., and Kranias, E. G. (2002) Mol. Cell. Biol. 22 4124–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tolstykh, T., Lee, J., Vafai, S., and Stock, J. B. (2000) EMBO J. 19 5682–5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hentschke, M., Wiemann, M., Hentschke, S., Kurth, I., Hermans-Borgmeyer, I., Seidenbecher, T., Jentsch, T. J., Gal, A., and Hubner, C. A. (2006) Mol. Cell. Biol. 26 182–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi, I., Aalkjaer, C., Boulpaep, E. L., and Boron, W. F. (2000) Nature 405 571–575 [DOI] [PubMed] [Google Scholar]
- 44.Romero, M. F., Fong, P., Berger, U. V., Hediger, M. A., and Boron, W. F. (1998) Am. J. Physiol. 274 F425–F432 [DOI] [PubMed] [Google Scholar]
- 45.Pushkin, A., Abuladze, N., Newman, D., Lee, I., Xu, G., and Kurtz, I. (2000) IUBMB Life 50 13–19 [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto, T., Shirayama, T., Sakatani, T., Takahashi, T., Tanaka, H., Takamatsu, T., Spitzer, K. W., and Matsubara, H. (2007) Am. J. Physiol. 293 H1254–H1264 [DOI] [PubMed] [Google Scholar]
- 47.Melvin, J. E., Park, K., Richardson, L., Schultheis, P. J., and Shull, G. E. (1999) J. Biol. Chem. 274 22855–22861 [DOI] [PubMed] [Google Scholar]
- 48.Takahashi, N., Chernavvsky, D. R., Gomez, R. A., Igarashi, P., Gitelman, H. J., and Smithies, O. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 5434–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gawenis, L. R., and Shull, G. E. (2005) Gastroenterol. 128 44 [Google Scholar]
- 50.Mattiazzi, A., Perez, N. G., Vila-Petroff, M. G., Alvarez, B., Camilion de Hurtado, M. C., and Cingolani, H. E. (1997) Am. J. Physiol. 272 H1131–H1136 [DOI] [PubMed] [Google Scholar]
- 51.Alvarez, B. V., Fujinaga, J., and Casey, J. R. (2007) Circ. Res. 89 1246–1253 [DOI] [PubMed] [Google Scholar]
- 52.Cingolani, H. E., and Camilion de Hurtado, M. C. (2002) Circ. Res. 90 751–753 [DOI] [PubMed] [Google Scholar]
- 53.Perez, N. G., Camilion de Hurtado, M. C., and Cingolani, H. E. (2001) Circ. Res. 88 376–382 [DOI] [PubMed] [Google Scholar]
- 54.Ennis, I. L., Alvarez, B. V., Camilion de Hurtado, M. C., and Cingolani, H. E. (1998) Hypertension 31 961–967 [DOI] [PubMed] [Google Scholar]
- 55.Pieske, B., and Houser, S. R. (2003) Cardiovasc. Res. 57 874–886 [DOI] [PubMed] [Google Scholar]
- 56.Baartscheer, A., Schumacher, C. A., Belterman, C. N. W., Coronel, R., and Fiolet, J. W. T. (2003) Cardiovasc. Res. 57 986–995 [DOI] [PubMed] [Google Scholar]
- 57.Baartscheer, A., Schumacher, C. A., van Borren, M. M. G. J., Belterman, C. N. W., Coronel, R., and Fiolet, J. W. T. (2003) Cardiovasc. Res. 57 1015–1024 [DOI] [PubMed] [Google Scholar]
- 58.MacLennan, D. H., and Kranias, E. G. (2003) Nat. Rev. 4 566–577 [DOI] [PubMed] [Google Scholar]
- 59.Brittsan, A. G., Carr, A. N., Schmidt, A. G., and Kranias, E. G. (2000) J. Biol. Chem. 275 12129–12135 [DOI] [PubMed] [Google Scholar]
- 60.Kadambi, V. J., Ponniah, S., Harrer, J. M., Hoit, B. D., Dorn, G. W., II, Walsh, R. A., and Kranias, E. G. (1996) J. Clin. Invest. 97 533–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Terracciano, C. M. N., De Souza, A. I., Philipson, K. D., and MacLeod, K. T. (1998) J. Physiol. 512 651–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adachi-Akahane, S., Lu, L., Frank, J. S., Philipson, K. D., and Morad, M. (1997) J. Gen. Physiol. 109 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yao, A., Su, Z., Nonaka, A., Zubair, I., Lu, L., Philipson, K. D., Bridge, J. H. B., and Barry, W. H. (1998) Circ. Res. 82 657–665 [DOI] [PubMed] [Google Scholar]
- 64.Solaro, R. J. (2001) Handbook of Physiology, Section 2: The Cardiovascular System, Vol. I, pp. 264–300, Oxford University Press, New York [Google Scholar]
- 65.Pathak, A., del Monte, F., Zhao, W., Schultz, J.-E., Lorenz, J. N., Bodi, I., Weiser, D., Hahn, H., Carr, A. N., Syed, F., Mavila, N., Jha, L., Qian, J., Marreez, Y., Chen, G., McGraw, D. W., Heist, E. K., Guerrero, J. L., DePaoli-Roach, A. A., Hajjar, R. J., and Kranias, E. G. (2005) Circ. Res. 96 756–766 [DOI] [PubMed] [Google Scholar]
- 66.Kirchhefer, U., Baba, H. A., Boknik, P., Breeden, K. M., Mavila, N., Bruchert, N., Justus, I., Matus, M., Schmitz, W., DePaoli-Roach, A. A., and Neumann, J. (2005) Cardiovasc. Res. 68 98–108 [DOI] [PubMed] [Google Scholar]
- 67.Sheehan, K. A., and Solaro, R. J. (2007) Am. J. Physiol. 293 R963–R973 [DOI] [PubMed] [Google Scholar]
- 68.Wu, J., Tolstykh, T., Lee, J., Boyd, K., Stock, J. B., and Broach, J. R. (2000) EMBO J. 19 5672–5681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoo, S., J.-S., Boylan, J. M., Brautigan, D. L., and Gruppuso, P. A. (2007) Arch. Biochem. Biophys. 461 186–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mumby, M. C. (2001) Sci. STKE PE1. [DOI] [PubMed]
- 71.Tikh, E. I., Fenton, R. A., Chen, J.-F., Schwarzschild, M. A., and Dobson, J. G. (2008) J. Cell. Physiol. 216 83–90 [DOI] [PubMed] [Google Scholar]
- 72.Liu, Q., and Hofmann, P. A. (2002) Am. J. Physiol. 283 H1314–H1321 [DOI] [PubMed] [Google Scholar]
- 73.Liu, Q., and Hofmann, P. A. (2003) Am. J. Physiol. 285 H97–H103 [DOI] [PubMed] [Google Scholar]