

Abstract
Helicobacter hepaticus strain 3B1 (H. hepaticus) contains a genomic island of ∼71 kb, HHGI1, with some of the common features shared among known bacterial pathogenicity islands. In this study, we characterized the pathogenic potential of HHGI1 by infecting B6.129-IL10tm1Cgn (IL10-/-) mice with an isogenic mutant (namely HhPAId1) lacking 19 predicted genes within HHGI1. In contrast to H. hepaticus (P < 0.001), HhPAId1did not cause typhlocolitis and hyperplasia in IL10-/- mice. Colonization levels of HhPAId1 were significantly higher in the cecum (P <0.007) and similar in the colon (P=0.27) when compared to H. hepaticus by 13 or 16 weeks post inoculation (WPI). The magnitude of the Th1-associated IgG2c response against HhPAId1 was less than that against H. hepaticus (P < 0.004). There was no significant difference in Th2-associated IgG1 responses against these two strains. Cecal and colonic mRNA levels of proinflammatory cytokines IFN-γ, TNF-α and IL-17a in the HhPAId1-infected mice were significantly lower than those in the H. hepaticus-infected mice (P <0.05) at 13 WPI. These results demonstrate that genes in the HHGI1 contribute to the pathogenicity of H. hepaticus, at least in part via up-regulation of proinflammatory mediators IFN-γ, TNF-α and IL-17a.
Keywords: H. HEPATICUS, ISOGENIC MUTANT, PATHOGENICITY ISLAND
1. Introduction
During microbial evolution, many bacterial genomes have acquired blocks of DNA-called “genomic islands” from other organisms by horizontal transfer (reviewed in [1]. These genomic islands can be divided into several subtypes based on their functionality, including ecological islands, saprophytic islands, symbiosis islands and pathogenicity islands (PAI) [1]. PAIs encode one or more virulence-associated factors and often have a G+C content distinct from the rest of the core genome [2]. Bacterial PAIs, such as the well characterized LEE in enteropathogenic Escherichia coli, cag in Helicobacter pylori and SaPI1 in Staphylococcus aureus, significantly contribute to the virulence of these pathogens [2].
Helicobacter hepaticus infection leads to chronic hepatitis, hepatocellular carcinoma and typhlocolitis in susceptible mouse strains [3, 4]. Recently, it has been demonstrated that this bacterium also contributes to the formation of cholesterol gallstones in C57L/J mice [5], colonic tumors in 129 Rag2-/- mice [6], and mammary tumors in Apcmin+ Rag2-/- mice [7]. However, our knowledge of virulence factors of this pathogen remains limited; the best characterized H. hepaticus virulence factor is cytolethal distending toxin (CDT) which is essential for persistent colonization, and plays an important role in the development of H. hepaticus-induced typhlocolitis in C57BL/6 IL10-/- mice [8] and hepatic dysplasia in male A/JCr mice [9]. A distinct genomic island of ∼71 kb (termed HHGI1) comprising 70 predicted genes was identified in the completely sequenced genome of H. hepaticus strain 3B1 (ATCC51449) [10]. HHGI1 displays several features of a bacterial PAI, including its relatively low G+C content as compared to the rest of the chromosome and a prophage P4-like integrase, and also like H. pylori cagPAI [11], contains several homologs (VirB10, VirB4, and VirD4) of components of the Agrobacterium tumefaciens type IV secretion system (T4SS). In addition, the presence of HHGI1 genes is highly variable among H. hepaticus isolates [10], a phenomenon also observed for the H. pylori cag PAI [11]. Importantly, male A/JCr mice infected with H. hepaticus strains lacking the entire HHGI1 (MIT 96-1809 isolated from mice originating in the Netherlands) or ∼62 out of 71 kb (MIT 96-284 from mice in Germany) developed less severe hepatitis than those infected with H. hepaticus 3B1 containing the intact HHGI1 [12]. These lines of evidence suggested that HHGI1 is a candidate PAI for H. hepaticus. In this study, we generated an isogenic mutant of H. hepaticus 3B1, in which a ∼20-kb portion of HHGI1 containing the VirB10 and VirB4 homologs was deleted. The effect of this deletion on colonization, pathogenicity and host proinflammatory responses was investigated in B6.129-IL10tm1Cgn mice.
2. Materials and Methods
2.1. Helicobacter hepaticus strains, growth media and conditions
H. hepaticus strain 3B1 (ATCC 51448) [3] was cultured on blood agar (Remel, Lexington, Kn) for 2-3 days under microaerobic conditions (10% H2, 10% CO2, 80% N2). Chloramphenicol (Cm)-resistant H. hepaticus mutants were selected on blood agar base supplemented with 10% horse blood and 10 mg/L of Cm.
2.2. Construction of isogenic mutants
In order to construct a mutant of H. hepaticus 3B1 where a block of HHGI1 genes (HH0250-HH0268) was deleted, two regions of approximately 2,000 bp were amplified by PCR, one 5′ of gene HH0250 with the primers hepI1P1fw (5′-cggggtaccTGTGGCTCATAAGGAGATCG-3′) and hepI1P1rv (ggaagatctATACCATTATA CCAAGCGACC) and a second one 3′ of the gene HH0268 with the primers hepI1P2fw (5′-ggaagatctTAACAGGAGTGGTAACACGG-3′) and hepI1P2rv (5′-cggggtaccAGCAGGTGC ATTGCCATTCC-3′). Capital letters in the primer sequences indicate homologous regions to the genome of H. hepaticus, bold letters KpnI-sites, and underlined letters BglII-sites. The PCR products were first digested with BglII, then one PCR-product was dephosphorylated and both PCR-products were ligated with each other (Fig.1). After cleanup, the ligation product was digested with KpnI and cloned into the KpnI site of pUC18 (pSUS2104). The chloramphenicol acetyltransferase (CAT) cassette of Campylobacter coli [13] was PCR-amplified from pBHpC8, digested with BamHI, and subsequently cloned into the compatible BglII sites created in pSUS2104, resulting in plasmid pSUS2105. Mutagenesis was performed by transformation of H. hepaticus with the mutant construct pSUS2105 by electroporation. Bacteria grown for 24 h on blood agar plates were harvested in electroporation buffer (10% glycerol), and washed two times by centrifugation at 5000 × g for 20 min. Bacteria were than resuspended in electroporation buffer. Bacterial suspension (80 μl) was mixed with 10-25 μg of dialysis-desalted DNA in a volume of 10 μl. . Electroporation was performed with electroporation cuvettes with a gap width of 0.1 cm (Biorad) and the following settings: a capacity of 25 μF, a voltage of 2.5 kV and a resistance of 2000 Ohm, resulting in a time constant between 4 and 4.5 sec. After electroporation, 500 μl of SOC-medium were added and the bacteria were incubated on non-selective blood agar plates for 1 day, followed by transfer of the bacteria to selective blood agar plates containing 10 mg/L Cm, and further incubated until Cm-resistant colonies appeared. These colonies were then propagated on new plates. Genetic characterization on chromosomal DNA isolated from the mutants was performed using PCR and sequencing.
Figure 1.

Construction of an isogenic deletion mutant within HHGI1. The hatched region denotes the genetic locus (HH0250 to HH0268, ∼21.7 kb) that was replaced with the Campylobacter coli chloramphemicol acetyltransferase gene (CAT) [13]. HH0252 and HH0246 in the deleted island segment display sequence similarity to Agrobacterium tumefaciens VirB10 and VirB4, respectively [10].
2.3. Experimental design for in vivo infection
Male Helicobacter spp.-free B6.129-IL10tm1Cgn (IL10-/-) mice (4 to 6-week-old) were originally purchased from the Jackson Laboratory (Bar Harbor, ME), rederived by embryo transfer, bred and housed in a specific pathogen-free (including Helicobacter spp.) facility. This facility is accredited by the Association for Accreditation and Assessment of Laboratory Animal Care, International. In the first study, the mice housed in static microisolator cages (6 for the control groups and 5 for the infection groups) were infected with H. hepaticus 3B1, or its PAI-deficient mutant (HhPAId1) in parallel with sham-dosed controls using Brucella broth for 16 weeks. A second study (5 mice per group) with a similar experimental design was performed to test reproducibility of the pathogenic potential of the mutant HhPAId1. In this experiment, necropsy was performed at 13 week post inoculation (WPI) due to deteriorating body conditions of the WT H. hepaticus-infected mice. For oral gavage, bacteria were cultured on blood agar, resuspended in Brucella broth, and adjusted to 109 organisms/ml as estimated by spectrophotometry at OD600nm. Mice received 0.2 ml of fresh inocula by gastric gavage every other day for three doses or sham dosed with Brucella broth.
H. hepaticus infection status was monitored using the Ready-To-Go PCR Bead system (GE Healthcare, Little Chalfont, England) and H. hepaticus-specific primers [14]. Immediately after CO2 euthanasia, blood was obtained and intestinal contents were removed by rinsing with sterile saline. Approximately 1-cm segments of cecum or colon were collected for RNA/DNA isolation. Tissues were frozen in liquid nitrogen immediately after sampling and stored at -70°C prior to use. Representative tissue sections were fixed in 10% buffered formalin for histology.
2.4. ELISA for serum IgG2c and IgG1 responses to H. hepaticus
Sera were obtained at necropsy from 10 IL-10-/- mice infected with HhPAId1 and 9 IL-10-/- mice infected with the WT H. hepaticus 3B1. Serum Th1-associated IgG2c and Th2-associated IgG1 responses to outer membrane antigens of H. hepaticus 3B1 were measured by ELISA as previously [15]. Antigen was coated on Immulon II plates at a concentration of 10 μg/ml with sera diluted 1:100. Biotinylated secondary antibodies included goat anti-mouse IgG (Southern Biotechnology Associates) and monoclonal anti-mouse antibodies produced by clones A85-1 and 5.7 (PharMingen) for detecting IgG1 and IgG2c, respectively. Incubation with extravidin peroxidase (Sigma) was followed by 2,2′-azinobis (3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt (ABTS®) substrate (Kirkegaard and Perry Laboratories, Gaithersburg, MD) for color development. Absorbance (or optical density) development at 405/562 nm was recorded by an ELISA plate reader (Dynatech MR7000, Dynatech Laboratories, Inc., Chantilly, VA).
2.5. Real-time quantification of H. hepaticus and selected cytokines in murine intestinal tissues
Chromosomal DNA from cultured bacteria was prepared using a High Pure PCR Template kit according to the manufacturer’s protocol (Roche Applied Science, Indianapolis, IN). Total DNA and RNA from cecum and colon were isolated using Trizol Reagents following the supplier’s procedure (Invitrogen). Quantification of H. hepaticus in each of the intestinal segments was determined by real-time quantitative PCR (Q-PCR) in the Prism Sequence Detection Systems 7700 (Applied Biosystems) as described elsewhere [16]. The copy numbers of H. hepaticus genomes were expressed per μg of mouse DNA whose quantity was determined by using Q-PCR and the 18S rRNA gene-based primers and probe (Applied Biosystems).
For cytokine mRNA quantification, 5 μg of total RNA from samples were converted into cDNA using High Capacity cDNA Archive kit following the supplier’s recommendation (Applied Biosystems). The cDNA levels for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IL-5, IL-13, IL-17a, IL-23a, TNFα, and IFNγ mRNA were measured by Q-PCR using commercial primers and probes (Applied Biosystems). Transcript levels were normalized to the endogenous control (GAPDH), and expressed as fold change compared with sham-dosed control mice using the Comparative CT method (Applied Biosystems User Bulletin no. 2).
2.6. Histopathology evaluation
The ileoceco-colic junction and colon were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin staining (H&E). Lesions were evaluated by two veterinary pathologists blinded to sample identity. The same scoring system was used as previously described [17].
2.7. Statistical analyses
The two-tailed Student’s t test or a nonparametric Mann-Whitney U test was used for comparing data on the colonization levels of H. hepaticus, cytokine mRNA levels and serology, or on histopathological scores. Values of P <0.05 are considered significant.
3. Results
3.1. Construction and characterization of an H. hepaticus HHGI1 partial deletion mutant
The HHGI1 island of H. hepaticus comprising the open reading frames HH0233-HH0302 presumably presents a pathogenicity island of H. hepaticus. This island is partially or completely missing in several clinical isolates of H. hepaticus [10] and we documented previously that two of these clinical isolates of H. hepaticus missing the HHGI1 island completely or partially induced a lower degree of hepatitis than a HHGI1-containing strain in A/JCr mice [12]. In order to verify that the HHGI1 island of H. hepaticus indeed is a pathogenicity island, a deletion mutant was constructed where the HHGI1 open reading frames HH0250-HH0268 were deleted. This part of the HHGI1 island was chosen because it comprises homologs to components of a T4SS, namely VirB10 (HH0252) and VirB4 (HH0260). The mutant was constructed by cloning approximately 2,000 bp of DNA on both sides of the chromosomal region to be deleted into pUC18, interspersed with a gene encoding CAT (Fig. 1). This construct (pSUS2105) was transformed into H. hepaticus 3B1 to generate a deletion mutant of HH0250-HH0268 by homologous recombination. The mutant called HhPAId1 was verified by an empty site PCR using primers binding 3′ and 5′ of the deleted region and by PCR using oligonucleotides binding in the deleted region (not shown). The mutant was examined microscopically and did not show any morphological differences compared to wild type bacteria. Also there were no differences in growth rate (not shown).
3.2. Partial deletion of HHGI1 did not reduce intestinal colonization levels of HhPAId1 compared to WT H. hepaticus
The successful colonization of WT H. hepaticus and HHGI1-defective HhPAId1 in the mice was monitored by fecal PCR at 4 and 8 WPI (data not shown). Colonization levels of H. hepaticus 3B1 and HhPAId1 for two infection experiments were combined and presented in Fig. 2. Deletion of the genetic locus containing HH0250 - HH0268 within the H. hepaticus HHGI1 did not decrease colonization levels of the mutant in the cecum and colon when compared to H. hepaticus 3B1. In fact, the colonization level of the mutant in the ceca was significantly higher (P = 0.007) than of H. hepaticus 3B1, whereas their levels were not significantly different in the colon.
Figure 2.
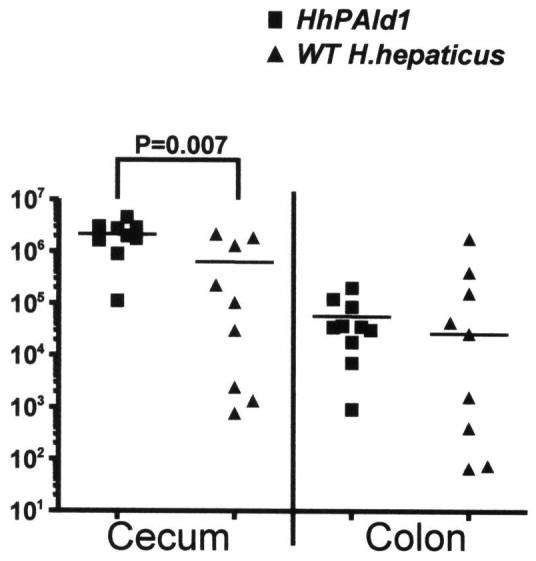
Colonization levels of HhPAId1 and WT H. hepaticus in the large bowel determined using Q-PCR. Numbers on the y axis represent the genome copies of H. hepaticus per μg mouse DNA.
3.3. HHGI-defective mutant lacks the ability to induce intestinal pathology
Mice infected with H. hepaticus 3B1 developed significantly more severe inflammation and hyperplasia in the cecocolic junction and the distal colon compared to mice infected with HhPAId1 (Fig. 3.1, P < 0.001). Seven of nine uninfected mice and 10 of 10 HhPAId1-infected mice had normal-appearing ceca and colons (not shown), with no development of typhlocolitis, hyperplasia or crypt atrophy after 13-16 WPI (Fig. 3.1 and 3.2). One mouse from the sham control group of each experiment also developed intestinal pathology comparable to the WT H. hepaticus-infected mice in the large bowel (Fig. 3.1); the development of these clinical presentations appeared to be spontaneous since H. hepaticus was not detected in the sham control mice by Q-PCR (data not shown). In contrast, in the WT H. hepaticus-infected group all 10 mice developed a moderate typhlitis, along with hyperplasia, crypt necrosis, mild crypt atrophy, early mild dysplastic glandular epithelium, submucosal inflammation and follicle formation and accompanying edema and ectatic lymphatic vessels (Fig. 3.2, C, D). Although lesions were most severe in the cecal-colic junction, the middle and distal colon had mild inflammation, occasional submucosal edema, and mild hyperplasia of the epithelium (not shown).
Figure 3.


Histopathological comparison among the mice infected with HhPAId1, WT H. hepaticus or sham dosed. 3.1. Pathological scores for the individual mice from the respective groups. P values, *< 0.05, **<0.01, *** <0.001. 3.2. H&E staining demonstrating representative pathological feaures. A nomal cecum from helicobacter-free male mouse. B. Normal cecum from a HhPAId1-infected mouse. C. Moderate diffuse typhlitis with glandular hyperplasia, mild glandular dysplasia, crypt atrophy, loss of goblet cells, a crypt abcess, and attenuated epithelium from H. hepaticus 3B1-infected mouse. D. Lower magnification of lesion in C; showing moderate mucosal inflammation and hyperplasia with submucosal inflammation and follicular formation along with severe submucosal edema and dilated lymphatic vessels. Bar = 100 μm in A-C, 250 μm in D.
3.4. Th1 or Th2-associated serum IgG isotype responses to H. hepaticus
The serological data combined from the two in vivo experiments demonstrated that the IL-10-/- mice infected with HhPAId1 or H. hepaticus 3B1 developed significantly higher levels of both Th1-associated IgG2c and Th2-associated IgG1 responses to H. hepaticus compared to the sham controls (Fig. 4, P <0.0001). In addition, the H. hepaticus - specific Th1-associated IgG2c response of the mice infected with H. hepaticus 3B1 was significantly higher than the mice infected with HhPAId1 (P <0.004), whereas there was no significant difference in the Th2-associated IgG1 responses to either infection (Fig. 4, P=0.14).
Figure 4.

Sera obtained at necropsy from 10 IL-10-/- mice infected with HhPAId1, and 9 IL-10-/- mice infected WT H. hepaticus. The Th1-associated IgG2c response to H. hepaticus antigens was lower in mice colonized by the mutant HhPAId1 (p<0.004). The Th2-associated IgG1 responses were equivalent (p=0.14). C represents uninfected control mice (n=10). Data represent mean ± standard error.
3.5. Partial deletion of HHGI1 diminished WT H. hepaticus-promoted host proinflammatory responses
Previous studies demonstrated that WT H. hepaticus infection is associated with Th1 cell-mediated immune responses and stimulates the production of TNF-α and IFN-γ [15, 18]. To dissect how the partial deletion of HHGI1 affects host immune responses, we measured cecal and colonic mRNA levels of 4 Th1/Th17 cytokines, including IL-17a, IL-23a, TNF-α and IFN-γ, as well as two Th2 cytokines, IL-5 and IL-13, in the second experiment (Fig. 5). H. hepaticus 3B1 infection significantly up-regulated mRNA levels of IL-17a (P = 0.028 for the cecum and <0.05 for the colon), TNF-α (P = 0.0003 for the cecum and P <0.05 for the colon) and IFN-γ (P = 0.0008 for the cecum and P=0.012 for the colon) when compared to the HHGI1-defective HhPAId1 infection; in contrast, there was no significant difference in the mRNA levels of IL-23a between the WT H. hepaticus- and the HhPAId1-infected groups (Fig. 5). The mRNA levels of cecal IL-17a were significantly higher in the both infected groups than the controls (Fig. 5). In spite of the notable higher levels on mean folds of the TNF-α and IFN-γ mRNA in the H. hepaticus 3B1-infected mice compared to the sham-dosed mice, they were statistically insignificant, which is due to the spontaneous inflammation that developed in one of four control mice, leading to the large standard deviation of the data for the control mice (Fig. 5). After removal of this mouse from the control group, the mRNA levels of cecal or colonic TNF-α and IFN-γ between the control mice and the mice infected with WT H. hepaticus but not with HhPAId1 were significantly different (P < 0.05, data not shown). For Th2 cytokines, H. hepaticus 3B1 infection induced significantly higher cecal mRNA levels of IL-5 and IL-13 than HhPAId1 infection or the sham control (Fig. 5, P < 0.05); the mRNA levels of these two cytokines in the colon were not significantly different between the groups.
Figure 5.

Relative mRNA levels of proinflammatory and inflammation-suppressive mediators determined by relative Q-PCR. The target mRMA was normalized to that of the “house-keeping: gene” Gapdh. Numbers represent mean fold change of the individual mRNA levels in reference to the control group (defined as 0 meaning no change) whose standard deviations of the individual cytokines are represented with the hatched boxes. Bars, standard deviations for the infection groups. P values when compared to the controls: * <0.05, **<0.01.
4. Discussion
The ∼71 kb genomic island HHGI1 present in the genome of H. hepaticus ATCC51449 displays many common features of bacterial PAIs and was proposed as a candidate PAI for H. hepaticus [10]. Our previous study demonstrating that H. hepaticus strains naturally lacking the entire or partial HHGI1 induced less severe chronic hepatitis in A/JCr mice provides supporting evidence for this proposal [12]. Here we demonstrate that the partial deletion of HHGI1 abolished the ability of H. hepaticus to induce typhlocolitis and hyperplasia in IL10-/- mice in contrast to H. hepaticus 3B1, linking HHGI1 to the pathogenicity of H. hepaticus. These collective data strongly indicate that HHGI1 is a pathogenicity island for H. hepaticus.
The region deleted from HHGI1 was not required for H. hepaticus colonization in the large bowel. This result is similar to the previous finding that the H. pylori cagPAI is dispensable for colonization in mice and piglets [19]. Interestingly, the mean colonization level of the HHGI1-deficient HhBac26 was significantly higher than that for H. hepaticus 3B1. The lower colonization density reached by H. hepaticus 3B1 compared to HhPAId1 in the cecum may result from up-regulation of proinflammatory mediators such as IFN-γ by H. hepaticus 3B1. A similar up-regulation of proinflammatory mediators has been demonstrated to promote clearance of H. pylori and Yersinia enterocolitica from mice [20-22]. These data are supported by our previous finding that ceca of hepatitis-resistant C57BL/6 mice contains more H. hepaticus organisms (∼100-fold) than chronic hepatitis/typhlocolitis-prone A/JCr mice [15, 23, 24].
Although the colonization levels of HhPAId1 in the cecum were higher than those of H. hepaticus 3B1, the H. hepaticus-specific Th1-associated IgG2c responses to the mutant were significantly lower than to WT H. hepaticus infection, suggesting that the gene products encoded by HHGI1 are important for causing proinflammatory Th1-type host responses to infection. Importantly, this isogenic mutant lost its ability to induce intestinal pathology and also did not up-regulate mRNA levels of IFN-γ and TNF-α when compared to WT H. hepaticus. IFN-γ and TNF-α are pleiotropic proinflammatory mediators involving multiple cellular functions. For example, TNF-α modulates a proinflammatory NF-κB pathway and apoptosis, and IFN-γ enhances natural killer cell and macrophage activity against microbial infection as well as regulation of B cell functions (reviewed in Ref. [25, 26]. It has been thoroughly documented that up-regulation of IFN-γ and TNF-α are often associated with H. hepaticus-induced diseases in susceptible murine models [7, 15, 18, 23, 27, 28]. In addition, neutralization of IFN-γ inhibited the development of H. hepaticus-induced typhlocolitis in C57BL/10 IL10-/- and C57BL/10 Rag-/- mice [18, 27]; treatment with anti-TNF-α antibody suppresses intestinal tumors induced by H. hepaticus infection in C57BL/6J Rag2-/-ApcMin/+ mice [7]. These findings indicate the importance of IFN-γ and TNF-α in the pathogenesis of H. hepaticus-induced diseases. It is worth noting that the mRNA levels of IL-5 and IL-13 were significantly higher in the WT H. hepaticus-infected ceca affected by severe inflammation compared to the HHGI1-deficient mutant-infected or sham dosed mice. These findings are likely due to compensatory host responses to counteract the induced tissue injuries and is consistent with a previous study reporting up-regulation of both Th1- and Th2-type cytokines in H. hepaticus-infected A/JCr mice [23]. Therefore, the results from this study ascertain that HHGI1 contributes to pathogenicity of H. hepaticus, at least in part, by up-regulating expression of IFN-γ and TNF-α and a Th1 response.
Recently, cytokines IL-23 and IL-17 have been demonstrated to play a pivotal role in innate immunity and T cell - mediated intestinal inflammation in immune-deficient mouse models [29] [28]. Neutralization of IL-23 or knockout of the IL-23 gene diminishes intestinal inflammation as well as elevated production of IL-17 induced by H. hepaticus infection or by adoptive transfer of effector CD4+ T cells in C57BL/6 Rag-/- mice [28]. Current evidence indicates that IL-23 is critical for IL-17-expressing Th17 function in vivo [30]. In this study, mRNA expression of cecal and colonic IL-17a was significantly increased by WT H. hepaticus infection compared to HhPAId1 infection (Fig. 5); however, no significant differences in mRNA levels of IL-23a were noted among the H. hepaticus 3B1-infected, the HhPAId1-infected and the sham control groups (Fig. 5). Intriguingly, the mRNA level of cecal IL-17 in the HhPAId1-infected mice was higher by 25-fold than that in the sham controls (Fig. 5), suggesting that additional factors from the remaining portion of HhPAId1 or other virulence properties are also involved the transcriptional up-regulation of IL-17a. We hypothesize that the H. hepaticus-associated up-regulation of the IL-17a mRNA is induced by an alternative pathway proposed by Weaver et al [30], in which IL-17 production could be induced directly by T cell receptor stimulation by bacterial antigens independent of IL-23. Further investigations are required for testing this hypothesis.
In summary, our data provided strong evidence supporting that HHGI1 is a H. hepaticus pathogenicity island [10]. Infection with this HhPAI mutant in other rodent models such as A/JCr mice for liver tumor development and Rag2-/- mice for progression of colitis to lower bowel cancer will further delineate virulence properties of HHGI1. This conclusion also opens an avenue for further characterizing virulence factors encoded by HHGI1 and comparing the pathogenic strategies utilized by different helicobacters (e.g, H. hepaticus versus H. pylori) to trigger inflammation and tumorigenesis in these respective hosts.
5. Acknowledgements
This project was financially supported by NIH grants R01 CA67529, P30ES02109 (J.G.F.) R01 DK52413 (DBS), and by grant SFB621/B8 from the Deutsche Forschungsgemeinschaft (S.S.) and by the PathoGenoMik initiative of the German Ministry for Education and Research (S.S. and C.J.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2001;2:376–381. doi: 10.1093/embo-reports/kve097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gal-Mor O, Finlay BB. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell. Microbiol. 2006;8:1707–1719. doi: 10.1111/j.1462-5822.2006.00794.x. [DOI] [PubMed] [Google Scholar]
- [3].Fox JG, Dewhirst FE, Tully JG, Paster BJ, Yan L, Taylor NS, Collins MJ, Jr., Gorelick PL, Ward JM. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J. Clin. Microbiol. 1994;32:1238–1245. doi: 10.1128/jcm.32.5.1238-1245.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rogers AB, Fox JG. Inflammation and Cancer. I. Rodent models of infectious gastrointestinal and liver cancer. Am. J. Physiol. 2004;286:G361–366. doi: 10.1152/ajpgi.00499.2003. [DOI] [PubMed] [Google Scholar]
- [5].Maurer KJ, Ihrig MM, Rogers AB, Ng V, Bouchard G, Leonard MR, Carey MC, Fox JG. Identification of cholelithogenic enterohepatic helicobacter species and their role in murine cholesterol gallstone formation. Gastroenterology. 2005;128:1023–1033. doi: 10.1053/j.gastro.2005.01.008. [DOI] [PubMed] [Google Scholar]
- [6].Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, Horwitz BH, Fox JG. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am. J. Pathol. 2003;162:691–702. doi: 10.1016/S0002-9440(10)63863-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, Horwitz BH, Fox JG, Erdman SE. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 2006;66:7395–7400. doi: 10.1158/0008-5472.CAN-06-0558. [DOI] [PubMed] [Google Scholar]
- [8].Young VB, Knox KA, Pratt JS, Cortez JS, Mansfield LS, Rogers AB, Fox JG, Schauer DB. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 2004;72:2521–2527. doi: 10.1128/IAI.72.5.2521-2527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ge Z, Rogers AB, Feng Y, Lee A, Xu S, Taylor NS, Fox JG. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 2007;9:2070–2080. doi: 10.1111/j.1462-5822.2007.00939.x. [DOI] [PubMed] [Google Scholar]
- [10].Suerbaum S, Josenhans C, Sterzenbach T, Drescher B, Brandt P, Bell M, Droge M, Fartmann B, Fischer HP, Ge Z, Horster A, Holland R, Klein K, Konig J, Macko L, Mendz GL, Nyakatura G, Schauer DB, Shen Z, Weber J, Frosch M, Fox JG. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. P. Natl. Acad. Sci. U.S.A. 2003;100:7901–7906. doi: 10.1073/pnas.1332093100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. P. Natl. Acad. Sci. U.S.A. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Boutin SR, Shen Z, Rogers AB, Feng Y, Ge Z, Xu S, Sterzenbach T, Josenhans C, Schauer DB, Suerbaum S, Fox JG. Different Helicobacter hepaticus strains with variable genomic content induce various degrees of hepatitis. Infect. Immun. 2005;73:8449–8452. doi: 10.1128/IAI.73.12.8449-8452.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang Y, Taylor DE. Chloramphenicol resistance in Campylobacter coli: nucleotide sequence, expression, and cloning vector construction. Gene. 1990;94:23–28. doi: 10.1016/0378-1119(90)90463-2. [DOI] [PubMed] [Google Scholar]
- [14].Shames B, Fox JG, Dewhirst F, Yan L, Shen Z, Taylor NS. Identification of widespread Helicobacter hepaticus infection in feces in commercial mouse colonies by culture and PCR assay. J. Clin. Microbiol. 1995;33:2968–2972. doi: 10.1128/jcm.33.11.2968-2972.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Whary MT, Morgan TJ, Dangler CA, Gaudes KJ, Taylor NS, Fox JG. Chronic active hepatitis induced by Helicobacter hepaticus in the A/JCr mouse is associated with a Th1 cell-mediated immune response. Infect. Immun. 1998;66:3142–3148. doi: 10.1128/iai.66.7.3142-3148.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ge Z, Feng Y, Whary MT, Nambiar PR, Xu S, Ng V, Taylor NS, Fox JG. Cytolethal distending toxin is essential for Helicobacter hepaticus colonization in outbred Swiss Webster mice. Infect. Immun. 2005;73:3559–3567. doi: 10.1128/IAI.73.6.3559-3567.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, Schauer DB. CD4+ CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res. 2005;65:3998–4004. doi: 10.1158/0008-5472.CAN-04-3104. [DOI] [PubMed] [Google Scholar]
- [18].Kullberg MC, Ward JM, Gorelick PL, Caspar P, Hieny S, Cheever A, Jankovic D, Sher A. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 1998;66:5157–5166. doi: 10.1128/iai.66.11.5157-5166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Eaton KA, Kersulyte D, Mefford M, Danon SJ, Krakowka S, Berg DE. Role of Helicobacter pylori cag region genes in colonization and gastritis in two animal models. Infect. Immun. 2001;69:2902–2908. doi: 10.1128/IAI.69.5.2902-2908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Autenrieth IB, Beer M, Bohn E, Kaufmann SH, Heesemann J. Immune responses to Yersinia enterocolitica in susceptible BALB/c and resistant C57BL/6 mice: an essential role for gamma interferon. Infect. Immun. 1994;62:2590–2599. doi: 10.1128/iai.62.6.2590-2599.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Autenrieth IB, Kempf V, Sprinz T, Preger S, Schnell A. Defense mechanisms in Peyer’s patches and mesenteric lymph nodes against Yersinia enterocolitica involve integrins and cytokines. Infect. Immun. 1996;64:1357–1368. doi: 10.1128/iai.64.4.1357-1368.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee CW, Rao VP, Rogers AB, Ge Z, Erdman SE, Whary MT, Fox JG. Wild-type and Interleukin-10-deficient regulatory T cells reduce effector T-cell-mediated gastroduodenitis in Rag2-/- Mice, but only wild-type regulatory T cells suppress Helicobacter pylori gastritis. Infect. Immun. 2007;75:2699–2707. doi: 10.1128/IAI.01788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Livingston RS, Myles MH, Livingston BA, Criley JM, Franklin CL. Sex influence on chronic intestinal inflammation in Helicobacter hepaticus-infected A/JCr mice. Comparative Med. 2004;54:301–308. [PubMed] [Google Scholar]
- [24].Myles MH, Livingston RS, Livingston BA, Criley JM, Franklin CL. Analysis of gene expression in ceca of Helicobacter hepaticus-infected A/JCr mice before and after development of typhlitis. Infect. Immun. 2003;71:3885–3893. doi: 10.1128/IAI.71.7.3885-3893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukocyte Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- [26].Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, De Smaele E, Tang WJ, D’Adamio L, Franzoso G. Gadd45β mediates the NF-κB suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004;6:146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- [27].Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, Sher A. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]