

Abstract
Defects in ankyrin underlie many hereditary disorders involving the mislocalization of membrane proteins. Such phenotypes are usually attributed to ankyrin's role in stabilizing a plasma membrane scaffold, but this assumption may not be accurate. We found in Madin-Darby canine kidney cells and in other cultured cells that the 25-residue ankyrin-binding sequence of α1-Na+-K+-ATPase facilitates the entry of α1,β1-Na+-K+-ATPase into the secretory pathway and that replacement of the cytoplasmic domain of vesicular stomatitis virus G protein (VSV-G) with this ankyrin-binding sequence bestows ankyrin dependency on the endoplasmic reticulum (ER) to Golgi trafficking of VSV-G. Expression of the ankyrin-binding sequence of α1-Na+-K+-ATPase alone as a soluble cytosolic peptide acts in trans to selectively block ER to Golgi transport of both wild-type α1-Na+-K+-ATPase and a VSV-G fusion protein that includes the ankyrin-binding sequence, whereas the trafficking of other proteins remains unaffected. Similar phenotypes are also generated by small hairpin RNA-mediated knockdown of ankyrin R or the depletion of ankyrin in semipermeabilized cells. These data indicate that the adapter protein ankyrin acts not only at the plasma membrane but also early in the secretory pathway to facilitate the intracellular trafficking of α1-Na+-K+-ATPase and presumably other selected proteins. This novel ankyrin-dependent assembly pathway suggests a mechanism whereby hereditary disorders of ankyrin may be manifested as diseases of membrane protein ER retention or mislocalization.
Keywords: spectrin, membrane protein, endoplasmic reticulum retention, Golgi, cytoskeleton, disease, endoplasmic reticulum
disorders involving the accumulation of misfolded proteins in the endoplasmic reticulum (ER) or their redirection within the secretory pathway are now well recognized. Many pathological conditions are also now recognized as failures of membrane protein trafficking or localization, either due to retention within the secretory pathway or by targeting to the wrong cellular compartment (for brief reviews, see Refs. 1 and 19). While such conditions frequently involve mutations in the transported proteins themselves, problems also arise from defects in protein processing or in associated cytosolic proteins that form the coats or scaffolds that mediate transport through the secretory pathway or anchor them to the membrane. One such scaffold is the spectrin-ankyrin skeleton.
Deletion of specific isoforms of ankyrin in mice leads to impaired accumulation of the voltage-gated Na+ channel in the sarcoplasmic reticulum of cardiac muscle, accounting for delayed cardiac repolarization and a vulnerability to lethal cardiac arrhythmias (31). Similar defects in human pedigrees account for the sudden death syndrome termed long-QT type 4 (32). Mutations in spectrin may also contribute to similar pathologies. Beyond the well-known spectrin (or ankyrin) mutations that cause hemolytic disease (15), mutations in βIII spectrin link to one variant of spinal-cerebellar ataxia type V (21), a condition most often associated with K+ or Ca2+ channel defects in Purkinje cell neurons (28). Similarly, autoantibodies to spectrin βIV, as seen in some paraneoplastic syndromes, disrupts the organization of Na+ channels at the nodes of Ranvier with neurological sequelae (6). Mutations in βIV spectrin in mice lead to deafness and a shivering phenotype (35). Total loss of βI spectrin in the ja/ja mouse, beyond a fatal hemolytic condition, also leads to altered α-Na+-K+-ATPase expression and disposition in skeletal muscle (48). In these conditions involving spectrin or ankyrin, proteins that typically act in unison, the pathology has been assumed (but never proven) to be a consequence of the loss of the stabilizing spectrin-ankyrin infrastructure at the plasma membrane with a consequential loss of membrane protein organization. Yet, in nonerythroid cells, spectrin and ankyrin are also associated with many intracellular organelles besides the plasma membrane and have a broader role than simply membrane stabilization (8). Spectrin can tether protein and lipid vesicles to the dynein-dynactin complex (20) and thereby enable the directed transport of organelles and vesicles along microtubules such as occurs with anterograde transport to the Golgi (40) or with retrograde traffic along the axon (33). Recombinant peptides of spectrin transfected into cultured cells also act in a dominant negative way to impair the delivery of α1-Na+-K+-ATPase to the Golgi (11), and similar peptides including those that block the interaction of spectrin with ankyrin impair CD45 delivery to the plasma membrane in Jurkat T cells (39). In the present study, we focused on ankyrin and examined whether ankyrin, like spectrin, might contribute not just to the stabilization of the plasma membrane but also to the biogenesis of specialized membrane domains by facilitating the passage of α1-Na+-K+-ATPase through the secretory pathway.
Previous studies have established that spectrins and ankyrins can be associated with the Golgi and with other organelles (2, 3, 11, 13, 14, 16, 43) and that (at least at the plasma membrane) α1-Na+-K+-ATPase binds directly to ankyrin (10, 23), predominately through a 25-residue sequence in its cytoplasmic domain (49). We termed this 25-residue sequence in α1- Na+-K+-ATPase “MAB” for “minimal ankyrin binding.” We found that deletion of this sequence from α1-Na+-K+-ATPase abrogates its ability to bind ankyrin, that expression of MAB as a soluble peptide in the cytosol of Madin-Darby canine kidney (MDCK) cells dominantly inhibits the transport of native α1-Na+-K+-ATPase from the ER to the Golgi, that the MAB sequence fused to vesicular stomatitis virus G protein (VSV-G) provides a positive ER to Golgi transport signal, and that proteins containing this 25-residue sequence require ankyrin for their efficient movement to the Golgi [as determined by both small hairpin (sh)RNA suppression of ankyrin expression as well as in semipermeabilized cells reconstituted with ankyrin-depleted vs. ankyrin-enriched cytosol]. These experiments identified a novel role for ankyrin in facilitating protein transport in the secretory pathway and defined a heretofore unknown mechanism relevant to the pathology of hereditary disorders of ankyrin characterized by misorting or ER retention of selected membrane proteins.
MATERIALS AND METHODS
Antibodies.
Rabbit anti-VSV-G polyclonal antibody was from Dr. John Rose (Yale University), anti-green fluorescent protein (GFP) murine monoclonal antibody was from Clontech (no. 632380), anti-Flag (M2) and anti-α1-Na+-K+-ATPase monoclonal antibodies were from Sigma, and anti-β1-Na+-K+-ATPase polyclonal antibody was a gift from Dr. Michael Caplan (Yale University). Antibodies to ankyrin were ankyrin G (Santa Cruz Biotechnology), monoclonal antibodies to ankyrin R (Santa Cruz Biotechnology and Research Diagnostics), and a polyclonal antibody to ankyrin R (7).
Expression constructs.
The tso45-VSV-G-enhanced (e)GFP construct (clone 328) was assembled by amplifying tso45-VSV-G cDNA using oligonucleotide 30305 (5′-CGGGATCCACCATGAAGTGCCTTTTGTAC-3′), which incorporated a BamHI site and a Kozak consensus sequence ahead of the ATG start codon, and antisense oligonucleotide 29886 (5′-CGGAATTCCTTTCCAAGTCGGTTCAT-3′), which removed the stop codon and provided an EcoRI site for cloning into pcDNA3.1 ZEO (Invitrogen). The VSV-G-pcDNA3.1 ZEO plasmid was digested with EcoRI, treated with Mung Bean nuclease, buffer exchanged, and digested with BamHI. This fragment was ligated into pEGFP-N1 (Clontech) that had been previously digested with BamHI, treated with Mung Bean Nuclease, buffer exchanged, and digested again with BglII. A similar strategy was used to make cytoplasmic domain-deleted tso45-VSV-G-pEGFP (clone 330) but using antisense oligonucleotide 55633 (5′-CGGAATTCATAAATACCAACTCGGAG-3′), which deleted the last 24 amino acids of VSV-G. tso45-VSV-MAB-pEGFP (clone 337) was assembled by amplifying MAB from a human testis marathon ready cDNA library (Clontech) using sense oligonucleotide 55628 (5′-CGGAATTCTCCTACTATCAAGAAGCTAA-3′) and antisense oligonucleotide 29889 (5′-CGGAATTCTTAATGTTCAATTTCTGCAGCGATGG-3′), both with 5′ EcoRI sites. The amplimer was digested with BstBI, blunt ended with Klenow enzyme, buffer exchanged, digested with EcoRI, and ligated with cytoplasmic domain-deleted VSV-G cDNA as a trimolecular ligation into pEGFP-3N. Several random mutants were prepared as negative controls using sequences that did not bestow transport but had equivalent amino acids in the cytoplasmic domain as MAB. The MAB sequence was inserted in frame to the COOH-terminal end of GFP or at the NH2-terminal end of GFP to express the MAB peptide alone. All constructs were shuttled from pEGFP vectors into pEYFP and pECFP vectors when needed to discriminate multiple chromophores.
Flag-tagged wild type (WT) and ΔMAB α1-Na+-K+-ATPase were prepared from rat α1-Na+-K+-ATPase cDNA in pGEM4 (9). Initially, the 5′-end was amplified by PCR with an oligonucleotide that incorporated a HinDIII site upstream of the ATG start codon (5′-GGAAGCTTGCCGCCACCATGGGGAAGGGGGTTGGACG-3′). The 3′-end of the cDNA was amplified with an antisense oligonucleotide (5′-GGAAGCTTTCACTTGTCATCGTCGTCCTTGTAGTCGTAGTAGGTTTCCTTCTCCACCCAGCC-3′) that replaced the stop codon with an in-frame Flag sequence followed by an exogenous stop codon and XbaI restriction site. After the modified ends had been ligated back into pGEM4, the entire cDNA was digested with HinDIII and XbaI and subcloned into pcDNA3. Deletion of MAB was accomplished by PCR with oligonucleotides designed to loop out the MAB sequence (sense 5′-CATAACTGGCTGTTTCTCCTACTATATTCGAAATGGAGAGAAGATGAGC-3′ and antisense 5′-GCTCATCTTCTCTCCATTTCGAATATAGTAGGAGAAACAGCCAGTTATG-3′). The resulting amplimer was then digested with HinDIII and BstBI and replaced into the WT ATPase pcDNA3 plasmid. All constructs were sequence verified at the Keck DNA sequence facility at Yale University.
Coexpression of tso45-VSV-MAB-eGFP with ankyrin shRNA.
shRNA pSM2c plasmids directed against human ankyrin R (5′-GAATATTCTGAAACTCATTCCC-3′) and ankyrin G (5′-CGGGACAGCATGATGATTGAA-3′) were purchased from Open Biosystems along with a control plasmid encoding nonsense shRNA and proprietary Arrest-in transfection reagent. Sequences that were constituitively expressed in all ankyrin transcripts were targeted so as to avoid selectively deleting just a subset of the many isoforms of ankyrin that arise by alternative splicing. Sequences were also chosen that were common to both human and Canis familiaris to facilitate knockdown in both HEK-293F and MDCK cell lines. It was not possible to find a sequence simultaneously compatible with humans, dogs, and monkeys, and, for that reason, this work was not performed in COS-7 cells. Several constructs were purchased for each ankyrin gene that fit these criteria, but only those that gave a significant and reproducible reduction in ankyrin expression by Western blot analysis were used in this study. For transient expression, a shRNA plasmid was cotransfected at an 8:1 molar ratio with tso45-VSV-eGFP constructs into either HEK or MDCK cells that had been grown for 24 h on MatTek poly-d-lysine-coated glass bottom culture dishes. Transfection was accomplished using Arrest-in transfection reagent according to the manufacturer's instructions. Live cells were viewed directly with a ×100 objective on an inverted microscope for up to 4 days after transfection. Cells were repeatedly incubated at 40°C for 1 h or more and then viewed at permissive temperature (32°C). After images had been captured, cells were either returned to 37°C for another day of growth or harvested in SDS buffer for Western blot analysis.
Cell preparation and visualization.
All work was carried out in cultured type I (high resistance) MDCK, COS-7, or HEK-293F cell lines. Methods for all lines were similar (as were the results). For immunohistochemistry, cells were grown on glass coverslips; washed three times with PBS; fixed with 1:1 methanol-acetone for 15 min at −20°C; rinsed with PBS; blocked with 1% normal goat serum, 2% BSA, and 2% nonfat dried milk for 30 min; incubated with primary antibody in 2% BSA-PBS at room temperature for 60 min (dilution of 1:2,000 for α1-Na+-K+-ATPase and 1:200 for Flag and ankyrin antibodies); washed with PBS three times for 15 min; incubated with Alexa fluor-conjugated secondary antibodies in 0.5% BSA-PBS for 30 min in the dark; washed with PBS three times for 15 min; and then mounted beneath one drop of mounting media containing 4′,6-diamidino-2-phenylindole from Vectashield. Live cell microscopy was performed on an Olympus IX70 inverted microscope using the TILL Photonics imaging system via a PCO Imaging charge-coupled device Sensicam. Cultured cells on glass coverslips were viewed mounted inside a temperature-controlled aluminum flow cell (Yale Machine Shop). Fixed cells were viewed using an Olympus Provis AX70 microscope with a Hamamatsu ORCA-ER digital camera controlled by Openlabs software by Improvision. Digital images were prepared for presentation by Photoshop on a Macintosh computer; manipulations beyond resizing and cropping were limited to minor adjustment of brightness and contrast.
Transient transfection.
Constructs were prepared using Qiagen Endotoxin-free DNA plasmid kits for transient transfection into either HEK, MDCK, or COS cells. Lipofectamine 2000 (10 μl) was diluted into 200 μl OptiMem and, after a 5-min incubation, combined with 2 μg plasmid DNA diluted into 200 μl OptiMem. The resulting mixture was then incubated another 20 min before being introduced dropwise to cultured cells.
Endoglycosidase H digestion.
COS-7 cells grown in p60 dishes for 24 h at ∼90% confluence were transiently transfected with one of the tso45-VSV-X-eGFP constructs. After 24 h, cells were incubated at 40°C overnight to load the ER. Cells were then shifted to 32°C, and soluble extracts prepared at various times using 300 μl/chamber of RIPA buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholic acid, and 1 mM PMSF]. The extract was cleared by centrifugation, and 3 μl antibody against VSV-G was added, allowed to bind overnight at 4°C, and precipitated with 25 μl prewashed protein A Dynabeads (Invitrogen) added to each tube for 1 h at room temperature. Precipitates were washed three times with PBS, resuspended in 40 μl of 0.5% SDS and 1% 2-mercaptoethanol, and incubated at 100°C for 10 min. Insoluble matter was precipitated, and 5 μl of 500 mM sodium citrate (pH 5.5) containing 5 μl endoglycosidase H (New England Biolabs) were added. Digestion was carried out for 6 h at 37°C, after which samples were analyzed by SDS-PAGE and Western blotted using anti-GFP antibody.
Immunoprecipitation and pulse chase.
Cells were grown to confluence, washed with PBS, and lysed with immunoprecipitation buffer [10 mM Tris·HCl (pH 7), 1% Nonidet P-40, 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, 2 mg/ml BSA, 0.5% deoxycholic acid, and protease inhibitor cocktail] for 20 min at 4oC by gentle rocking. Lysates were decanted and spun at 6,000 g for 5 min to remove debris and precleared for 60 min at 4°C with 25 μl nonimmune rabbit serum plus 200 μl protein A-Sepharose (50% slurry) for every 2 ml of lysate. For precipitation, 2 μl of 2 mg/ml antibody solution were used for 4 h at 4°C. To measure the relative lifetime of newly synthesized Flag-α1-Na+-K+-ATPase or the mutant that lacked the MAB sequence, MDCK and COS cells were transfected with a 1:1 mixture of α/β-Na+-K+-ATPase with or without the MAB domain in p60 tissue culture dishes as described above. The next day, cells were washed three times with PBS and incubated with labeling media consisting of methionine-free DMEM supplemented with 10% FBS that had been dialyzed overnight against PBS and spiked with 20 μl/p60 dish of [l-35S]-methionine [cell-labeling grade, >600Ci (22.2TBq)/mmol, Perkin Elmer Life and Analytical Sciences]. After 1 h, labeling media were removed, and cells were washed three times with PBS and replaced with normal media. Cells were harvested into 1 ml of RIPA buffer and snap frozen in liquid nitrogen at three different time points after the pulse: 1, 12, and 60 h. The cell lysate was then cleared at 21,000 g, and the supernatant was mixed with 2 μl rabbit anti-Flag polyclonal antibody (Sigma). After 1 h, 10 μl Ultralink protein A/G (Pierce) was added to the lysate, and, another hour after that, the beads were spun at 1,000 g, washed three times with RIPA buffer, analyzed by SDS-PAGE, and transferred to an Immobilon polyvinylidene difluoride membrane for autoradiography.
Microinjection of Sar1 mutant peptide.
To ascertain the effect of Sar1 on exit from the ER, COS-7 cells were transiently transfected with tso45-VSV-G-eGFP or tso45-VSV-MAB-eGFP, incubated at 40°C for 6 h, and then microinjected with recombinant Sar1a(H79G) following established methods (36, 41). Microinjections used a semiautomatic system employing Transjector 55246 and Micromanipulator 5171 (Eppendorf, Hamburg, Germany). Needles were pulled from 1.2-mm-diameter glass capillaries (Clark Electromedical Instruments, Reading, UK) using a P-97 needle puller (Sutter Instruments, Novato, CA). Purified Sar1a (H79G), prepared recombinantly with a NH2-terminal His tag, was injected at 1 μg/ml with mouse IgG at 1 mg/ml serving as the carrier protein and injection marker. To induce ER exit and transport, cells were incubated at permissive temperature (32°C) for 45 min. Cells were fixed for 15 min with 4% paraformaldehyde in PBS. After being washed in PBS, injected cells were visualized by incubation for 1 h with anti-mouse IgG conjugated to rhodamine (Molecular Probes). Both tso45-VSV-G-eGFP and tso45-VSV-MAB-eGFP were detected by GFP autofluorescence.
Semi-intact cell transport assay.
ER to Golgi transport assays using semi-intact cells followed established methods (38). COS-7 cells grown on glass coverslips were transfected with ts045-VSV-G-eGFP or ts045-VSV-MAB-eGFP, cultured at 40°C for 3 h to accumulate G protein in the ER, washed on ice with KHM buffer [110 mM potassium acetate, 20 mM HEPES, (pH 7.2), and 2 mM magnesium acetate], and permeabilized with 40 μg/ml digitonin in KHM buffer for 5 min. Liver cytosol (100–500 μg) was prepared from freshly harvested rat livers using a Teflon tissue grinder in 25 mM HEPES (pH 7.2) and 125 mM potassium acetate and collected as supernatant from a 100,000-g sedimentation. This cytosol was devoid of ankyrin R and ankyrin G by Western blot analysis. Cytosol was supplemented with an ATP-regenerating solution [25 mM HEPES (pH7.2), 75 mM potassium acetate, 2.5 mM magnesium acetate, 1.8 mM CaCl2, and 1 mM ATP containing 2 μg/ml of either BSA or ankyrin R purified from red blood cells (39)] and incubated with permeabilized cells for 20 min at 32°C. Cells were thereupon fixed as above, and the distribution of G protein was detected by GFP autofluorescence.
RESULTS
The MAB sequence is critical for proper α1-Na+-K+-ATPase folding and ER export.
The ankyrin-binding region of α1-Na+-K+-ATPase (MAB) is a 25-residue sequence highly conserved only in Na+-K+-ATPases (49). It is predicted to lie just within the cytoplasmic domain [based on homology modeling with the three-dimensional structure of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA); Fig. 1A ; also see the dynamic computer models in supplemental Fig. S-1].1 However, given that the MAB sequence per se appears to be an additional sequence not present in other P-type ATPases, it was hoped that it might be deleted without adverse consequences on the overall integrity of α1-Na+-K+-ATPase. Thus, as a first step to determining its role in the trafficking of α1-Na+-K+-ATPase, a mutant of α1-Na+-K+-ATPase was prepared that had deleted MAB residues and incorporated the Flag epitope tag at its COOH terminus (α1-Na+-K+-ATPaseΔMAB-Flag). Flag-tagged WT α1-Na+-K+-ATPase served as the control. These constructs were transiently coexpressed with a construct encoding WT β1-Na+-K+-ATPase in cultured MDCK cells, and the distribution of α1-Na+-K+-ATPase was monitored by indirect immunofluorescence with antibodies to Flag and by immunoprecipitation (Fig. 1, B and C). In these experiments, it was helpful to coexpress β-Na+-K+-ATPase because the α- and β-ATPase subunits assemble in the ER and exit efficiently only as a complex (4). Upon transient expression, Flag-tagged WT α1-Na+-K+-ATPase moved to the plasma membrane and became largely Triton X-100 insoluble in a fashion similar to that of unlabeled WT protein. Conversely, deletion of the MAB sequence in α1-Na+-K+-ATPaseΔMAB-Flag led to enhanced Triton X-100 extractability and its accumulation in a cytoplasmic compartment. At short times after transfection (≈12 h), α1-Na+-K+-ATPaseΔMAB-Flag appeared in an ER-like pattern; at longer time periods (>24 h), α1-Na+-K+-ATPaseΔMAB-Flag shifted to discrete cytosolic organelles, presumably lysosomes or aggresomes. While the nature of these compartments was not further explored, it was apparent that the deletion of MAB from α1-Na+-K+-ATPase impaired its transport to the plasma membrane. Pulse chase experiments also confirmed that the lifetime of the newly synthesized MAB-less protein (α1-Na+-K+-ATPaseΔMAB-Flag) was reduced by nearly threefold at 12 h in MDCK cells compared with WT protein (α1-Na+-K+-ATPase-Flag; supplemental Fig. S-2).
Fig. 1.
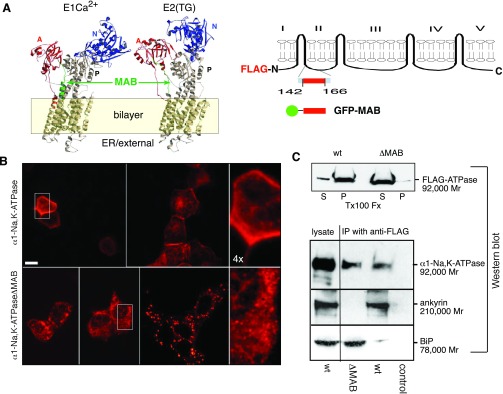
The minimal ankyrin binding (MAB) sequence is critical for proper α1-Na+-K+-ATPase folding and endoplasmic reticulum (ER) export. A: predicted location of MAB based on the structure of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA). Both α1-Na+-K+-ATPase and SERCA are P-type membrane ATPases closely related in sequence. The crystal structure of SERCA is known for both its Ca2+-bound (E1) (46) and Ca2+-unbound (E2) state stabilized with the inhibitor thapsigargin (22). The overall structure of α1-Na+-K+-ATPase is likely to be similar. While SERCA does not have a sequence directly comparable with MAB, the approximate location of MAB can be predicted (green residues) based on an overall sequence alignment between α1-Na+-K+-ATPase and SERCA. Note that MAB appears adjacent to the cytoplasmic face of the bilayer and that the conformation of MAB is predicted to change between the E1 and E2 states, as does the conformation of the N (blue), A (red), and P cytoplasmic domains and the exposed portions of the pump in the external (ER luminal) domain. In supplemental Figs. S-1A and S-1B, a computer simulation of the transition between the E1 and E2 states is shown, highlighting the putative conformational changes involving the MAB sequence. GFP, green fluorescent protein. B: indirect immunofluorescent analysis of the distribution of Flag-tagged α1-Na+-K+-ATPase (top row) and α1-Na+-K+-ATPaseΔMAB (bottom) after transient expression in Madin-Darcy canine kidney (MDCK) cells, as detected by the H1 anti-Flag antibody. Note that whereas the distribution of wild-type (WT) Flag-tagged α1-Na+-K+-ATPase was largely on the plasma membrane after 12–24 h in culture, the ΔMAB construct remained cytosolic. At early time points (≈12 h), it assumed an ER-like distribution (bottom), whereas after ≈24 h, it was often punctate but still cytosolic. Separate pulse chase experiments (supplemental Fig. S-2) indicated that the MAB-deleted protein degraded nearly three times more rapidly than the WT protein. Bar = 10 μm; bottom right and top right images are enlarged ×4. C: Triton X-100 (Tx100) extractability of Flag-α1-Na+-K+-ATPase and ΔMAB ATPase. While >90% of the WT protein existed in the pellet (P) fraction, nearly all Flag-α1-Na+-K+-ATPase was soluble. D: MDCK cultures as above were immunoprecipitated with anti-Flag antibody and Western blotted as indicated. Note that whereas WT α1-Na+-K+-ATPase precipitated ankyrin R, the ΔMAB variant did not, but instead associated with BiP. IP, immunoprecipitation.
This result is significant only in that if α1-Na+-K+-ATPase had continued to move to the plasma membrane after deletion of the MAB sequence, one could exclude a priori a role for MAB in the secretory pathway. However, given the observed result, it was of interest to distinguish whether the failure to transport in MAB-deleted mutants was attributable to simply misfolding of the protein versus a specific effect of MAB. Immunoprecipitation with antibodies to Flag confirmed that the ankyrin-binding capacity of α1-Na+-K+-ATPase was abrogated by the deletion of MAB (Fig. 1C). As expected, WT α1-Na+-K+-ATPase-Flag bound ankyrin in MDCK cells, whereas α1-Na+-K+-ATPaseΔMAB-Flag did not. However, MAB-deleted α1-Na+-K+-ATPase retained a high level of binding to the immunoglobulin-binding protein BiP, an ER chaperone that monitors the quality of folding of several membrane proteins in the ER, including α1-Na+-K+-ATPase (4). Thus, while the deletion of the MAB sequence from α1-Na+-K+-ATPase eliminates its ankyrin-binding capacity, this deletion also must impair the folding of the protein. Its retention in the ER can thus be accounted for by its retention by ER chaperones.
Cytosolic MAB peptide acts in trans to block ER exit of α,β-Na+-K+-ATPase.
To avoid uncertainties as above associated with the mutation of α1-Na+-K+-ATPase, all subsequent work focused on systems with either WT α1-Na+-K+-ATPase or on well-characterized peptide models. MDCK cells were transfected with a construct encoding the 25-residue sequence of MAB alone joined at either its NH2- or COOH-terminal end to eGFP. The distribution of native α1-Na+-K+-ATPase was monitored by indirect immunofluorescence. Results were comparable regardless of which end of MAB was fused to eGFP. Earlier work has demonstrated that this peptide binds directly to ankyrin in vitro (49). Transfected cells expressing eGFP-tagged MAB yielded a dramatic phenotype; they were larger, misshapen, and accumulated significant amounts of α1-Na+-K+-ATPase in a cytosolic compartment that appeared to be the ER, with relatively little accumulation at the plasma membrane (Fig. 2A). Adjacent cells in these cultures that did not express eGFP-MAB displayed normal patterns of α1-Na+-K+-ATPase distribution. Immunoprecipitation of eGFP-MAB from these cells with an antibody to GFP confirmed that the MAB peptide was binding to ankyrin in vivo (Fig. 2B). Surprisingly, immunoprecipitation with an antibody to BiP demonstrated that the eGFP-MAB peptide also caused native α1-Na+-K+-ATPase to now retain BiP, whereas in the absence of eGFP-MAB (as in Fig. 1), little steady-state binding of α1-Na+-K+-ATPase to BiP could be detected (Fig. 2C). Consistent with its apparent ER distribution, most of the of native α1-Na+-K+-ATPase remained Triton X-100 extractable (57 ± 10%) in cultures expressing high levels of eGFP-MAB vs. <10% soluble in parallel cultures transfected with eGFP alone (Fig. 2D). Finally, transport inhibition was further demonstrated by the lack of Golgi processing of the β-Na+-K+-ATPase subunit in cells transfected with the eGFP-MAB construct. β-Na+-K+-ATPase is glycosylated as it passes through the Golgi, yielding an increase in apparent molecular weight by Western blot analysis. There was a persistence of core-glycosylated β-Na+-K+-ATPase in eGFP-MAB-transfected cultures (Fig. 2E). The level of impairment in Golgi processing of β-Na+-K+-ATPase was highly correlated with the extent of eGFP-MAB transfection. When only ≈10% of the cells in the culture were transfected (as in Fig. 2A), only a small increase in core- and nonglycosylated β-Na+-K+-ATPase was detected. However, with high levels of eGFP-MAB transfection (≈90%), the dramatic impact of eGFP-MAB on blockade of the glycosylation was readily apparent, as shown in Fig. 2E. Collectively, these findings indicate that the ankyrin-binding peptide MAB, acting in trans in the cytoplasm, blocked the transport of native α1-Na+-K+-ATPase and its associated subunit β-Na+-K+-ATPase to the Golgi and induced a persistent association of α1-Na+-K+-ATPase with BiP. Unlike the experiments shown in Fig. 1, no modifications were made to α1-Na+-K+-ATPase. Thus, presumably due to competition for cytosolic ankyrin, MAB caused WT α1-Na+-K+-ATPase to hang in the ER and, surprisingly, to retain BiP. There was no effect of cytosolic MAB on the transport of WT VSV-G protein (see below) or E-cadherin (data not shown).
Fig. 2.

Cytosolic MAB peptide blocks the ER to Golgi movement of α1-Na+-K+-ATPase. A: MDCK cells were transfected with either enhanced (e)GFP vector alone (control) or with MAB-GFP and stained for either GFP or α1-Na+-K+-ATPase. Note the normal distribution of α1-Na+-K+-ATPase in eGFP-transfected cells, whereas it was largely cytoplasmic in cells expressing MAB-GFP. The same results were obtained when GFP was on the NH2-terminus of MAB. Bar = 10 μm. B: MDCK cell lysates, transfected as above, were immunoprecipitated with antibodies to GFP and Western blotted with antibodies to ankyrin G (AnkG; which is most abundant in these cells). Note that the MAB-eGFP peptide bound ankyrin, whereas eGFP alone did not. C: MDCK cells were immunoprecipitated for BiP and then Western blotted for α1-Na+-K+-ATPase. Note the retention of α1-Na+-K+-ATPase by BiP in cultures expressing MAB-GFP. D: Triton X-100 extraction of MDCK cells after a high level of MAB-GFP transfection. The distribution of α1-Na+-K+-ATPase in the supernatant (s) or in the 30,000-g pellet (p) was compared. Typically, with high levels of transfection efficiency, over half (57% in this experiment) of α1-Na+-K+-ATPase becomes detergent soluble. E: Western blot of β-Na+-K+-ATPase from MDCK cells with a high (≈90%) level of MAB-GFP transfection. This level of transfection was much higher than in the cells shown in A. Note the abundance of nonglycosylated and core-glycosylated β-Na+-K+-ATPase in MAB-GFP-expressing cells versus the fully mature form in cells transfected only with eGFP [control (cntrl)].
The MAB sequence is a positive Sar1-dependent ER to Golgi transport signal.
Protein trafficking between the ER and Golgi may be controlled by both positively acting exit signals and by the blockage of negatively acting retention signals. To determine which type of signal MAB conferred on α1-Na+-K+-ATPase, use was made of the well-characterized protein trafficking pathway of VSV-G. A model system was established in which the COOH-terminal 24 residues of VSV-G were replaced with 29 residues of α1-Na+-K+-ATPase that incorporated the 25-residue MAB sequence along with 4 flanking residues. Control constructs based on well-established studies in which the cytoplasmic domain of VSV-G was deleted or replaced with random sequences were also prepared (as shown in Fig. 3A). The results presented employed the temperature-sensitive variant tso45-VSV-G with eGFP fused to its COOH-terminus. The tso45 mutant of VSV-G is misfolded and retained in the ER at 40°C but quickly refolds and acquires transport competency after a shift to permissive temperature (32°C). Many previous studies have exhaustively established the utility of tso45-VSV-G-eGFP for studying the earliest steps in the secretory pathway (40) and have demonstrated that VSV-G mutants lacking their cytoplasmic domain fold and trimerize normally at permissive temperature but still fail to exit the ER due to their loss of a critical di-acidic motif resident in the cytoplasmic domain (34). Comparable results have also been obtained with constructs lacking the eGFP tag. MDCK, COS-7, and HEK cells were used in these experiments; all gave comparable results. Following transfection with tso45-VSV-G-eGFP, the G protein quickly moved to the Golgi within 15 min after cells had been returned to a permissive temperature and thereafter to the plasma membrane, indicating efficient transport through the secretory pathway (Fig. 3B; also see supplemental QT-movies that demonstrate the real-time dynamics of each protein in its movement from the ER to the Golgi in supplemental Figs. S-3 to S-6). Constructs lacking the cytoplasmic domain (supplemental Fig. S-4) or those with random sequences (Fig. 3B and supplemental Fig. S-5) showed very little Golgi accumulation even at extended times. Conversely, when the cytoplasmic domain was replaced with MAB (tso45-VSV-MAB-eGFP), the VSV-MAB protein moved efficiently to the Golgi, albeit with somewhat slower kinetics compared with WT VSV-G (Fig. 3B; see also Fig. 5 and supplemental Fig. S-6). There also appeared to be less concentration of tso45-VSV-MAB-eGFP visible in the transport intermediates that were so prominent with tso45-VSV-G-eGFP, suggesting that while it transports efficiently, it may not concentrate to the same degree in the transport carriers as does WT G protein. Finally, although not directly related to the role of ankyrin in ER-Golgi trafficking, it was observed that at very long time periods after transfection (>120 min; data not shown), tso45-VSV-G-eGFP appeared on the plasma membrane surface, whereas tso45-VSV-MAB-eGFP did not. Instead, the MAB-containing protein appeared to circulate in the cell in post-Golgi transport carriers, rapidly moving to the plasma membrane and back, but failing to incorporate. A similar observation could also be discerned from the results shown in Fig. 5, where tso45-VSV-MAB-eGFP protein failed to associate to any significant degree with the plasma membrane, even in the control cultures. We do not know the genesis of this interesting effect and whether it relates to the lack of available ankyrin at the plasma membrane face that is not already ligand bound or whether additional targeting determinants are required. This will be a topic for future study.
Fig. 3.

MAB is a positive ER exit signal when substituted for the cytoplasmic domain of vesicular stomatitis virus G protein (VSV-G). A: comparison of cytoplasmic domain sequences of the expressed constructs. The five residues closest to the putative transmembrane segment were preserved in all constructs. The tso45-VSV-MAB-eGFP construct (VSV-MAB) replaced the remainder of the native G protein sequence with MAB from α1-Na+-K+-ATPase and included the four native residues of α1-Na+-K+-ATPase that flank MAB. All constructs were fused at their COOH termini to eGFP via short linker sequences. B: time lapse video frames of the expressed constructs in COS-7 cells at permissive temperature (32°C). Note that both VSV-G and VSV-MAB efficiently moved to the Golgi from the ER, whereas the construct with a random cytoplasmic domain sequence did not. The trafficking of VSV-tailless was indistinguishable from the random sequence (shown in supplemental figures). C: inhibition of VSV-G and VSV-MAB transport to the Golgi by Sar1a (H79G). HEK cells transfected with either VSV-G or VSV-MAB were microinjected with Sar1a (H79G) (a dominant negative inhibitor of COPII coat function) and examined at 25 or 50 min, respectively, after transfer to permissive temperature. In uninjected cells or in sham-injected cells (not shown), there was a normal accumulation of either VSV-G or VSV-MAB in the Golgi (arrows). In cells receiving Sar1a (H79G), recognizable by their accumulation of diffuse cytosolic immunofluorescence from coinjected mouse IgG, accumulation of either VSV-G or VSV-MAB in the Golgi was inhibited.
The same experiments were then repeated with cells transfected with the above VSV-G constructs and microinjected with recombinant Sar1a (H79G), a mutant Sar1 that lacks GTPase activity and thus remains blocked in its GTP form. This protein dominantly inhibits vesicle budding from ER exit sites and COPII coat function (36). As expected, Sar1a (H79G) blocked the movement of tso45-VSV-G-eGFP to the Golgi; it also blocked the movement of tso45-VSV-MAB-eGFP to the Golgi (Fig. 3C). Neighboring cells that were not injected with Sar1a (H79G) served as controls; in these cells, Golgi accumulation of both VSV-G and VSV-MAB was apparent (Fig. 3C, arrows). Thus, at least one mechanism decoding the MAB signal is Sar1 and presumably COPII dependent.
Transport of VSV-MAB but not VSV-G to the Golgi is blocked by cytosolic MAB.
A comparison of the time course for the attainment of endoglycosidase H resistance for VSV-G (tso45-VSV-G-eGFP vs. tso45-VSV-MAB-eGFP) revealed that while both moved efficiently to the Golgi at permissive temperatures, the WT G protein moved more quickly (half-time = 14.3 ± 2.4 vs. 47.9 ± 44.1 min; Fig. 4A). Constructs with absent or random sequences in the cytoplasmic domain transitioned to the Golgi very inefficiently. However, the most dramatic difference between the trafficking of WT G protein and the G fusion protein carrying MAB was apparent in cells that also expressed eCFP-MAB in the cytoplasm (Fig. 4B). Whereas cytoplasmic eCFP-MAB had no discernable effect on the transport of tso45-VSV-G-eGFP, it completely blocked the efficient transfer of tso45-VSV-MAB-eGFP to the Golgi, reducing it to a rate indistinguishable from that of truncated or random sequence variants (for purposes of comparison, the kinetics of tso45-VSV-MAB-eGFP in the presence of eCFP-MAB are plotted along with the kinetics of VSV-G lacking its cytoplasmic domain in Fig. 4B, showing that there no discernable difference). Thus, whereas both VSV-G and VSV-MAB proteins require Sar1 for their ER exit, they are distinguished by their different transport rates and distinct vulnerabilities (one inhibitable by MAB, the other not). These results also preclude a trivial explanation of the effects of cytosolic MAB as simply a toxic effect, since its blockage of ER to Golgi trafficking is highly protein selective.
Fig. 4.

VSV-G and VSV-MAB exit the ER by distinguishable mechanisms. A: COS-7 cells transfected with each of the four constructs shown in Fig. 3 were sampled at various times after transfer to permissive temperature, and the level of endoglycosidase H (endo-H) resistance was determined (a measure of their arrival in the medial Golgi). The half-time (±2 SD) for ER to Golgi transit as determined by least squares fitting to a zero-order kinetic model for VSV-G was 14.3 ± 2.4 vs. 47.9 ± 44.1 min for VSV-MAB. The (−) lane shows results at time 0 with cells not digested with endo-H. B: COS-7 cells were transfected with yellow fluorescent protein (YFP)-labeled tso45-VSV-G or tso45-VSV-MAB and were also cotransfected with MAB-cyan fluorescent protein (CFP). The rate of accumulation of endo-H resistance was then measured as in A. For purposed of comparison, also plotted from A are the data from VSV-G-GFP and data from the VSV construct containing only a random cytoplasmic domain. Note that while the presence of the soluble MAB-CFP peptide had no effect on the trafficking of VSV-G, it reduced the rate of ER to Golgi transfer of VSV-MAB to a level indistinguishable from the random-tail variant.
The MAB-dependent ER-Golgi transport signal is decoded by ankyrin R.
While the MAB sequence was first identified based on its ankyrin-binding capacity, its role in the earliest stages of the secretory pathway could relate to properties that are independent of ankyrin. To examine this possibility, MDCK and HEK cells were cotransfected with either tso45-VSV-G-eGFP or tso45-VSV-MAB-eGFP and with shRNA designed to block ankyrin expression. Two shRNA plasmids were employed: one designed to block ankyrin G expression, and the other designed to block ankyrin R. These are the only two ankyrin isoforms expressed in MDCK cells as detected by RT-PCR (data not shown). The shRNAs selected were designed to target a constitutive exon expressed in all alternative transcripts of ankyrin G or ankyrin R (see materials and methods). After 48–72 h in culture, cells were moved to permissive temperature (32°C), and the efficiency of transfer of each VSV-G construct to the Golgi was evaluated by live cell microscopy and indirect immunofluorescence (Fig. 5, A and B). Based on Western blot analysis of the transiently transfected cultures, the overall levels of both ankyrin R and ankyrin G were suppressed by over 50% in these experiments. The suppression of ankyrin G had no effect on the trafficking of either tso45-VSV-G-eGFP or tso45-VSV-MAB-eGFP (Fig. 5B; see also supplemental Figs. S-7 to S-10, in which the real-time dynamics of the trafficking are shown). Randomized shRNA was without effect. However, suppression of ankyrin R selectively blocked the movement of tso45-VSV-MAB-eGFP protein to the Golgi but did not affect the movement of tso45-VSV-G-eGFP, indicating that the overall secretory pathway (at least as it pertains to the movement of VSV-G) remained intact despite the shRNA inhibition of ankyrin R.
Fig. 5.
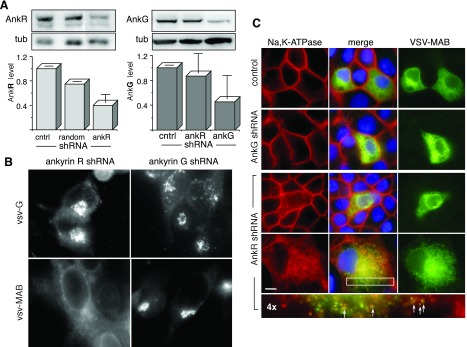
Small hairpin (sh)RNA suppression of ankyrin R (AnkR) blocks VSV-MAB and α1-Na+-K+-ATPase transport to the Golgi. A: Western blot of cultured WT HEK cells (control) transfected with nonsense shRNA (random) or with shRNA designed to block either ankyrin G or ankyrin R. These are the only two ankyrins expressed in HEK and MDCK cells as determined by RT-PCR analysis. In transiently transfected cultures, overall ankyrin G or ankyrin R levels were suppressed overall by >50% compared with WT cells [normalized to constant tubulin (Tub)]. Two ankyrin G bands at ≈210 and ≈130 k Mr were present in the blots; the 130-k band was the most abundant and is shown here. The reductions in ankyrin G (210 k Mr) were comparable. B: immunofluorescent vital micrographs of HEK cells with suppressed ankyrin R or ankyrin G transfected with either tso45-VSV-G-eGFP (VSV-G) or with tso45-VSV-MAB-eGFP (VSV-MAB) photographed at 25 or 50 min, respectively, after transition to permissive temperature. Note the persistent ER distribution of VSV-MAB in ankryin R-suppressed cells. C: MDCK cells cotransfected with VSV-MAB (tso45-VSV-MAB-eGFP) and shRNA for ankyrin G or ankyrin R, stained for α1-Na+-K+-ATPase and VSV-MAB, and photographed at steady state under permissive conditions. Control cells were transfected with VSV-MAB alone. The fourth row from the top represents ankryin R-suppressed cells at partial confluence; other images are at established confluence. The bottom micrograph is the boxed area magnified ×4. Note that α1-Na+-K+-ATPase remained cytosolic in ankyrin R-suppressed cells and that both α1-Na+-K+-ATPase and VSV-MAB concentrated into the same punctate dispersed spots (arrows) in ankyrin R-suppressed cells. Bar = 10 μm.
To determine if these results also applied to WT α1-Na+-K+-ATPase, cultures of MDCK cells were doubly transfected as above with shRNA against either ankyrin R or ankyrin G and with tso45-VSV-MAB-eGFP. The ratio of shRNA plasmid to the VSV-MAB plasmid was 8:1 in these experiments, assuring that cells marked by the expression of tso45-VSV-MAB-eGFP also expressed shRNA. Cells were grown to confluence at permissive temperature and stained for VSV-MAB (tso45-VSV-MAB-eGFP) and α1-Na+-K+-ATPase (Fig. 5C). As before, the suppression of ankyrin G had no effect on either the appearance of MDCK cells or on the distribution of α1-Na+-K+-ATPase or tso45-VSV-MAB-eGFP. Conversely, suppression of ankyrin R caused the accumulation of α1-Na+-K+-ATPase in the cytoplasm and blocked it from accumulating at the plasma membrane. This was particularly apparent in partially confluent cells (Fig. 5C, bottom row), where both α1-Na+-K+-ATPase and tso45-VSV-MAB-eGFP accumulated into punctate vesicular bodies dispersed throughout the cytoplasm (arrows). These punctate structures did not stain with antibodies to Sec31B, a COPII component (44).
The ankyrin dependence of proteins containing MAB was also confirmed in a separate series of experiments by evaluating in semipermeabilized HEK cells the transfer of tso45-VSV-G-eGFP versus tso45-VSV-MAB-eGFP to the Golgi (Fig. 6). The loss of cytosol upon cell permeabilization halted COPII coat formation and the exit of all proteins from the ER. Restoration of ankyrin-deficient cytosol (containing all COPII components) activated the transport of WT VSV-G protein from the ER to the Golgi; this transport was not affected by the addition of ankyrin R to the cytosol (Fig. 6, A and B, arrows). In such preparations, a centralized Golgi was not reliably reestablished but instead appeared as dispersed Golgi fragments (38). Conversely, the addition of ankyrin-deficient cytosol did not restore the transport of VSV-MAB (tso45-VSV-MAB-eGFP) to the Golgi; MAB-containing VSV-G protein remained dispersed in the ER (Fig. 6C). However, the transport of tso45-VSV-MAB-eGFP was rescued by the addition to the cytosol of ankyrin R (purified from human erythrocytes). Ankyrin R is thus required for decoding the MAB-based ER to Golgi signal.
Fig. 6.

Ankyrin R-enriched cytosol is required for the ER to Golgi transport of VSV-MAB in semipermeabilized cells. COS-7 cells transfected with either VSV-G (tso45-VSV-G-eGFP) or VSV-MAB (tso45-VSV-MAB-eGFP) were permeabilized with digitonin and incubated with either rat liver cytosol alone or with the same cytosol augmented by the addition of ankyrin R prepared from erythrocytes. As prepared, the rat liver cytosol was devoid of ankyrin immunoreactivity. In the absence of added cytosol, both proteins remained in the ER and displayed an ER distribution by indirect immunofluorescence (not shown). A: when transfected cells were incubated at permissive temperature in the presence of added rat liver cytosol, VSV-G moved efficiently from the ER to ER exit sites and to Golgi remnants. B: the presence of added ankyrin R in the cytosol did not affect the movement of VSV-G to the Golgi. C: distribution of VSV-MAB at permissive temperature in the presence of cytosol. Note its ER distribution and lack of transport to the Golgi remnants. D: the addition of purified ankyrin R to the cytosol restored trafficking of VSV-MAB to the Golgi remnants. In all micrographs, arrows highlight the concentration of VSV protein in transitional ER and Golgi remnants. Bar = 10 μm.
DISCUSSION
These results establish that ankyrin facilitates the passage of α1-Na+-K+-ATPase through the secretory pathway, at least to the Golgi, and that the 25-residue ankyrin-binding sequence in α1-Na+-K+-ATPase is responsible for this activity. This sequence thus bestows a novel activity previously unassociated with ER to Golgi trafficking in that it is decoded by ankyrin R, an adapter protein that typically links the spectrin-based cytoskeleton to specific integral membrane proteins (8). This conclusion is supported by several direct lines of evidence: 1) the MAB sequence, expressed as a cytosolic peptide, binds ankyrin and acts in trans to block the delivery of α,β-Na+-K+-ATPase to the Golgi; 2) the MAB sequence, when incorporated into VSV-G fusion peptides (VSV-MAB) that otherwise do not exit the ER, bestows efficient transport to the Golgi; 3) VSV-G and VSV-MAB have distinct ER to Golgi trafficking mechanisms, since VSV-MAB exits the ER with slower kinetics and is selectively blocked by cytosolic MAB, whereas VSV-G is not blocked; 4) depletion of ankyrin R, but not ankyrin G, blocks VSV-MAB but not VSV-G transport to the Golgi; 5) when their transport to the Golgi is suppressed by the loss of ankyrin R, both α1-Na+-K+-ATPase and VSV-MAB (tso45-VSV-MAB-eGFP) are shunted together into a vesicular compartment that is devoid of COPII coat components; and 6) in semipermeabilized cells, cytosol lacking ankyrin R restores the transport of VSV-G to the Golgi but not the transport of VSV-MAB and the addition of ankyrin R purified from erythrocytes rescues the transport deficiency of VSV-MAB.
These results extend previous studies that identified a role for spectrin in the movement of α-Na+-K+-ATPase to the plasma membrane, based on the observation that βI spectrin peptides that preserve spectrin's NH2-terminal domain but delete its ankyrin-binding domain can also disrupt α,β-Na+-K+-ATPase transport to the plasma membrane (11). Two mechanisms by which spectrin (and now ankyrin) might influence the secretory pathway have been hypothesized. One envisions a conventional role for spectrin and ankyrin as organelle stabilizers (i.e., a “Golgi mesh”) (8, 26); the other proposes a more active role for spectrin and ankyrin in forming a tether linking transport intermediates in a cargo-specific fashion to motors of active transport [the “spectrin-ankyrin-adapter protein tethering system” (8)]. Given that spectrin and ankyrin dynamically associate with many organelles and membranes in higher organisms, and that by tethering cargo proteins they and their carrier membranes may be stabilized, the two hypotheses are not mutually exclusive. Two models of how ankyrin (and spectrin) might participate in ER to Golgi trafficking have been envisioned (Fig. 7A). In model 1, α1-Na+-K+-ATPase exits the ER together with its paired β-Na+-K+-ATPase subunit in conventional COPII-coated vesicular-tubular carriers. After the COPII coat is shed in the intermediate compartment, ankyrin binds to α1-Na+-K+-ATPase, protecting it from capture by ADP-ribosylation factor 1 (ARF1)-activated COPI-mediated retrograde pathways returning to the ER. The contribution of ankyrin to anterograde movement of a protein could thus arise either by the blockage of a novel ER retrieval signal resident in MAB or by linkage of α,β-Na+-K+-ATPase (and its carrier) to spectrin and thereby facilitating anterograde delivery to the Golgi via spectrin's attachment to dynein-dynactin (20, 33). By this model, there is nascent assembly of a spectrin-ankyrin cytoskeleton on vesicular-tubular clusters within the intermediate compartment; this skeletal assembly then moves with α,β-Na+-K+-ATPase to the Golgi. The apparent retention of α1-Na+-K+-ATPase in the ER in the absence of ankyrin R would thus be the consequence of a futile cycle of exit (via COPII carriers) and return via retrograde pathways [or shunting into a membrane-bounded degradation pathway, as has been observed for class I major histocompatibility proteins (24)]. This model, with nascent assembly of a spectrin-ankyrin skeleton late in the intermediate compartment and its persistence on the Golgi, is similar to the observed association of cis-Golgi matrix proteins such as GM130 and GRASP65 with incoming tubulovesicular carriers late in the intermediate compartment, before they reach and integrate with the cis-Golgi (29). This model would also account for the Sar1 and ankyrin R dependencies of α1-Na+-K+-ATPase trafficking as well as for the transient presence of both ankyrin and spectrin on the Golgi (8). The persistent binding of BiP to α1-Na+-K+-ATPase whenever ankyrin R function is blocked is also consistent with observations that misfolded VSV-G undergoes a futile recycling between the ER, intermediate compartment, and cis-Golgi accompanied by BiP (18).
Fig. 7.

Two possible mechanisms of ankyrin action on the trafficking of α,β-Na+-K+-ATPase. In model 1, α,β-Na+-K+-ATPase collects in a COPII-coated carrier (1) and is extruded from the ER. Vesicular carriers fuse in the intermediate compartment to form vesicular-tubular clusters (VTCs) (2). Coincident with this process, α,β-Na+-K+-ATPase is stabilized in the VTC by its association with a nascent ankyrin-spectrin scaffold, preventing its return to the ER via COPI-mediated processes and facilitating the release of luminal chaperones (BiP) (3). Anterograde transport to the cis-Golgi is facilitated by a spectrin ankyrin-mediated linkage of the α,β-Na+-K+-ATPase-enriched carrier to dynein-dynactin (4), enabling efficient transport along microtubules to the cis-Golgi (5). In model 2, ankyrin with or without spectrin binds to α,β-Na+-K+-ATPase while it is still within the transitional ER (1), retarding scission of the Sar1-induced nascent vesicular-tubular carrier. Extrusion of the α,β-Na+-K+-ATPase-enriched carrier to VTCs (2), its protection from retrograde pathways (3), and its subsequent transport to the cis-Golgi is facilitated as in model 1 (4 and 5). In either model, an absence of ankyrin binding impairs the efficiency of anterograde transport.
Alternatively, a second, albeit less probable, model (Fig. 7, model 2) of MAB action that cannot be excluded proposes that ankyrin acts directly on the tubules forming out of the ER under the influence of Sar1/COPII. Coincident with the extrusion of the α,β-Na+-K+-ATPase-loaded vesicular/tubular carrier, the assembly of a nascent peripheral skeleton triggers the release of BiP and enhances the capture of α1-Na+-K+-ATPase into the forming tubular/vesicular carrier in a fashion similar to that proposed for the secretion of collagen fibrils (30). By this model, the α,β-Na+-K+-ATPase-ankyrin assembly, with or without spectrin, may be too large to be fully accommodated into the highly structured COPII coatomer, delaying scission of the vesicle and favoring the formation of larger more pleomorphic COPII-associated carriers. Large pleomorphic carriers have been identified as a putative alternative ER exit pathway (30), and similar structures are well recognized in post-Golgi sorting pathways (27). While we do not favor this hypothesis, in future work it will be interesting to compare the morphology of VSV-G- versus VSV-MAB-loaded cargo carriers budding from the ER to see if they can be morphologically distinguished.
Other features of this ankyrin-dependent ER to Golgi pathway deserve mention. It is noteworthy that this pathway, at least for α1-Na+-K+-ATPase in MDCK cells, appears to be exclusively ankyrin R dependent. The loss of ankyrin R cannot be complemented by ankyrin G, which is abundant in renal epithelial cells including MDCK and HEK cells (3, 12, 37). We do not understand the genesis of this exquisite requirement for ankyrin R, but note that ankyrin R has been found on the Golgi (3), as has βI spectrin (2, 11), and that in other studies only βI spectrin peptides block α1-Na+-K+-ATPase trafficking in MDCK cells (11) or CD45 trafficking (also an ankyrin-binding protein) in T lymphocytes (39).
A second surprising observation is that ankyrin binding is apparently required for the release of α1-Na+-K+-ATPase from BiP. Both ankyrin and MAB (when expressed as just the free peptide) exist as soluble proteins in the cytoplasm; BiP is resident in the ER lumen. Thus, the docking of ankyrin to the cytoplasmic domain of α1-Na+-K+-ATPase must transmit a signal across the ER membrane. One signal may be an induced lateral concentration of α1-Na+-K+-ATPase or a change in its oligomerization (although neither has been observed), events typically conducive to ER export. A more interesting process may be an induced transmembrane conformational change in the structure of α1-Na+-K+-ATPase that follows ankyrin binding. While the three-dimensional structure of α1-Na+-K+-ATPase remains unknown, the structure of a closely related member in the P-type ATPase family, SERCA, has been solved both with Ca2+ (E2 state) (46) and without Ca+2 (E1 state) (22). These structures are shown in Fig. 1A and in supplementary figures. While SERCA does not contain a sequence or structure comparable with MAB (49), the alignment of the overall sequence of α1-Na+-K+-ATPase with SERCA indicates that MAB would approximately correspond to the position of residues 107–129 in SERCA (highlighted in green in Fig. 1A). Of note is the considerable conformational change that this region undergoes as SERCA binds and releases Ca2+. Major conformational changes that span the membrane and involve the luminal domain also accompany these transitions. If α1-Na+-K+-ATPase behaves in a similar fashion, then a mechanism for transmitting the state of ankyrin binding across the membrane would be intrinsic to this class of proteins and would constitute a plausible mechanism by which BiP association may be regulated by ankyrin binding. Parenthetically, this putative relationship between structural transition and ankyrin binding might also be a mechanism by which ankyrin binding could modulate the transport characteristics of the pump, such as has been observed with the neuronal Na+ channel Nav1.6 (42). Looked at another way, the observed linkage between ankyrin binding and BiP release might constitute an important quality control mechanism, assuring that molecules of α1-Na+-K+-ATPase defective in ankyrin binding cannot enter the secretory pathway.
Finally, it is fair to ask how widespread is the participation of ankyrin (or spectrin) in the secretory pathway. While we cannot answer this question definitively, it seems likely that many membrane proteins may be involved. Ankyrin itself is a multifunctional protein characterized by a diverse gene family with many alternative transcripts, capable of binding a wide array of integral proteins via its repeating 33-residue ankyrin motif (5). Disruption of spectrin or ankyrin in either cultured cells or in genetically modified animals leads to a variety of pathologies that can be attributed to failures of membrane protein trafficking or sorting, including α,β-Na+-K+-ATPase (this study and Ref. 11), CD45 and CD3 in T lymphocytes (39), the anion channel AE1 in 293 cells (17), the voltage-dependent Na+ channel in the brain and muscle (25, 50), and SERCA, the inositol 1,4,5-trisphosphate receptor, and the ryanodine receptor in striated muscle, the heart, and lymphocytes (47). It is also important to note that with the exception of homozygous loss of the αII or βII spectrin alleles (M. C. Stankewich, unpublished observations, and Ref. 45), deletion of any of the other spectrin or ankyrin genes are compatible with overtly normal mammalian development and maturation. These proteins are also not found in some primitive eukaryotic species, and, in particular, Saccharomyces cerevisiae, where the most fundamental mechanisms of the secretory pathway are represented. Thus, it appears that the participation of ankyrin or spectrin in the secretory pathway may represent a vertebrate adaptation as a “quality of life” issue but is not essential for life since primitive pathways can at least partially compensate for the loss of their function. Presumably this feature accounts for the difficulty in identifying their role in the secretory pathway by simple genetic screens, and perhaps is why their deletion or mutation typically causes (except in red blood cells, where no compensation is possible) late-onset or degenerative disease such as type 4 long QT cardiac arrhythmias (ankyrin deficiency) (32), spinocerebellar ataxia type 5 (βIII spectrin mutation) (21), or deaf quivering mice (βIV spectrin mutation) (35). In future work, it will be important to closely examine these conditions and other instances of spectrin or ankyrin deficiency to identify the full repertoire of pumps, channels, and receptors that depend on ankyrin and/or spectrin to chauffer them through the secretory pathway and to maintain them at their sites of physiological action.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL-28560, R01-DK-43812, and P01-DK-55389 (to J. S. Morrow).
Supplementary Material
Acknowledgments
The authors thank Drs. Graham Warren and Miljan Simonovic for helpful discussions and other assistance and Dr. Mimi Liu for assistance with the earliest phases of this study.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Supplemental material for this article is available at the American Journal of Physiology-Cell Physiology website.
REFERENCES
- 1.Aridor M, Balch WE. Integration of endoplasmic reticulum signaling in health and disease. Nat Med 5: 745–751, 1999. [DOI] [PubMed] [Google Scholar]
- 2.Beck KA, Buchanan JA, Malhotra V, Nelson WJ. Golgi spectrin: identification of an erythroid beta-spectrin homolog associated with the Golgi complex. J Cell Biol 127: 707–723, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck KA, Buchanan JA, Nelson WJ. Golgi membrane skeleton: identification, localization and oligomerization of a 195 kDa ankyrin isoform associated with the Golgi complex. J Cell Sci 110: 1239–1249, 1997. [DOI] [PubMed] [Google Scholar]
- 4.Beggah A, Mathews P, Beguin P, Geering K. Degradation and endoplasmic reticulum retention of unassembled alpha- and beta-subunits of Na,K-ATPase correlate with interaction of BiP. J Biol Chem 271: 20895–20902, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Bennett V, Chen L. Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol 13: 61–67, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Berghs S, Ferracci F, Maksimova E, Gleason S, Leszczynski N, Butler M, De Camilli P, Solimena M. Autoimmunity to beta IV spectrin in paraneoplastic lower motor neuron syndrome. Proc Natl Acad Sci USA 98: 6945–6950, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cianci CD, Giorgi M, Morrow JS. Phosphorylation of ankyrin down-regulates its cooperative interaction with spectrin and protein 3. J Cell Biochem 37: 301–315, 1988. [DOI] [PubMed] [Google Scholar]
- 8.De Matteis MA, Morrow JS. Spectrin tethers and mesh in the biosynthetic pathway. J Cell Sci 113: 2331–2343, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Devarajan P, Gilmore-Hebert M, Benz EJ Jr. Differential translation of the Na,K-ATPase subunit mRNAs. J Biol Chem 267: 22435–22439, 1992. [PubMed] [Google Scholar]
- 10.Devarajan P, Scaramuzzino DA, Morrow JS. Ankyrin binds to two distinct cytoplasmic domains of Na,K-ATPase alpha subunit. Proc Natl Acad Sci USA 91: 2965–2969, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devarajan P, Stabach PR, De Matteis MA, Morrow JS. Na,K-ATPase transport from endoplasmic reticulum to Golgi requires the Golgi spectrin-ankyrin G119 skeleton in Madin Darby canine kidney cells. Proc Natl Acad Sci USA 94: 10711–10716, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devarajan P, Stabach PR, Mann AS, Ardito T, Kashgarian M, Morrow JS. Identification of a small cytoplasmic ankyrin (AnkG119) in the kidney and muscle that binds beta I sigma spectrin and associates with the Golgi apparatus. J Cell Biol 133: 819–830, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fath KR, Trimbur GM, Burgess DR. Molecular motors and a spectrin matrix associate with Golgi membranes in vitro. J Cell Biol 139: 1169–1181, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fucini RV, Navarrete A, Vadakkan C, Lacomis L, Erdjument-Bromage H, Tempst P, Stamnes M. Activated ADP-ribosylation factor assembles distinct pools of actin on golgi membranes. J Biol Chem 275: 18824–18829, 2000. [DOI] [PubMed] [Google Scholar]
- 15.Gallagher PG, Forget BG. Spectrin genes in health and disease. Semin Hematol 30: 4–20, 1993. [PubMed] [Google Scholar]
- 16.Godi A, Santone I, Pertile P, Devarajan P, Stabach PR, Morrow JS, Di Tullio G, Polishchuk R, Petrucci TC, Luini A, De Matteis MA. ADP ribosylation factor regulates spectrin binding to the Golgi complex. Proc Natl Acad Sci USA 95: 8607–8612, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomez S, Morgans C. Interaction between band 3 and ankyrin begins in early compartments of the secretory pathway and is essential for band 3 processing. J Biol Chem 268: 19593–19597, 1993. [PubMed] [Google Scholar]
- 18.Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol 126: 41–52, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirose S A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res 70, Suppl 1: S206–S217, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Holleran EA, Ligon LA, Tokito M, Stankewich MC, Morrow JS, Holzbaur EL. Beta III spectrin binds to the Arp1 subunit of dynactin. J Biol Chem 276: 36598–36605, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, Stevanin G, Durr A, Zuhlke C, Burk K, Clark HB, Brice A, Rothstein JD, Schut LJ, Day JW, Ranum LP. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet 38: 184–190, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Jensen AM, Sorensen TL, Olesen C, Moller JV, Nissen P. Modulatory and catalytic modes of ATP binding by the calcium pump. EMBO J 25: 2305–2314, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jordan C, Puschel B, Koob R, Drenckhahn D. Identification of a binding motif for ankyrin on the alpha-subunit of Na+,K+-ATPase. J Biol Chem 270: 29971–29975, 1995. [DOI] [PubMed] [Google Scholar]
- 24.Kamhi-Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ. A novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell 12: 1711–1723, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lacas-Gervais S, Guo J, Strenzke N, Scarfone E, Kolpe M, Jahkel M, De Camilli P, Moser T, Rasband MN, Solimena M. BetaIVsigma1 spectrin stabilizes the nodes of Ranvier and axon initial segments. J Cell Biol 166: 983–990, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lorra C, Huttner WB. The mesh hypothesis of Golgi dynamics. Nat Cell Biol 1: E113–E115, 1999. [DOI] [PubMed] [Google Scholar]
- 27.Luini A, Ragnini-Wilson A, Polishchuck RS, De Matteis MA. Large pleiomorphic traffic intermediates in the secretory pathway. Curr Opin Cell Biol 17: 353–361, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Margolis RL The spinocerebellar ataxias: order emerges from chaos. Curr Neurol Neurosci Rep 2: 447–456, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Marra P, Maffucci T, Daniele T, Tullio GD, Ikehara Y, Chan EK, Luini A, Beznoussenko G, Mironov A, De Matteis MA. The GM130 and GRASP65 Golgi proteins cycle through and define a subdomain of the intermediate compartment. Nat Cell Biol 3: 1101–1113, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Mironov AA, Mironov AA Jr, Beznoussenko GV, Trucco A, Lupetti P, Smith JD, Geerts WJ, Koster AJ, Burger KN, Martone ME, Deerinck TJ, Ellisman MH, Luini A. ER-to-Golgi carriers arise through direct en bloc protrusion and multistage maturation of specialized ER exit domains. Dev Cell 5: 583–594, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Mohler PJ, Bennett V. Ankyrin-based cardiac arrhythmias: a new class of channelopathies due to loss of cellular targeting. Curr Opin Cardiol 20: 189–193, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421: 634–639, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Muresan V, Stankewich MC, Steffen W, Morrow JS, Holzbaur EL, Schnapp BJ. Dynactin-dependent, dynein-driven vesicle transport in the absence of membrane proteins: a role for spectrin and acidic phospholipids. Mol Cell 7: 173–183, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura N, Balch WE. A di-acidic signal required for selective export from the endoplasmic reticulum. Science 277: 556–558, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Parkinson NJ, Olsson CL, Hallows JL, McKee-Johnson J, Keogh BP, Noben-Trauth K, Kujawa SG, Tempel BL. Mutant beta-spectrin 4 causes auditory and motor neuropathies in quivering mice. Nat Genet 29: 61–65, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Pepperkok R, Lowe M, Burke B, Kreis TE. Three distinct steps in transport of vesicular stomatitis virus glycoprotein from the ER to the cell surface in vivo with differential sensitivities to GTP gamma S. J Cell Sci 111: 1877–1888, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Peters LL, John KM, Lu FM, Eicher EM, Higgins A, Yialamas M, Turtzo LC, Otsuka AJ, Lux SE. Ank3 (epithelial ankyrin), a widely distributed new member of the ankyrin gene family and the major ankyrin in kidney, is expressed in alternatively spliced forms, including forms that lack the repeat domain. J Cell Biol 130: 313–330, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plutner H, Davidson HW, Saraste J, Balch WE. Morphological analysis of protein transport from the ER to Golgi membranes in digitonin-permeabilized cells: role of the P58 containing compartment. J Cell Biol 119: 1097–1116, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pradhan D, Morrow J. The spectrin-ankyrin skeleton controls CD45 surface display and interleukin-2 production. Immunity 17: 303–315, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature 389: 81–85, 1997. [DOI] [PubMed] [Google Scholar]
- 41.Seemann J, Jokitalo EJ, Warren G. The role of the tethering proteins p115 and GM130 in transport through the Golgi apparatus in vivo. Mol Biol Cell 11: 635–645, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shirahata E, Iwasaki H, Takagi M, Lin C, Bennett V, Okamura Y, Hayasaka K. Ankyrin-g regulates inactivation gating of the neuronal sodium channel, nav1.6. J Neurophysiol 96: 1347–1357, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Siddhanta A, Radulescu A, Stankewich MC, Morrow JS, Shields D. Fragmentation of the Golgi apparatus. A role for beta III spectrin and synthesis of phosphatidylinositol 4,5-bisphosphate. J Biol Chem 278: 1957–1965, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Stankewich MC, Stabach PR, Morrow JS. Human Sec31B: a family of new mammalian orthologues of yeast Sec31p that associate with the COPII coat. J Cell Sci 119: 958–969, 2006. [DOI] [PubMed] [Google Scholar]
- 45.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science 299: 574–577, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature 405: 647–655, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Tuvia S, Buhusi M, Davis L, Reedy M, Bennett V. Ankyrin-B is required for intracellular sorting of structurally diverse Ca2+ homeostasis proteins. J Cell Biol 147: 995–1008, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams MW, Resneck WG, Kaysser T, Ursitti JA, Birkenmeier CS, Barker JE, Bloch RJ. Na,K-ATPase in skeletal muscle: two populations of beta-spectrin control localization in the sarcolemma but not partitioning between the sarcolemma and the transverse tubules. J Cell Sci 114: 751–762, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Devarajan P, Dorfman AL, Morrow JS. Structure of the ankyrin binding domain of α-Na,K-ATPase. J Biol Chem 273: 18681–18684, 1998. [DOI] [PubMed] [Google Scholar]
- 50.Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143: 1295–1304, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.