

Abstract
Factor VIII (FVIII) is a multi-domain protein that is important in the clotting cascade. Its deficiency causes Hemophilia A, a bleeding disorder. The unfolding of protein domains can lead to physical instability such as aggregation, and hinder their use in replacement therapy. It has been shown that the aggregation of rFVIIII is initiated by small fluctuations in the protein’s tertiary structure (Grillo et al., 2001, Biochemistry 40:586–595). We have investigated the domain(s) involved in the initiation of aggregation using circular dichroism (CD), size exclusion chromatography (SEC), fluorescence anisotropy, domain specific antibody binding, and clotting activity studies. The studies indicated that aggregation may be initiated as a result of conformational change in the C2 domain encompassing the lipid-binding region (2303–2332). The presence of O-phospho-L-Serine (OPLS), which binds to the lipid-binding region of FVIII, prevented aggregation of the protein.
Keywords: recombinant human FVIII (rFVIII), physical instability, multi-domain, lipid-binding region, hemophilia A, inhibitor development
INTRODUCTION
Hemophilia A is a bleeding disorder caused by a deficiency of Factor VIII (FVIII), a large multi-domain protein that functions as a cofactor in the blood coagulation cascade.1 The severity of the bleeding disorder varies among patients depending upon the degree of deficiency of FVIII. Cloning of the FVIII gene, together with advances in biotechnology and protein engineering, have made it feasible to manufacture recombinant human FVIII (rFVIII) in large quantities for the treatment of hemophilia A and for structural characterization.2–4 Recombinant FVIII is structurally and functionally similar to plasma derived FVIII. The protein is composed of six domains: A1, A2, B, A3, C1, and C2. Detailed investigations of the expression of FVIII in autologous systems5 have shown that N-linked glycosylation occurs in the endoplasmic reticulum, followed by transport of the protein to the Golgi apparatus. Inside the Golgi, FVIII is subject to further processing, involving modification of N- and O-linked oligosaccharides. The protein undergoes intracellular proteolysis at Arg1313 and Arg1648 which appear within Arg-X-X-Arg motifs in the B-domain.6 This results in the disruption of the covalent linkage of the heavy chain (A1, A2, and B) and light chain (A3-C1-C2) of FVIII, causing the protein to exist as heterodimers.5 The two polypeptide chains are held together by a metal ion linkage between the A1 and A3 domains.7,8 Thus the native protein consists of multiple polypeptides ranging in weight from 80 to 210000 kDa.5
A large protein is often organized into distinct domains that play a critical role in its structure and function.9,10 The folding/unfolding behavior of these individual domains, and the interactions between them, may coordinate various functions in multi-domain proteins. Furthermore, the modular assembly of multi-domain proteins often results in complex unfolding characteristics and the existence of intermediate species which may lead to physical instability including a tendency to aggregate.11
The physical phenomenon of aggregation can have profound impacts on the stability of therapeutic proteins. Not only does a loss of activity occur, but also the presence of aggregates has been shown to elicit enhanced antibody mediated immune responses which may have pathological consequences.12 It has been observed that ~15%–30% of hemophilic patients develop antibodies that inhibit the activity of the administered protein, thus complicating therapy.13,14 It has been reported recently that rFVIII is susceptible to aggregation due to minor structural alterations in tertiary structure.15 In this study, we built upon the observations of Grillo et al.15 and provide insight into the domains involved in the conformational change(s) which lead to aggregation of rFVIII. Results from this study indicate that unfolding of rFVIII is a complex process in which conformational changes occurring in the C2 domain, encompassing the lipid-binding region (2303–2332) of FVIII, may be at least partly responsible for initiation of aggregation.
EXPERIMENTAL PROCEDURES
Materials
FVIII deficient plasma was purchased from Trinity Biotech (Co Wicklow, Ireland). Full-length rFVIII expressed in Chinese Hamster Ovary (CHO) was provided by Baxter Biosciences, Duarte, CA. A plasmid containing the cDNA of human B-domain deleted FVIII (BDDFVIII) was a gift from Dr. Rita Sarkar, University of Pennsylvania. Cos-7 cells were purchased from ATCC (Manassas, VA). Recombinant human B-domain deleted FVIII (rBDDFVIII) was purified chromatographically, and characterized by SDS–PAGE and SEC (data not shown). The samples were stored at −70°C and care was taken to avoid repeated freeze-thawing of the sample. Monoclonal antibodies ESH 4 and ESH 8 were obtained from American Diagnostica, Inc. (Greenwich, CT). O-phospho-L-serine (OPLS) was obtained from Sigma (St. Louis, MO). Buffer salts were purchased from Fisher Scientific (Fair Lawn, NJ) or Sigma and used without further purification.
Expression and Purification of rBDDFVIII
rBDDFVIII was expressed transiently by transfecting COS-7 cells with a plasmid containing pMT2 vector as the backbone and cDNA of human BDDFVIII as the insert. The plasmid pMT2-BDDFVIII was obtained as a gift from Dr. Rita Sarkar (University of Pennsylvania, PA). The transfected cells were cultured in serum free media. The protein was purified from the media by a two step ion-exchange chromatography procedure as described earlier.16 Briefly, the factor VIII-containing media was loaded on a HiPrep 16/10 SP FF column (Amersham Biosciences, Piscataway, NJ) and eluted using a linear gradient of 100 to 650 mM NaCl in HEPES-CaCl2 buffer. Fractions containing rBDDFVIII were loaded on a Resource Q column (Amersham Biosciences) and eluted with a 200 mM to 1 M NaCl linear gradient in the HEPES/Tris-CaCl2 buffer. The purity of the isolated protein was assessed by silver stained SDS–polyacrylamide gel electrophoresis and size exclusion chromatography (SEC) using a Biosep SEC-S-4000 (Phenomenex, Torrance, CA).
High Performance Size Exclusion Chromatography
High performance (HP) SEC was performed using a Biosep SEC-S-4000 4.6 × 300 mm column. The analytical column was maintained at 20°C using a Shimadzu CT0-10AC column oven. The chromatography system consisted of a Waters 510 HPLC Pump, a Rheodyne injector with a 50 μL PEEK sample loop and a Hitachi F1050 fluorescence detector. Excitation and emission wavelengths were set at 285 and 335 nm, respectively, to monitor the elution of the protein. Gel filtration was carried out under isocratic conditions at a flow rate of 0.4 mL/min using an aqueous buffer consisting of 25 mM Tris (or 10 mM MOPS), 5 mM CaCl2, and 300 mM NaCl, pH 7.0. For unfolding studies, the protein was heated at 60°C/h; samples were withdrawn at various temperatures, stored on ice, and analyzed within 3 h. The ability of the chromatographic conditions to resolve aggregated versus native protein was evaluated by analyzing mixtures containing various amounts of the two different forms. It was found that the column could resolve and detect the existence of as little as 1%–2% aggregated material (by weight) in the presence of native protein (data not shown).
Circular Dichroism Experiments
CD spectra were acquired with a JASCO J-715 spectropolarimeter calibrated with d10 camphor sulfonic acid. The stock solution used to prepare the samples had a specific activity of 2466 IU (~0.5 mg/mL). The protein concentration used was typically ~20 μg/mL, which corresponds to a specific activity of ~98.6 IU/mL. Path-length of the quartz cuvette used was 1 cm. Samples were scanned from 255 to 208 nm for secondary structural analysis. The CD spectra of the protein were corrected by subtracting the spectrum of the buffer alone. Since the CD spectra of samples containing aggregated protein may be distorted by light scattering and/or absorption flattening, correction factor was applied as described earlier.17 The unfolding of the protein was followed by monitoring the ellipticity at 215 nm over the temperature range of 20–80°C at a heating rate of 60°C/h, with a 2 min holding time at every 5°C. The temperature scans were performed with a Peltier 300 RTS unit, and the profiles were generated using the software provided by the manufacturer. The data were represented as δθ (change in ellipticity), as a function of temperature. This representation of the data was chosen to account for any variations in starting protein concentrations. δθ was calculated as θt−θnative, where θnative is the ellipticity of the protein in the native state and θt is the ellipticity at any given temperature. θnative was estimated by computing the mean ellipticity at initial temperatures. The transition temperature (Tm), for each unfolding profile was determined by fitting the data to a sigmoid function using WinNonlin (Pharsight Corporation, Mountainview, CA):
where Yobserved is the ellipticity at 215 nm at any give temperature T, Ynative is the spectral value for the native state, Tm is the transition temperature, and gamma (γ) is the cooperativity function. The magnitude of spectral change is denoted by Δm and is defined as (Ynative−Yunfolded), where Yunfolded is the spectral value for the unfolded state.
Steady State Fluorescence Anisotropy Studies
Temperature-dependent steady-state emission anisotropy (r) measurements were carried out at a heating rate of 60°C/h with a PTI fluorometer (Photon Technology International, Lawrenceville, NJ) equipped with a xenon arc lamp, Peltier unit, and motorized Glan-Thompson polarizing prisms having large apertures. Studies were performed over the temperature range of 20–80°C with a holding time of 1–2 min every 5°C. The concentration of the protein was typically ~5 or 20 μg/mL. Path-length of the quartz cuvette used was 1 cm. The G-factor, which is the ratio of the sensitivities of the detection system for vertically and horizontally polarized light, was determined for each sample at 20°C and was held fixed over the temperature range of study. The excitation (Ex) wavelength was set at 280 nm and emission (Em) was monitored at 335 nm. Ex and Em slit widths were set at 4 nm. The data is presented as percent (%) change in (r) as a function of temperature. The percent change in anisotropy was computed as
where rn is the anisotropy of the protein in its native state (i.e., at 20°C) and rt is the anisotropy at any given temperature.
Biological Activity Assay of rFVIII
rFVIII clotting activity was determined using a one-stage activated partial thromboplastin time (APTT) assay18 with micronized silica or platelin L reagent (BioMerieux, Durham, NC) as activator and FVIII deficient plasma as the substrate. The APTT assay was performed using a COAG-A-MATE coagulation analyzer (Organon Teknika Corporation, Durham, NC). Briefly, rFVIII was added to FVIII deficient plasma and the clotting time was monitored. The activity of the rFVIII was then obtained from a calibration curve constructed using the clotting times determined from various dilutions of a lyophilized reference concentrate of known activity.
Sandwich Enzyme-Linked Immunosorbent Assay (ELISA)
Conformational changes in different domains of rFVIII were followed by sandwich ELISA as described previously.19 Nunc-Maxisorb 96-well plates were coated with capture monoclonal antibody ESH4, an antibody that recognizes epitope 2303–2332,20 which is the lipid binding region21 or with 8860, an antibody that recognizes an epitope in the A2 domain. Fifty microliter of an antibody at 5 μg/mL in carbonate buffer (0.2 M, pH 9.4) was incubated in each well overnight at 4°C. The plates were then washed 10 times with 100 μL of phosphate buffer (PB; 10 mM Na2HPO4, 1.8 mM KH2PO4, 14 mM NaCl, 2.7 mM KCl, and 0.02% NaN3) containing 0.05% Tween 20 (PBT). Non-specific protein binding sites on the plastic’s adsorptive surface were blocked by incubating 200 μL of PB buffer containing 1% bovine serum albumin (PBA) for 2 h at room temperature. The plates were washed 10 times with PBT followed by the addition of 50 μL of 100 ng/mL of rFVIII (native protein or protein heated to various temperatures) in PBA and incubated at 37°C for 1 h. The plates were washed 10 times with PBT and incubated with 50 μL of a 1 μg/mL solution of biotinylated monoclonal antibody ESH8 (a probe antibody that recognizes epitope region 2248–2285 in the C2 domain22) and 50 μL of a 1:1000 dilution of avidin-alkaline phosphatase conjugate, both in PBA, at room temperature for 1 h. The plates were washed 10 times with PBT and 100 μL of 1 mg/mL p-nitrophenyl phosphate solution in diethanolamine buffer (consisting of 1 M diethanolamine, 0.5 mM MgCl2). The plates were incubated at room temperature for 30 min and the reaction was quenched by adding 100 μL of 3 N NaOH. The alkaline phosphatase reaction product was determined by absorbance at 405 nm using a Spectramax plate reader (Molecular Devices Corporation, Sunnyvale, CA). For temperature-dependent studies, rFVIII was heated from 20 to 47°C at 60°C/h, held at the indicated temperatures for ~2 min, and cooled to room temperature prior to analysis by ELISA.
Statistical Analysis
Data was analyzed by ANOVA using Analyst Application of SAS (SAS Institute, Inc., Cary, NC). Dunnetts post hoc multiple comparison test was used to detect significant differences (p <0.05) in antibody binding relative to the control.
RESULTS AND DISCUSSION
Temperature-Induced Unfolding of rFVIII
Thermal stress has been widely used as a perturbant to probe the relationship between protein folding and stability.17,23,24 Therefore, we investigated temperature induced unfolding of rFVIII to gain insight into the mechanisms of aggregation. Unfolding was investigated by acquiring far-UV CD spectra (Figure 1A, top panel) of solutions of FVIII heated over the temperature range of 20 to 80°C at a rate of 60°C/h. At 20°C, a broad negative band at 215 nm suggested that the protein existed predominantly in a β-sheet conformation (Figure 1A, bottom panel, ^). As the temperature was increased from 20 to 50°C, there were no significant changes in the far-UV CD spectrum, indicating that the secondary structure of the protein was unaltered. At temperatures >50°C however, the negative ellipticity at 215 nm increased progressively with temperature, suggesting an increase in β-sheet content (Figure 1A, bottom panel, ○). The mid-point of the transition (Tm), defined as the temperature at which a 50% change in ellipticity was observed at 215 nm, was ~60°C. At 75–80°C, CD spectra of the protein displayed a bathochromic shift in the negative band and a positive band below 210 nm,25 as a result of aggregate formation (Figure 1A bottom panel, ▾). This is consistent with the formation of intermolecular β-strands observed with FTIR spectroscopy.15
Figure 1.


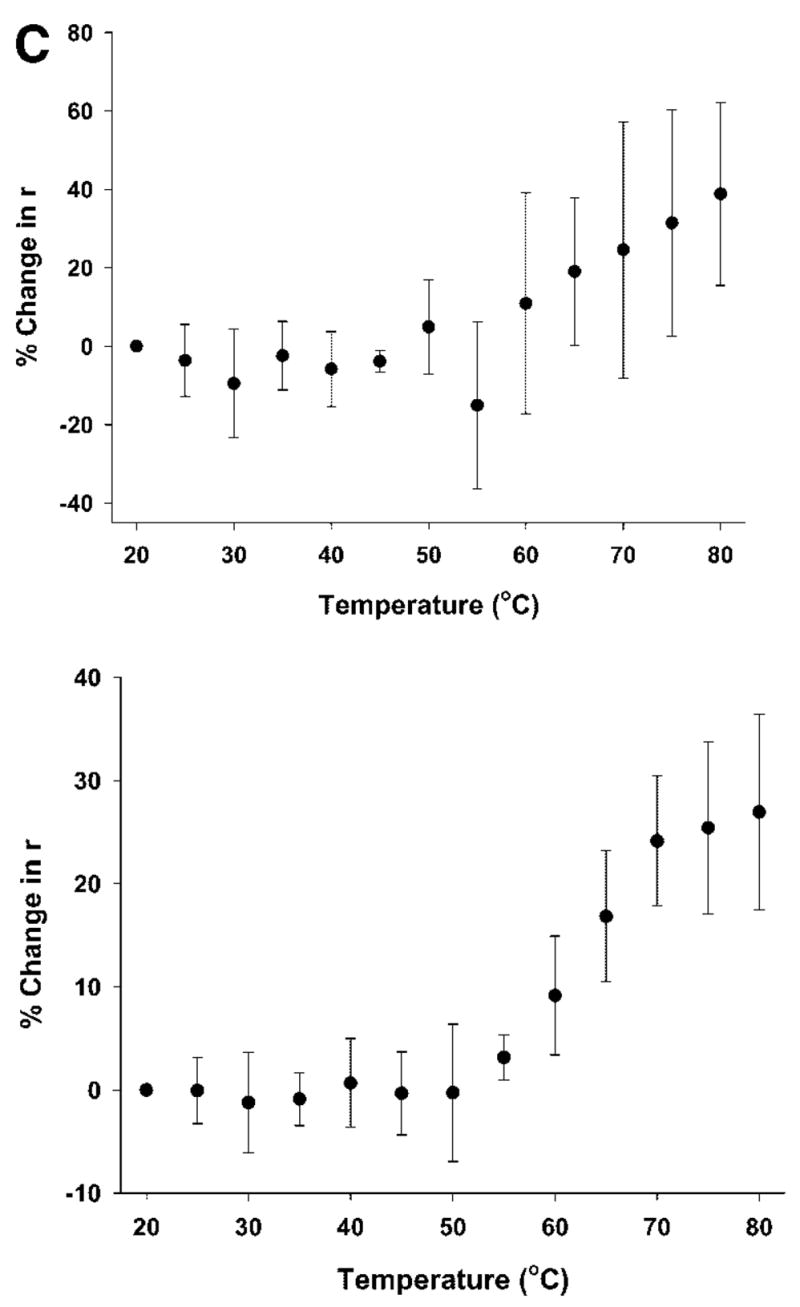
(A) Top panel: Change in the ellipticity of rFVIII (in 300 mM NaCl, 25 mM Tris, 5 mM CaCl2, pH 7.0 or in 300 mM NaCl, 10 mM MOPS, 5 mM CaCl2, pH 7.0 monitored at 215 nm, as a function of temperature: the secondary structure transition of rFVIII was monitored at 215 nm while heating the protein at a rate of 60°C/h. The protein concentration was ~20 μg/mL. Bottom panel: CD spectra of rFVIII after heating to various temperatures (●, 20°C; ○, 60°C; ▼, 80°C; ▽, 20°C, after cooling). (B) Size exclusion chromatographic profiles of rFVIII (in 300 mM NaCl, 25 mM Tris, 5 mM CaCl2, pH 7.0) heated to various temperatures at a heating rate of 60°C/h. The elution times are indicated in minute. The time scale on the abscissa is shown for the chromatogram at 25°C and is the same for SEC runs at other temperatures. Arrows indicates peak containing aggregates that coexist with the native protein. The temperature of the column was maintained at 20°C. (C) Steady-state fluorescence anisotropy of rFVIII (in 300 mM NaCl, 25 mM Tris, 5 mM CaCl2, pH 7.0, n = average of five measurements from three independent experiments ±SD, top panel, or in 300 mM NaCl, 10 mM MOPS, 5 mM CaCl2, pH 7.0, n = average of nine measurements from a representative experiment ±SD, bottom panel) as a function of temperature. The change in fluorescence anisotropy of rFVIII was monitored over the temperature range of 20–80°C at a heating rate of 1°C/min. The protein concentration was ~5 or 20 μg/mL.
The unfolding of rFVIII was also monitored using SEC analysis. Figure 1B shows SEC profiles of rFVIII heated at the same rate as the CD studies over the temperature range of 25–75°C. The native protein (at 25°C) eluted as a single broad peak at a retention time of ~6.56 min, whereas aggregated protein eluted in the void volume with a retention time of ~5.1–5.2 min. SEC analysis did not show any change in rFVIII heated to temperatures up to 45°C, consistent with the CD studies. For protein heated to 55°C however, two peaks were observed, one that eluted at ~5.64 min and the other at ~6.50 min. The peak at ~5.64 min probably consists of small aggregates. Under similar conditions, an increase in negative ellipticity at 215 nm was observed in the CD studies, suggesting an increase in the β-sheet content of the protein. This was consistent with previously reported results.15
When rFVIII was heated to 60°C (at 60°C/h, with a 2 min holding time) prior to SEC analysis, the broad peak at ~6.50 min (attributed to the native protein) disappeared, and a single peak was observed at ~5.32 min. Although SEC does not provide a high resolution separation of the aggregates, the retention time of the peak presumably containing aggregates was found to vary with temperature (indicated by arrows); the elution time decreased from ~5.64 min for protein heated at 55°C, to ~5.32 min for protein heated at 60°C, and ~5.22 min for protein heated at 65°C. The observed decrease in elution time (and corresponding decrease in peak width) with increasing temperature was interpreted as an increase in the size of the aggregates. In the case of protein heated to 75°C, the retention time of the peak decreased to ~5.16 min, and was comparable to the retention time of maximally-aggregated protein, which elutes in the void volume at ~5.1–5.2 min.
The observed decrease in retention time with increasing temperature was interpreted as an increase in the size of the aggregates. We hypothesize that the increase in the size of the aggregates with increasing temperature results from the incorporation of additional protein molecules into existing pool of aggregates. Based on the exclusion limit of the column used, the final aggregated form of the protein consists of at least six protein molecules. CD spectroscopy suggests the proteins in these aggregates are stabilized by intermolecular molecular β-strands (Figure 1A, bottom panel, ▾). It was not possible, however, to ascertain whether the aggregates were differentially enriched in FVIII light or heavy chains.
It has been shown that the heavily glycosylated B-domain is not critical for activity26 and that deletion of the B-domain minimizes the heterogeneity of the native protein due to reduced intracellular processing. To investigate whether heterogeneity of the native protein contributes to the initiation of temperature-dependent aggregation, studies were also carried out with a recombinant form of human FVIII in which the B-domain was deleted (rBDDFVIII). SEC indicated that the truncated protein was also susceptible to aggregation (data not shown) which is consistent with published reports.27 Therefore this type of heterogeneity does not appear to influence aggregation.
Steady state fluorescence anisotropy was used to determine whether changes in the molecular environment of Trp residues occurred in conjunction with the temperature-dependent transitions observed by CD and SEC. The anisotropy of the protein would be expected to change upon unfolding/aggregation as a result of changes in the fluorescence decay time, rotational correlation time, or both. Figure 1C shows the percent change in anisotropy (r) as a function of temperature as the protein was heated at 60°C/h. The anisotropy increased over the temperature range in which the transition was observed. Since the change in r is the weighted sum of the relative fluorescence intensities of unfolded and aggregated protein combined, the increase in r values suggests the existence of aggregated protein, particularly at temperatures >50°C.
Based on the CD, SEC, and fluorescence anisotropy studies described above of molecular events associated with thermally induced structural alterations of rFVIII, the initiation of the unfolding transition coincides with the formation of small aggregates. As the transition proceeds, the size of these aggregates increases and a final aggregated state is formed at elevated temperatures. The shape and the magnitude of the transition may reflect the relative percentages of the native and aggregated forms of the protein, which coexist during the transition.
The presence of aggregates in the unfolding pathway of a protein has been shown to contribute to irreversibility of the main transition.28 To investigate the reversibility of the transition, unfolding and refolding of the protein was carried out at a rate of 60°C/h. The protein was heated to 80°C, cooled to 20°C, and analyzed by CD. The refolding profile of the protein indicated that neither a native-like structure (Figure 1A, bottom panel, ▽) nor activity (data not shown) was recovered from the unfolded state. These results suggest not only that the transition was irreversible, but also that aggregation may be the cause of this irreversibility.
It is appropriate to mention here that the temperature induced unfolding studies were carried out in Tris buffer (containing 300 mM NaCl, 25 mM Tris, 5 mM CaCl2, pH 7). This buffer was chosen for its low metal binding capacity29 and is of particular relevance here, since the heavy and light chains of FVIII are held together by a metal ion (calcium or copper) that is critical for the activity of the protein. The high temperature coefficient of Tris1 however, will result in changes in pH at elevated temperatures. Thus, the observed changes in protein structure could be influenced by both temperature and pH. Therefore, thermal studies were also performed in MOPS buffer (containing 300 mM NaCl, 10 mM MOPS, 5 mM CaCl2, pH 7) in which the temperature coefficient is much lower (ΔpKa/°C = −0.006) and the metal ion binding capacity is higher than Tris.29 As shown in Figure 1A (top panel), the unfolding profile of rFVIII in MOPS buffer was similar to that observed in Tris buffer and consistent with previously published results.15 SEC (data not shown) and anisotropy (Figure 1C, bottom panel) profiles in MOPS buffer were also comparable. Based on these results, it is unlikely that changes in pH or differences in metal ion binding play a critical role in the initiation of aggregation. Rather, conformational change(s) over the temperature range of 45–50°C (prior to the main transition) are probably directly involved.
Conformational Changes Involved in the Initiation of Aggregation
Although proteins subjected to thermal stress can aggregate from fully unfolded state,31 this event usually occurs from only partially unfolded conformational states.32,33 Most often these latter states are molten-globule like states in which the protein retains much of its secondary structural features while losing most of its tertiary interactions. Such partially unfolded states have increased apolar character are thus more likely to aggregate than the native or unfolded states. To investigate whether formation of related states are responsible for the aggregation of rFVIII, far-(Figure 2A) and near-UV (Figure 2B) CD spectra were acquired in the temperature range of 45–50°C, conditions under which the aggregation process is initiated. Neither the far- nor near-UV CD showed any substantial change on heating the protein from 20 to 45°C, conditions under which the aggregation process is initiated. This suggested that the formation of molten globule like states is not likely to be involved in the initiation of aggregation of this protein and is consistent with what has been observed for other proteins such as recombinant human interferon gamma (rhIFN-γ)34 and recombinant human granulocyte colony stimulating factor (rhGCSF).35 It is important to note that our data does not rule out the involvement of transient molten globule like states in aggregation, which were not detectable with the techniques used in our studies.
Figure 2.

(A) Far-UV and (B) near-UV CD spectra of rFVIII (in 300 mM NaCl, 25 mM Tris, 5 mM CaCl2, pH 7.0) at the indicated temperatures. The protein concentration was ~20 μg/mL (far-UV CD) or ~0.5 mg/mL (near-UV CD).
To investigate the nature of the conformational changes that lead to aggregation, antibodies against specific protein domains were used in a sandwich ELISA system, which determines the binding affinity of a pair of antibodies. The ESH 4 antibody which recognizes an epitope comprised of residues 2303–2332 in the C2 domain,20 which encompasses the phospholipid-binding region21 or the 8860 antibody which recognizes an epitope in the A2 domain was used as the capture antibody to immobilize the protein. In both cases, the ESH 8 antibody, which recognizes an epitope comprised of residues 2248–2285 of the C2 domain22 was used as the probe antibody. Antibody binding to rFVIII at 20°C was used as a baseline to determine the relative binding observed at higher temperatures.
When ESH 4 and ESH 8 were used, antibody binding decreased significantly when the protein was heated to 37, 42.5 and 47°C (Figure 3A). Under these conditions, the protein retained its activity at the indicated temperatures (Figure 3A, inset). The reduction in antibody binding (~15%–20%) could be due to reduced affinity of the antibody as a result of conformational changes in the epitope region (2303–2332), or a reduction in the number of protein molecules in their native or native-like states as a consequence of aggregation. Aggregation could be ruled out, however, based on the temperature dependent (Figure 1B) SEC studies which indicate an absence of aggregates at temperatures below 50°C. Studies using 8860 and ESH 8 under identical conditions showed that the protein binding was not significantly affected as the temperature was increased to 47°C (Figure 3B). Thus, the ELISA studies suggest that the conformation of the C2 domain encompassing the lipid-binding region is probably altered as the temperature is increased from 20 to 47°C (see footnote).
Figure 3.

Antibody binding to rFVIII as determined by sandwich ELISA following the heating of rFVIII to the indicated temperatures. (A) Binding of ESH 4 (2303–2332) and ESH 8 (2248–2285) and (B) binding of 8860 (A2 specific) and ESH 8 (C2 domain 2248–2285) to rFVIII as determined by sandwich ELISA following the heating of rFVIII (in 300 mM NaCl, 25 mM Tris, 5 mM CaCl2, pH 7.0) at 60°C/h to the indicated temperatures. Inset: clotting time (measure of rFVIII activity) as measured by APTT assay. Statistical analysis of the data (*p <0.05) was carried out as described in the Experimental Procedure section.
The antibody binding experiments suggest the involvement of the lipid-binding domain in the initiation of the aggregation process. Because of the indirect nature of the experimental system employed, there are several possible explanations for this observation, some of which were discussed above. To further test this hypothesis, SEC (Figure 4) was carried out following incubation of the protein at 45°C in the presence of OPLS, which is known to bind to the lipid-binding region in the C2 domain.36 The rationale was that if conformational changes in the lipid-binding region are indeed involved in the initiation of the aggregation event, then a ligand capable of binding to this region might inhibit these changes and/or interfere with the aggregation process.
Figure 4.

Time-dependent changes in the SEC profile of rFVIII (300 mM NaCl, 25 mM Tris, 5 mM CaCl2, pH 7.0) in the presence and absence of OPLS. (a) Native protein at 20°C before heating; (b) protein heated at 45°C for 120 min; (c) protein heated at 45°C for 120 min in the presence of 10 mM OPLS. The elution time is indicated in minute.
In the absence of OPLS, SEC revealed the existence of smaller aggregates upon incubating rFVIII at 45°C for 2 h (Figure 4). Inclusion of 10 mM OPLS prevented the formation of these smaller aggregates, further supporting the involvement of the region containing residues 2303–2332 in aggregation. The precise mechanism by which OPLS interferes with the formation of small aggregates is unclear. It could involve either preferential binding of the ligand to the native state or shifting of the equilibrium towards the more compact native state, away from an aggregation competent species. A common approach used to determine the effect of a ligand on the thermodynamic stability of the native state involves determining the free energy of two-state unfolding (ΔGN-D) in the absence and presence of the ligand using chaotropes such as guanidine hydrochloride (GdnHCl).37,38 However, intrinsic fluorescence studies of rFVIII reported by Grillo et al.15 as a function of GdnHCl indicates the existence of multiple transitions in the folding/unfolding reaction. Therefore, it was not possible to ascertain the free energy change following binding of OPLS with rFVIII. Nevertheless, the equilibrium dissociation constant for the OPLS-rFVIII interaction was estimated to be ~5.7 μM by fluorescence spectroscopy which suggests that the interaction of the ligand with the protein is weak. Hence we do not anticipate OPLS to increase the thermodynamic stability of the native state significantly, although it interfered with the formation of intermolecular beta strands at elevated temperatures. Details of the interaction of OPLS with rFVIII will be published elsewhere (in press). The structural details of the C2 domain obtained from crystallography and site-directed mutagenesis indicate the presence of four hydrophobic loops (Figure 5).39–41 Thus, one possibility is that conformational alterations in these hydrophobic regions may participate in the initiation of aggregation. This observation is consistent with the fact that the lipid-binding region is also known to contribute to the binding of von Willebrand factor (vWf) in plasma. FVIII circulates as a non-covalent complex with vWf. One role of vWF may then be to shield this sticky membrane-binding region.41 Of course, our data does not rule out the involvement of other domains in the aggregation process.
Figure 5.
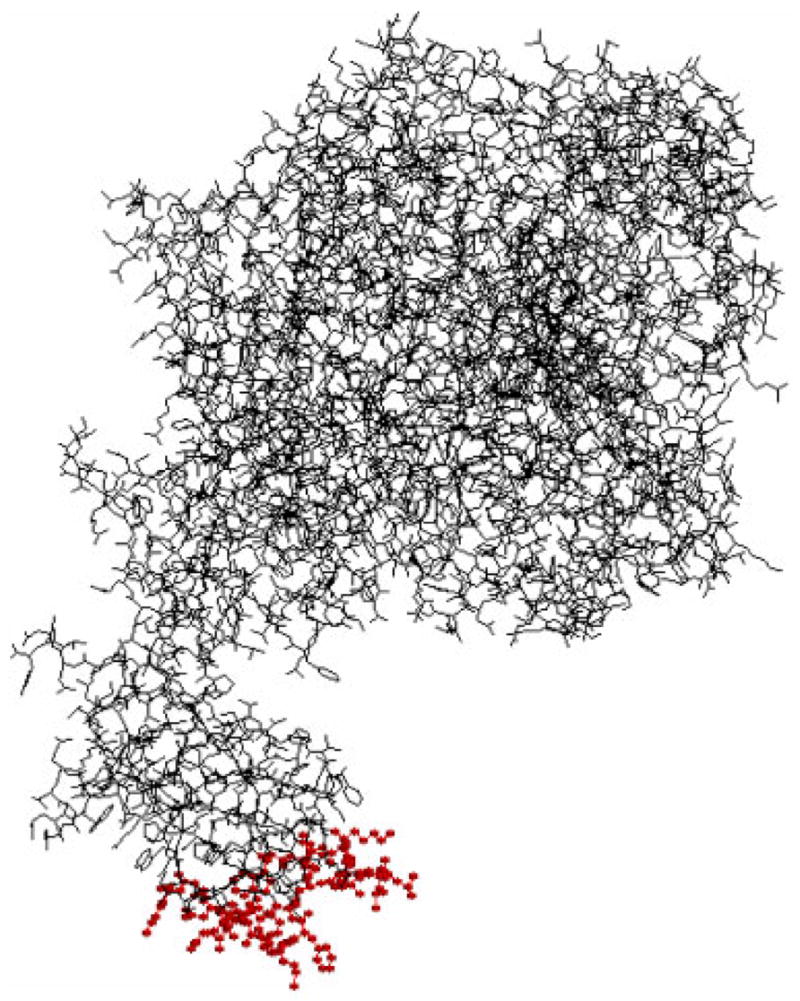
Molecular model of FVIII showing the proposed hydrophobic loops (represented as ball and stick in red) in the C2 domain: 2196–2201, 2222–2227, 2249–2255, and 2313–2315. The figure was reconstructed using Rasmol (Ver. 2.7.2.1) based on the coordinates available at http://europium.csc.mrc.ac.uks. [Color figure can be seen in the online version of this article, available on the website, www.interscience.wiley.com.]
The mechanisms by which proteins aggregate have both important practical and pharmaceutical implications. It is clear that the mechanisms of aggregation are complex. For proteins such as rhIFN-γ and rhGCSF aggregation can occur even under solution conditions that are considered to be physiological. 31 In many such cases, the rate-limiting step appears not to be the generation of extensively unfolded states. In the case of rhIFN-γ, aggregation has been shown to occur through the formation of a transiently expanded conformational species, whose surface area is greater than the native state by only 9%. 34 Similarly, native rhGCSF has been shown to aggregate through the formation of a monomeric transition state in which the surface area increase was only 15%.35 In the case of rFVIII, neither complete unfolding of the protein nor the generation of partially unfolded states of the protein appears to be a prerequisite for the protein to aggregate. Rather, aggregation appears to involve subtle conformational changes in the C2 domain encompassing the lipid-binding region.
Acknowledgments
This work was supported by NHLBI, National Institute of Health grant R01 HL-70227 02 to S.V.B. K.R. was supported by an unrestricted fellowship to the Department of Pharmaceutical Sciences (University at Buffalo) from Pfizer, Inc., Groton, CT. The authors thank the Pharmaceutical Sciences Instrumentation Facility of the University at Buffalo (SUNY) for the use of the Circular Dichroism spectropolarimeter, which was obtained through Shared Instrumentation Grants RR13665 from the National Center for Research Resources, National Institutes of Health. We thank Dr. J.P. Balthasar for providing access to the plate reader. The authors thank Dr. Geoffrey Kemball-Cook for the coordinates to generate the molecular model described in Figure 5.
Abbreviations
- APTT
activated partial thromboplastin time
- CD
circular dichroism
- ELISA
enzyme-linked immunosorbent assay
- GdnHCl
guanidine hydrochloride
- OPLS
O-phospho-L-Serine
- PB
phosphate buffer
- PBT
phosphate buffer containing tween
- PBA
phosphate buffer containing albumin
- rFVIII
recombinant human Factor VIII
- rBDDFVIII
recombinant human B-domain deleted FVIII
- rhGCSF
recombinant human granulocyte colony stimulating factor
- rhIFN-γ
recombinant human interferon gamma
- SEC
size exclusion chromatography
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- vWf
von Willebrand Factor
Footnotes
The pH decreases approximately by 0.03 units/°C from 5 to 25°C and by 0.025 units/°C from 25 to 37°C. Over the temperature range of 20–45°C, the pH was approximately estimated to change from 7.0 to 6.6. Over this pH range, the protein has been shown to be stable.30
References
- 1.Larner AJ. The molecular pathology of haemophilia. Q J Med. 1987;63:473–491. [PubMed] [Google Scholar]
- 2.Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM. Characterization of the human factor VIII gene. Nature. 1984;312:326–330. doi: 10.1038/312326a0. [DOI] [PubMed] [Google Scholar]
- 3.Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orr EC, Amphlett GW, Foster BW, Coe ML, Knutson GJ, Fass DN, Hewick RN. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984;312:342–347. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- 4.Vehar GA, Keyt B, Eaton D, Rodriguez H, O’Brien DP, Rotblat F, Oppermann H, Keck R, Wood WI, Harkins RN, Tuddenham EGD, Lawn D, Capon DJ. Structure of human factor VIII. Nature. 1984;312:337–342. doi: 10.1038/312337a0. [DOI] [PubMed] [Google Scholar]
- 5.Kaufman RJ, Wasley LC, Dorner AJ. Synthesis, processing, and secretion of recombinant human factor VIII expressed in mammalian cells. J Biol Chem. 1988;263:6352–6362. [PubMed] [Google Scholar]
- 6.Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92:3983–3996. [PubMed] [Google Scholar]
- 7.Foster PA, Zimmerman TS. Factor VIII structure and function. Blood Rev. 1989;3:180–191. doi: 10.1016/0268-960x(89)90015-5. [DOI] [PubMed] [Google Scholar]
- 8.Fay PJ. Factor VIII structure and function. Thromb Haemost. 1993;70:63–67. [PubMed] [Google Scholar]
- 9.Jaenicke R. Stability and folding of domain proteins. Prog Biophys Mol Biol. 1999;71:155–241. doi: 10.1016/s0079-6107(98)00032-7. [DOI] [PubMed] [Google Scholar]
- 10.Vermeer AW, Norde W. The thermal stability of immunoglobulin: Unfolding and aggregation of a multi-domain protein. Biophys J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Creighton TE. Protein folding. New York: W.H. Freeman and Co; 1999. p. xix.p. 547. [Google Scholar]
- 12.Braun A, Kwee L, Labow MA, Alsenz J. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon alpha (IFN-alpha) in normal and transgenic mice. Pharm Res. 1997;14:1472–1478. doi: 10.1023/a:1012193326789. [DOI] [PubMed] [Google Scholar]
- 13.Lollar P. Molecular characterization of the immune response to factor VIII. Vox Sang. 2002;83:403–408. doi: 10.1111/j.1423-0410.2002.tb05342.x. [DOI] [PubMed] [Google Scholar]
- 14.Saenko EL, Ananyeva NM, Kouiavskaia DV, Khrenov AV, Anderson JA, Shima M, Qian J, Scott D. Haemophilia A: Effects of inhibitory antibodies on factor VIII functional interactions and approaches to prevent their action. Haemophilia. 2002;8:1–11. doi: 10.1046/j.1365-2516.2002.00579.x. [DOI] [PubMed] [Google Scholar]
- 15.Grillo AO, Edwards KL, Kashi RS, Shipley KM, Hu L, Besman MJ, Middaugh CR. Conformational origin of the aggregation of recombinant human factor VIII. Biochemistry. 2001;40:586–595. doi: 10.1021/bi001547t. [DOI] [PubMed] [Google Scholar]
- 16.Doering C, Parker ET, Healey JF, Craddock HN, Barrow RT, Lollar P. Expression and characterization of recombinant murine factor VIII. Thromb Haemost. 2002;88:450–458. [PubMed] [Google Scholar]
- 17.Balasubramanian SV, Bruenn J, Straubinger RM. Liposomes as formulation excipients for protein pharmaceuticals: A model protein study. Pharm Res. 2000;17:344–350. doi: 10.1023/a:1007561308498. [DOI] [PubMed] [Google Scholar]
- 18.Over J. Methodology of the one-stage assay of Factor VIII (VIII:C) Scand J Haematol Suppl. 1984;41:13–24. doi: 10.1111/j.1600-0609.1984.tb02764.x. [DOI] [PubMed] [Google Scholar]
- 19.Purohit VS, Ramani K, Kashi RS, Durrani MJ, Kreiger TJ, Balasubramanian SV. Topology of factor VIII bound to phosphatidylserine-containing model membranes. Biochim Biophys Acta. 2003;1617:31–38. doi: 10.1016/j.bbamem.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Saenko EL, Scandella D. The acidic region of the factor VIII light chain and the C2 domain together form the high affinity binding site for von willebrand factor. J Biol Chem. 1997;272:18007–18014. doi: 10.1074/jbc.272.29.18007. [DOI] [PubMed] [Google Scholar]
- 21.Foster PA, Fulcher CA, Houghten RA, Zimmerman TS. Synthetic factor VIII peptides with amino acid sequences contained within the C2 domain of factor VIII inhibit factor VIII binding to phosphatidylserine. Blood. 1990;75:1999–2004. [PubMed] [Google Scholar]
- 22.Scandella D, Gilbert GE, Shima M, Nakai H, Eagleson C, Felch M, Prescott R, Rajalakshmi KJ, Hoyer LW, Saenko E. Some factor VIII inhibitor antibodies recognize a common epitope corresponding to C2 domain amino acids 2248 through 2312, which overlap a phospholipid-binding site. Blood. 1995;86:1811–1819. [PubMed] [Google Scholar]
- 23.Tsai PK, Volkin DB, Dabora JM, Thompson KC, Bruner MW, Gress JO, Matuszewska B, Keogan M, Bondi JV, Middaugh CR. Formulation design of acidic fibroblast growth factor. Pharm Res. 1993;10:649–659. doi: 10.1023/a:1018939228201. [DOI] [PubMed] [Google Scholar]
- 24.Remmele RL, Jr, Nightlinger NS, Srinivasan S, Gombotz WR. Interleukin-1 receptor (IL-1R) liquid formulation development using differential scanning calorimetry. Pharm Res. 1998;15:200–208. doi: 10.1023/a:1011902215383. [DOI] [PubMed] [Google Scholar]
- 25.Kendrick BS, Cleland JL, Lam X, Nguyen T, Randolph TW, Manning MC, Carpenter JF. Aggregation of recombinant human interferon gamma: Kinetics and structural transitions. J Pharm Sci. 1998;87:1069–1076. doi: 10.1021/js9801384. [DOI] [PubMed] [Google Scholar]
- 26.Toole JJ, Pittman DD, Orr EC, Murtha P, Wasley LC, Kaufman RJ. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci USA. 1986;83:5939–5942. doi: 10.1073/pnas.83.16.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fatouros A, Osterberg T, Mikaelsson M. Recombinant factor VIII SQ—inactivation kinetics in aqueous solution and the influence of disaccharides and sugar alcohols. Pharm Res. 1997;14:1679–1684. doi: 10.1023/a:1012163309468. [DOI] [PubMed] [Google Scholar]
- 28.Galisteo ML, Mateo PL, Sanchez-Ruiz JM. Kinetic study on the irreversible thermal denaturation of yeast phosphoglycerate kinase. Biochemistry. 1991;30:2061–2066. doi: 10.1021/bi00222a009. [DOI] [PubMed] [Google Scholar]
- 29.Derrick TS, Kashi RS, Durrani M, Jhingan A, Middaugh CR. Effect of metal cations on the conformation and inactivation of recombinant human factor VIII. J Pharm Sci. 2004;93:2549–2557. doi: 10.1002/jps.20167. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Kelner DN. Correlation of rFVIII inactivation with aggregation in solution. Pharm Res. 2003;20:693–700. doi: 10.1023/a:1023271405005. [DOI] [PubMed] [Google Scholar]
- 31.Chi EY, Krishnan S, Randolph TW, Carpenter JF. Physical stability of proteins in aqueous solution: Mechanism and driving forces in non-native protein aggregation. Pharm Res. 2003;20:1325–1336. doi: 10.1023/a:1025771421906. [DOI] [PubMed] [Google Scholar]
- 32.Manning MC, Patel K, Borchardt RT. Stability of protein pharmaceuticals. Pharm Res. 1989;6:903–918. doi: 10.1023/a:1015929109894. [DOI] [PubMed] [Google Scholar]
- 33.Fink AL. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 34.Kendrick BS, Carpenter JF, Cleland JL, Randolph TW. A transient expansion of the native state precedes aggregation of recombinant human interferon-gamma. Proc Natl Acad Sci USA. 1998;95:14142–14146. doi: 10.1073/pnas.95.24.14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnan S, Chi EY, Webb JN, Chang BS, Shan D, Goldenberg M, Manning MC, Randolph TW, Carpenter JF. Aggregation of granulocyte colony stimulating factor under physiological conditions: Characterization and thermodynamic inhibition. Biochemistry. 2002;41:6422–6431. doi: 10.1021/bi012006m. [DOI] [PubMed] [Google Scholar]
- 36.Gilbert GE, Drinkwater D. Specific membrane binding of factor VIII is mediated by O-phospho-L-serine, a moiety of phosphatidylserine. Biochemistry. 1993;32:9577–9585. doi: 10.1021/bi00088a009. [DOI] [PubMed] [Google Scholar]
- 37.Shirley BA. Protein stability and folding: Theory and practice. Totowa, N.J: Humana Press; 1995. pp. 177–190. [Google Scholar]
- 38.Zhang Y, Roy S, Jones LS, Krishnan S, Kerwin BA, Chang BS, Manning MC, Randolph TW, Carpenter JF. Mechanism for benzyl alcohol-induced aggregation of recombinant human interleukin-1 receptor antagonist in aqueous solution. J Pharm Sci. 2004;93:3076–3089. doi: 10.1002/jps.20219. [DOI] [PubMed] [Google Scholar]
- 39.Pratt KP, Shen BW, Takeshima K, Davie EW, Fujikawa K, Stoddard BL. Structure of the C2 domain of human factor VIII at 1.5 A resolution. Nature. 1999;402:439–442. doi: 10.1038/46601. [DOI] [PubMed] [Google Scholar]
- 40.Stoilova-McPhie S, Villoutreix BO, Mertens K, Kemball-Cook G, Holzenburg A. 3-Dimensional structure of membrane-bound coagulation factor VIII: Modeling of the factor VIII heterodimer within a 3-dimensional density map derived by electron crystallography. Blood. 2002;99:1215–1223. doi: 10.1182/blood.v99.4.1215. [DOI] [PubMed] [Google Scholar]
- 41.Gilbert GE, Kaufman RJ, Arena AA, Miao H, Pipe SW. Four hydrophobic amino acids of the factor VIII C2 domain are constituents of both the membrane-binding and von Willebrand factor-binding motifs. J Biol Chem. 2002;277:6374–6381. doi: 10.1074/jbc.M104732200. [DOI] [PubMed] [Google Scholar]