

Adeno-associated virus type 6, a human DNA virus that is being developed as a vector for gene therapy, has been crystallized in a form suitable for structure determination at about 3.2 Å resolution.
Keywords: adeno-associated viruses, parvoviruses
Abstract
Adeno-associated viruses are being developed as vectors for gene therapy and have been used in a number of clinical trials. Vectors to date have been based on the type species AAV-2, the structure of which was published in 2002. There is growing interest in modulating the cellular tropism and immune neutralization of AAV-2 with variants inspired by the properties of other serotypes. Towards the determination of a structure for AAV type 6, this paper reports the high-yield production, purification, crystallization and preliminary diffraction studies of infectious AAV-6 virions. The crystals diffracted to 3.2 Å resolution using synchrotron radiation. The most promising crystal form belonged to space group R3 and appeared to be suitable for initial structure determination.
1. Introduction
Adeno-associated viruses (AAVs) are 4.7 kb single-stranded DNA viruses that depend on helper viruses such as adenoviruses for replication (Berns, 1996 ▶). AAV infections do not present clinical symptoms beyond those of the helper-virus infection and their main interest is in the development of safe and effective vectors for in vivo gene therapy, for which they are a leading candidate (Carter, 2006a ▶,b ▶). 12 serotypes have been characterized to date, of which at least four appear to be primarily human, with the others infecting nonhuman primates although human cells are often also susceptible (Schmidt et al., 2008 ▶; Mori et al., 2004 ▶). AAV-6 is a variant of AAV-1 and these two serotypes are among the closest relatives to the type species AAV-2 (Rutledge et al., 1998 ▶). Most of the development of AAV transducing vectors has focused on AAV-2, for which the biology is most developed. However, there are challenges with AAV-2 vectors, including variability in transduction efficiencies according to cell type, reduced transduction rates after prior vector exposure and the large titers required in some transduction protocols (Baker, 2003 ▶), although progress is being made in several of these areas, including elevating transduction efficiency with self-complementary vectors (Kwon & Schaffer, 2007 ▶; McCarty, 2008 ▶). There is growing interest in exploiting the different tropisms, immunological responses and transduction efficiencies of different human and nonhuman serotypes and chimeric constructs thereof (Choi et al., 2005 ▶). In support of such efforts, following the atomic structure of the type species AAV-2 (Xie et al., 2002 ▶), several other AAVs have been crystallized and structures are emerging for the nonhuman forms (Kaludov et al., 2003 ▶; DiMattia et al., 2005 ▶; Miller et al., 2006 ▶; Nam et al., 2007 ▶). An alternative approach of rationally engineering changes to the capsid would be furthered by an understanding of how biological function is modulated through presumably more subtle structural differences between the human forms that are most related in sequence. This motivates the current study of AAV-6.
The capsid is an important determinant of the cellular specificity of the virus. Understanding the structural basis of receptor attachment will facilitate the development of vectors based on other serotypes to transduce cells that are resistant to AAV-2 infection. The primary receptor for AAV-2 on the cell surface is heparan sulfate proteoglycan, as is also the case for AAV-3 (∼87% sequence identity), but with lower affinity (Rabinowitz et al., 2002 ▶). AAV-4 and AAV-5, which are ∼58% identical to AAV-2 and to each other, are primarily simian viruses and bind sialic acid (Davidson et al., 2000 ▶). Although AAV-1 is much closer to AAV-2 (∼83% identity), it does not bind heparan sulfate (Rabinowitz et al., 2002 ▶). Intriguingly, the capsid of AAV-6, which differs from AAV-1 at only six sites, binds heparan sulfate, albeit with lower affinity than AAV-2 (Wu et al., 2006 ▶). It appears that a substitution of a single lysine near the proposed heparin-binding site accounts for the difference in heparin binding and in vector-transduction specificities (Xie et al., 2002 ▶; Kern et al., 2003 ▶; Wu et al., 2006 ▶).
Another biological property with implications for both fundamental virology and gene-therapy applications is the antigenicity of the capsid. Infection by wild-type AAV results in the production of neutralizing antibodies. 50–80% of adults are seropositive, with antibodies against AAV-2 being the predominant serotype (Blacklow et al., 1968 ▶; Parks et al., 1970 ▶). The presence of high-titer neutralizing antibodies is likely to severely decrease transduction levels, as seen in animal models upon re-administration of AAV-2 vectors (Halbert et al., 1997 ▶). It was shown that vectors produced from AAV-3 and AAV-6 differed from AAV-2 vectors in host range and serological reactivity (Rutledge et al., 1998 ▶). With each AAV serotype eliciting a distinct humoral response, one could imagine how alternative vectors based on different natural or artificial serotypes might allow high transduction rates to be maintained in those sensitized by prior exposure to the virus either naturally or in prior gene therapy (Choi et al., 2005 ▶).
2. Results and discussion
2.1. Production and purification
Historically, the greatest obstacle to AAV structure determination has been the production of sufficient amounts of pure material. The atomic structure of AAV-2 was the first structure to be elucidated of an animal satellite virus, a virus that propagates only through co-infection with a helper virus which kills the cells in which AAV must be grown. The AAV-2 structure became possible only after methods for propagating from an infectious clone (Laughlin et al., 1983 ▶) had been optimized and scaled up (Xie et al., 2004 ▶). An alternative is to use non-infectious virus-like particles produced though baculovirus expression of a capsid gene (Miller et al., 2006 ▶). Here, advantage is taken of the availability of the pAAV-6 infectious clone (Rutledge et al., 1998 ▶). AAV-6 was produced following the AAV-2 protocol (Xie et al., 2004 ▶). Briefly, a primary inoculum was produced by transfection with 10 µg of six T225 flasks of 80% confluent monolayers of cultured HeLa cells of the pAAV-6 plasmid, followed shortly thereafter by infection with 2.5 × 107 pfu ml−1 (final concentration) adenovirus 2 helper virus. After 48–72 h, this primary culture was then freeze–thawed three times and the resulting crude lysate or ‘primary inoculum’ was used to infect a 1–2 l suspension of HeLa cells at 0.6 × 106 cells ml−1.
The entire culture (cells, debris and medium) was harvested 48–72 h after the secondary infection and then treated with trypsin (0.0125% final concentration) and sodium deoxycholate (0.5% final concentration). AAV-6 was further purified by three runs of cesium chloride ultracentrifugation (Xie et al., 2004 ▶). AAV-6 yields from 1 l HeLa suspension cultures were 1.5–2.0 mg, similar to those obtained for AAV-2.
Preparations of AAV-6 in the laboratory were alternated with those of other infectious serotypes. There was a danger of cross-contamination and solving the wrong virus structure. Diagnostic tests were of two types: restriction digests of the starting plasmids and application of a multiplexed PCR assay to the purified virus. The restriction digests distinguished between AAV-2 and AAV-6 plasmids through the generation of different-length DNA fragments after digests with BglII and PstI for pAV2 and NruI for pAAV-6. The purified AAV was subjected to a multiplexed PCR assay capable of detecting sub-picogram levels of contaminating AAV-2 or AAV-3 in an AAV-6 preparation (Mitchell et al., 2006 ▶). It is based on the use of differentially annealing serotype-specific primers targeted to variable regions of the capsid-coding region of the genome.
The purity of the virus after three cesium-gradient ultracentrifugations was assessed in several ways. The purity from contaminating proteins or from the presence of empty (DNA-free) virions was assessed by the absorbance ratio OD280/OD260, which attained levels within 0.01 AU of the expected 0.72. Spectrometry was also used to monitor for the presence of aggregates using the reduced wavelength-dependent transmission that would result from Rayleigh scattering between 200 and 600 nm.
Protein purity was also assayed by LDS–PAGE. Samples were denatured by heating to 343 K in 2% NuPage LDS (Invitrogen) for 10 min. Denaturing acrylamide gels (4–12% NuPage bis-tris gradient) were loaded with 1–3 µg sample per lane or overloaded with 10 µg sample in order to detect contaminant proteins. Gels were stained with Coomassie Blue R-250 or G-250 (Sigma, EZ Blue) or silver nitrate (Xie et al., 2004 ▶). Gel electrophoresis is quantitative because the known ratios of the capsid proteins VP1, VP2 and VP3 provide an internal standard for calibrating overloaded to regular lanes. There was no single protein contaminant at levels greater than 1%.
2.2. Concentration and crystallization
AAV-6 is more soluble in low-salt buffers than AAV-2 and could therefore be concentrated and desalted by centrifugal ultrafiltration using Microcon-100 devices (Amicon Bioseparations Inc.). ∼400 µl purified AAV-6 (at ∼1 mg ml−1) in the CsCl from ultracentrifugation was loaded into the Microcon-100 after pre-wetting the membrane with 10 µl 3.3 M CsCl. Ultrafiltration was performed in a bench-top microcentrifuge (Eppendorf 5415 D) at 2000 rev min−1 and 277 K for ∼40–45 min. Once ∼330–340 µl filtrate had been removed, 380 µl HM buffer (100 mM HEPES, 50 mM MgCl2, 0.03% sodium azide, pH 7.3) was added to the sample. This step was repeated twice. About ∼60 µl final retentate was collected by inverting the filter unit at 1000 rev min−1 for 2 s. Aggregates were removed by centrifugation at 16 000g for 10 min. Concentration was determined spectrophotometrically at 280 nm (using a Nanodrop spectrophotometer and a calculated ∊280 of 4.54 ml mg−1 cm−1) from a 2 µl aliquot immediately before crystallization.
Crystallization conditions were tested close to those that had been successful with AAV-2 (Xie et al., 2004 ▶), namely AAV-6 concentrations in the range 1.5–5.0 mg ml−1 and precipitant (PEG 6000) concentrations of 1.0–5.0% in the reservoir. All trials were performed at room temperature in a buffer of 100 mM HEPES pH 7.2–7.3, 50 mM MgCl2, 0.03% sodium azide with or without 25% glycerol cryoprotectant (in both the drop and the reservoir). Initial screening took place in ‘batch’ mode with hanging drop and reservoir at the same (equilibrium) precipitant concentrations. Optimization was attempted by conventional vapor diffusion, with drops consisting of 2 µl 3.5–8 mg ml−1 sample plus 2 µl precipitant (4.8–5.6% PEG 6000) solution equilibrated against 800 µl precipitant reservoir solution. However, the best crystals (Fig. 1 ▶) were obtained in 2–3 weeks using the hanging-drop ‘batch’ mode, with drops containing 1 µl sample at 5.0 mg ml−1 plus 1 µl 5% PEG, yielding a precipitant concentration equal to the reservoir at 2.5% PEG 6000. The highest resolution diffraction (see below) was obtained using crystals from one of the three virus preparations that yielded crystals. No high-quality crystals were obtained in the presence of glycerol, in contrast to AAV-2 where 25% glycerol improved the quality.
Figure 1.

Crystals of AAV-6. (a) As grown in HM buffer by vapor diffusion, with a longest dimension of 0.6 mm. (b) As mounted in a cryo-loop (longest dimension of 0.2 mm); this is the crystal from which the reported data were collected.
2.3. Data collection and processing
Diffraction data were collected on the biohazard-level 2 F1 beamline at the Cornell High Energy Synchrotron Source (CHESS) with λ = 0.9186 Å. Prior to flash-freezing, crystals were cryoprotected using a single-step 1 h soak in a 30% glycerol solution made up in 6% PEG 6000 precipitant and HM buffer. Diffraction images were collected using a side-by-side dual Quantum 4 CCD detector (ADSC, Poway, California, USA) with an oscillation angle of 0.3°. The crystal-to-detector distance was 500 mm and the exposure time was 180 s. The measured intensities were indexed, integrated and scaled with the HKL v.1.97.9 program package (Otwinowski & Minor, 1997 ▶).
Diffraction quality varied for the seven crystals tested. Three crystals that diffracted to only 7 Å resolution and one that diffracted to 3.5 Å resolution but with a twinned lattice were not considered further. Of three crystals showing visible diffraction to about 3.0 Å resolution, two indexed as primitive orthorhombic with unit-cell parameters a = 354.8, b = 363.9, c = 371.9 Å and a completeness of 46.6%. The R merge of 19% is no worse than for many other usable virus data sets, but was surpassed by another crystal. The more fully characterized crystal processed as rhombohedral R3, with unit-cell parameters a = b = c = 252.9 Å, α = β = γ = 61.4° and R merge = 7.9% (100–3.0 Å; Table 1 ▶). Radiation damage limited data collection to 55 frames and completeness to 30.8%, but there are many examples of symmetrical virus structures that have been solved with even lower completeness.
Table 1. Data-processing statistics.
Values in parentheses are for the 3.43–3.33 Å resolution shell.
| Space group | R3 |
| Unit-cell parameters (Å, °) | |
| Hexagonal setting | a = b = 258.4, c = 613.0 |
| Rhombohedral setting | a = b = c = 252.9, α = β = γ = 61.4 |
| No. of observations | 96330 |
| Unique reflections | 91552 |
| Resolution (Å) | 100–3.0 |
| Rmerge† (%) | 7.9 (26.7) |
| 〈I〉/〈σ(I)〉 | 4.5 (1.5) |
| Completeness (%) | 30.8 (30.9) |
R
merge = 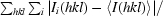
 , where Ii(hkl) is the ith observation of a symmetry equivalent of reflection hkl.
, where Ii(hkl) is the ith observation of a symmetry equivalent of reflection hkl.
Challenges had to be overcome in the data processing. Maintenance of a ten-pixel spot separation with such large unit cells requires a large crystal-to-detector distance. To increase the high-resolution coverage, a dual-detector system was used (Fig. 2 ▶). The detectors were placed asymmetrically so that the direct beam fell on one, detector A, while for detector B the beam location was not directly visible. Prior calibration of the beam coordinates (−67.5, 87.25 mm) using small-molecule diffraction was not sufficiently precise, even through the detectors were held in juxtaposition by a carousel. Symptoms in the processing of B frames included predictions that deviated increasingly from spot positions after about 7.5° of crystal rotation and an combined R merge of >30% for A-frame and B-frame data that greatly exceeded that of the A-frame data alone (6%). In retrospect, we know that positional refinement of the detector parameters converged upon an incorrect solution for the B panel that was frame-shifted by one lattice unit. This was revealed through a search procedure in which the detector parameters were refined following translation by one of six nearest-neighbor lattice vectors in the panel B two-dimensional diffraction image. Scaling with the A-frame data gave R merge > 30% for all translations except the correct one, where beam coordinates of (−69.80, 87.60 mm) led to an R merge of 7.8%. The error of 2.3 mm in the initial inferred beam position had, it was presumed, originated during refinement of the indexing of the small-molecule calibration data, where an adjustment of beam location compensated for inaccuracy in another covariant parameter. For protein crystals, this precision of calibration might suffice, because refinement should converge if the error is less than half the lattice unit. However, with the fine spot separation of the large unit cell of a virus, the error was sufficient to cause mis-indexing so the detector parameters were outside the convergence radius of refinement.
Figure 2.

Diffraction images of AAV-6. To maximize the coverage of high-resolution scattering while maintaining adequate spot separation, a dual-detector system was used at a long crystal-to-detector distance. The beam-stop shadow can be seen on detector B (top), but the beam coordinates relative to detector A (bottom) can only be inferred indirectly from the indexing: a challenge with the virus’ fine spot separation.
2.4. Rotation function
Particle orientations were determined by computing both ordinary and icosahedrally locked self-rotation functions using the program GLRF (Tong & Rossmann, 1997 ▶). The initial 14 and 10 Å resolution ordinary rotation function showed most of the expected fivefold, threefold and twofold icosahedral noncrystallographic symmetry (NCS) axes (Fig. 3 ▶ a), but the peak heights were uneven and extended into ripples. The corresponding locked rotation function at 10 Å gave two neighboring solutions. Although noisy, an ordinary (slow) rotation-function search at 6.0–3.0 Å was free from the low-resolution ripples. As the locked rotation function is calculated in progressively higher resolution shells the second peak weakens, leaving only a single solution at 6.0–3.0 Å. These rotation-function calculations are subject to truncation artifacts from the incomplete data (31%). The data were collected in a single sweep from a single frozen crystal, differing from many earlier partial data sets of viruses, which were merged from a few frames from each of dozens of randomly oriented crystals. Thus, the truncation effects are more systematic because there are regions of reciprocal space that were not sampled. The resolution-dependence is heightened because in a dual-detector configuration there is an unseen disk of reciprocal space where reflections would fall on the inactive area between the two detectors. This excludes a greater proportion of low-resolution data, such that the completeness of the AAV-6 data at low resolution is ∼3/4 of that at higher resolution.
Figure 3.

Rotation functions. (a) Spherical polar projection (XZK, lower hemisphere) of the ordinary rotation function at 14 Å, showing the expected six icosahedral fivefolds. (b) The corresponding locked rotation function at 6.0–3.0 Å resolution. (c) The location of the symmetry elements implied by the locked rotation-function solution, shown in the same projection as (a) with pentagons representing fivefolds, triangles representing threefolds and ovals representing twofolds.
The constraints of packing a ∼250 Å diameter parvovirus into a 253 Å R3 unit cell limit the unit-cell contents to one virus with an icosahedral axis aligned along the crystallographic threefold. The center of the virus can be placed at the origin of the unit cell without need for a translation-function search.
3. Conclusion
A structure determination of the AAV-6 capsid to 3.2 Å resolution is in progress using the data described above and molecular-replacement phases calculated from the known AAV-2 structure refined using the 20-fold noncrystallographic symmetry of AAV-6. Several aspects of the data collection were in retrospect sub-optimal, as discussed above, but an initial determination is being attempted because with the combined challenges of propagation, crystallization and multiple crystal forms, assurance was wanted that the efforts with this crystal form would pay dividends. The 20-fold NCS appears to be sufficient to compensate for the incompleteness of the diffraction data and to support an initial structure determination. All indications are that this structure will be adequate to reveal differences in the surface structure between the AAV serotypes that are important to their capsid-mediated host interactions. The structure of AAV-6 is therefore likely to provide insights into the mediation of viral cell tropism and immune neutralization issues, as discussed in §1, that are central to the successful exploitation of AAV for gene-therapy treatments of genetically based diseases.
Acknowledgments
The authors would like to thank Thayumanasamy Somasundaram, Weishu Bu and the staff at CHESS who helped with data collection. CHESS is supported by the NSF and NIH/NIGMS via NSF award DMR-0225180 and the MacCHESS resource is supported by NIH/NCRR award RR-01646. The research was supported by the National Institutes of Health R01-GM66875 (MSC) and a predoctoral fellowship from the American Heart Association, Florida and Puerto Rico affiliate 0515201B (HMO).
References
- Baker, A. H. (2003). Mol. Ther.7, 433–434. [DOI] [PubMed]
- Berns, K. I. (1996). Fields Virology, edited by B. N. Fields, D. M. Knipe & P. M. Howley, pp. 1017–1041. Philadelphia: Raven.
- Blacklow, N. R., Hoggan, M. D. & Rowe, W. P. (1968). J. Natl Cancer Inst.40, 319–327. [PubMed]
- Carter, B. J. (2006a). Parvoviruses, edited by J. R. Kerr, S. F. Cotmore, M. E. Bloom, R. M. Linden & C. R. Parrish, pp. 497–498. London: Edward Arnold.
- Carter, B. J. (2006b). Parvoviruses, edited by J. R. Kerr, S. F. Cotmore, M. E. Bloom, R. M. Linden & C. R. Parrish, pp. 499–510. London: Edward Arnold.
- Choi, V. W., McCarty, D. M. & Samulski, R. J. (2005). Curr. Gene Ther.5, 299–310. [DOI] [PMC free article] [PubMed]
- Davidson, B. L., Stein, C. S., Heth, J. A., Martins, I., Kotin, R. M., Derksen, T. A., Zabner, J., Ghodsi, A. & Chiorini, J. A. (2000). Proc. Natl Acad. Sci. USA, 97, 3428–3432. [DOI] [PMC free article] [PubMed]
- DiMattia, M., Govindasamy, L., Levy, H. C., Gurda-Whitaker, B., Kalina, A., Kohlbrenner, E., Chiorini, J. A., McKenna, R., Muzyczka, N., Zolotukhin, S. & Agbandje-McKenna, M. (2005). Acta Cryst. F61, 917–921. [DOI] [PMC free article] [PubMed]
- Halbert, C. L., Standaert, T. A., Aitken, M. L., Alexander, I. E., Russell, D. W. & Miller, A. D. (1997). J. Virol.71, 5932–5941. [DOI] [PMC free article] [PubMed]
- Kaludov, N., Padron, E., Govindasamy, L., McKenna, R., Chiorini, J. A. & Agbandje-McKenna, M. (2003). Virology, 306, 1–6. [DOI] [PubMed]
- Kern, A., Schmidt, K., Leder, C., Muller, O. J., Wobus, C. E., Bettinger, K., Von der Lieth, C. W., King, J. A. & Kleinschmidt, J. A. (2003). J. Virol.77, 11072–11081. [DOI] [PMC free article] [PubMed]
- Kwon, I. & Schaffer, D. V. (2007). Pharm. Res.25, 489–499. [DOI] [PMC free article] [PubMed]
- Laughlin, C. A., Tratschin, J. D., Coon, H. & Carter, B. J. (1983). Gene, 23, 65–73. [DOI] [PubMed]
- McCarty, D. M. (2008). Mol. Ther.16, 1648–1656. [DOI] [PubMed]
- Miller, E. B., Gurda-Whitaker, B., Govindasamy, L., McKenna, R., Zolotukhin, S., Muzyczka, N. & Agbandje-McKenna, M. (2006). Acta Cryst. F62, 1271–1274. [DOI] [PMC free article] [PubMed]
- Mitchell, D. A., O’Donnell, J., Hare, J. T. & Chapman, M. S. (2006). J. Virol. Methods, 136, 277–282. [DOI] [PubMed]
- Mori, S., Wang, L., Takeuchi, T. & Kanda, T. (2004). Virology, 330, 375–383. [DOI] [PubMed]
- Nam, H. J., Lane, M. D., Padron, E., Gurda, B., McKenna, R., Kohlbrenner, E., Aslanidi, G., Byrne, B., Muzyczka, N., Zolotukhin, S. & Agbandje-McKenna, M. (2007). J. Virol.81, 12260–12271. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Parks, W., Boucher, D., Melnick, J., Taber, L. & Yow, M. (1970). Infect. Immun.2, 716–722. [DOI] [PMC free article] [PubMed]
- Rabinowitz, J. E., Rolling, F., Li, C., Conrath, H., Xiao, W., Xiao, X. & Samulski, R. J. (2002). J. Virol.76, 791–801. [DOI] [PMC free article] [PubMed]
- Rutledge, E., Halbert, C. & Russell, D. (1998). J. Virol.72, 309–319. [DOI] [PMC free article] [PubMed]
- Schmidt, M., Voutetakis, A., Afione, S., Zheng, C., Mandikian, D. & Chiorini, J. A. (2008). J. Virol.82, 1399–1406. [DOI] [PMC free article] [PubMed]
- Tong, L. & Rossmann, M. G. (1997). Methods Enzymol.276, 594–611. [PubMed]
- Wu, Z., Asokan, A., Grieger, J. C., Govindasamy, L., Agbandje-McKenna, M. & Samulski, R. J. (2006). J. Virol.80, 11393–11397. [DOI] [PMC free article] [PubMed]
- Xie, Q., Bu, W., Bhatia, S., Hare, J., Somasundaram, T., Azzi, A. & Chapman, M. S. (2002). Proc. Natl Acad. Sci. USA, 99, 10405–10410. [DOI] [PMC free article] [PubMed]
- Xie, Q., Hare, J., Turnigan, J. & Chapman, M. S. (2004). J. Virol. Methods, 122, 17–27. [DOI] [PubMed]