

Abstract
The PI3K/PTEN/Akt/mTOR pathway plays critical roles in the regulation of cell growth. The effects of this pathway on drug resistance and cellular senescence of breast cancer cells has been a focus of our laboratory. Introduction of activated Akt or mutant PTEN constructs which lack lipid phosphatase [PTEN(G129E)] or lipid and protein phosphatase [PTEN(C124S)] activity increased the resistance of the cells to the chemotherapeutic drug doxorubicin, and the hormonal drug tamoxifen. Activated Akt and PTEN genes also inhibited the induction of senescence after doxorubicin treatment; a phenomenon associated with unrestrained proliferation and tumorigenesis. Interference with the lipid phosphatase domain of PTEN was sufficient to activate Akt/mTOR/p70S6K as MCF-7 cells transfected with the mutant PTEN gene lacking the lipid phosphatase activity [PTEN(G129E)] displayed elevated levels of activated Akt and p70S6K compared to empty vector transfected cells. Cells transfected with mutant PTEN or Akt constructs were hypersensitive to mTOR inhibitors when compared with the parental or empty vector transfected cells. Akt-transfected cells were cultured for over two months in tamoxifen from which tamoxifen and doxorubicin resistant cells were isolated that were >10-fold more resistant to tamoxifen and doxorubicin than the original Akt-transfected cells. These cells had a decreased induction of both activated p53 and total p21Cip1 upon doxorubicin treatment. Furthermore, these cells had an increased inactivation of GSK-3β and decreased expression of the estrogen receptor-α. In these drug resistant cells, there was an increased activation of ERK which is associated with proliferation. These drug resistant cells were hypersensitive to mTOR inhibitors and also sensitive to MEK inhibitors, indicating that the enhanced p70S6K and ERK expression was relevant to their drug and hormonal resistance. Given that Akt is overexpressed in greater than 50% of breast cancers, our results point to potential therapeutic targets, mTOR and MEK. These studies indicate that activation of the Akt kinase or disruption of the normal activity of the PTEN phosphatase can have dramatic effects on activity of p70S6K and other downstream substrates and thereby altering the therapeutic sensitivity of breast cancer cells. The effects of doxorubicin and tamoxifen on induction of the Raf/MEK/ERK and PI3K/Akt survival pathways were examined in unmodified MCF-7 breast cells. Doxorubicin was a potent inducer of activated ERK and to a lesser extent Akt. Tamoxifen also induced ERK. Thus a consequence of doxorubicin and tamoxifen therapy of breast cancer is the induction of a pro-survival pathway which may contribute to the development of drug resistance. Unmodified MCF-7 cells were also sensitive to MEK and mTOR inhibitors which synergized with both tamoxifen and doxorubicin to induce death. In summary, our results point to the key interactions between the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways in regulating chemotherapeutic drug resistance/sensitivity in breast cancer and indicate that targeting these pathways may prevent drug and hormonal resistance.
Keywords: Conventional therapy, targeted therapy, signal transduction inhibitors, breast cancer
Introduction
Breast cancer ranks as the second most common cause of cancer death among women in the United States. Only lung cancer, which results primarily from cigarette smoking, induces more cancer deaths in women in the USA. Approximately 1 in 7 women in the United States will be diagnosed with breast cancer during her lifetime (Jemal et al., 2004). Over 210,000 new cases of breast cancer are diagnosed in the United States each year (Centers for Disease Control and Prevention, 2006). Breast cancer is the cause of death of over 40,000 women in the United States each year. Many drugs have been demonstrated to extend survival of breast cancer patients. Anticancer agents frequently used to treat breast cancer include chemotherapeutic drugs such as methotrexate, 5-fluorouracil (5-FU), cyclophosphamide, anthracyclines, taxanes, monoclonal antibodies such as trastuzumab, hormonal based therapeutics such as tamoxifen and aromatase inhibitors. Mechanisms by which these agents inhibit breast cancer progression vary from drug to drug. While these drugs are the mainstay of chemo, immuno and hormonal therapy of breast cancer, a common problem with these treatments is the development of drug resistance. Breast cancer cells can become drug resistant by multiple mechanisms which include: increased expression of membrane transporters which transport the toxic drug out of the cell or modify/detoxify the drug, increased expression of signaling and anti-apoptotic pathways as well as other mechanisms which allow the cells to grow in the presence of the drug. This manuscript will discuss some of the mechanisms by which the altered expression of key signaling and apoptotic pathways may lead to breast cancer drug resistance and how targeting these pathways may result in the suppression of neoplastic growth.
Overview of the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways and their roles in breast cancer
Mutations may occur in breast and other cancer cells which result in elevated expression or constitutive activation of various growth factor receptors such as the epidermal growth factor receptor (EGFR or c-Erb-B1) or the related c-Erb-B2 receptor. The PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways are often activated by mutations/amplifications in these and other growth factor receptors. These pathways are also regulated by upstream Ras which is mutated in 20 to 30% of human cancers. The PI3K and B-Raf kinases are also activated by mutation and in particular in breast cancer (Hollestelle et al., 2007). Many of the events elicited by the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways have direct effects on survival and proliferative pathways. Aberrant regulation of these pathways can contribute to uncontrolled cell growth and lead to malignant transformation. Effective targeting of these pathways may result in suppression of cell growth and death of malignant cells. An overview of the effects of PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways on downstream signaling pathways leading to growth and the prevention of apoptosis in breast cancer is presented in Figure 1.
Figure 1. Overview of PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK Pathways.

The PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK PI3K/PTEN/Akt/mTOR and pathways are regulated by upstream growth factor and mitogen receptors as well as Ras and various kinases. Many kinases serve to phosphorylate S/T and Y residues on Raf and S/T residues on Akt. Some of the Raf phosphorylation events serve to enhance its activity (shown by a black P in a white circle) whereas others serve to inhibit its activity (shown by a white P in a black circle. Moreover there are phosphatases such as PP2A, PTEN, SHIP and PHLPP, which remove phosphates on certain regulatory residues on Raf, MEK, ERK, PtdIns(3,4,5)P3 and Akt as well as other signaling molecules. The PI3K/Akt pathway is also activated after receptor ligation. Akt and ERK have many downstream targets which serve to regulate cell growth and apoptosis. Some of the downstream transcription factors regulated by these pathways are indicated in diamond shaped outlines.
The PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways are also activated by many growth factors and cytokines which are important in driving proliferation and preventing apoptosis of breast cells (Blalock et al., 1999, Lee et al., 2002, Steelman et al., 2004, McCubrey et al., 2005, McCubrey et al., 2007). After growth receptor ligation, Shc, a Src homology (SH)-2 (SH2)-domain containing protein, becomes associated with the c-terminus of the growth factor receptor (Matsuguchi et al., 1994, Inhorn et al., 1995, Okuda et al., 1999). Shc recruits the GTP-exchange complex growth factor receptor bound-2 (Grb2)/son of sevenless exchange (Sos) proteins resulting in the loading of membrane bound Ras with GTP (Tauchi et al., 1994, Lanfrancone et al., 1995). Ras:GTP then recruits Raf to the membrane where it becomes activated, likely via a Src-family tyrosine (Y) kinase (Karin et al., 1994, Lange-Carter et al., 1994, Marais et al., 1995). Raf is responsible for phosphorylation of the mitogen associated/extracellular regulated kinase-1 (MEK1) (Xu et al., 1995, Marais et al., 1997, Mason et al., 1999). MEK1 phosphorylates extracellular regulated kinases 1 and 2 (ERKs 1 and 2) on specific threonine (T) and Y residues (Xu et al., 1995, Marais et al., 1997, Mason et al., 1999). Activated ERK1 and ERK2 serine (S)/T kinases phosphorylate and activate a variety of substrates. The number of potential substrates of the ERK kinases has been speculated to be more than several hundred. The 90 kDa ribosomal six kinase 1 p90Rsk1 is one such substrate (Deng et al., 1994, Davis et al., 1995, Xing et al., 1996, Cardone et al., 1998, Coutant et al., 2002, Iijima et al., 2002, Allan et al., 2003, Blalock et al., 2003). p90Rsk1 can activate the cyclic-AMP response element binding protein (CREB) transcription factor (Xing et al., 1996). Moreover, ERK can translocate to the nucleus and phosphorylate additional transcription factors such as Elk1, CREB and Fos which bind promoters of many genes, including growth factor and cytokine genes important in stimulating the growth and survival of multiple cell types including breast cells (Deng et al., 1994, Wang et al., 1994, Davis., 1995, Thomas et al., 1997, Robinson et al., 1998, McCubrey et al., 2000, Aplin et al., 2001, Tresini et al., 2001, Eblen et al., 2001, Adachi et al., 2002, Ponti et al., 2002, Fry et al., 2002). The Raf/MEK/ERK pathway can also modulate the activity of many proteins involved in apoptosis including: Bcl-2, Bad, Bim, myeloid cell leukemia-1 (Mcl-1), caspase 9, and survivin (Deng et al., 2001, Carter et al., 2003, Jia et al., 2003, Troppmair et al., 2003, Ley et al., 2003, Weston et al., 2003, Domina et al., 2004, Harada et al., 2004, Marani et al., 2004, Gelinas et al., 2006, McCubrey et al., 2007).
Growth factor/cytokine receptor ligation also leads to rapid activation of phosphatidylinositol 3-kinase (PI3K) (Martelli et al., 2007). Class IA PI3Ks are heterodimeric proteins which consist of 85-kDa regulatory and 110-kDa catalytic subunits. The p85 regulatory subunit contains: a SH2 domain, that recognizes phosphorylated (p) Y (pY) residues, an iSH2 domain (a rigid tether for p110), a conserved domain related to sequences present in the Break Point Cluster Region (BCR) gene and a SH3 domain which are found in proteins that interact with other proteins and mediate assembly of specific protein complexes via binding to proline-rich motifs (Yu et al., 1996, Fu et al., 2004). The 110-kDa PI3K Cass 1A catalytic subunit contains a p85 binding domain, a Ras binding domain, a kinase domain and a helical domain (Chang et al., 2003, Steelman et al., 2004). Cytokine stimulation often creates a PI3K binding site on the cytokine receptor. The p85 subunit SH2 domain associates with this site (Rao et al., 1995, Drexler et al., 1996, Chang et al., 2003). The p85 subunit is then phosphorylated which leads to activation of the p110 catalytic subunit. This often occurs at the inner leaflet of the cytoplasmic membrane, although there are important roles of PI3K in the nuclear membranes which have been reviewed recently (Martelli et al., 2006, Evangelisti et al., 2007, Cocco et al., 2007).
The class IA regulatory subunits recruit the p110 catalytic subunit to pY residues in receptors, adaptor proteins and other molecules. This localizes the class IA p110 subunits in the membranes where their lipid substrates reside. The adaptor/regulatory subunits act to localize PI3K to the plasma membrane by the interaction of their SH2 domains with pY residues in activated receptors. They also serve to stabilize p110 and to limit its activity.
The preferred substrate for class I PI3K, which is the class of PI3K discussed in this manuscript, in vivo is phosphatidylinositol 4,5 bisphosphate [PtdIns(4,5)P2] which is phosphorylated to yield phosphatidylinositol 3,4,5 trisphosphate [PtdIns(3,4,5)P3]. PtdIns(3,4,5)P3 serves as an anchor for pleckstrin homology (PH) domain-containing proteins, such as Akt or phosphoinositide-dependent protein kinase-1 (PDK-1). PDK1 then phosphorylates an Akt T regulatory residue. Akt is also a member of a multi-gene family (Akt-1, Akt-2 and Akt-3) and is also called protein kinase B (PKB). Depending on the Akt isoform, PDK1 can phosphorylate Akt on T 308/309/305. A second kinase phosphorylates Akt on a S regulatory residue (S 473/474/472) (Songyang et al., 1997, Troussasrd et al., 2003, Xu et al., 2003, Persad et al., 2003). The identity of the second kinase which phosphorylates Akt has remained elusive. Integrin linked kinase (ILK), PDK1, rictor-mTOR complex (see below) and Akt autophosphorylation have all been suggested to be responsible for phosphorylation of Akt at the second S phosphorylation site (Noguchi et al., 2007). The PH domain leucine-rich repeat protein phosphatase (PHLPP) dephosphorylates S473 on Akt which induces apoptosis and inhibits tumor growth (Gao et al., 2005)
Akt has been postulated to phosphorylate over nine thousand proteins (Lawlor et al., 2001, Hay, 2005). Thus Akt is clearly a critical growth regulatory switch. Akt can transduce an anti-apoptotic signal by phosphorylating downstream target proteins involved in the regulation of cell growth [e.g., glycogen synthase kinase-3β (GSK-3β), apoptotic signal kinase (ASK1), Bim, Bad, MDM-2, p21Cip1, X-linked inhibitor of apoptosis (XIAP) and the Foxo3a transcription factor] (Songyang et al., 1997, del Peso et al., 1997, Scheid et al., 1998, Nakae et al., 1999, Brunet et al., 1999, Mayo et al., 2000, Medema et al., 2000, Dijkers et al., 2000, Mayo et al., 2001, Gottlieb et al., 2002, Zhou et al., 2002, Dan et al., 2004, Qi et al., 2006). Phosphorylated Foxo3a loses its ability to induce Fas, p27Kip1, Bim, Noxa, and Puma gene transcription (You et al., 2006, Obexer et al., 2007). Akt also phosphorylates I-κB kinase (I-κK), which subsequently phosphorylates inhibitory subunit (I-κB) which binds nuclear transcription factor kappa light chain in B cells (NF-κB) transcription factor, resulting in its ubiquitination and subsequent degradation in proteosomes (Ozes et al., 1999, Romashkova et al., 1999, Mayo et al., 2000, Madrid et al., 2000, Howe et al., 2002, Howe et al., 2004, Hu et al., 2004, Shishodia et al., 2004). Disassociation of I-κB from NF-κB enables NF-κB to translocate into the nucleus to promote gene expression. The PI3K/Akt pathway can also phosphorylate and activate CREB which regulates transcription of Mcl-1 and B cell leukemia-2 Bcl-2 (Du et al., 1998, Wang et al., 1999, Arcinas et al., 2001). The PI3K/Akt pathway also regulates the mammalian target of rapamycin (mTOR) and ribosomal protein kinases such as p70 ribosomal six kinase (p70S6K) (Mahalingam et al., 1996, Dufner et al., 1999, Romanelli et al., 1999, Harada et al., 2001, Edinger et al., 2004, Panwalker et al., 2004, Jonassen et al., 2004, Ma et al., 2005) (see Below). It is worth noting that Akt can cause the activation of specific substrates (e.g., IκKα and CREB) or may mediate the inactivation of other proteins (e.g., Raf-1, B-Raf [by the Akt related kinase serum glucocorticoid kinase (SGK)], p21Cip-1, Bim, Bad, procaspase-9, Foxo3a, and GSK-3β). An overview of the effects of the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways on apoptotic and cell cycle cascades is presented in Figure 2.
Figure 2. Effects of PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK Pathways on Apoptotic Circuity.

Growth factors can induce multiple signal transduction pathways which can effect the expression of apoptotic molecules by transcriptional and post-transcriptional mechanisms. The effects of the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways are often counterbalanced by JNK and GSK-3β which can serve to promote apoptosis.
mTOR is a 289-kDa S/T kinase. It regulates translation in response to nutrients/growth factors by phosphorylating components of the protein synthesis machinery, including p70S6K and eukaryotic initiation factor (eIF)-4E binding protein (4EBP-1), the latter resulting in release of the translation initiation factor eIF-4E, allowing eIF-4E to participate in the assembly of a translational initiation complex (Wendel et al., 2004). p70S6K, which can also be directly activated by PDK-1, phosphorylates the 40S ribosomal protein, S6, leading to active translation of mRNAs (Giles et al., 2005). Integration of a variety of signals (mitogens, growth factors, hormones) by mTOR assures cell cycle entry only if nutrients and energy are sufficient for cell duplication (Fingar et al., 2004, Tokunaga et al., 2004). Therefore, mTOR controls multiple steps involved in protein synthesis, but importantly enhances production of key molecules such as c-Myc, cyclin D1, p27Kip1, and retinoblastoma protein (pRb) (Martin et al., 2004). mTOR also controls the translation of hypoxia-inducible transcription factor-1α (HIF-1α) mRNA. HIF-1α upregulation leads to increased expression of angiogenic factors such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) (Witzig et al., 2006). Moreover, HIF-1α regulates the glycolytic pathway by controlling the expression of glucose-sensing molecules including glucose transporter (Glut) 1 and Glut3 (Mobasheri et al., 2005).
By regulating protein synthesis, p70S6K and 4E-BP1 also control cell growth and hypertrophy, which are important processes for neoplastic progression. Akt-mediated regulation of mTOR activity is a complex multi-step phenomenon. Akt inhibits tuberous sclerosis 2 (TSC2 or hamartin) function through direct phosphorylation (Manning et al., 2002). TSC2 is a GTPase-activating protein (GAP) that functions in association with the putative tuberous sclerosis 1 (TSC1 or tuberin) to inactivate the small GTPase protein Rheb (Ras homolog enriched in brain) (Zhang et al., 2003). TSC2 phosphorylation by Akt represses GAP activity of the TSC1/TSC2 complex, allowing Rheb to accumulate in a GTP-bound state. Rheb-GTP then activates, through a mechanism not yet elucidated, the protein kinase activity of mTOR when complexed with the Raptor (Regulatory associated protein of mTOR) adaptor protein and mLST8, a member of the Lethal-with-Sec-Thirteen gene family, first identified in yeast (Hay, 2005). The mTOR/Raptor/mLST8 complex (mTORC1) is sensitive to rapamycin and, importantly, inhibits Akt via a negative feedback loop which involves, at least in part, p70S6K (Hay, 2005). The relationship between Akt and mTOR is further complicated by the existence of the mTOR/Rictor (Rapamycin-insensitive companion of mTOR/mLST8 complex (mTORC2), which displays rapamycin-insensitive activity. The mTORC2 complex has been found to directly phosphorylate Akt on S473 in vitro and to facilitate T308 phosphorylation. Thus, the mTORC2 complex might be the elusive PDK-2 which phosphorylates Akt on S473 in response to growth factor stimulation (Hresko et al., 2005). Akt and mTOR are linked to each other via ill-defined positive and negative regulatory circuits, which restrain their simultaneous hyperactivation through a mechanism involving p70S6K and PI3K. Assuming that equilibrium exists between these two complexes within the cell, when the mTORC1 complex is formed, it could antagonize the formation of the mTORC2 complex and reduce Akt activity (Hresko et al., 2005). Thus, at least in principle, inhibition of the mTORC1 complex could result in Akt hyperactivation. This is one complication associated with rapamycin treatment (see below).
The PI3K/PTEN/Akt/mTOR pathway is negatively regulated by phosphatases. PTEN (phosphatase and tensin homologue deleted on chromosome ten) is primarily a lipid phosphatase that removes the 3-phosphate from the PI3K lipid product PtdIns (3,4,5)P3 to produce PtdIns (4,5)P2 which prevents Akt activation (Chang et al., 2003, Li et al., 1997., Steck et al., 1997, Li et al., 1997, Steelman et al., 2004, Shaw et al., 2006). PTEN is also a protein phosphatase (Steelman et al., 2004, Mahimainathan et al., 2004, Raftopoulou et al., 2004). Two other phosphatases, SH2 domain-containing inositol 5′phosphatase (SHIP)-1 and SHIP-2, remove the 5-phosphate from PtdIns(3,4,5)P3 to produce PtdIns(3,4)P2 (Damen et al., 1996, Kavanaugh et al., 1996, Lioubin et al., 1996, Muraille et al., 1999, Taylor et al., 2000). Mutations in these phosphatases, which eliminate their activity, can lead to tumor progression. Consequently, the genes encoding these phosphatases are referred to as anti-oncogenes or tumor suppressor genes.
Signaling pathways and breast cancer
Breast cancer originates from genetic causes. Mutated or amplified genes are either inherited or occur sporadically. Hereditary breast cancer accounts for only about 10% of all breast cancer cases and generally results from lack of a tumor suppressor gene as opposed to gain of an oncogene. Approximately 45% of hereditary breast cancer is attributable to mutations in breast cancer associated gene-1 (BRCA1) and an additional 45% is attributable to mutation in BRCA2 (Centers for Disease Control and Prevention, 2006). Other tumor suppressor genes implicated in hereditary breast cancer include p53 and PTEN (Steelman et al., 2004). The p53 tumor suppressor is a transcription factor involved in cell cycle regulation and DNA damage repair. Germline p53 mutation is present in approximately 50% of patients with Li-Fraumeni syndrome (LFS), which is a multicancer familial syndrome that includes adrenocortical carcinoma, brain tumors, leukemia, and osteosarcomas in addition to early onset breast cancer. Breast cancer attributable to germline p53 mutation in the absence of LFS is rare. Germline PTEN mutation is present in approximately 80% of patients with Cowden syndrome (CS) (Navolanic et al., 2003, Steelman et al., 2004). This disease, which is also known as multiple hamartoma syndrome, is another familial syndrome that includes many different types of cancer conditions including early onset breast cancer. Mutations have been reported to occur at PTEN in breast cancer in varying frequencies (5-21%) (Rhei et al., 1997, Singh et al., 1998, Feilotter et al., 1999, DeGraffenried et al., 2004, Hollestelle et al., 2007). Loss of heterozygosity (LOH) at PTEN is probably more common (30%) (Singh et al., 1998). PTEN promoter methylation leads to low expression. In one study, 26% of primary breast cancers had low PTEN levels (Singh et al., 1998). Low PTEN levels have been correlated with lymph node metastases and poor prognoses (Singh et al., 1998, DeGraffenried et al., 2004, Garcia et al., 2004, Tsutsui et al., 2005). Mutations at certain residues of PTEN, which are associated with CS, affect the ubiquitination of PTEN and prevent nuclear translocation. These mutations leave the phosphatase activity intact (Trotman et al., 2007). Inhibition of PTEN activity leads to centromere breakage and chromosome instability (Shen et al., 2007). PTEN expression may also be silenced in the absence of obvious genetic mutations. Disruption of PTEN activity by various genetic mechanisms could have vast effects on different processes affecting the sensitivity of breast cancers to diverse therapeutic approaches.
Sporadic breast cancer accounts for the remaining 90% of all breast cancer cases. The PI3K p110 catalytic subunit is mutated in approximately 25% of breast cancer specimens and these mutations frequently result in activation of its kinase activity (Bader et al., 2005, Kang et al., 2005, Engleman et al., 2006, Vogt et al., 2006, Tokunaga et al., 2006, Hollestelle et al., 2007) (See below). Somatic mutation of p53 is associated with many cancers and exists in approximately 20% of sporadic breast cancer cases. In contrast, somatic mutation of BCRA1 or BCRA2 is rare in breast cancer patients. Another important cause of sporadic breast cancer is amplification/overexpression of HER2 which occurs in approximately 25 to 30% of breast cancer. Expression and activity of downstream signal transduction cascades, such as the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways, change as a result of these mutations. Akt and ERK are frequently activated in breast cancer specimens (Hollestelle et al., 2007, Greenman et al., 2007). Thus the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways are therapeutic targets in breast cancer.
Association of genes that regulate signal transduction pathways with breast cancer implies an important role of these pathways in this disease. Perhaps the best example of this is association of HER2 gene amplification with breast cancer. While a normal breast cell possesses 20,000 to 50,000 HER2 molecules, amplification of this gene can increase levels of HER2 up to 2,000,000 molecules per cell (Chang et al., 2003, Shelton et al., 2005a). Overexpression of HER2 in breast cancers is linked to comedo forms of ductal carcinoma in situ (DCIS). DCIS is present in approximately 90% of the patients with HER2 amplification. HER2 overexpression will lead to increased expression of both the Raf/MEK/ERK and PI3K/Akt pathways. An overview of some of the mutations which can result in activation of the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK cascades is presented in Figure 3.
Figure 3. Sites of Gene Mutation/Amplification which Can Result in Activation of the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK Cascades.
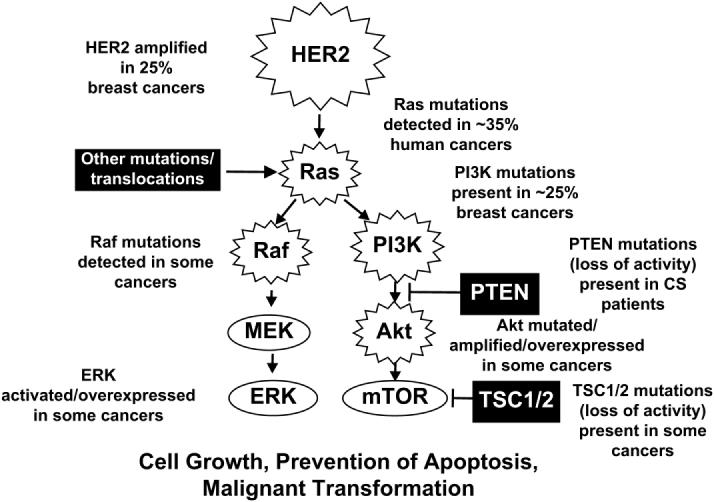
Amplification of HER2 is observed in 25 to 30% of breast cancer patients which can result in activation of both the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK cascades. Ras is mutated in approximately 35% of human cancers. B-Raf and Raf-1 are also mutated in certain human cancers. The PI3K/PTEN/Akt/mTOR pathway is also activated in breast cancer due to mutations at PI3K, PTEN and Akt as well as mutations at upstream receptors. Abnormal Akt activation is associated with a poorer prognosis for breast cancer and resistance to chemo-hormonal therapy.
Effective targeting of signal transduction pathways activated by mutations and gene amplification may be an effective means to limit cancer growth and metastasis (See below). The PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK and PI3K/Akt pathways can be activated by mutations/amplifications of upstream growth factor receptors.
Mutations that lead to the expression of constitutively-active Ras proteins have been observed in approximately 20 to 30% of human cancers (Flotho et al., 1999, Stirewalt et al., 2001). These are often point mutations which alter key residues that affect Ras activity, although amplification of Ras is also detected in some tumors. Mutations that result in increased Ras activity also perturb the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK kinase cascades.
In a recent survey of 40 breast cancer lines, many were mutated at components of either the PI3K/PTEN/Akt/mTOR or Raf/MEK/ERK pathways (Hollestelle et al., 2007). 36% were mutated at PIK3CA (PI3K p110 subunit gene), 21% at PTEN (with PTEN either mutation or no protein present), 13% were mutated at K-RAS, 5% at H-RAS, 3% at N-RAS, and 10% at B-RAF. In other studies it has been shown that the PI3K p110 catalytic subunit (PIK3CA) is mutated in: approximately 25% of breast, 32 % of colorectal, 27% of brain, 25% of gastric, 4% of lung cancers (Samuels et al., 2004; Kang et al., 2005, Vogt et al., 2006). These mutations frequently result in activation of its kinase activity. A recent report indicated that Akt-1 is mutated in: 8% of breast, 6% of colorectal and 2% of ovarian cancers examined in that study (Carpten et al., 2007). This mutation results in a lysine (K) substitution for glutamic acid (E) at amino acid 17 (E17K) in the PH domain. Cells with this Akt1 mutation have not been observed to have mutations at PI3K, a similar scenario is also frequently observed with Ras and B-Raf mutations (Greenman et al., 2007). The Akt mutation alters the electrostatic interactions of Akt which allows it to form new hydrogen bonds with the natural phosphoinositol ligand (Carpten et al., 2007). The PH domain mutation confers many different properties to the Akt-1 gene. Namely, the mutant Akt-1 gene: 1) has an altered PH domain conformation, 2) is constitutively active, 3) has an altered cellular distribution as it is constitutively associated with the cell membrane, 4) morphologically transforms Rat-1 tissue culture cells and 5) interacts with c-Myc to induce leukemia in Eμ-Myc mice (Eμ = enhancer of immunoglobulin M (μ) gene, Myc = Myc oncogene originally isolated in avian myelocytomatosis virus) (Carpten et al., 2007). This PH domain mutated Akt-1 gene does not alter its sensitivity to ATP competitive inhibitors, but does change its sensitivity to allosteric kinase inhibitors (Carpten et al., 2007). These results demonstrate that targeting the kinase domain of Akt may not be sufficient to suppress the activity of various Akt genes which have mutations in the PH domain (Carpten et al., 2007). It will be important to determine if this Akt-1 mutation (E17K) is also present in other solid and non solid tumors as well as its overall mutation frequency in human cancer. In summary, mutations do occur at key components of the PI3K/PTEN/Akt/mTOR and Ras/Raf/MEK/ERK pathways in breast cancer and targeting of these pathways may be important therapeutic approach, either by themselves as monotherapy or combination methods with various chemo-hormonal- and antibody treatments.
Furthermore as stated previously, components of these pathways may be activated either by upstream mutations or by other genetic/epigenetic mechanisms. Activated Akt is often upregulated in breast cancer cells and its overexpression has been associated with a poor prognosis (Clark et al., 2002, DeGraffenried et al., 2004, Tsutui et al., 2005, Kirkegaard et al., 2005, Tokunaga et al., 2006). However, this may actually render the breast cancer cells sensitive to Akt as well as downstream mTOR inhibitors. The formation of the rapamycin sensitive mTORC1 complex in the drug resistant breast cancer cells which overexpress activated Akt may be different than in drug sensitive breast cells which do not overexpress activated Akt. In cells which express activated Akt, Akt should phosphorylate TSC-2 resulting in its inactivation. mTORC1 complex is then formed and downstream p70S6K and 4E-BP1 are phosphorylated, allowing the disassociation of eIF-4E, ribosome biogenesis and protein synthesis. In contrast, in the absence of constitutive Akt and ERK activation, which is the case in the drug sensitive cell, this complex should not be formed. Rapamycin targets this complex; hence the cells which constitutively-express activated Akt cells are more sensitive to rapamycin than the cells which do not. In the cells which do not constitutively-express activated Akt, this complex should be transiently assembled after growth factor treatment. In contrast, the assembly of the rapamycin-insensitive mTORC2 complex (Rictor, mTOR, mLST8) should be lower in the cells that constitutively express activated Akt than in those cells that do not as there is equilibrium between the mTORC1 and mTORC2 complexes. The significance of these complex biochemical signaling events is that breast cancer cells which overexpress activated Akt or lack PTEN expression have an Achilles heel with regards to therapeutic intervention as they are highly sensitive to rapamycin treatment. Breast cancer may also be sensitive to Raf/MEK inhibitors as ERK plays critical roles in the phosphorylation of TSC-2 and p70S6K (see below).
We have observed that ERK is activated after tamoxifen and doxorubicin treatment of the MCF-7 breast cancer cell line. Furthermore, drug resistant derivative cell lines have increased levels of constitutively active ERK. These results are important with regards to the sensitivity of breast cancer cells to MEK inhibitors. ERK can phosphorylate and contribute to the inactivation of TSC-2. Akt can also phosphorylate TSC-2, at a different residue, which leads to its inactivation. This also leads to mTOR activation. Inhibition of TSC-2 phosphorylation by PI3K/Akt and Raf/MEK inhibitors may make cells more sensitive to chemo-, hormonal-, and immunotherapies.
Aberrant regulation of apoptosis may contribute to breast cancer and subsequent drug resistance
Cell death following cytotoxic drug treatment is generally apoptotic as opposed to necrotic. Many chemotherapeutic drugs induce apoptosis by activating the intrinsic cell death pathway which involves cytochrome c release and activation of the apoptosome-catalyzed caspase cascade. During apoptosis, activation of caspase family cysteine proteases occurs. Although various cytotoxic drugs differ in their mechanisms of action, each ultimately relies upon built in apoptotic machinery to elicit cell death (Deng et al., 2001, Harada et al., 2001, Navolanic et al., 2003, Chang et al., 2003, Steelman et al., 2004, Martelli et al., 2007). Caspase family cysteine proteases are responsible for proteolytic cleavage of cellular proteins carboxyl terminal to aspartate residues.
In a study involving 46 breast cancer patients, 75% were determined to lack caspase-3 mRNA transcripts and protein expression (Devarajan et al., 2002). The MCF-7 cell line has a mutation in caspase-3 and is deficient in certain aspects of apoptosis. In this respect, MCF-7 cells are a stringent model to investigate breast cancer apoptosis (Liang et al., 2001). Caspase-9 can be activated in MCF-7 cells which can result in the sequential activation of caspases-7 and -6 (Twiddy et al., 2006). Caspase-7 is activated by the apoptosome complex and forms a XIAP-caspase-7 complex. This XIAP-caspase-7 complex is more stable in MCF-7 cells due to the absence of functional caspase-3. ERK activity maintains XIAP levels; however the mechanism by which this occurs is unknown. Resistance to chemotherapeutic drugs induced by the Raf/MEK/ERK pathway may be due in part to prolonged XIAP and caspase 9 expression which prevents caspase 7 from exerting its effects on apoptosis (Dan et al., 2004, Gardai et al., 2004). ERK phosphorylates caspase-9, which inhibits its activation. Negative regulation of caspases by ERK represents a mechanism by which Raf/MEK/ERK pathway activation prevents apoptosis. Raf/MEK inhibitors may affect caspase 9 activation and XIAP levels and promote apoptosis of breast cancer cells (see below).
Therapeutic targeting of the PI3K/Akt/PTEN/mTOR and Raf/MEK/ERK pathways
Small molecule inhibitors such as Imatinib have proven effective in the treatment of chronic myeloid leukemia (CML) and certain other cancers which proliferate in response to translocated BCR-ABL, mutant platelet derived growth factor receptor (PDGF-R) and c-Kit genes (Lee et al., 2002, Hochhaus et al., 2002, Navolanic et al., 2003, Steelman et al., 2004, Barnes et al., 2005, Hollestelle et al., 2007) such as gastro-intestinal stromal tumors (GIST). Non small cell lung carcinomas (NSCLC) which have mutations in EGFR are sensitive to EGFR inhibitors (Lynch et al., 2004, Sordella et al., 2004, Pao et al., 2004, Shelton et al., 2005, Pao et al., 2005, Sequist et al., 2005, McCubrey et al., 2007). Currently, these cancers are the poster children of targeted therapies as their growth can be successfully inhibited and dramatic effects observed in cancer patients. Certain antibodies have been developed such as Herceptin which effectively target HER2 those breast cancer patients (25 to 30%) which overexpress HER2 due to genetic amplification and other mechanisms (Shelton et al., 2005a).
PI3K, PDK, Akt, and mTOR inhibitors have been and are being developed. mTOR inhibitors have been used for many years as immunosuppressive drugs in kidney transplant patients. A side effect of mTOR inhibitors is the inhibition of a negative feed back pathway which can result in Akt activation (Weber et al., 2004). A diagram illustrating the sites of action and other inhibitors is presented in Figure 4.
Figure 4. Targeting Signal Transduction Pathways in Breast Cancer.

Potential sites of action of small molecular weight inhibitors and cytotoxic antibodies are indicated. In some cases inhibitors will suppress growth, apoptotic and cell cycle regulatory pathways. This diagram serves to illustrate the concept that targeting PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways can have dramatic effects on many growth regulatory molecules. Proteins inactivated by S/T phosphorylation induced by the PI3K/PTEN/Akt/mTOR and Raf/MEK/ERK pathways are shown in black circles with white P in a black circle.
PI3K can be inhibited with many inhibitors including: LY294002, Wortmannin, KN309, SF1124 and PX-866. Novel PI3K inhibitors have been developed. PWT-458 is a pegylated-17-hydroxywortmannin which inhibits PI3K and has been shown to suppress glioma, NSCLC and renal cell carcinoma in xenograft models (Yu et al., 2005). PWT-458 augmented the anticancer effects of paclitaxel and pegylated rapamycin in certain xenograft models.
PDK1 can be inhibited with OSU-03012 and Celecoxib. Akt can be inhibited with many inhibitors including; A-443654, API-2, PX316, Curcumin, Rotenone (Deguelin), and Perifosine. Perifosine is an oral bioactive novel alkylphospholipid that inhibits Akt. mTOR can be inhibited by rapamycin and modified rapamycins (CCI-779, RAD001 and AP23573). Initially mTOR inhibitors showed much promise as PTEN is often deleted, mutated or silenced in various tumors by many different mechanisms. However, it has been recently determined that the mTOR pathway has a complicated feed-back loop which actually involves suppression of Akt, hence mTOR inhibitors would be predicted to activate Akt in some cells. Due to the complicated Akt/mTOR pathway, with many inhibitory phosphorylation events, an alternative therapeutic approach will be to develop strategies which simultaneously inhibit both Akt and mTOR activity. As pointed out previously, recently mutations in the PH domain of Akt have been detected in certain human cancers. Cells harboring these mutations may be refractory to Akt inhibitors which target the kinase domain but may be more sensitive to inhibitors which target the PH domain (Carpten et al., 2007). The role of overexpression of the different Akt isoforms in cancer prognosis is controversial (Kornblau et al., 2006, Tamburini et al., 2007). Some studies have suggested that elevated Akt expression is associated with a poor prognosis in breast (Kirkegaard et al., 2005) and hematopoietic (Kornblau et al., 2006) cancers. Controversially it has been observed recently that constitutive PI3K/Akt activation is a favorable prognostic factor in AML patients (Tamburini et al., 2007) as Akt activation may keep a high percentage of the cells in S phase which renders them sensitive to chemotherapeutic drugs. These and other studies document the complexity of enhanced Akt expression which may make certain cells more sensitive or resistant to certain anti-cancer therapies.
Recent evidence has highlighted that mTOR can also be activated by Raf/MEK/ERK (Mukherjee et al., 2005, Shimada et al., 2006). This may well be another relevant cross-talk between the PI3K/PTEN/Akt/mTOR and Ras/Raf/MEK/ERK pathways and offer a further rationale for treatments combining drugs which inhibit both signaling networks.
Raf and MEK inhibitors have been developed and some are in clinical trials (Hochhaus et al., 2002, Navolanic et al., 2003, Steelman et al., 2004, Hollestelle et al., 2007, Tortora et al., 2007). We have determined that a consequence of doxorubicin and hormonal treatment of some breast, hematopoietic and prostate cancer cell lines is the induction of ERK (McCubrey et al., 2007). Eliminating this deleterious side-effect of these compounds with Raf and MEK inhibitors may enhance the ability of chemo- and hormonal based therapies to kill drug resistant cancer.
Certain Raf inhibitors have been developed which are small molecule competitive inhibitors of the ATP-binding site of Raf protein. These inhibitors (e.g., L-779,450, ZM 336372, Bay 43-9006, a.k.a Sorafenib) bind the Raf kinase domain and therefore prevent its activity. Some Raf inhibitors may affect a single Raf isoform (e.g., Raf-1), others may affect Raf proteins, which are more similar (Raf-1 and A-Raf), while other pan Raf inhibitors may affect all three Raf proteins (Raf-1, A-Raf and B-Raf). We have observed that the L-779,450 inhibitor suppresses the effects of A-Raf and Raf-1 more than the effects of B-Raf (Shelton et al., 2003). Like many Raf inhibitors, L-779,450 is not specific for Raf; it also inhibits the closely related p38MAPK. Likewise, Sorafenib inhibits other kinases besides Raf (e.g., VEGF-II Receptor, PDGF-R, Kit, Flt-3, and Fms) and is more appropriately referred to as a multi-kinase inhibitor. Knowledge of the particular Raf gene mutated or overexpressed in certain tumors may provide critical information regarding how to treat the patient as some cancers which overexpress a particular Raf gene may be more sensitive to inhibition by agents which target that particular Raf protein. Inhibition of certain Raf proteins might prove beneficial while suppression of other Raf proteins under certain circumstances might prove detrimental. Thus the development of unique and broad-spectrum Raf inhibitors may prove useful in cancer therapy. Some “Raf” inhibitors (Sorafenib) are approved for the treatment of certain cancers (e.g., renal cell carcinoma) (Heimbrook et al., 1998, Hall-Jackson et al., 1999, Lyons et al., 2001, Blagosklonny 2002, Shelton et al., 2003, Tortora et al., 2007).
Currently it is believed that MEK1 is not frequently mutated in human cancer. Recently, there was a report that MEK1 and MEK2, as well as B-Raf, are mutated in some patients with cardio-facio cutaneous syndrome (Rodriguez-Viciana et al., 2006). Aberrant expression of MEK1 is observed in many different cancers due to the activation of the Raf/MEK/ERK pathway by upstream kinases (e.g., BCR-ABL) and growth factor receptors (e.g., EGFR, Fms, Flt-3, PDGFR) as well as other unknown mechanisms. Specific inhibitors to MEK have been developed (PD98059, U0126, PD184352 (a.k.a., CI1040), PD-0325901, Array MEK inhibitors [ARRY-142886 and others]) (Tortora et al., 2007). The successful development of MEK inhibitors may be due to the relatively few phosphorylation sites on MEK involved in activation/inactivation. MEK inhibitors differ from most other kinase inhibitors as they do not compete with ATP binding which confers a very high specificity (Delaney et al., 2002). MEK inhibitors are very specific and do not inhibit many different protein kinases including p38MAPK and JNK (Davies et al., 2000). The crystal structures of MEK1 and MEK2 have been determined as ternary complexes with ATP and PD184352 and have revealed that both MEK1 and MEK2 have unique inhibitor binding sites located on a hydrophobic pocket adjacent to but not overlapping with the ATP binding site (Ohren et al., 2004). Furthermore, effective targeting of MEK1,2 is highly specific as ERK1,2 are the only well-described downstream targets. An advantage of targeting the Raf/MEK/ERK cascade is that it can be targeted without knowledge of the precise genetic mutation, which results in its aberrant activation. This is important as the nature of the critical mutation(s), which leads to the drug resistant growth of at many breast cancers, is not currently known. An advantage of targeting MEK is that the Raf/MEK/ERK pathway is a convergence point where a number of upstream signaling pathways can be blocked with the inhibition of a single kinase (MEK). MEK inhibitors such as ARRY-142886 (AZD6244) are also being evaluated to treat hematopoietic malignancies such as multiple myeloma (Tai et al., 2006, Wang et al., 2006, Yeh et al., 2007).
To our knowledge, no small molecular weight ERK inhibitors have been developed yet, however, inhibitors to ERK could prove very useful as ERK can phosphorylate many targets (Rsk, c-Myc, Elk, and at least 150 more). There are at least 2 ERK molecules regulated by the Raf/MEK/ERK cascade, ERK1 and ERK2. Little is known about the different in vivo targets of ERK1 and ERK2. However ERK2 has been postulated to have pro-proliferative effects while ERK1 may have anti-proliferative effects (Mazzucchelli et al., 2002). Development of specific inhibitors to ERK1 and ERK2 might eventually prove useful in the treatment of certain diseases.
Combination therapies to enhance toxicity and effectiveness of breast cancer therapy
Classical chemotherapy often remains the most used anti-cancer therapy for many different types of cancers including breast cancer. Drugs such as doxorubicin and taxol are effective in the treatment of many cancers, even though in some cases drug resistance does develop after prolonged treatment. Doxorubicin and taxol target cellular events such as DNA replication and cell division which are downstream of the targets of signal transduction pathway inhibitors. Thus by combining classical chemotherapy with targeted therapy, it may be possible to enhance toxicity while lowering the effective concentrations of classical chemotherapeutics necessary for effective elimination of the particular tumor.
Recent studies have indicated that the effectiveness of certain antibody based therapies (e.g., Herceptin, which targets HER2) may be greatly enhanced by inclusion of mTOR inhibitors. These observations were obtained in both preclinical studies performed in tissue culture and xenograft models and are being further evaluated in phase 2 clinical trials (Wang et al., 2007). The cytotoxic effects of Herceptin can also be improved by addition of an inhibitor such as Lapatinib which targets both EGFR and HER2 (Konecny et al., 2006). Many ongoing clinical trials are examining the effectiveness of targeting the PI3K/Akt/mTOR and other pathways. These and the other studies we have described document the promise of targeting the PI3K/PTEN/Akt/mTOR pathway in human health care. Furthermore, combining classical chemo and hormonal therapy with PI3K, Akt and mTOR inhibitors is also a very attractive therapeutic option under intense investigation.
We have investigated the effects of combining classical chemo- or hormonal therapies with signal transduction inhibitors in suppressing the growth of breast cancer cells. Treatment of breast cancer cells with MEK or mTOR inhibitors with doxorubicin, paclitaxel or tamoxifen resulted in a synergistic response documenting the effectiveness of classical chemo- hormonal with targeted therapies (see below).
Materials & Methods
Cell Culture
MCF-7 cells were derived from a human breast adenocarcinoma (Soule et al., 1973). MCF-7 cells have an epithelial morphology, are adherent, and form a monolayer in culture. These cells express both estrogen receptor (ER)α and ERβ (Brooks et al., 1973, Enmark et al, 1997, Fuqua et al., 1999, Leygue et al., 1999, Fasco et al., 2000, Poola et al., 2000, Yoshida et al., 2000, Lespagnard et al., 2001, Poola et al., 2002) and (WT) PTEN (Li et al., 1998, Lu et al., 1999, Weng et al., 1999, Clark et al., 2002). MCF-7 cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA).
Cell culture medium for MCF-7 cells consisted of 10% (v/v) heat inactivated fetal bovine serum (FBS) (CellGrow-Mediatech, Herndon, VA), 2 mM L-glutamine (Invitrogen, Carlsbad, CA), 100 µg/ml streptomycin (Invitrogen), and 100 units/L penicillin G (Invitrogen) in Roswell Park Memorial Institute-1640 (RPMI 1640) medium (Invitrogen).
PTEN(WT), PTEN(C124S), and PTEN(G129E)Plasmids
A plasmid encoding human WT PTEN [PTEN(WT)] was generated by inserting the corresponding cDNA into the multiple cloning site of pEGFP-C2 (Leslie et al., 2000). Mutated PTEN cDNAs were inserted into the multiple cloning site of pEGFP-C2 to generate plasmids encoding various PTEN mutants (Leslie et al., 2000). These PTEN mutants include: PTEN(C124S) and PTEN(G129E). PTEN(C124S) and PTEN(G129E) each differ from PTEN(WT) by substitution of a single amino acid. PTEN(C124S) lacks both lipid and protein phosphatase activities (Myers et al., 1997, Leslie et al., 2000). PTEN(G129E) lacks lipid phosphatase activity yet retains protein phosphatase activity (Leslie et al., 2000). These PTEN mutants were originally described in CS breast cancer patients and postulated to function as dominant negative (DN) mutations (Liaw et al., 1997, Weng et al., 2001).
Transfection of MCF-7 cells with PTEN Constructs
5 × 105 MCF-7 cells were plated into 6-well cell culture plates (BD Biosciences, Mountainview, CA) one day prior to transfection. Cells were rinsed with Opti-MEM medium (Invitrogen) to remove FBS. Cells were transfected with 10 µg of DNA with Lipofectin (Invitrogen) as described by the manufacturer. 48 hrs after transfection, selection medium (RPMI + 10% FBS + 2 mg/ml G418) (Geneticin, Invitrogen, Carlsbad, CA) was added to isolate stably transfected cells. Cells were provided with fresh selection medium every three days. Mock transfections were performed by replacing DNA with 10 µl of Tris-EDTA buffer. Mock transfected MCF-7 cells did not generate viable colonies in the presence of selection medium. The number of MCF-7 colonies present after incubation in selection medium for 3 weeks was determined. The mean number of MCF-7 colonies present and corresponding standard deviation was calculated from 3 replicate wells for each plasmid. The transfections were repeated 4 times and averaged together.
Analysis of Sensitivity to Doxorubicin and Rapamycin
Cells were seeded in 96-well cell culture plates (BD Biosciences) at a density of 5,000 cells/well in 100 µl/well of phenol red free RPMI-1640 containing 5% charcoal stripped FBS (CS-FBS). Cell culture plates were incubated for 1 day to permit cells to adhere to the bottom of each well. A 100 µl dose of treatment medium (doxorubicin or rapamycin) was added to each well the day after cells were initially seeded. Cell culture plates were incubated at 37 °C until extent of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, Sigma, St. Louis, MO) reduction in each well was quantified. Absorbance of each well was measured at 530 nm with a FL600 microplate fluorescence reader (Bio-Tek Instruments, Winooski, VT).
The mean and corresponding standard deviation of normalized adjusted absorbance was calculated from 8 replicate wells for each drug concentration and duration of incubation in order to investigate effects of the drug and ectopic gene expression on cell proliferation rate. Inhibitory concentration 50% (IC50) is defined in this context as the concentration of the drug that causes MCF-7 cells to proliferate at a rate that is half as rapid as cells incubated in the absence of the drug.
Apoptosis Analysis
The extent of apoptosis in the PTEN transfected cells after exposure to different concentrations of doxorubicin was estimated by determining the percentage of sub-G1 cells by flow cytometric analysis after PI staining of the cells. Results were analyzed by the ModFit LT 3.1 software program (Verity Software, Topsham, ME).
Clonogenic Assays
MCF-7 cells were collected and seeded in 6-well cell culture plates at densities of 1000 and 2000 cells/well (3 wells at each density). Cell culture plates were incubated for 1 day at 37 °C in a cell culture incubator to permit cells to adhere to the bottom of each well. Cell culture plates were incubated for 3-4 weeks at 37 °C in a cell culture incubator. Cells were provided with fresh treatment containing 100 nM doxorubicin, 100 nM 4-hydroxy tamoxifen (4HT, Sigma), the combination of doxorubicin and 4HT or no supplement every 3 days. Cells were rinsed with PBS at the end of the 3 week treatment period. Rinsed cells were fixed in 100% (v/v) methanol (Sigma) for 10 minutes at room temperature then dried for 10 minutes at room temperature. Fixed cells were incubated in Giemsa stain (Sigma) for 5 minutes at room temperature. Stained cells were rinsed with water then dried. The number of colonies present in each well was counted. Colonies consisted of at least 50 cells. The mean number of colonies and corresponding standard deviation was calculated from 3 replicate wells for each condition.
Cellular Senescence Assay
When cells undergo senescence, they express senescence associated (SA) β-galactosidase (SA-β-gal). Senescent cells can be stained by a SA-β-gal senescence assay. 25,000 cells were seeded in each well of a 6 well plate containing an etched cover slip at different concentrations of doxorubicin and incubated for 4 days. SA-β-gal hydrolyzes X-gal (5-bromo-4-chloro-3-indolyl b-D-galactopyranoside) into a colorless galactose and 4-chloro-3-brom-indigo which forms an intense blue precipitate. SA-β-gal activity was detected at pH 6.0 as lysosomal β-gal has a pH optimum of 4.0. Senescent cells contained a dark blue stain in the perinuclear area, and displayed altered morphology. Fixed cells were photographed with a Nikon Eclipse E600 microscope under bright field settings.
Western blot analysis
MCF/EGFPc2 and MCF/PTEN(G129E) cells were collected after trypsination and plated in 100 mm tissue culture plates and cultured in RPMI + 10% FBS. After allowing the cells to adhere for 24 hrs, the monolayers were washed twice with PBS and then cultured with phenol red free RPMI 1640 containing 0.5% CS-FBS (to avoid endogenous estrogenic like compounds and reduce serum growth factors) for 24 hrs. Cells were then stimulated with 10% FBS for the indicated time intervals and protein lysates prepared as previously described (Shelton et al., 2005b). Western blots were performed with antibodies specific for phospho and total Akt, Ask-1, p70S6K, ERK and Hsp70, as previous described (Shelton et al., 2005b). Antibodies used in this study were purchased from Cell Signaling (Beverly, MA).
Results and discussion
Increased PTEN expression suppresses colony formation of MCF-7 cells
MCF-7 cells were transfected with PTEN plasmids encoding WT, C124S, G129E or the empty vector EGFP-C2 (Leslie et al., 2000) to determine how signals transduced by this pathway affect breast cancer cell proliferation. The number of colonies present after transfection with plasmids encoding the PTEN mutants C124S or G129E or the empty vector EGFP-C2 were not statistically different. However, a decrease in the number of colonies was observed after transfection with a WT PTEN plasmid in comparison to transfection with the other plasmids. Thus increased expression of WT PTEN decreased the number of colonies isolated directly after selection in medium containing G418.
Effects of PTEN mutants on growth and induction of apoptosis in the presence of doxorubicin
The proliferation of pools of MCF-7 cells stably transfected with WT, C124S, G129E PTEN genes was compared with empty vector EGFP-C2 transfected cells, in the presence of different concentrations of doxorubicin over a 4 day time period in 96 well plates at 5000 cells/well. The growth of PTEN WT and EGFP-C2 transfected cells were similar in medium containing 0, 125 and 500 nM doxorubicin and no growth was observed at 2000 nM doxorubicin. Thus in stably transduced pools of PTEN (WT) and empty vector transfected cells, which had been isolated and expanded in culture for over one month, there was no difference in growth rates while there were differences in the initial colony formation efficiencies. In contrast, the growth PTEN mutant C124S and G129E cells differed from EGFP-C2 cells both in the absence and presence of doxorubicin. In the absence of doxorubicin, the mutant PTEN transfected cells reached lower saturation densities and proliferated at slower rates, while at higher doxorubicin concentrations, the mutant PTEN transfected cells proliferated to higher levels than either the EGFP-C2 or WT PTEN transfected cells. Thus, altering the levels of the PTEN mutant phosphatases changed the growth properties of MCF-7 cells both in the presence and absence of doxorubicin, but did not affect the colony formation efficiencies in the absence of doxorubicin.
The effects of the mutant and WT PTEN genes on the doxorubicin IC50s were determined. While the IC50S for the PTEN WT and empty vector transfected cells in the presence of doxorubicin were similar, the mutant PTEN transfected cells had higher IC50s for doxorubicin. The IC50s for doxorubicin in the C124S and G129E mutant PTEN transfected cells were 8.6 and 6.2-fold higher than empty vector or PTEN WT transfected cells. Statistical analysis revealed that the IC50 in the lipid and protein phosphatase deficient PTEN mutant transfected cells (C124S) was higher than in the PTEN mutant (G129E) transfected cells which just lacked PTEN phosphatase activity.
The effects of the PTEN mutants on the suppression of apoptosis were determined. MCF-7 cells transfected with WT PTEN, the various PTEN mutants or the empty EGFP-c2 vector were treated with different concentrations of doxorubicin and the extent of apoptosis measured by determining the percentage of sub-G1 cells after PI staining followed by flow cytometric analysis. Less apoptotic cells were observed in MCF-7 cells transfected with the PTEN mutants than in the PTEN WT or empty vector transfected cells. Furthermore less apoptotic cells were recovered from the MCF-7 cells transfected with the PTEN mutant lacking the lipid and protein phosphatase activity than the mutant lacking just lipid phosphatase activity. Thus altering the PTEN phosphatase activity decreased the induction of apoptosis by doxorubicin.
Mutant PTEN genes alter colony formation in medium containing doxorubicin or tamoxifen
The effects of inheritance of WT and mutant PTEN genes on colony formation in the presence and absence of chemotherapeutic (doxorubicin) and hormonal (tamoxifen [4HT]) based drugs were examined. The PTEN mutants had a higher cloning efficiency in medium containing 4HT than the PTEN (WT) or EGFP-C2 transfected cells. The lipid and protein phosphatase deficient mutant had a higher cloning efficiency in the presence of doxorubicin than the empty vector transfected cells. Thus the mutant PTEN gene altered the colony forming ability in the presence of drugs used to treat cancer patients.
Induction of cellular senescence in breast cancer cells after doxorubicin treatment
When certain cells are treated with chemotherapeutic drugs, they undergo cellular senescence. Drug resistance is often associated with an inhibition of cellular senescence (Ellmore et al., 2005, Lehmann et al., 2007). When cells undergo senescence, they express SA-β-gal. Senescent cells can be visualized by a SA-β-gal senescence assay.
In the absence of doxorubicin, very little senescence was observed after a four day incubation period in the different PTEN transfected cells. In contrast in the MCF-7 cells transfected with the PTEN mutations resulting in loss of lipid or lipid and protein phosphatase activity, increased resistance to chemotherapeutic drug induced cellular senescence was observed. The C124S mutation deactivates both the lipid and protein phosphatase activity of PTEN, and conferred the strongest resistance to drug induced senescence. The G129E mutation disengaged only the lipid phosphatase activity, and also bestowed drug resistance, but still yielded more senescent cells upon doxorubicin treatment than the C124S mutation. Senescent cells flattened out and assumed a much wider shape in a 2-D plane. Photographs documenting the difference in the induction of cellular senescence in the empty vector and PTEN mutant C124S transfected cells after doxorubicin treatment are presented in Figure 5. In summary, both the protein phosphatase, as well as the lipid phosphatase activities of PTEN were important with regards to chemosensitivity in terms of both colony formation and inhibition of cellular senescence.
Figure 5. Induction of Cellular Senescence by Doxorubicin in the Presence and Absence of DN PTEN.

MCF-7 cells transfected with the empty vector (EGFP-c2) as well as various PTEN constructs were seeded into 6 well plates containing cover slips and different concentrations of doxorubicin and cultured for 4 days. Induction of cellular senescence was determined by a β-galactosidase assay with X-gal as a substrate. In this figure, photographs of MCF/EGFP-c2 and MCF/PTEN(C124S) cells cultured in the presence and absence of 100 nM doxorubicin and subsequently stained for β-galactosidase activity are presented. Fixed cells were photographed under a Nikon Eclipse E600 microscope under bright field settings.
Effects of mutant PTEN expression on downstream signal transduction pathways important in breast cancer
The effects of inheritance of phosphatase deficient PTEN genes on signal transduction pathways were determined. Akt activation was monitored as decreased PTEN expression could result in Akt activation. Apoptotic signal kinase-1 (Ask-1) phosphorylation was also examined as it lies downstream of Akt. Phosphorylation of Ask-1 results in its ubiquitination and subsequent proteosomal degradation. p70S6K phosphorylation was monitored as it lies downstream of mTOR, is an important regulator of protein synthesis and its activation is a reflection of functional activation of this pathway. Activation of ERK was also examined as it is an important kinase implicated in proliferation and prevention of apoptosis and its activity can be regulated by Akt.
Upon serum stimulation of EGFP-C2 transfected cells, activated Akt was detected after one hr, however, its levels then decreased at later time points up to 24 hrs. In contrast, the levels of total Akt protein remained constant. Thus serum treatment, led to a transient increase in phosphorylated Akt. In contrast, activated Akt was detected after one hr of stimulation of mutant PTEN transfected cells and remained elevated throughout the time course. Thus the mutant PTEN transfected cells expressed activated Akt for prolonged time periods after serum treatment.
Higher levels of phosphorylated Ask-1 were detected in the mutant PTEN transfected than in empty vector transfected cells. Phosphorylated and ubiquitinated Ask-1 may contribute to the apoptotic resistance of the mutant PTEN transfected cells.
The levels of phosphorylated p70S6K increased in empty vector transfected cells 8 and 24 hrs after serum addition. Phosphorylated p70S6K was detected with phosphospecific antibodies which detect phosphorylation of p70S6K at either T389 or T421 and S424. In contrast, relatively equal levels of total p70S6K were detected. The levels of phosphorylated p70S6K were low after serum deprivation of mutant PTEN transfected cells but increased rapidly after serum addition (0.5 hr) and remained elevated over the time course. Thus the mutant PTEN transfected cells expressed activated p70S6K earlier than the empty vector transfected cells.
Phosphorylated ERK was detected 4 hrs after FBS stimulation of empty vector transfected cells and remained elevated for 24 hrs. In contrast, activated ERK was detected constitutively in the mutant PTEN transfected cells. Thus disruption of PTEN activity results in alterations in Akt, Ask-1, p70S6K and ERK activation. These results document the dramatic effects that altered PTEN expression can have on downstream anti-apoptotic and growth related signaling
Mutant PTEN genes confer an Achilles' heel to drug resistant breast cancer cells
It has been reported that altering Akt or PTEN levels will change the sensitivity of breast cancer cells to rapamycin (DeGraffenried et al., 2004). However, the relationships between PTEN, rapamycin and chemotherapeutic drug resistance in breast cancer are not clearly elucidated. Thus we examined the effects of the mutant PTEN genes on the sensitivity of the breast cancer cells to the combination of doxorubicin and rapamycin treatment. The cells transfected with the mutant PTEN genes were more sensitive to rapamycin than the cells transfected with WT or the empty EGFPc2 vector. The effects of rapamycin were seen over a broad dose range (1 to 1000 nM). Rapamycin also synergistically suppressed (decreased) the IC50 for doxorubicin in the cells transfected with the PTEN mutants.
In this manuscript, the effects of introduction of WT and mutant PTEN genes on the clonogenicity and sensitivity of MCF-7 cells to doxorubicin, 4HT and rapamycin have been examined. Introduction of additional copies of WT PTEN into PTEN-positive MCF-7 cells, inhibited initial colony formation in the absence of doxorubicin or rapamycin when compared to MCF-7 cells transfected with mutant PTEN or the control EGFPc2 plasmid. While the WT PTEN gene reduced the colony forming ability in the absence of doxorubicin or rapamycin, it did not alter the growth rate or sensitivity to either doxorubicin or rapamycin in the pools of stably transfected PTEN WT cells which were isolated and expanded in the presence of G418. The two different assays differed in the time frames of the experiments; the initial colony formation assays were performed over a 3 to 4 week period while the growth assays were performed over a 5 day time period on the stably transfected cells. Furthermore, once the cells are selected to grow, they may be able to survive in the presence of higher levels of PTEN activity. Alternatively increased PTEN levels may be suppressed in the cells which survive and proliferate. These two possibilities are a common occurrence in gene transfer experiments and are under further investigation.
Introduction of PTEN mutants lacking lipid and protein or lipid phosphatase activity enhanced the growth of the breast cancer cells in the presence of chemotherapeutic drugs. Interfering with the phosphatase activity of PTEN also resulted in higher levels of activated Akt detected in the serum and hormone deprived samples and prolonged detection of activated Akt after FBS stimulation in the breast cancer cells transfected with PTEN mutant than in the empty vector transfected cells. Akt has been reported to slow the growth rate of certain cells as well as increase the cell size (Jin et al., 2005). The phosphatase deficient PTEN transfected cells, which displayed elevated Akt signaling proliferated slower than the empty vector transfected cells. Phosphatase deficient PTEN mutants also decreased the sensitivity of MCF-7 cells to doxorubicin and increased their sensitivity to rapamycin when compared with the empty vector transfected cells.
Inheritance of a PTEN mutant lacking both lipid and protein phosphatase activity conferred more resistance to doxorubicin than the lipid phosphatase deficient PTEN mutant, indicating that interfering with both lipid and protein phosphatase activities of PTEN were important in terms of therapeutic resistance. Also rapamycin exerted a greater degree of inhibition of the doxorubicin IC50 in cells transfected with the lipid and protein phosphatase deficient PTEN gene than in the cells transfected with the lipid phosphatase PTEN mutant.
PTEN activity is critically involved in regulation of downstream Akt activity which is important in drug sensitivity. Altering normal PTEN activity by some PTEN mutations may lead to drug resistance. Augmented Akt activity may increase resistance to doxorubicin. In fact, published reports (Clark et al., 2002, Tokunaga et al., 2006) and our own preliminary data indicate that ectopic Akt expression increases the resistance of MCF-7 breast cancer cells to doxorubicin and tamoxifen.
These results are relevant to potential cancer therapies as the PTEN gene is frequently mutated in human cancer. Mutations occur which either delete the PTEN gene or alter its activity. Sometimes these mutations actually make the cells sensitive to Akt and mTOR inhibitors as the growth of the cells becomes dependent upon elevated Akt levels (DeGraffenried et al., 2006). Furthermore, epigenetic mechanism may silence PTEN transcription and transcriptional mechanisms may alter the splicing of PTEN mRNA transcripts. Restoration of WT PTEN activity or inhibition of Akt activity are key strategies to prevent chemoresistance. PI3K, PDK, Akt and downstream mTOR inhibitors have been developed and evaluated in clinical trials. Unfortunately, PI3K (LY294002) and Akt (A-443654) inhibitors tend to have associated toxicity and specificity problems (Luo et al., 2005., Martelli et al., 2007). This may not be that surprising as this pathway regulates numerous pathways involved in cell cycle progression, growth and apoptosis as well as glucose homeostasis.
Some point mutations eliminating PTEN phosphatase activity such as the C124S and G129E, without gene deletion, may result in PTEN genes which serve as DN mutants suppressing the normal PTEN activity encoded by the remaining endogenous WT allele. These PTEN mutants may serve to stimulate chemo- and hormonal-resistance of certain tumors. Identification of such mutations may become important in breast cancer therapy as it has been shown that the loss of PTEN activity is associated with Herceptin resistance (Nagata et al., 2007). Only one allele of PTEN may be mutated in many human cancers. Interesting both the C124S and G129E PTEN mutations were originally observed in CS patients and postulated to act as DN mutations in human cancer and other diseases (Lian et al., 1997).
While inhibition of the lipid phosphatase activity of PTEN was sufficient to induce resistance of the cells to doxorubicin, inhibition of the protein phosphatase activity of PTEN further increased chemoresistance indicating that the protein targets of PTEN are also important in its effects as a tumor suppressor. Further elucidation of these targets of PTEN may aid in the identification of suitable targets for therapeutic intervention. Fak and Shc are known targets of the protein phosphatase activity of PTEN (Steelman et al., Li et al., 1997, Li et al., 1997, Nimwegen et al., 2006, Tamura et al., 1998). Elucidating the protein substrates of the PTEN phosphatase could reveal important targets for therapeutic intervention.
The PTEN phosphatase deficient mutant transfected cells were also more resistant to the induction of cellular senescence induced by doxorubicin. While differences in the induction of cellular senescence in the PTEN WT and mutant transfected cells were not observed in the absence of doxorubicin, the PTEN mutant cells, which expressed higher levels of activated Akt than the PTEN WT cells were more resistant to the induction of cellular sensescence mediated by doxorubicin. Thus altering the PTEN genetic status in human cancer may inhibit the ability of chemotherapeutic drugs to induce cellular senescence and allow the outgrowth of resistant clones which result in metastasis and death of the cancer patient.
Summary
Breast cancer is a common disease which affects women and men in the prime of their lives. Unfortunately there is no common magic bullet which can effectively cure all breast cancers. Breast cancer arises from many diverse genetic mechanisms. Frequently breast cancer becomes resistant to various therapies and residual, dormant, senescent, drug resistant breast cancer cells resume their growth and often metastize to other sites. At this point, therapeutic options are limited and we desperately need more effective treatments. This chapter has focused on the key roles that the PI3K/PTEN/Akt/mTOR pathway plays in breast cancer drug resistance and induction of cellular sensecence. Furthermore, breast cancer cells which proliferate in response to this pathway may have an Achilles heel and are highly sensitive to mTOR inhibitors.
Acknowledgements
JAM, WHC, RAF and LSS were supported in part by a grant from the NIH (R01098195). JB was supported in part from the Deutsche Krebshilfe (106125). AMM, MM and AT were supported in part from grants from Associazione Italiana Ricerca sul Cancro (AIRC Regional grants). AMM was also supported in part by a grant from the CARISBO Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi T, Kar S, Wang M, Carr BI. Transient and sustained ERK phosphorylation and nuclear translocation in growth control. J Cell Physiol. 2002;192:151–159. doi: 10.1002/jcp.10124. [DOI] [PubMed] [Google Scholar]
- Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 by phosphorylation at Thr125 by ERK MAP kinase. Nature Cell Biol. 2003;5:647–654. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- Aplin AE, Stewart SA, Assoian RK, Juliano RL. Integrin-mediated adhesion regulates ERK nuclear translocation and phosphorylation of Elk-1. J Cell Biol. 2001;153:273–282. doi: 10.1083/jcb.153.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcinas M, Heckman CA, Mehew JW, Boxer LM. Molecular mechanisms of transcriptional control of bcl-2 and c-myc in follicular and transformed lymphoma. Cancer Res. 2001;61:5202–5206. [PubMed] [Google Scholar]
- Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–929. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- Barnes G, Bulusu VR, Hardwick RH, Carroll N, Hatcher H, Earl HM, Save VE, Balan K, Jamieson NV. A review of the surgical management of metastatic gastrointestinal stromal tumours (GISTs) on imatinib mesylate (Glivec trade mark) Int J Surg. 2005;3:206–212. doi: 10.1016/j.ijsu.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV. Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002;16:455–462. doi: 10.1038/sj.leu.2402415. [DOI] [PubMed] [Google Scholar]
- Blalock WL, Navolanic PM, Steelman LS, Shelton JG, Moye PW, Lee JT, Franklin RA, Mirza A, McMahon M, White MK, McCubrey JA. Requirement for the PI3K/Akt pathway in MEK1-mediated growth and prevention of apoptosis: identification of an Achilles heel in leukemia. Leukemia. 2003;17:1058–1067. doi: 10.1038/sj.leu.2402925. [DOI] [PubMed] [Google Scholar]
- Blalock WL, Weinstein-Oppenheimer C, Chang F, Hoyle PE, Wang X-Y, Algate PA, Franklin RA, Oberhaus SM, Steelman LS, McCubrey JA. Signal transduction, cell cycle regulatory, and anti-apoptotic pathways regulated by IL-3 in hematopoietic cells: possible sites for intervention with anti-neoplastic drugs. Leukemia. 1999;13:1109–1166. doi: 10.1038/sj.leu.2401493. [DOI] [PubMed] [Google Scholar]
- Brooks SC, Locke ER, Soule HD. Estrogen receptor in a human cell line (MCF-7) from breast carcinoma. J Biol Chem. 1973;1248:6251–6253. [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MHT, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Carter BZ, Milella M, Tsao T, McQueen T, Schober WD, Hu W, Dean NM, Steelman LS, McCubrey JA, Andreeff M. Regulation and targeting of antiapoptotic XIAP in acute myeloid leukemia. Leukemia. 2003;17:2081–2089. doi: 10.1038/sj.leu.2403113. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention . Centers for Disease Control and Prevention. Atlanta, GA: 2006. Cancer: Symptoms of breast cancer. http://www.cdc.gov/cancer/breast/basic_info/symptoms.htm. [Google Scholar]
- Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- Chang F, Steelman LS, Shelton JG, Lee JT, Navolanic PN, Blalock WL, Franklin RA, McCubrey JA. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway. Int J Oncol. 2003;22:469–480. [PubMed] [Google Scholar]
- Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1:707–17. [PubMed] [Google Scholar]
- Cocco L, Follo MY, Faenza I, Bavelloni A, Billi AM, Martelli AM, Manzoli L. Nuclear inositide signaling: An appraisal of phospholipase C beta1 behavior in myeloblastic and leukemia cells. Adv Enzyme Regul. 2007;47 doi: 10.1016/j.advenzreg.2006.12.003. in press. [DOI] [PubMed] [Google Scholar]
- Coutant A, Rescan C, Gilot D, Loyer P, Guguen-Guillouzo C, Baffet G. PI3K-FRAP/mTOR pathway is critical for hepatocyte proliferation whereas MEK/ERK supports both proliferation and survival. Hepatology. 2002;36:1079–1088. doi: 10.1053/jhep.2002.36160. [DOI] [PubMed] [Google Scholar]
- Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triposphate 5-phosphatase. Proc Natl Acad Sci USA. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan HC, Sun M, Naneko S, Feldman RI, Nicosia SV, Wang HG, Tsang BK, Cheng JQ. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP) J Biol Chem. 2004;279:5405–5412. doi: 10.1074/jbc.M312044200. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Transcriptional regulation by MAP kinases. Mol Reprod Dev. 1995;4:459–467. doi: 10.1002/mrd.1080420414. [DOI] [PubMed] [Google Scholar]
- Davis RJ, Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- DeGraffenried LA, Fulcher L, Friedrichs WE, Grunwald V, Ray RB, Hidalgo M, Hidalgo M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Annals of Oncology. 2004;15:1510–1516. doi: 10.1093/annonc/mdh388. [DOI] [PubMed] [Google Scholar]
- del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Delaney AM, Printon JA, Chen H, Faumann EB, Dudley DT. Identification of a novel mitogen-activated protein kinase kinase activation domain recognized by the inhibitor PD 184352. Mol Cell Biol. 2002;22:7593–7602. doi: 10.1128/MCB.22.21.7593-7602.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng T, Karin M. c-Fos transcriptional activity stimulated by H-Ras-activated protein kinase distinct from JNK and ERK. Nature. 1994;371:171–175. doi: 10.1038/371171a0. [DOI] [PubMed] [Google Scholar]
- Deng X, Kornblau SM, Ruvolo PP, May WS., Jr. Regulation of Bcl2 phosphorylation and potential significance for leukemic cell chemoresistance. J Natl Cancer Inst Monogr. 2001;28:30–37. doi: 10.1093/oxfordjournals.jncimonographs.a024254. [DOI] [PubMed] [Google Scholar]
- Deng X, Xiao L, Lang W, Gao F, Ruvolo P, May WS., Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem. 2001;276:23681–23688. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, Sapino A, Zhang F, Sharma D, Yang XH, Tora AD, Mehta K. Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene. 2002;21:8843–8851. doi: 10.1038/sj.onc.1206044. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27Kip1. Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domina AM, Vrana J, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23:5301–5315. doi: 10.1038/sj.onc.1207692. [DOI] [PubMed] [Google Scholar]
- Drexler HG. Expression of the FLT-3 receptor and response to FLT3 ligand by leukemic cells. Leukemia. 1996;10:588–599. [PubMed] [Google Scholar]
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- Dufner A, Anjelkovic M, Burgering BMT, Hemmings B, Thomas G. Protein kinase B localization and activation and eukaryotic translational initiation factor 4E-binding protein phosphorylation. Mol Cell Biol. 1999;19:4525–4534. doi: 10.1128/mcb.19.6.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eblen ST, Catling AD, Assanah MC, Weber MJ. Biochemical and biological functions of the N-terminal, noncatalytic domain of extracellular signal-regulated kinase 2. Mol Cell Biol. 2001;21:249–259. doi: 10.1128/MCB.21.1.249-259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. An activated mTOR mutant supports growth factor-independent, nutrient-dependent cell survival. Oncogene. 2004;23:5654–5663. doi: 10.1038/sj.onc.1207738. [DOI] [PubMed] [Google Scholar]
- Elmore LW, Xu D, Dumur C, Holt SE, Gewirtz DA. Evasion of a single-step, chemotherapy-induced senescence in breast cancer cells: implications for treatment response. Clin Cancer Res. 2005;11:2637–2643. doi: 10.1158/1078-0432.CCR-04-1462. [DOI] [PubMed] [Google Scholar]
- Engleman JA, Luo J, Canley LC. The evolution phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, Nordenskjöld M, Gustafsson JA. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab. 1997;82:4258–4265. doi: 10.1210/jcem.82.12.4470. [DOI] [PubMed] [Google Scholar]
- Evangelisti C, Bortul R, Falà F, Tabellini G, Goto K, Martelli AM. Nuclear diacylglycerol kinases: emerging downstream regulators in cell signaling networks. Histol Histopathol. 2007;22:573–579. doi: 10.14670/HH-22.573. [DOI] [PubMed] [Google Scholar]
- Fasco MJ, Keyomarsi K, Arcaro KF, Gierthy JF. Expression of an estrogen receptor alpha variant protein in cell lines and tumors. Mol Cell Endocrinol. 2000;166:156–169. [PubMed] [Google Scholar]
- Feilotter HE, Coulon V, McVeigh JL, Boag AH, Dorion-Bonnet F, Duboue B, Latham WC, Eng C, Mulligan LM, Longy M. Analysis of the 10q23 chromosomal region and the PTEN gene in human sporadic breast carcinoma. Br J Cancer. 1999;79:718–723. doi: 10.1038/sj.bjc.6690115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- Flotho S, Valcamonica S, Mach-Pascual G, Schmahl L, Corral J, Ritterbach H, Hasle M, Arico A, Biondi CM. RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML) Leukemia. 1999;13:32–37. doi: 10.1038/sj.leu.2401240. [DOI] [PubMed] [Google Scholar]
- Fry TJ, Mackall CL. Interleukin-7: from bench to clinic. Blood. 2002;99:3892–3904. doi: 10.1182/blood.v99.11.3892. [DOI] [PubMed] [Google Scholar]
- Fu Z, Aronoff-Spencer E, Wu H, Gerfen GJ, Backer JM. The iSH2 domain of PI 3-kinase is a rigid tether for p110 and not a conformational switch. Arch Biochem Biophys. 2004;432:244–251. doi: 10.1016/j.abb.2004.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuqua SA, Schiff R, Parra I, Friedrichs WE, Su JL, McKee DD, Slentz-Kesler K, Moore LB, Willson TM, Moore JT. Expression of wild-type estrogen receptor beta and variant isoforms in human breast cancer. Cancer Res. 1999;59:5425–8. [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Garcia JM, Silva J, Pena C, Garcia V, Rodriguez R, Cruz MA, Cantos B, Provencio M, Espana P, Bonilla R. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer. 2004;41:117–124. doi: 10.1002/gcc.20062. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, Whitlock BB, Xiao YQ, Bratton DB, Henson PM. Oxidants inhibit ERK/MAPK and prevent its ability to delay neutrophil apoptosis downstream of mitochondrial changes and at the level of XIAP. J Biol Chem. 2004;279:44695–44703. doi: 10.1074/jbc.M405313200. [DOI] [PubMed] [Google Scholar]
- Gëlinas C, White E. BH3-only proteins in control: specificity regulates MCL-1 and BAK-mediated apoptosis. Genes & Development. 2006;19:1263–1268. doi: 10.1101/gad.1326205. [DOI] [PubMed] [Google Scholar]
- Giles FJ, Albitar M. Mammalian target of rapamycin as a therapeutic target in leukemia. Curr Mol Med. 2005;5:653–661. doi: 10.2174/156652405774641034. [DOI] [PubMed] [Google Scholar]
- Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299–1303. doi: 10.1038/sj.onc.1205181. [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Jackson CA, Eyers PA, Cohen P, Goedert M, Boyle FT, Hewitt N, Plant H, Hedge P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem & Biol. 1999;6:559–568. doi: 10.1016/s1074-5521(99)80088-x. [DOI] [PubMed] [Google Scholar]
- Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. P70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule Bad. Proc Natl Acad Sci USA. 2001;98:9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci USA. 2004;101:15313–15317. doi: 10.1073/pnas.0406837101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–18311. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Heimbrook DC, Huber HE, Stirdivant SM, Claremon D, Liverton N, Patrick DR, Selnick H, Ahern J, Conroy R, Drakas R, Falconi N, Hancock P. Identification of potent, selective kinase inhibitors of Raf. Proc Amer Assoc Cancer Res Annual Meeting. 1998;39:558. [Google Scholar]
- Hochhaus A, McCubrey JA, Killmann NMB. Spotlight imatinib: a model for signal transduction inhibitors. Leukemia. 2002;16:1205–1206. [Google Scholar]
- Hollestelle A, Elstrodt F, Nagel JHA, Kallemeijn WW. Phosphatidylinositol-3-OH kinase or Ras pathway mutations in human breast cancer cell lines. Mol Cancer Res. 2007;5:195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- Howe CJ, LaHair MM, McCubrey JA, Franklin RA. Redox regulation of the CaM-kinases. J Biol Chem. 2004;279:44573–44581. doi: 10.1074/jbc.M404175200. [DOI] [PubMed] [Google Scholar]
- Howe CJ, LaHair MM, Maxwell JA, Lee JT, Robinson PJ, Rodriguezz-Mora O, McCubrey JA, Franklin RA. Participation of the calcium/calmodulin-dependent kinases in hydrogen peroxide-induced Ikappa B phosphorylation in human T lymphocytes. J Biol Chem. 2002;277:30469–30476. doi: 10.1074/jbc.M205036200. [DOI] [PubMed] [Google Scholar]
- Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Iijima Y, Laser M, Shiraishi H, Willey CD, Sundaravadivel B, Xu L, McDermott PJ, Kuppuswamy D. c-Raf/MEK/ERK pathway controls protein kinase C-mediated p70S6K activation in adult cardiac muscle cells. J Biol Chem. 2002;277:23065–23075. doi: 10.1074/jbc.M200328200. [DOI] [PubMed] [Google Scholar]
- Inhorn RC, Carlesso N, Durstin M, Frank DA, Griffin JD, Inhorn RC, Carlesso N, Durstin M, Frank DA, Griffin JD. Identification of a viability domain in the granulocyte/macrophage colony-stimulating factor receptor beta-chain involving tyrosine-750. Proc Natl Acad Sci USA. 1995;92:8665–8669. doi: 10.1073/pnas.92.19.8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Tiwar RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ. Cancer statistics. CA Cancer J Clin. 2004;54:829. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- Jia W, Yu C, Rahmani M, Krystal G, Sausville EA, Dent P, Grant S. Synergistic antileukemic interactions between 17-AAG and UCN-01 involve interruption of RAF/MEK- and AKT-related pathways. Blood. 2003;102:1824–1832. doi: 10.1182/blood-2002-12-3785. [DOI] [PubMed] [Google Scholar]
- Jin J, Woodgett JR. Chronic activation of protein kinase Bbeta/Akt2 leads to multinucleation and cell fusion in human epithelial kidney cells: events associated with tumorigenesis. Oncogene. 2005;18:5459–5470. doi: 10.1038/sj.onc.1208704. [DOI] [PubMed] [Google Scholar]
- Jonassen AK, Mjos OD, Sack MN. p70S6 kinase is a functional target of insulin activated Akt cell-survival. Biochem Biophys Res Commun. 2004;315:160–165. doi: 10.1016/j.bbrc.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Kang S, Bader AG, Zhao L, Vogt PK. Mutated PI 3-kinases: cancer targets on a silver platter. Cell Cycle. 2005;4:578–581. doi: 10.4161/cc.4.4.1586. [DOI] [PubMed] [Google Scholar]
- Karin M, Minden A, Lin A, McMahon M, Lange-Carter C, Derijard B, Davis RJ, Johnson GL. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266:1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- Kavanaugh WM, Pot DA, Chin SM, Deuter-Reinhard M, Jefferson AB, Norris FA, Masiarz FR, Cousens LS, Majerus PW, Williams LT. Multiple forms of an inositol polyphosphate 5-phosphatase form signaling complexes with Shc and Grb2. Curr Biol. 1996;6:438–445. doi: 10.1016/s0960-9822(02)00511-0. [DOI] [PubMed] [Google Scholar]
- Kirkegaard T, Witton CJ, McGlynn LM, Tovey S, Dunne B, Lyon A, Bartlett JM. AKT activation predicts outcome in breast cancer patients treated with tamoxifen. J. Pathol. 2005;207:139–146. doi: 10.1002/path.1829. [DOI] [PubMed] [Google Scholar]
- Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, Keith BR, Gilmer TM, Berger M, Podratz KC, Slamon DJ. Activity of the dual kinase inhibitor lapatinib ( GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66:1630–9. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- Kornblau SM, Womble M, Qiu YH, Jackson CE, Chen W, Konopleva M, Estey EH, Andreeff M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108:2358–2365. doi: 10.1182/blood-2006-02-003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfrancone L, Pelicci G, Brizzi MF, Aronica MG, Casciari C, Giuli S, Pegoraro L, Pawson T, Pelicci PG, Arouica MG. Overexpression of Shc proteins potentiates the proliferative response to the granulocyte-macrophage colony-stimulating factor and recruitment of Grb2/Sos and Grb2/p140 complexes to the beta receptor subunit. Oncogene. 1995;10:907–917. [PubMed] [Google Scholar]
- Lange-Carter CA, Johnson GL. Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science. 1994;265:1458–1461. doi: 10.1126/science.8073291. [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- Liaw D, Marsh DJ, Li J, Dahia PLM, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature Genetics. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- Lee JT, Jr, McCubrey JA. Targeting the Raf kinase cascade in cancer therapy—novel molecular targets and therapeutic strategies. Expert Opin Ther Targets. 2002;6:659–678. doi: 10.1517/14728222.6.6.659. [DOI] [PubMed] [Google Scholar]
- Lee JT, Jr, McCubrey JA. The Raf/MEK/ERK signal transduction cascade as a target for chemotherapeutic intervention. Leukemia. 2002;16:486–507. doi: 10.1038/sj.leu.2402460. [DOI] [PubMed] [Google Scholar]
- Lehmann BD, McCubrey JA, Jefferson HS, Paine MS, Chappell WH, Terrian DM. A dominant role for p53-dependent cellular senescence in radiosensitization of human prostate cancer cells. Cell Cycle. 2007;6:595–605. doi: 10.4161/cc.6.5.3901. [DOI] [PubMed] [Google Scholar]
- Leslie NR, Gray A, Pass I, Orchiston EA, Downes CP. Analysis of the cellular functions of PTEN using catalytic domain and C-terminal mutations: differential effects of C-terminal deletion on signaling pathways downstream of phosphoinositide 3-kinase. Biochem J. 2000;346:827–833. [PMC free article] [PubMed] [Google Scholar]
- Lespagnard L, Gancberg D, Maaroufi Y, Leclercq G, Larsimont D, Verhest A. In situ amplification of oestrogen receptor alpha mRNA in breast cancer cell lines and tumours. Mol Pathol. 2001;54:197–199. doi: 10.1136/mp.54.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Leygue E, Dotzlaw H, Watson PH, Murphy LC. Expression of estrogen receptor beta1, beta2, and beta5 messenger RNAs in human breast tissue. Cancer Res. 1999;59:1175–1179. [PubMed] [Google Scholar]
- Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998;58:5667–5672. [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Liang Y, Yan C, Schor NF. Apoptosis in the absence of caspase 3. Oncogene. 2001;20:6570–6578. doi: 10.1038/sj.onc.1204815. [DOI] [PubMed] [Google Scholar]
- Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR. P150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes & Devel. 1996;10:1084–1095. doi: 10.1101/gad.10.9.1084. [DOI] [PubMed] [Google Scholar]
- Luo Y, Shoemaker AR, Liu X, Woods KW, Thomas SA, deJong R, Han EK, Li T, Stoll VS, Powlas JA, Oleksijew A, Mitten MJ, Shi Y, Guan R, McGonigal TP, Klinghofer V, Johnson EF, Leverson JD, Bouska JJ, Mamo M, Smith RA, Gramling-Evans EE, Zinker BA, Mika AK, Nguyen PT, Oltrsdorf T, Rosenberg SH, Giranda VL. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol. Cancer Ther. 2005;4:977–986. doi: 10.1158/1535-7163.MCT-05-0005. [DOI] [PubMed] [Google Scholar]
- Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, Zinner R, Hung MC, Steck P, Siminovitch K, Mills GB. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–7045. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Bannigan BW, Harris PL, Haserlat SM, Supko JB, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocrine-Related Cancer. 2001;8:219–225. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the Rel A/p65 subunit of NF-kappaB. Mol Cell Biol. 2000;20:1626–1638. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam M, Templeton D. Constitutive activation of S6 kinase by deletion of amino-terminal autoinhibitory and rapamycin sensitivity domains. Mol. Cell Biol. 1996;16:405–413. doi: 10.1128/mcb.16.1.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahimainathan L, Choudhury GG. Inactivation of platelet-derived growth factor receptor by the tumor suppressor PTEN provides a novel mechanism of action of the phosphatase. J Biol Chem. 2004;279:15258–15268. doi: 10.1074/jbc.M314328200. [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Mason CS, Marshall CJ. Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J Biol Chem. 1997;272:4378–4383. doi: 10.1074/jbc.272.7.4378. [DOI] [PubMed] [Google Scholar]
- Marani M, Hancock D, Lopes R, Tenev T, Downward J, Lemoine NR. Role of Bim in the survival pathway induced by Raf in epithelial cells. Oncogene. 2004;23:2431–2441. doi: 10.1038/sj.onc.1207364. [DOI] [PubMed] [Google Scholar]
- Martelli AM, Tazzari PL, Evangelisti C, Chiarini F, Blalock WL, Billi AM, Manzoli L, McCubrey JA, Cocco L. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin module for acute myelogenous leukemia therapy: from bench to bedside. Current Medicinal Chem. 2007;14:2009–2023. doi: 10.2174/092986707781368423. [DOI] [PubMed] [Google Scholar]
- Martelli AM, Faenza I, Billi AM, Manzoli L, Evangelisti C, Fala F, Cocco L. Intranuclear 3′-phosphoinositide metabolism and Akt signaling: new mechanisms for tumorigenesis and protection against apoptosis? Cell Signal. 2006;18:1101–1107. doi: 10.1016/j.cellsig.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Martin DE, Soulard A, Hall MN. TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell. 2004;119:969–979. doi: 10.1016/j.cell.2004.11.047. [DOI] [PubMed] [Google Scholar]
- Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999;18:2137–2148. doi: 10.1093/emboj/18.8.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuguchi T, Salgia R, Hallek M, Eder M, Druker B, Ernst TJ, Griffin JD. Shc phosphorylation in myeloid cells is regulated by granulocyte macrophage colony-stimulating factor, interleukin-3, and steel factor and is constitutively increased by p210BCR/ABL. J Biol Chem. 1994;269:5016–5021. [PubMed] [Google Scholar]
- Mayo LD, Donner DB. A phopphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Baldwin AS. The transcription factor NF-kappaB: control of oncogenesis and cancer therapy resistance. Biochem Biophys Acta. 2000;1470:M55–62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouysségur J, Brambilla R. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Lee JT, Steelman LS, Blalock WL, Moye PW, Chang F, Pearce M, Shelton JG, White MK, Franklin RA, Pohnert SC. Interactions between the PI3K and Raf signaling pathways can result in the transformation of hematopoietic cells. Cancer Detect. & Prevent. 2005;25:375–393. [PubMed] [Google Scholar]
- McCubrey JA, May WS, Duronio V, Mufson A. Serine/threonine phosphorylation in cytokine signal transduction. Leukemia. 2000;14:9–21. doi: 10.1038/sj.leu.2401657. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Mobasheri A, Richardson S, Mobasheri R, Shakibaei M, Hoyland JA. Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol. 2005;20:1327–1338. doi: 10.14670/HH-20.1327. [DOI] [PubMed] [Google Scholar]
- Mukherjee R, Bartlett JMS, Krishna NS, Underwood MA, Edwards J. Raf-1 expression may influence progression to androgen insensitive prostate cancer. The Prostate. 2005;64:101–107. doi: 10.1002/pros.20211. [DOI] [PubMed] [Google Scholar]
- Muraille E, Pesesse X, Kuntz C, Erneux C. Distribution of the src-homology-2-domain-containing inositol 5-phosphatase SHIP-2 in both non-haemopoietic and haemopoietic cells and possible involvement in SHIP-2 in negative signaling of B-cells. Biochem J. 1999;342:697–705. [PMC free article] [PubMed] [Google Scholar]
- Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem. 1999;274:15982–15985. doi: 10.1074/jbc.274.23.15982. [DOI] [PubMed] [Google Scholar]
- Navolanic PM, Steelman LS, McCubrey JA. EGFR family signaling and its association with breast cancer development and resistance to chemotherapy. Int J Oncol. 2003;22:237–252. [PubMed] [Google Scholar]
- Nimwegen MJ, Huigsloot H, Camier A, Tijdens IB, van de Water B. Focal adhesion kinase and protein kinase B cooperate to suppress doxorubicin-induced apoptosis of breast tumor cells. Mol Phamacol. 2006;70:1330–1339. doi: 10.1124/mol.106.026195. [DOI] [PubMed] [Google Scholar]
- Noguchi M, Ropars V, Roumestand C, Suizu F. Proto-oncogene TCL1: more than just a coactivator for Akt. The FASEB Journal. 2007;21:1–12. doi: 10.1096/fj.06-7684com. [DOI] [PubMed] [Google Scholar]
- Obexer P, Geiger K, Ambros PF, Meister B, Ausseriechner MJ. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ. 2007;14:534–547. doi: 10.1038/sj.cdd.4402017. [DOI] [PubMed] [Google Scholar]
- Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, Yan C, McConnell P, Spessard C, Banotai C, Mueller WT, Delaney A, Omer C, Sebolt-Leopold J, Dudley DT, Leung IK, Flamme C, Warmus J, Kaufman M, Barrett S, Tecle H, Hasemann CA. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–1197. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- Okuda K, Foster R, Griffin JD. Signaling domains of the beta c chain of the GM-CSF/IL-3/IL-5 receptor. Ann NY Acad Sci. 1999;872:305–313. doi: 10.1111/j.1749-6632.1999.tb08474.x. [DOI] [PubMed] [Google Scholar]
- Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LLM, Donner DB. NF-kappaB activation by tumor necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- Panwalkar A, Verstovsek S, Giles FJ. Mammalian target of rapamycin inhibition as therapy for hematologic malignancies. Cancer. 2004;100:657–666. doi: 10.1002/cncr.20026. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor mutations are common in lung cancers from “never smokers” are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS. Med. 2005;2:57–61. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad S, Dedhar S. The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metastasis Rev. 2003;22:375–384. doi: 10.1023/a:1023777013659. [DOI] [PubMed] [Google Scholar]
- Ponti C, Gibellini D, Boin F, Melloni E, Manzoli FA, Cocco L, Zauli G, Vitale M. Role of CREB transcription factor in c-fos activation in natural killer cells. Eur J Immunol. 2002;32:3358–3365. doi: 10.1002/1521-4141(200212)32:12<3358::AID-IMMU3358>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Poola I, Abraham J, Baldwin K. Identification of ten exon deleted ERbeta mRNAs in human ovary, breast, uterus and bone tissues: Alternate splicing pattern of estrogen receptor beta mRNA is distinct from that of estrogen receptor alpha. FEBS Lett. 2002;516:133–138. doi: 10.1016/s0014-5793(02)02521-8. [DOI] [PubMed] [Google Scholar]
- Poola I, Koduri S, Chatra S, Clarke R. Identification of twenty alternatively spliced estrogen receptor alpha mRNAs in breast cancer cell lines and tumors using splice targeted primer approach. J Steroid Biochem Mol Biol. 2000;72(5):249–258. doi: 10.1016/s0960-0760(00)00033-9. [DOI] [PubMed] [Google Scholar]
- Qi XJ, Wildey GM, Howe PH. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J Biol Chem. 2006;281:813–823. doi: 10.1074/jbc.M505546200. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303:1179–1181. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- Rao P, Mufson RA. A membrane proximal domain of the human interleukin-3 receptor beta c subunit that signals DNA synthesis in NIH 3T3 cells specifically binds a complex of Src and Janus family tyrosine kinases and phosphatidylinositol 3-kinase. J Biol Chem. 1995;270:6886–6893. doi: 10.1074/jbc.270.12.6886. [DOI] [PubMed] [Google Scholar]
- Rhei E, Kang L, Bogomolniy F, Federici MG, Borgen PI, Boyd J. Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in primary breast carcinomas. Cancer Res. 1997;57:3657–3659. [PubMed] [Google Scholar]
- Robinson MJ, Stippec SA, Goldsmith E, White MA, Cobb MH. A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr Biol. 1998;21:141–1150. doi: 10.1016/s0960-9822(07)00485-x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Romanelli A, Martin KA, Toker A, Bleinis J. p70 S6 Kinase is regulated by protein kinase Cζ and participates in a phosphoinositide 3-kinase-regulated signaling complex. Mol Cell Biol. 1999;19:2921–2928. doi: 10.1128/mcb.19.4.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romashkova JA, Makarov SS. NF-kappaB is a target of Akt in anti-apoptotic PDGF signaling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JKV, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Duronio V. Dissociation of cytokine-induced phosphorylation of Bad and activation of PKB/akt: involvement of MEK upstream of Bad phosphorylation. Proc Natl Acad Sci USA. 1998;95:7439–7444. doi: 10.1073/pnas.95.13.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Haber DA, Lynch TJ. Epidermal growth factor receptor mutations in non-small cell lung cancer; predicting clinical response to kinase inhibitors. Clin Cancer Res. 2005;11:5668–5670. doi: 10.1158/1078-0432.CCR-05-1055. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signaling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Shelton JG, Steelman LS, Lee JT, Knapp SL, Blalock WL, Moye PW, Franklin RA, Pohnert SC, Mirza AM, McMahon M, McCubrey JA. Effects of the RAF/MEK/ERK and PI3K/AKT signal transduction pathways on the abrogation of cytokine-dependence and prevention of apoptosis in hematopoietic cells. Oncogene. 2003;22:2478–2492. doi: 10.1038/sj.onc.1206321. [DOI] [PubMed] [Google Scholar]
- Shelton JG, Moye PW, Steelman LS, Blalock WL, Lee JT, Franklin RA, McMahon M, McCubrey JA. Differential effects of kinase cascade inhibitors on neoplastic and cytokine-mediated cell proliferation. Leukemia. 2003;17:1765–1782. doi: 10.1038/sj.leu.2403052. [DOI] [PubMed] [Google Scholar]
- Shelton JG, Steelman LS, Abrams SL, Bertrand FE, Franklin RA, McMahon M, McCubrey JA. The epidermal growth factor receptor as a target for therapeutic intervention in numerous cancers--What's genetics got to do with it? Expert Opinion Therap Targets. 2005a;9:1009–1030. doi: 10.1517/14728222.9.5.1009. [DOI] [PubMed] [Google Scholar]
- Shelton JG, Steelman LS, Abrams SL, White ER, Akula SM, Franklin RA, Bertrand FE, McMahon M, McCubrey JA. Conditional EGFR promotes cell cycle progression and prevention of apoptosis in the absence of autocrine cytokines. Cell Cycle. 2005b;4:822–830. doi: 10.4161/cc.4.6.1724. [DOI] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Shimada A, Taki T, Tabuchi K, Tawa A, Horibe K, Tsuchida M, Hanada R, Tsukimoto I, Hayashi Y. KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8:21): a study of the Japanese Childhood AML. Cooperative Study Group. Blood. 2006;107:1806–1809. doi: 10.1182/blood-2005-08-3408. [DOI] [PubMed] [Google Scholar]
- Shishodia S, Aggarwal BB. Nuclear factor-kappaB activation mediates cellular transformation, proliferation, invasion angiogenesis and metastasis of cancer. Cancer Treat Res. 2004;119:139–173. doi: 10.1007/1-4020-7847-1_8. [DOI] [PubMed] [Google Scholar]
- Singh B, Ittmann MM, Krolewski JJ. Sporadic breast cancers exhibit loss of heterozygosity on chromosome segment 10q23 close to the Cowden disease locus. Genes Chromosome Cancer. 1998;21:166–171. [PubMed] [Google Scholar]
- Songyang Z, Baltimore D, Cantley LC, Kaplan DR, Franke TF. Interleukin 3-dependent survival by the Akt protein kinase. Proc Natl Acad Sci USA. 1997;94:11345–11350. doi: 10.1073/pnas.94.21.11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst. 1973;51:1409–1416. doi: 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Steelman LS, Bertrand FE, McCubrey JA. The complexity of PTEN: mutation, marker and potential target for therapeutic intervention. Expert Opinion Ther Targets. 2004;8:537–550. doi: 10.1517/14728222.8.6.537. [DOI] [PubMed] [Google Scholar]
- Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, Radich JP. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97:3589–3595. doi: 10.1182/blood.v97.11.3589. [DOI] [PubMed] [Google Scholar]
- Tai YT, Fulciniti M, Hideshima T, Song W, Leiba M, Li XF, Rumizen M, Burger P, Morrison A, Podar K, Chauhan D, Tassone P, Richardson P, Munshi NC, Ghobrial I, Anderson KC. Inhibition of ERK1/2 activity by the EMR1/2 inhibitor AZD6244 (ARRY-142886) induces human multiple myeloma cell apoptosis in the bone marrow microenvironment: a new therapeutic strategy for MM. Blood. 2006;108:3463. [Google Scholar]
- Tamburini J, Elie C, Bardet V, Chapuis N, Park S, Broët P, Cornillet-Lefebvre P, Lioure B, Ugo V, Blanchet O, Ifrah N, Witz F, Dreyfus F, Mayeux P, Lacombe C, Bouscary D. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood. 2007;110:1025–1028. doi: 10.1182/blood-2006-12-061283. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Tauchi T, Boswell HS, Leibowitz D, Broxmeyer HE, Tauchi T, Boswell HS, Leibowitz D, Broxmeyer HE. Coupling between p210bcr-abl and Shc and Grb2 adaptor proteins in hematopoietic cells permits growth factor receptor-independent link to ras activation pathway. J Exp Med. 1994;179:167–175. doi: 10.1084/jem.179.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor V, Wong M, Brandts C, Reilly L, Dean NM, Cowsert LM, Moodie S, Stokoe D. 5′phospholipid phosphatase SHIP-2 causes protein kinase B inactivation and cell cycle arrest in glioblastoma cells. Mol Cell Biol. 2000;20:6860–6871. doi: 10.1128/mcb.20.18.6860-6871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RS, Tymms MJ, McKinlay LH, Shannon MF, Seth A, Kola I. ETS1, NFkappaB and AP1 synergistically transactivate the human GM-CSF promoter. Oncogene. 1997;23:2845–2855. doi: 10.1038/sj.onc.1201125. [DOI] [PubMed] [Google Scholar]
- Tokunaga E, Kimura Y, Mashino K, Oki E, Kataoka A, Ohno S, Morita M, Kakeji Y, Baba H, Maehara Y. Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer. 2006;13:137–144. doi: 10.2325/jbcs.13.137. [DOI] [PubMed] [Google Scholar]
- Tokunaga C, Yoshino K, Yonezawa K. mTOR integrates amino acid- and energy-sensing pathways. Biochem Biophys Res Commun. 2004;313:443–446. doi: 10.1016/j.bbrc.2003.07.019. [DOI] [PubMed] [Google Scholar]
- Tortora G, Bianco R, Daniele G, Ciardiello F, McCubrey JA, Ricciardi MR, Ciuffreda L, Cognetti F, Tafuri A, Milella M. Overcoming resistance to molecularly targeted anticancer therapies: rational drug combinations based on EGFR and MAPK inhibition for solid tumors and hematological malignancies. Drug Resistance Updates. 2007 doi: 10.1016/j.drup.2007.03.003. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresini M, Lorenzini A, Frisoni L, Allen RG, Cristofalo VJ. Lack of Elk-1 phosphorylation and dysregulation of the extracellular regulated kinase signaling pathway in senescent human fibroblast. Exp Cell Res. 2001;269:287–300. doi: 10.1006/excr.2001.5334. [DOI] [PubMed] [Google Scholar]
- Troppmair J, Rapp UR. Raf and the road to cell survival: a tale of bad spells, ring bearers and detours. Biochem Pharmacol. 2003;66:1341–1345. doi: 10.1016/s0006-2952(03)00483-0. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troussard AA, Mawji NM, Ong C, Mui A, St-Arnaud R, Dedhar S. Conditional knock-out of integrin-linked kinase demonstrates an essential role in protein kinase B/Akt activation. J Biol Chem. 2003;278:22374–22378. doi: 10.1074/jbc.M303083200. [DOI] [PubMed] [Google Scholar]
- Tsutsui S, Inoue H, Yasuda K, Suzuki K, Higashi H, Era S, Mori M. Reduced expression of PTEN protein and its prognostic implications in invasive ductal carcinoma of the breast. Oncology. 2005;68:398–404. doi: 10.1159/000086981. [DOI] [PubMed] [Google Scholar]
- Twiddy D, Cohen GM, MacFarlane M, Cain K. Casape-7 is directly activated by the ∼700-kDa apoptosome complex and is released as a stable XIAP-caspase-7 ∼200-kDa complex. J Biol Chem. 2006;281:3876–3888. doi: 10.1074/jbc.M507393200. [DOI] [PubMed] [Google Scholar]
- Vogt PK, Bader AG, Kang S. Phosphoinositide 3-kinase: from viral oncoprotein to drug target. Virology. 2006;344:131–138. doi: 10.1016/j.virol.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Wang CY, Bassuk AG, Boise LH, Thompson CB, Bravo R, Leiden JM. Activation of the granulocyte-macrophage colony-stimulating factor promoter in T cells requires cooperative binding of Elf-1 and AP-1 transcription factors. Mol Cell Biol. 1994;14:1153–1159. doi: 10.1128/mcb.14.2.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Boerner SA, Winkler JD, LoRusso PM. Clinical experience of MEK inhibitors in cancer therapy. Biochim. et. Biophysica. Acta. 2006 doi: 10.1016/j.bbamcr.2006.11.009. In Press. [DOI] [PubMed] [Google Scholar]
- Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF. The antiapoptotic gene Mcl-1 is upregulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol. 1999;19:6195–6206. doi: 10.1128/mcb.19.9.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, Chan JLK, Li W. Rapamycin together with herceptin significantly increased anti-tumor efficacy compared to either alone in ErbB2 over expressing breast cancer cells. Int J Can. 2007;121:2911–2918. doi: 10.1002/ijc.22606. [DOI] [PubMed] [Google Scholar]
- Weber MJ, Gioeli D. Ras signaling in prostate cancer progression. J Cell Biochem. 2004;91:13–25. doi: 10.1002/jcb.10683. [DOI] [PubMed] [Google Scholar]
- Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- Weng LP, Smith WM, Dahia PL, Ziebold U, Gil E, Lees JA, Eng C. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999;59:5808–5814. [PubMed] [Google Scholar]
- Weng LP, Brown JL, Eng C. PTEN coordinates G1 arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Human Molecular Genetics. 2001;10:599–604. doi: 10.1093/hmg/10.6.599. [DOI] [PubMed] [Google Scholar]
- Weston CR, Balmanno K, Chalmers C, Hadfield K, Molton SA, Ley R, Wagner EF, Cook SJ. Activation of ERK1/2 by deltaRaf-1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene. 2003;22:1281–1293. doi: 10.1038/sj.onc.1206261. [DOI] [PubMed] [Google Scholar]
- Witzig TE, Kaufmann SH. Inhibition of the phosphatidylinositol 3-kinase/mammalian target of rapamycin pathway in hematologic malignancies. Curr Treat Options Oncol. 2006;7:285–294. doi: 10.1007/s11864-006-0038-1. [DOI] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME. Coupling of the Ras-MAPK pathway to gene activation by Rsk2, a growth factor regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- Xu S, Robbins D, Frost J, Dang A, Lange-Carter C, Cobb MH. MEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen-activated protein kinase. Proc Natl Acad Sci USA. 1995;92:6808–6812. doi: 10.1073/pnas.92.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Ma DZ, Wang LY, Su JM, Zha XL. Transforming growth factor-beta1 stimulated protein kinase B serine-473 and focal adhesion kinase tyrosine phosphorylation dependent on cell adhesion in human hepatocellular carcinoma SMMC-7721 cells. Biochim Biophys Res Commun. 2003;312:388–396. doi: 10.1016/j.bbrc.2003.10.130. [DOI] [PubMed] [Google Scholar]
- Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, Marlow A, Hurley B, Lyssikatos J, Lee PA, Winkler JD, Koch K, Wallace E. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Eguchi H, Nakachi K, Tanimoto K, Higashi Y, Suemasu K, Iino Y, Morishita Y, Hayashi S. Distinct mechanisms of loss of estrogen receptor alpha gene expression in human breast cancer: Methylation of the gene and alteration of trans-acting factors. Carcinogenesis. 2000;21:2193–2201. doi: 10.1093/carcin/21.12.2193. [DOI] [PubMed] [Google Scholar]
- You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Mao X, Li WX. Inhibition of the PI3K pathway sensitizes fludarabine-induced apoptosis in human leukemic cells through an inactivation of MAPK-dependent pathway. Biochem Biophys Res Commun. 2005;331:391–397. doi: 10.1016/j.bbrc.2005.03.182. [DOI] [PubMed] [Google Scholar]
- Yu J, Wjasow C, Backer JM. Regulation of the p85/p110alpha phosphatidylinositol 3′-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J Biol Chem. 1996;273:30199–30203. doi: 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Hung MC. Novel targets of Akt, p21(Cip1.WAF1), and MDM2. Semin Oncol. 2002;29:62–70. doi: 10.1053/sonc.2002.34057. [DOI] [PubMed] [Google Scholar]