

Abstract
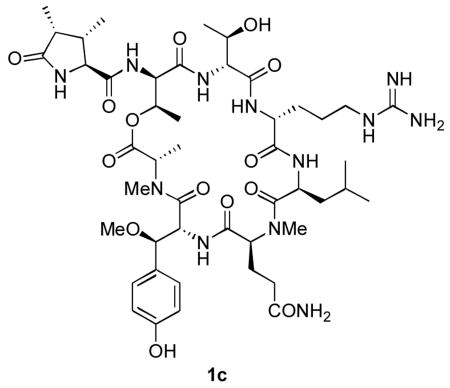
The cytotoxic, cyclic heptadepsipeptide natural product callipeltin B was synthesized on a solid-phase support in 15% overall yield. Comparison of the 1H NMR spectra of three synthetic isomers with those of callipeltin B confirmed the configurational reassignment of its threonine residues as D-allothreonine and the assignment of the configuration of its β-methoxytyrosine residue as (2R, 3R).
The cyclic depsipeptide callipeltin B (1) was isolated from the Lithistid sponge callipelta sp. and structurally characterized by Minale and coworkers in 1996.1 Along with the related cyclic depsipeptide callipeltin A,2 1 possesses a 22-membered macrolactone composed of the unique, non-proteinogenic amino acids β-methoxytyrosine (β-MeOTyr) and (3S,4R)-3,4-dimethyl-L-pyroglutamic acid (DiMePyroGlu) as well as several D- and N-methylated amino acids. Both callipeltins A and B showed broad-spectrum cytotoxicity against a number of tumor cell lines, including several drug-resistant cell lines,1 and callipeltin A shows potent anti-HIV activity.2
Although the initially published structure of callipeltin B (1a) has never been revised, revisions to the structure of the closely related cyclic depsipeptide callipeltin A have strongly implied a need for a structural revision of 1. Specifically, D'Auria and coworkers, after isolating and characterizing two smaller, linear peptidic fragments of callipeltin A that they termed callipeltins D and E, suggested that one of the residues initially identified as L-threonine should be reassigned as D-allothreonine.3 Such a change thus implied that the structure of callipeltin B should be reassigned as 1b, in which the configuration of C-21 is R.
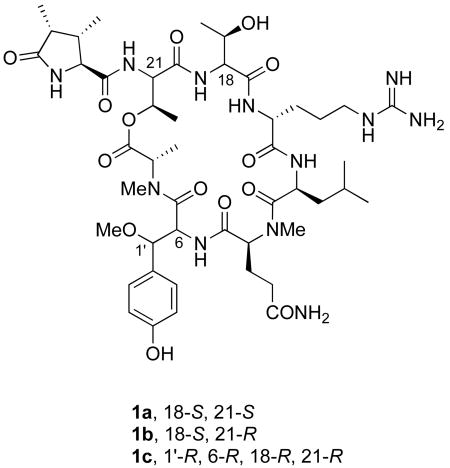
More recently, Bifulco and D'Auria have published computational studies that suggest that both of the residues in callipeltin A initially assigned as L-threonine by Minale and coworkers are actually D-allothreonine.4 At the same time, the configuration of the β-MeOTyr residue of callipeltin A, which could not be determined in Minale's original structural study, was determined to be (2R, 3R) by D'Auria and co-workers, using a combination of synthesis and degradation studies.5 Both of these structural reassignments were confirmed in our laboratory by the synthesis and spectral correlation of two diastereomers of callipeltin E.6 Taken together, these studies implied that the structure of callipeltin B should be further revised to 1c, in which the configuration of C-18 has been changed and those of C-1′ and C-6 are assigned as R. Herein we wish to report the synthesis of the three candidate structures 1a-c and the confirmation of 1c as the structure of callipeltin B by 1H NMR correlation.
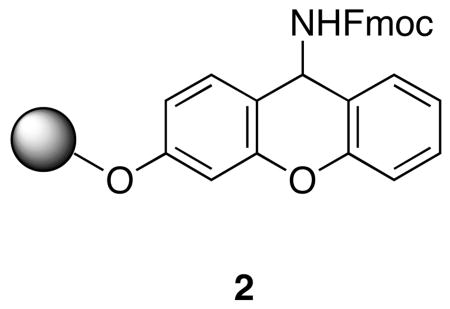
A solid phase synthetic strategy was chosen to expedite the synthesis of 1, its analogues and related depsipeptides. To permit macrocyclization on a solid support, it was decided to anchor the sidechain of the N-methylglutamine residue to a peptide amide synthesis resin, thereby leaving available both N- and C-termini for macrocyclization by amide bond formation. The Tentagel-based TG Sieber amide resin7 (2) was chosen for the relatively low amounts of TFA (1-5% in CH2Cl2) needed for efficient cleavage of the peptide, thereby minimizing the possibility of acid-catalyzed β-MeOTyr decomposition during cleavage from the resin. A standard Fmoc-based, C-to-N peptide synthesis approach thus required the use of allyl-based protecting groups for N- and C- termini immediately prior to macrocyclization. All sidechain protecting groups were selected for their ability to be removed either during the mildly acidic cleavage from the Sieber resin or a subsequent catalytic hydrogenation.
The initial plan to close the macrocycle at the MeGln/β-MeOTyr peptide bond, permitting a C-to-N synthesis from the MeGln residue, had to be abandoned because of spontaneous diketopiperazine formation upon deprotection of the Leu-MeGln dipeptide; instead, it was decided to close the macrocycle at the β-MeOTyr/N-methylalanine peptide bond, despite the known problem of slower acylation of secondary amines. Additionally, an initial plan to install the N-terminal (3S,4R)-3,4-dimethyl-L-pyroglutamic acid after macrocyclization was revised because of rapid O to N transacylation upon deprotection of the N-terminal Fmoc group. As a result, it was decided to install the (3S,4R)-3,4-dimethyl-L-pyroglutamic acid residue prior to forming the ester bond to prevent transacylation.
All of the constituent residues needed to make 1a-c were either commercially available or had been previously synthesized. In addition to D'Auria's synthesis of β-MeOTyr,5 several others had been published;8 several different syntheses of DiMePyroGlu had also been reported.9 The syntheses of 1a-c began with the deprotection and acylation of 2 (Scheme 1) with the cyclic anhydride of Fmoc-N-methylglutamic acid (3),10 followed by activation of the resin-bound acid with HATU and coupling with (2R, 3R)-O-benzyl-β-methoxytyrosine allyl ester (4)7c to afford a resin-bound dipeptide. All deprotection and coupling reactions were monitored by removal of an aliquot of 1-2 mg of resin, cleavage of the peptide from the resin and analysis of the crude cleavage mixture by reverse phase HPLC and MALDI-MS. It was found that use of HATU with no added HOAt afforded complete consumption of starting material with no epimerization evident by HPLC analysis. Sequential installation of Fmoc-Leu, Fmoc-D-Arg(NO2), Fmoc-D-aThr(THP) and Fmoc-D-aThr using coupling conditions optimized for each step proceeded without incident to afford the resin-bound hexapeptide 5 in >95% purity as determined by HPLC analysis. For the syntheses of 1a and 1b, protected L-threonine was substituted in this sequence for the second, or both, D-allothreonine residues. Additionally, in the syntheses of 1a and 1b, Fmoc-D-Arg(Z,Z) was substituted for Fmoc-D-Arg(NO2).
Scheme 1.

Solid Phase Synthesis of 1c
Deprotection of the N-terminal Fmoc afforded a β-amino alcohol that could be selectively N-acylated with the N-hydroxysuccinimide ester of (3S,4R)-3,4-dimethyl-L-pyroglutamic acid (6). Esterification of the hydroxyl with alloc-N-methylalanine using the sulfonyl nitrotriazole reagent MSNT11 in conjunction with N-methylimidazole afforded a resin-bound, protected heptadepsipeptide in high purity. Palladium-catalyzed deprotection of N- and C-termini followed by macrolactamization using PyAOP to minimize epimerization of the activated β-methoxytyrosine afforded the resin-bound macrocycle in roughly 80% purity as judged by HPLC analysis of the crude deprotection mixture. Removal of the cyclized product from the resin with 2% TFA-CH2Cl2 and complete deprotection by catalytic transfer hydrogenation afforded 1c, after purification by reverse phase HPLC, in 15% overall yield. The isomeric depsipeptides 1a and 1b were obtained in 15% and 14% overall yields, respectively.
Because of the unavailability of a sample of natural callipeltin B, the correlation of synthetic 1a-c with callipeltin B involved the comparison of spectroscopic data. Comparison of the 1H NMR of 1c with that of callipeltin B showed no significant differences between the two apart from the absence of several exchangeable amide protons in CD3OD.12 On the other hand, the 1H NMR spectra of 1a and 1b showed considerable differences from that of callipeltin B; in fact, the spectra of 1a and 1b showed evidence of conformational heterogeneity at ambient temperature. On the basis of these comparisons, we now conclude that the correct structure of callipeltin B is 1c. Current studies are underway to synthesize analogues of 1c and several related cyclic depsipeptides using the methodology described herein.
In summary, the synthesis of the cyclic depsipeptide natural product callipeltin B has been accomplished, thereby providing confirmation of the recent configurational reassignment of the previously assigned L-threonine residues as D-allothreonine and the recent configurational assignment of the β-methoxytyrosine residue as (2R, 3R). This synthesis illustrates the facility with which even complex natural products can be synthesized on solid supports. In addition, the expeditious solid-phase synthesis of 1 opens the door for the rapid synthesis of analogues to explore structure-function relationships and for the synthesis of other, more complex cyclic depsipeptides.
Supplementary Material
Complete experimental details and spectroscopic details for 1 and the novel protected amino acids used in its synthesis. A tabulated comparison of 1H NMR spectra of 1 and callipeltin B and reproduced spectra of both are also provided. This information is available free of charge via the Internet at http :// pubs.acs.org.
Acknowledgments
We wish to thank Prof. Maria Valeria D'Auria and Dr. Kirk Gustafson for supplying spectral information and for helpful discussions. We also thank Selçuk Çalimsiz for his assistance and to both him and Minoo Sedighi for their synthesis of dimethylpyroglutamic acid for this project. We gratefully acknowledge the National Institutes of Health (AI-50888) for financial support of this work.
References
- 1.D'Auria MV, Zampella A, Gomez-Paloma L, Minale L. Tetrahedron. 1996;52:9589. [Google Scholar]
- 2.Zampella A, D'Auria MV, Gomez-Paloma L, Casapullo A, Minale L, Debitus C, Henin Y. J Am Chem Soc. 1996;118:6202. [Google Scholar]
- 3.Zampella A, Randazzo A, Borbone N, Luciani S, Trevisi L, Debitus C, D'Auria MV. Tetrahedron Lett. 2002;43:6163. [Google Scholar]
- 4.Bassarello C, Zampella A, Monti MC, Gomez-Paloma L, D'Auria MV, Riccio R, Bifulco G. Eur J Org Chem. 2006:604. [Google Scholar]
- 5.Zampella A, D'Orsi R, Sepe V, Casapullo A, Monti MC, D'Auria MV. Org Lett. 2005;7:3585. doi: 10.1021/ol0513600. [DOI] [PubMed] [Google Scholar]
- 6.Çalimsiz S, Morales Ramos ÁI, Lipton MA. J Org Chem. 2006;71:6351. doi: 10.1021/jo060351h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sieber P. Tet Lett. 1987;28:2107. [Google Scholar]
- 8.(a) Okamoto N, Hara O, Makino K, Hamada Y. J Org Chem. 2002;67:9210. doi: 10.1021/jo0258352. [DOI] [PubMed] [Google Scholar]; (b) Hansen DB, Wan XB, Carroll PJ, Joullie MM. J Org Chem. 2005;70:3120. doi: 10.1021/jo047876z. [DOI] [PubMed] [Google Scholar]; (c) Oku N, Krishnamoorthy R, Benson AG, Ferguson RL, Lipton MA, Phillips LR, Gustafson KR, McMahon JB. J Org Chem. 2005;17:6842. doi: 10.1021/jo0508853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Okamoto N, Hara O, Makino K, Hamada Y. Tetrahedron: Asymmetry. 2001;12:1353. [Google Scholar]; (b) Acevedo CM, Kogut EF, Lipton MA.Tetrahedron 200157635318159221 [Google Scholar]
- 10.The regioselectivity of opening of 3 was established by cleavage of the acylation product from the resin and HPLC correlation with authentic samples of Fmoc-Nα-methylglutamine and the C-terminal carboxamide of Fmoc-Nα-methylglutamic acid. Exclusive attack on the less hindered carbonyl by the sterically hindered nitrogen of the Sieber linker was found by reverse phase HPLC analysis.
- 11.Blankemeyer-Menge B, Nimtz M, Frank R. Tet Lett. 1990;31:1701. [Google Scholar]
- 12.The only differences outside of the amide proton region noted in the comparison were three singlets present in the 1H NMR of the natural product (at δ ∼2.08, 2.1 and 2.2) that were not found in the spectrum of 1c, nor were included in the tabulated data in Ref. 1; additionally, a singlet at δ 2.85 was found in the 1H NMR of 1c that didn't correspond to any resonance in callipeltin B and might possibly result from a small amount of DMF present in the sample.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental details and spectroscopic details for 1 and the novel protected amino acids used in its synthesis. A tabulated comparison of 1H NMR spectra of 1 and callipeltin B and reproduced spectra of both are also provided. This information is available free of charge via the Internet at http :// pubs.acs.org.