

Abstract
Cyclization of 1,5-dienes bearing nucleophilic traps with electrophilic trisphosphine pincer ligated Pt(II) complexes results in the formation of a polycyclic Pt-alkyl via an Pt(η2-alkene) intermediate. With electron rich triphosphine ligands an equilibrium between the Pt(η2-alkene) and Pt-alkyl was observed. The position of the equilibrium was sensitive to ligand basicity, conjugate acid strength, solvent polarity, and ring size. In cases where the ligand was electron poor and did not promote retro-cyclization, then kinetic products adhering to the Stork-Eschenmoser postulate were observed (E-alkenes give trans-ring junctions). When retrocyclization was rapid, then alternative thermodynamic products resulting from multi-step rearrangements were observed (cis [6,5]-bicycles). Under both kinetic and thermodynamic conditions remote methyl substituents led to highly diastereoselective reactions. In the case of trienol substrates, long-range asymmetric induction from a C-ring substituent was considerably attenuated and only modest diastereoselectivity was observed (~2:1). The data suggests that for a tricyclization, the long-range stereocontrol results from diastereo-selecting interactions that develop during the organization of the nascent rings. In contrast, the bicyclization diastereoselectivities result from reversible cascade cyclization.
Introduction
The stereoselective cascade cyclization of polyprenoids into multicyclic steroid-like structures stands as one of the crowning achievements in organic chemistry.i The state of the art synthetic methods reflect the sum of enormous efforts aimed at understanding the rules for controlling the efficiency and selectivity of the non-enzymatic cation-olefin cascade responsible for ring construction. The Stork-Eschenmoser postulate (SEP) was one of the key early findings that enabled the ring junction stereochemistry to be predicted from the starting olefin geometry; E-alkenes give trans ring junctions and Z-alkenes give cis ring junctions.ii,iii The postulate is a manifestation of a favorable anti addition of electrophile and nucleophile across the original alkene (e.g. Scheme 1). Ultimately, however, the stereochemical outcome of such cascade cyclizations is explainable by evaluating the role that neighboring group participation plays on the kinetics of the cation-olefin reaction;iii the key being that coordination of a stabilizing group (Lewis base, alkene, etc) to a carbenium ion simultaneously advances the cyclization reaction coordinate.iv Interaction of an electron rich neighboring group on a developing carbenium ion (i.e. stabilizing it) lowers the punitive enthalpic cost of localizing a full positive charge on the center in question at the cost of heightened entropic organization, while simultaneously preserving the stereochemical information (i.e. E vs Z) stored in the alkene.
Scheme 1.
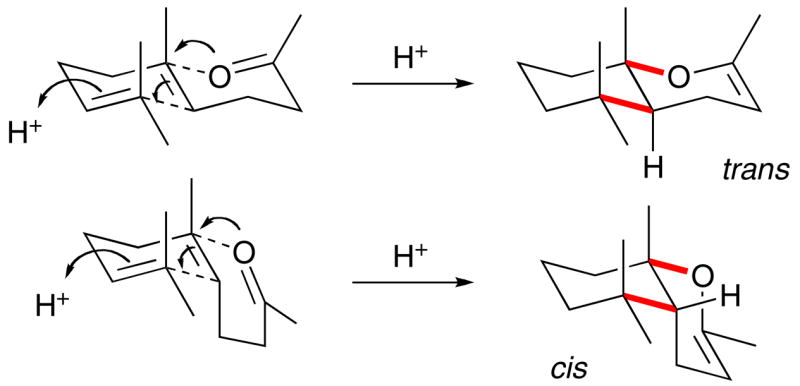
The ability of the neighboring group to engage the developing carbenium ion also effects the efficiency and stereospecificity of cyclization reactions.v When a good participant is available, the reactions tend to be more favorable and selective since they avoid free carbenium ions and stepwise pathways that can erode stereospecificity.
Another factor influencing the step-wise versus concerted cyclization manifolds is the stability of the initiating carbenium ion or reacting group (oxo-carbenium ion, allyl cation, etc). When the initiator is highly reactive, it does not need the stabilizing influence of a neighboring group to react with the internal alkene, which leads to free carbenium ions and less selective, possibly non-stereospecific step-wise reactions.vi On the other hand, when the initiating groups are less reactive, then the beneficial effect of a stabilizing group (alkene or terminating protic group) on ΔG‡ can be significant and this favors concerted, stereospecific reactions.
The debate over step-wise versus concerted reactions is also germane to the discussion of long-range stereocontrol. Numerous examples demonstrate that stereocenters remote from the point of initiation can influence the cascade selectivity (e.g. eq 1).vii These results were most easily rationalized by invoking the influence of the substituent on the ordered conformation of the polyene in a concerted cyclization (i.e. pseudo-equatorial vs. pseudo-axial substituent in a nascent polycycle). Alternatively, a reversible cascade cyclization could account for the selectivity as the retrocyclization provides a means for error correction and access to thermodynamic products. Bartlett in his authoritative 1982 review concluded, primarily using free energy arguments, that long range stereocontrol in sterol cyclizations could not come from a reversible cyclization scenario and that the only logical means for achieving these remarkable effects was through the preorganization that accompanies a concerted cyclization.1c,viii
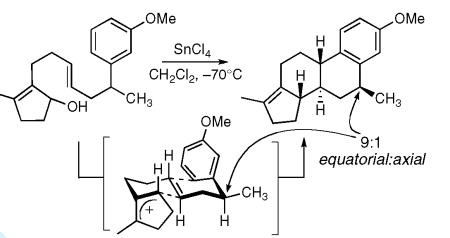 |
(1) |
Carbophilic transition metal complexes are also known to activate terminal alkenes for cascading cation-olefin reactions.ix In most of these cases the Stork-Eschenmoser postulate holds and polyprenoids provide polycyclic structures through chair-like transition structures. Recent advances include oxidative polycyclizations using PdCl2 catalysts in combination with benzoquinone,x,xi (pyridyl-bisphosphine)PdII dications,xii (trisphosphine)PtII dications,xii and Hg(II) mediated polyprenoid cyclizations.xiii In addition to transition metals, several important developments have occurred in the area of chiral electrophilic halonium ionsxiv and chiral Brønsted-Lewis acid catalystsxv for promoting enantioselective cation-olefin polycyclizations. Unlike H+, Hg(II), and X+ (X= I, Br, Cl), the carbophilic Pd(II) and Pt(II) Lewis acids have a strong preference for coordinating and activating the least substituted alkene, which provides a strong chemodirecting effect in initiating the cation-olefin cyclization of polyene substrates.xvi
Recent efforts in our laboratory have shown that pincer ligated Pd(II)- and Pt(II)-dications serve as excellent initiators of cation-olefin multicyclizations when the terminus is monosubstituted.xvii The pincer ligands efficiently block migratory deinsertion pathways resulting in stable polycyclic organometallic products (e.g. eq 2). In the course of evaluating the effect of ligand electronics on the cyclization reaction, it was discovered that ligand basicity not only affected the kinetics of cyclization, but also the thermodynamics. With electron rich ligands, an equilibrium between cyclized and acyclic (η2-alkene) structures was discovered and the position of this equilibrium was responsive to ligand electronics.
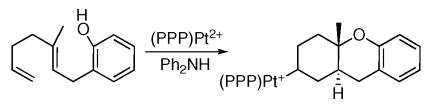 |
(2) |
Given the potential role of reversibility on the stereoselectivity of biomimetic cation-olefin cascade reactions, a study was initiated to establish its role on the diastereoselectivity of Pt-mediated cyclization reactions. We report herein, the effect that solvent, ligand basicity, ring strain, and ring size have on the position of the cyclized/acyclic equilibrium. These data were then utilized to establish conditions wherein retrocyclization was operative and could be utilized to measure the effect of remote substituents on the kinetic and thermodynamic cyclization diastereoselectivity.
Results
Activation Conditions
The Pt(II)-dications for this investigation were generated by one of two means. For complexes bearing more electron donating tridentate ligands (EtPPPEt, tBuPPPEt and PPPEt, see Chart 1) the dication was preferably generated via protonolysis of the Pt-Me precursor using [Ph2NH2][BF4].xix A typical activation procedure involved the addition of 10 equivalents of [Ph2NH2][BF4] to a solution of [(RPPPR′)Pt-Me][BF4] and dienyl substrate in the appropriate solvent; at the point of initiation, the reaction thus contained 9 equiv of [Ph2NH2][BF4] and one equiv of Ph2NH. For complexes bearing the more electron poor PPP and EtPPP ligands (Chart 1), the preferred mode of activation was with AgBF4 and the diiodide [(RPPPR′)Pt-I][I] precursor, followed by the addition of one equiv of base (Ph2NH or Ph2NMe). Activation via halide abstraction was possible with the more electron rich Pt complexes, however, this approach required the addition of two equivalents of acetone to act as a weak labile ligand.xviii
Chart 1.

Cyclization Reactions
Previous work on the conversion of 1,6-dienes into bicyclopropanes by (PPP)Pt(II) dications indirectly revealed that Pt-mediated C-C bond forming carbenium ion generation was both rapid and reversible.xvii This observation also suggested a similar possibility in cascade cyclization processes. The first evidence for this behavior surfaced while monitoring the cyclization of 10 equiv of a 2:1 E:Z mixture of 1,5-dienyl sulfonamide (E:Z)-1 by (EtPPPEt)Pt2+ in CH2Cl2 (see Chart 1). Instead of the expected smooth conversion to a Pt-alkyl, an equilibrium between the starting η2-alkene adduct and expected product was observed (eq 3).xix The identity of these complexes was readily apparent from the characteristic JPt-P of their central phosphorusxx (Pt(η2-alkene), A, JPt-P = 2762 Hz; Pt-alkyl, B, JPt-P = 1281 Hz).xxi The presence of uncyclized η2-alkene adduct was unexpected because all previous cyclizations of dienyl substrates with intramolecular traps had been rapid and proceeded to completion. After one hour, an A:B ratio of ~1:2 was observed by 31P NMR,xxii and although decomposition occurred over extended times, an equilibrium constant could be measured at early times (~60). xxiii
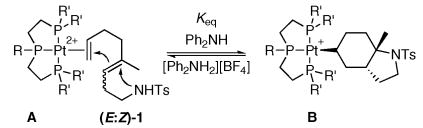 |
(3) |
Solvent and Ligand Effects on Keq
From this starting point, an investigation into the factors controlling the equilibrium position was undertaken.xxiv With [(EtPPPEt)Pt][BF4]2 and (E:Z)-1 as the probe, Keq decreases with increasing polarity, presumably by the favorable solvation of the dicationic Pt(η2-alkene) complex over the monocationic Pt-alkyl (Table 1), though a solvent effect on the acid strength may also contribute. The poor solubility of [Ph2NH2][BF4] limited the accessible solvents. Nitromethane (Keq = 0.68, Figure 1) was selected for further analysis since Pt-olefin complex A was favored (Figure 1).
Table 1.
Solvent effects on the cyclization of (E:Z)-1 with (EtPPPEt)Pt2+ a
| Entry | Solvent | Keq (A:B)b | ΔG (kcal/mol) |
|---|---|---|---|
| 1 | CH2Cl2c | 60 (1:2) | −2 |
| 2 | ClCH2CH2Clc | 110 (1:3) | −2.8 |
| 3 | EtNO2 | 3.2 (4:1) | −0.69 |
| 4 | MeNO2 | 0.68 (14:1) | 0.23 |
Conditions: [Pt] = 0.027 M, [Ph2NH2][BF4] = 0.27 M, [1] = 0.27 M, 25(1)° C (see Supplementary Material).
Relative concentrations determined by 31P NMR. Average of three measurements.
[Pt] = 0.012 M.
Figure 1.
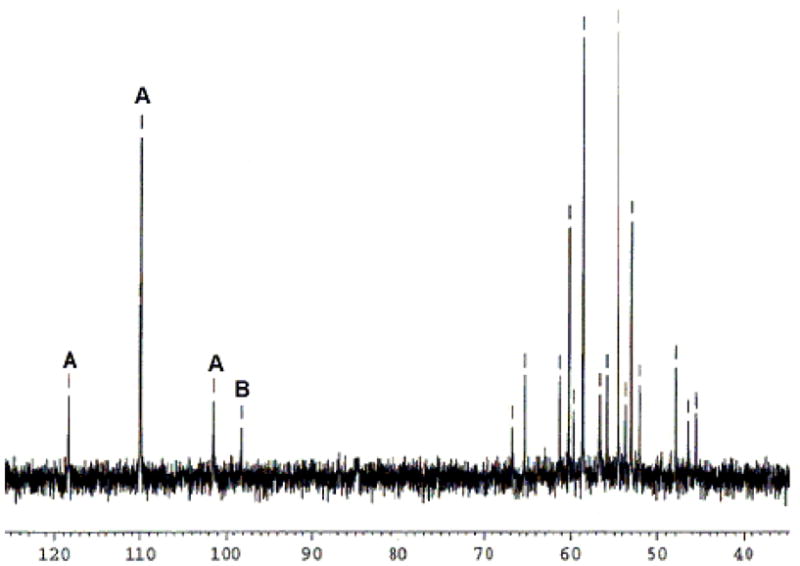
31P NMR of the cyclization of 1 with (EtPPPEt)Pt2+ in MeNO2.
Since previous reports with the more electron poor PPP ligands had not indicated the potential for either reversibility, or stepwise cyclization of polyenes, the effect of ligand basicity on the cyclization equilibrium was explored.xxv Table 2 shows the results of modifying the donor properties of the phosphine substituents and the consequent metal electrophilicity.xxvi Not surprisingly, more electrophilic complexes shifted the cyclization equilibrium towards the monocationic Pt-alkyl species, a result of increased activation of the η2-alkene form and the favorable pairing of the less basic ligand with the more donating alkyl. While no Pt(η2-alkene) species (<5%) was detected for the most electrophilic case ((PPP)Pt2+), the detectable limit of reversibility was observed using EtPPP as the tridentate ligand (Keq = 1100; Figure S3).
Table 2.
| Entry | Ligand | Keq (A:B)b | ΔG (kcal/mol) |
|---|---|---|---|
| 1 | EtPPPEt | 0.68 (14:1) | 0.23 |
| 2 | tBuPPPEt | 7.2 (2:1) | −1.2 |
| 3 | PPPEt | 14 (1.2:1) | −1.6 |
| 4 | EtPPP | 1100 (1:10) | −4.1 |
| 5 | PPP | >4200 (1:>20) | < −4.9 |
Reaction conditions: [Pt] = 0.027 M, [Ph2NH2][BF4] = 0.27 M, [1] = 0.27 M, 25(1)°C (see Supplementary Material).
Relative concentrations determined by 31P NMR. Average of three measurements.
Based on the cyclization reaction, it was anticipated that stronger bases would drive the equilibrium towards the cyclized product and that stronger conjugate acids would promote retrocyclization to the Pt(η2-alkene) complex. Experiments to confirm this were limited to bulky diaryl ammonium acids since the conjugate bases of smaller, more electron rich ammonium acids ligate the metal and poison the complex. With (EtPPPEt)Pt2+, the Keq shifts from 0.68 (A:B = 14:1) using [Ph2NH2][BF4] (Table 1), to 11 (A:B = 1.4:1) using [Ph2NMeH][BF4], consistent with the notion that a stronger base such as Ph2NMe helps promote the cyclization.xxvii Adding base or acid to a preequilibrated system also shifted the ratios of A and B in the expected fashion. Adding 5 equivalents of Ph2NH to a mixture where A predominated (entry 4, Table 1) decreased the A:B ratio from 14:1 to almost 2:1. Likewise, 5 equivalents of [Ph2NH2][BF4] shifted the equilibrium to the left; 1:10 to 1:4 (A:B).xxviii
It was anticipated that ring strain in the cyclized organic moiety would also effect the equilibrium in a predictable manner. To investigate this factor, the dienyl sulfonamide E-2, which should produce a less strained 6,6 ring structure, was subjected to reversible conditions using (EtPPPEt)Pt2+. The less strained aza-decalin structure did indeed shift the equilibrium towards the cyclized complex 3 (Keq = 1.0; eq 4),xxix however the magnitude of the shift was less than expected based on generic ring strain measures (6.3 kcal/mol for cis-hydrindan versus -1.9 kcal/mol for trans-decalin).xxx
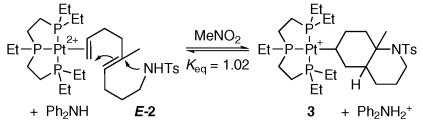 |
(4) |
Stereocontrol in Reversible Polycyclizations
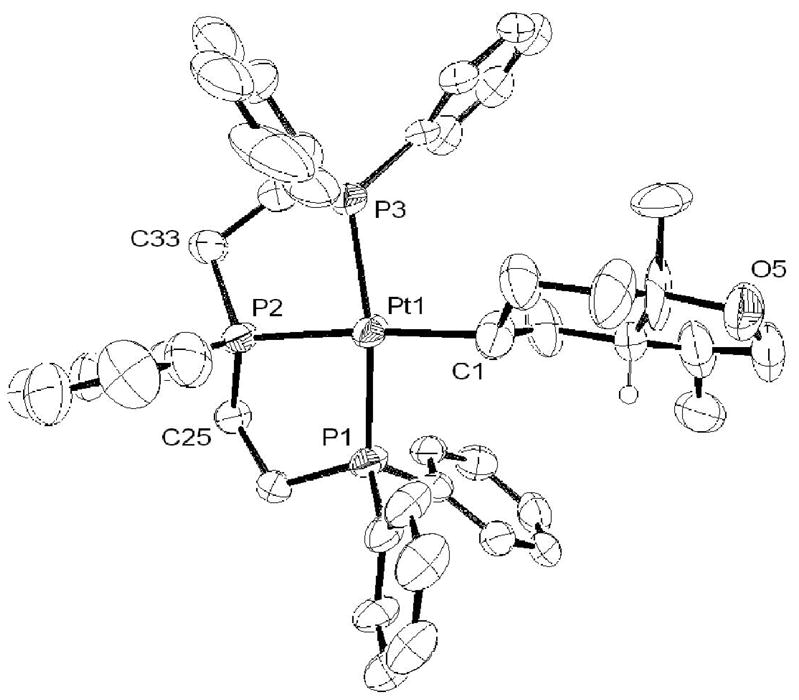
As discussed above, the PPP complex smoothly reacts with 1 equivalent of (E:Z)-1 to generate alkyl complex 4 with no trace of η2-alkene (eq 5). This complex was isolated as its BF4− salt and characterized by X-ray crystallography. The ORTEP in Figure 2 clearly shows a cis ring juncture for the bicyclic fragment,xxxi which contrasts the trans bicyclic products typically obtained from catalytic and stoichiometric cyclizations of E-alkenes.xxxii To rule out the possibility of a selective cyclization of the minor Z isomer present in the 10 equivalents of (E:Z)-1 (which directly leads to the observed product via the SEP), E-1 was synthesized and cyclized (Scheme 2). By 31P NMR the Pt-alkyl obtained from this reaction was identical to 4, indicating that both stereoisomers of 1 converge to a single product. This result clearly conflicts with the notion of a concerted cyclization that follows the Stork-Eschenmoser postulate, both because only one product is obtained and because it requires an intermediate(s) capable of converging the E and Z isomers of 1 into a single stereoisomer of 4 (vide infra).
Figure 2.
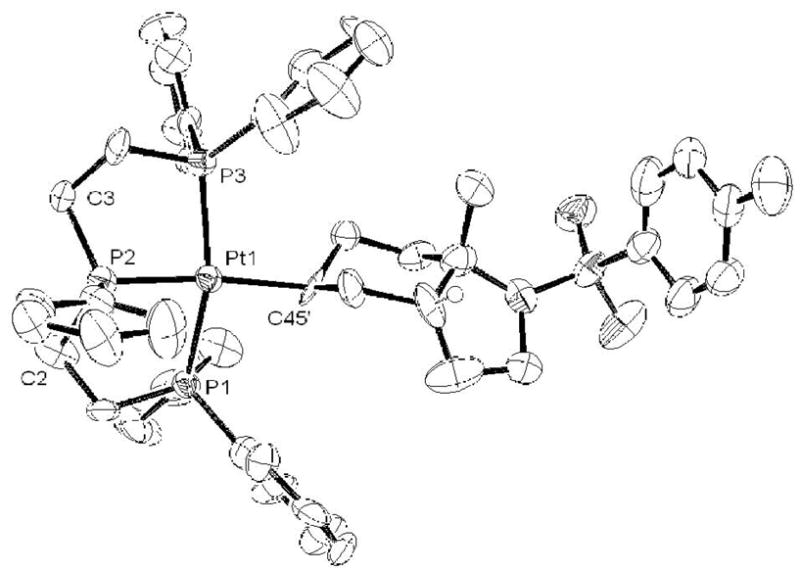
ORTEP representation of 4. Hydrogen atoms and BF4− counter ion removed for clarity. Selected bond lengths (Å): Pt-P1 = 2.269(4), Pt-P2 = 2.247(3), Pt-P3 = 2.308(4), Pt-C45 = 2.150(1). Selected bond angles (deg): P1-Pt-P2 = 84.51(16), P2-Pt=P3 = 84.63(16), P1-Pt-C45 = 92.2(4), P3-Pt-C45 = 100.6(4), C3-P2-C2 = 117.0(6).
Scheme 2.
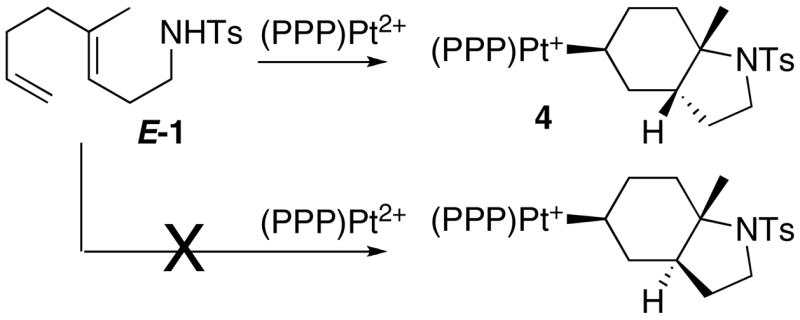
 |
(5) |
The primary alcohol version of E-1, E-5, was also examined as a substrate using the (PPP)Pt2+ complex obtained by HNTf2 activation of [(PPP)Pt-Me][BF4]. In the presence of 1 equivalent of Ph2NMe, compound E-5 cyclized smoothly to a single alkyl product. Cleavage of the bicycle using NaBH4 generated the cis-fused octahydrobenzofuran 6, a product corresponding to a Pt-alkyl that again cannot be the primary cyclization product of E-5 since the SEP predicts a trans ring junction (eq 6). Similar to the cyclization of E-1, the isolated product is the thermodynamically more stable cis-[6,5] ring junction.
 |
(6) |
To investigate how existing stereocenters, especially those on remote positions of the polyene would influence the diastereoselectivity of the cyclization reactions, several readily available dienyl and trienyl alcohols were synthesized.xxxiii The 1,5-dienyl alcohol E-7, bearing a methyl group β to the trapping –OH, was synthesized as shown in Scheme 3. It underwent rapid cyclization (<20 min) with (PPP)Pt2+ (Scheme 4) to generate one cationic (PPP)Pt-alkyl product (8) by 31P NMR (89.6 ppm, JPt-P = 1316 Hz), with no evidence for the intermediacy of other compounds.xxxiv As expected from the previous results with (PPP)Pt2+ and 1, no Pt(η 2-alkene) was observed in the 31P NMR. In contrast to the above cases, however, crystallographic (X-ray) analysis revealed that 8 had a trans ring juncture and a pseudo equatorially disposed methyl group on the furan ring (Figure 3). The product thus appears to be that predicted from a concerted, chair-like transition state that positions the methyl group in a pseudoequatorial position (AA, Scheme 4). The observed 3,5 trans relationshipxxxv indicates efficient 1,6-stereoinduction, and since one would expect no diastereofacial preference in the alkene coordination step, it also indicates that the BB diastereomer either does not cyclize or it rapidly reverts back to starting material upon doing so.xxxvi
Scheme 3.

Scheme 4.

Figure 3.
ORTEP representation of 8. Hydrogen atoms and BF4− counter ion omitted for clarity. Selected bond lengths (Å): Pt-P1 = 2.262(3), Pt-P2 = 2.290(3), Pt-P3 = 2.290(3), Pt-C1 = 2.158 (11). Selected bond angles (deg): P1-Pt-P2 = 84.80(10), P2-Pt-P3 = 83.15(10), P1-Pt-C1 = 90.6(4), P3-Pt-C1= 101.7(4), C27-P2-C33 = 112.2(5).
An illuminating observation was made when E-7 was reacted with [(EtPPP)Pt][BF4]2 under the standard AgBF4 activation conditions. Instead of a smooth conversion to a single alkyl product (or an equilibrium mixture of η2-alkene and alkyl), the 31P NMR spectrum indicated, after 3 hours at RT, a mixture of Pt(η2-alkene) at 100.4 ppm (JPt-P = 2904 Hz) and two Pt-alkyl species at 90.9 (JPt-P = 1241 Hz) and 90.2 ppm (JPt-P = 1239 Hz) in a 1:1 ratio. Over time, this mixture converged to a single Pt-alkyl (9) at 90.9 ppm (Figure 4) with no trace of the η2-alkene.
Figure 4.
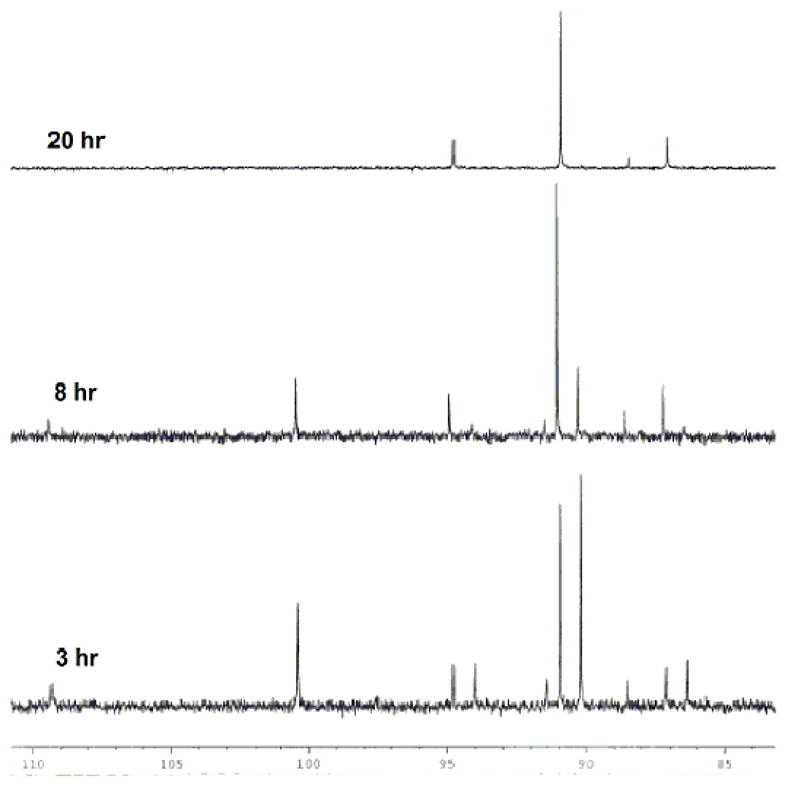
31P NMR stack plot of the cyclization of E-7 with (EtPPP)Pt2+.
Upon completion of this reaction, 9 was isolated and crystallographically characterized. As shown in Figure 5, the bicyclic fragment has the cis ring junction, and is thus similar to 4 and 6. As in the case of 4, 9 has Pt in an equatorial position on the cyclohexyl A-ring and the heteroatom in a 1,4-trans relationship (see Figure 2 for comparison).xxxvii Additionally similar to 4 and 6, the stereochemical outcome cannot be explained by a simple rearrangement (vide infra). When this reaction was stopped after three hours and treated with NaBH4, it was determined (by GC) that the early forming product was the same trans isomer obtained by cleavage of the cycloalkyl in 8 (see Supporting Information).
Figure 5.
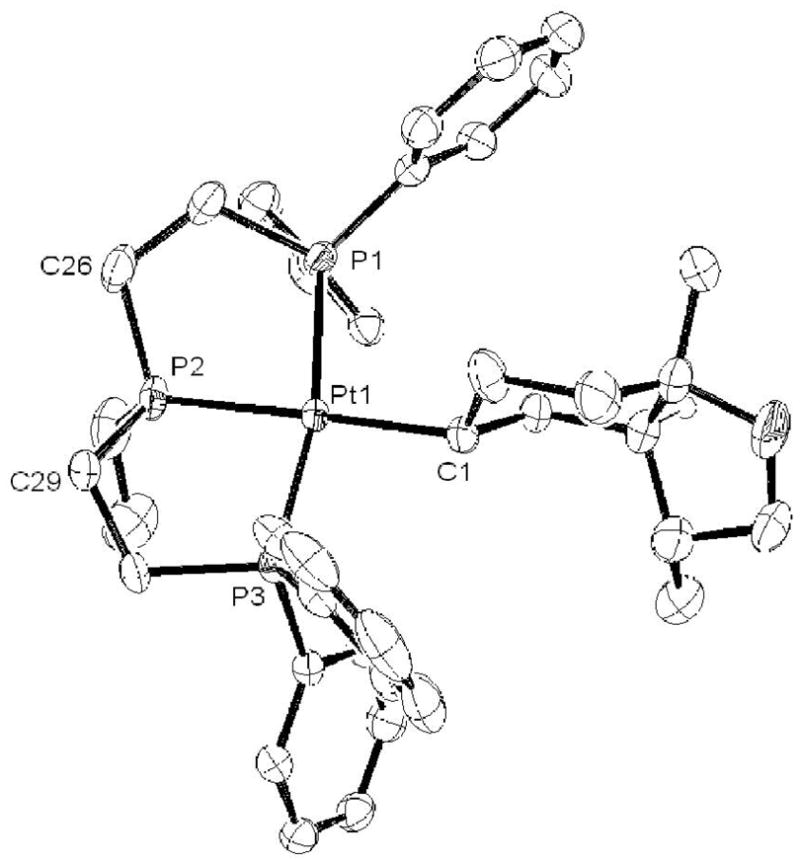
ORTEP representation of 9 with hydrogen atoms and BF4− counter ion omitted for clarity. Selected bond lengths (Å): Pt-P1 = 2.3032(8), Pt-P2 = 2.3016(8), Pt-P3 = 2.2722(8), Pt-C1 = 2.134(3). Selected bond angles (deg): 101.07(9), P3-Pt-C1 = 90.04(9), C26-P2-C29 = 110.85(18).
To ascertain if the stereoselective reactivity would extend to a tricyclization, the trienol E,E-10, bearing a –Me substituent β to the trapping –OH on the nascent C-ring, was examined (Scheme 5). The trienol could be efficiently obtained by a sequence of dihydrofuran metallation/alkylation followed by Ni-catalyzed ring opening.xxxviii A priori, one expects a larger driving force for the complete cyclization of a tricycle as compared to E-7. The ΔHcyclization for E,E-10 is relatively straightforward (~20 kcal/mol more negative than E-7), and although ΔScyclization will certainly be more negative, it is difficult to estimate by how much.id In any case, one can reasonably predict a more negative free energy for a tricyclization than a bicyclization, which would then tend to favor alkyl products over the Pt(η2-alkene) provided that the reaction is kinetically feasible.
Scheme 5.

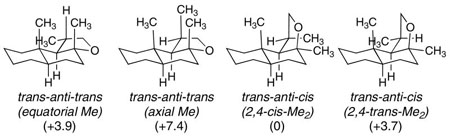
Scheme 6 shows the reaction of E,E-10 with the (EtPPPEt)Pt2+, (EtPPP)Pt2+, and (PPP)Pt2+ cyclization initiators. 31P NMR analysis of the combination of (EtPPPEt)Pt2+ and E,E-10 under standard reaction conditions showed that although the Pt(η2-alkene) complex forms normally, it does not cyclize. Ten additional equivalents of Ph2NMe pushes the reaction to ~90% Pt-alkyl and 10% Pt(η2-alkene), however, additional amine did not complete the reaction. Adding 10 equivalents of [Ph2NHMe][BF4] to this solution did not change the ratio of Pt-alkyl to Pt(η2-alkene), suggesting a poorly behaved, but static mixture (c.f. E-7).xxxix In contrast, the cyclization of E,E-10 occurred smoothly when the more electron withdrawing EtPPP and PPP ligands were used. Since the 31P NMR for the (PPP)Pt-alkyls were broad, the cyclization diastereoselectivity was determined by first cleaving the tricycle with NaBH4 and then analyzing by GC. This protocol yielded a 2:1 ratio of diastereomers that were inseparable by column chromatography, but whose structures could be ascertained by careful NMR analysis. These data are most consistent with the major diastereomer having the trans-anti-trans core with the C-ring methyl substituent adopting a pseudo-equatorial orientation, while the minor was most consistent with the trans-anti-trans tricycle having 3 axial methyl groups (Scheme 7).xl
Scheme 6.
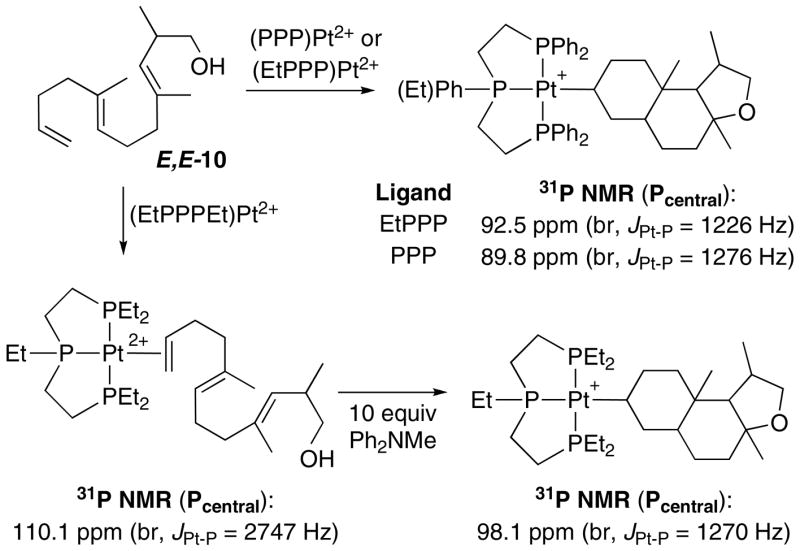
Scheme 7.

Discussion
Reversible Polycyclizations Using a 1,5-Dienyl Sulfonamide
The reversible cyclization of 1 proved to be a useful tool for establishing the effect of reaction conditions and metal complex electronics on the thermodynamics of cation-olefin cyclization. Taken together, the data paint an internally consistent picture wherein acid/base strengths, ring strain, ligand basicity, Pt-electrophilicity and solvent effects all have a predictable effect on the driving force for cascade cyclization. Not surprisingly, cyclization was favored by less polar solvents, more electrophilic metals, stronger bases, and a less strained bicyclic product. The data quantitatively describe the magnitude of these effects while guiding the choice of conditions to establish a reversible cyclization reaction for examining the effect of existing stereogenic centers on cation-olefin polycyclization diastereoselectivities.
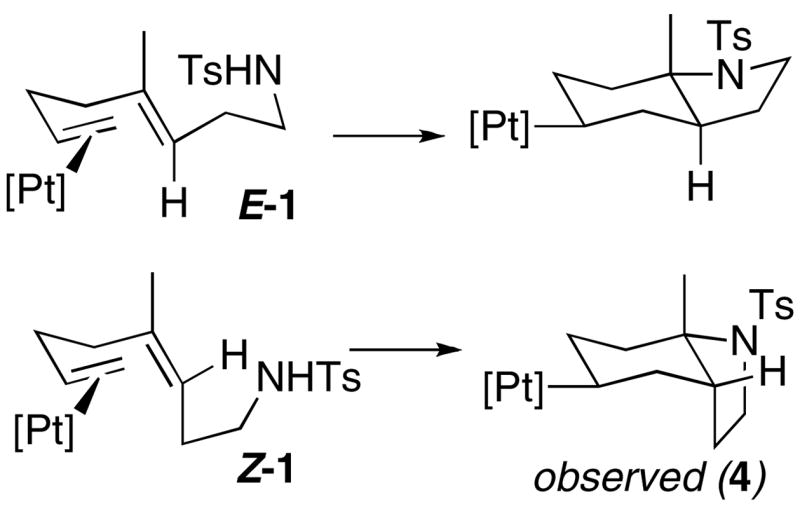
Since (E:Z)-1 is a 2:1 mixture of E and Z isomers, generating two diastereomeric Pt-alkyls, one with a trans ring juncture and one with a cis ring juncture, should be possible (Scheme 8). Only one Pt-alkyl was observed by 31P NMR, suggesting that either Z-1 cyclized directly to 4 (Scheme 8) or that E-1 first cyclizes to a trans product and then subsequently isomerizes to the observed product, or both. The smooth conversion of E-1 to 4 (Scheme 2) suggests that the former case is not dominant. Since Z-1 does not build up in the 1H NMR of the E-1 reaction, this eliminates the possibility of a reversible precyclization E/Z isomerization. It is important to note that isomerization of the trans product in Scheme 8 to 4 is not as simple as acid-promoted ionization of the C-N bond and recoordination to the opposite face, as this scenario provides an unobserved diastereomer where the [Pt] and N have a 1,4-cisoid arrangement instead of 1,4-transoid.
Scheme 8.
Mechanistically revealing was the set of experiments carried out on the methylated compound E-7. Under conditions that minimized retro-cyclization (electron poor PPP ligand), a single product with a trans ring junction was observed. We favor the hypothesis wherein this is the “kinetic” product, and that it proceeds through the most favorable chair-pseudo-chair transition state I, which places the B-ring methyl group in a pseudo equatorial orientation (8, Scheme 9). We qualify the term “kinetic” in this case because under irreversible conditions one should observe some of the less favored axial methyl product (from BB, Scheme 4) since diastereofacial control on η2-alkene formation should be low. If this product does form, it reverts to starting material and eventually traps at 8; nevertheless, we consider this chair-chair product the kinetic product and will refer to it as such (Scheme 9). We suspect that this chair-chair arrangement leads to the kinetic product in each of the cases studied herein, though as outlined below it may convert to a more stable isomer under certain conditions.
Scheme 9.

When the triphosphine is more electron rich than PPP, a scenario conducive to retrocyclization, the kinetic product converts to the more stable cis product 9. Since 9 does not result from a simple ionization of the C-O bond and recoordination to the opposite face of the carbenium ion (leading to III), a more complex sequence of steps is needed to invert the C3 stereocenter. One potential mechanism is outlined in Scheme 9, the keys steps being a retrocyclization of 8 and re-cyclization via the boat transition state IV to V, which then ionizes and recoordinates to form 9.xli By invoking a boat transition state followed by C-O fragmentation and reformation it is possible to access a cis ring junction from an E-alkene, and thus avoid the constraints of the SEP. The potential for boat-like transition states was previously hinted at in the cyclization of a diene-phenol by (PPP)Pt2+ (eq 2). The diastereoselectivity in this reaction was 96:4, and >99:1 after cleavage of the bicycle with NaBH4, a result that was interpreted as being due to competing chair-chair and boat-chair transition structures.xii,xlii,xliii The importance of a B-ring boat transition state is well established in sterol biosynthesis.i In the present case IV is higher in energy than I and is normally non-competitive when retrocyclization is slow.
A similar sequence of steps was likely responsible for the formation of 4 and the Pt-alkyl that resulted from the cyclization of E-5. Less obvious was why E-5 converted to the cis product using (PPP)Pt2+ while E-7 did not. It is possible that the single methyl group on the THF ring reduced the rate of ring opening (an attenuated Thorpe-Ingold effect), with a concomitant lowering of the Pt(η2-alkene) concentration such that the kinetic product is temporally stabilized. This question will require more experimentation.
While a good case can be made for reversibility affecting the diastereoselectivity of bicyclization reactions, the same was not true for the tricyclization of E,E-10. Interestingly, under standard reaction conditions, the more electron rich ligand EtPPPEt was incapable of initiating the cyclization, though it could with 10 equivalents of the stronger base Ph2NMe. With more electron poor ligands, however, a smooth conversion to alkyl products ensued and the diastereoselectivity could be ascertained by cleavage and GC analysis (2:1 dr). Careful NMR analysis of the diastereomer mixture pointed to products of the chair-anti-chair conformation, with the mixture occurring at the beta carbon of the THF ring (pseudo-axial and -equatorial). Although modest, this 2:1 selectivity represents 1,10-stereoinduction,viii and since the reaction is apparently irreversible,xliv it requires a different rate of cyclizing the two diastereomeric acyclic activated complexes (CC and DD), consistent with Bartlett’s assertion that long-range stereocontrol in sterol cyclizations comes from developing intra-ring interactions in the organizing assembly.id Additionally consistent with a concerted cyclization was the formation of trans-anti-trans products, as reversibility in the C-O bond forming step would access the more stable [6.6.5]-trans-anti-cis products.xlv

The marked difference in reactivity between the phosphine ligands in bi- vs. tricyclization was intriguing. In the former case, the ligands can each catalyze the cyclization, and the observed effect of ligand basicity is on the position of the equilibrium. In the tricyclization case, however, the electron rich EtPPPEt cannot initiate cyclization (standard conditions), while the less basic EtPPP and PPP ligands do so smoothly. This difference can be understood by noting that the bicyclization is not a true cation-olefin reaction since no developing carbenium ion reacts with another alkene, though the activated terminal alkene certainly behaves like one. The developing positive charge at C4 is stabilized by the hydroxyl center, either with or without the aid of base (EE).xxvii Since an alcohol will be a good neighboring group for stabilizing such a species, the reaction coordinate may proceed forward without needing to build up a large positive charge. On the other hand, B-ring formation in the tricyclization requires a genuine cation-olefin reaction, and since an alkene is a poorer ligand for the developing carbenium ion (FF), the degree of stabilization imparted by the neighboring group will be significantly lessened.xlvi Moving the reaction coordinate forward in a tricyclization thus requires the buildup of more positive charge on C4 to engage the alkene, which concomitantly requires a more electrophilic complex.

Systematically modifying the electrophilicity of the initiating Pt(η2-alkene) complex in the cation-olefin cyclization provided the means for examining how the cyclization thermodynamics responded. Not surprisingly, the more electrophilic the initiating group the more favorable the cyclization. When the cyclization was reversible high diastereoselectivities were observed, suggesting that this mechanism is indeed effective at error correcting and promoting long-range transfer of stereochemical information.xlvii Moreover, when the cyclizations are reversible, alternative reaction pathways are explored that may access more stable products not otherwise accessible under the kinetic control of a typical cascade cyclization that is constrained by the SEP. The ability of pincer-ligated platinum(II) complexes to achieve this enables the formation of cis-ring fused bicyclic complexes that are not predicted by the SEP. Tricyclizations, however, were too favorable to find conditions where the η2-alkene form was stable enough to enable retrocyclization and electrophilic enough to initiate the forward cascade cyclization.
Supplementary Material
Tables and figures for equilibrium studies including sample 31P NMR spectra, full experimental procedures, and X-ray structural data for 4, 8 and 9. This material is free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Institutes of Health Institute of General Medicine for support of this research. We thank Dr. Peter White for X-ray structural determinations; correspondence regarding them should be directed to his attention at UNC Chapel Hill (pwhite@unc.edu). We also thank Dr. Marc ter Horst (UNC Chapel Hill NMR Facility) and Dr. Tony Ribeiro (Duke University NMR Center) for extensive help with NMR analysis on the mixture of diastereomers produced from the cyclization and subsequent cleavage of 10.
References
- i.(a) Yoder RA, Johnston JN. Chem Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sutherland JK. In: In Comprehensive Organic Synthesis. Trost BM, editor. Vol. 1. Pergamon Press; 1991. pp. 341–377. [Google Scholar]; (b) Wendt KU, Schultz GE, Corey EJ, Liu DR. Angew Chem Int Ed. 2000;39:2812–2833. [PubMed] [Google Scholar]; (c) Bartlett PA. In: In Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; New York: 1984. pp. 341–409. [Google Scholar]
- ii.(a) Stork G, Burgstahler AW. J Am Chem Soc. 1955;77:5068–5077. [Google Scholar]; (b) Eschenmoser A, Ruzika L, Jeger O, Arigoni D. Helv Chim Acta. 1955;38:1890–1904. [Google Scholar]
- iii.For an English translation and historical perspective on the 50th Anniversary of the Eschenmoser, Ruzika, Jeger and Arigoni 1955 paper, see: Eschenmoser A, Arigoni D. Helv Chim Acta. 2005;88:3011–3050.
- iv.(a) Johnson WS, Bailey DM, Owyang R, Bell RA, Jacques B, Crandall JK. J Am Chem Soc. 1964;86:1959–1966. [Google Scholar]; (b) Bartlett PD, Clossen WD, Cogdell TJ. J Am Chem Soc. 1965;87:1308–1314. [Google Scholar]; (c) Poulter CD, King CR. J Am Chem Soc. 1982;104:1422–1424. [Google Scholar]
- v.(a) Bartlett PA. In: In Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; New York: 1984. pp. 410–454. [Google Scholar]; (b) Yoder RA, Johnston JN. Chem Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vi.(a) Harding KE, Leopold EJ, Hudrlik AM, Johnson WS. J Am Chem Soc. 1974;96:2540–2549. [Google Scholar]; (b) Nishizawa M, Takenaka H, Hayashi Y. J Am Chem Soc. 1985;107:522–523. [Google Scholar]
- vii.(a) Marinus B, Groen MB, Zeelen FJ. J Org Chem. 1978;43:1961–1964. [Google Scholar]; (b) Hart DJ. J Am Chem Soc. 1980;102:397–398. [Google Scholar]; (c) Hart DJ. J Org Chem. 1981;46:367–373. [Google Scholar]; (d) Johnson WS, Berner D, Dumas DJ, Nederlof PJR, Welch J. J Am Chem Soc. 1982;104:3508–3510. [Google Scholar]
- viii.Radical cascade cyclizations can also show remarkable long-range stereoinduction, see: Heinemann C, Demuth M. J Am Chem Soc. 1999;121:4894–4895.For a review of stereoselective radical (and transition metal-catalyzed) multi-cyclizations see: Malacria M. Chem Rev. 1996;96:289–306. doi: 10.1021/cr9500186.
- ix.Chianese AR, Lee SJ, Gagné MR. Angew Chem Int Ed. 2007;46:4042–4059. doi: 10.1002/anie.200603954.Hahn C. Chem Eur J. 2004;10:5888–5899. doi: 10.1002/chem.200400550. and references within.
- x.Koh JH, Mascarenhas C, Gagné MR. Tetrahedron. 2004;60:7405–7410. [Google Scholar]
- xi.Prior to our own efforts in this area were discoveries by the Overman group detailing the utility of catalytic PdCl2(RCN)2 to accelerate Cope-like rearrangements. Mechanistic studies pointed to cyclogenerated carbenium ions via chair-like transition structures. See for example: Overman LE, Renaldo AF. J Am Chem Soc. 1990;112:3945–3949. and references therein.For earlier stoichiometric examples, see Lutz RP. Chem Rev. 1984;84:205–247. and references therein.
- xii.Koh JH, Gagné MR. Angew Chem Int Ed. 2004;43:3459–3461. doi: 10.1002/anie.200453913. [DOI] [PubMed] [Google Scholar]
- xiii.(a) Kang SH, Kim M. J Am Chem Soc. 2003;125:4684–4685. doi: 10.1021/ja029777d. [DOI] [PubMed] [Google Scholar]; (b) Nishizawa M, Yadav VK, Skwarczynski M, Imagawa TH, Sugihara T. Org Lett. 2003;5:1609–1611. doi: 10.1021/ol034201u. [DOI] [PubMed] [Google Scholar]
- xiv.Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]
- xv.(a) Ishibashi H, Ishihara K, Yamamoto H. J Am Chem Soc. 2004;126:11122–11123. doi: 10.1021/ja0472026. [DOI] [PubMed] [Google Scholar]; (b) Ishihara K, Ishibashi H, Yamamoto H. J Am Chem Soc. 2002;124:3647–3655. doi: 10.1021/ja0124865. [DOI] [PubMed] [Google Scholar]
- xvi.(a) Hegedus LS. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 4. Pergamon Press; Elmsford, NY: 1991. pp. 551–569. [Google Scholar]; (b) Hegedus LS. Transition Metals in the Synthesis of Complex Organic Molecules. University Science Books; Mill Valley, California: 1994. pp. 199–236. [Google Scholar]
- xvii.(a) Kerber WD, Gagné MR. Org Lett. 2005;7:3379–3381. doi: 10.1021/ol051277c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kerber WD, Koh JH, Gagné MR. Org Lett. 2004;6:3013–3015. doi: 10.1021/ol048780u. [DOI] [PubMed] [Google Scholar]; (c) See also footnote xb.
- xviii.Addition of acetone generated a Pt(II)-acetone adduct which was stable in solution and was readily displaced by dienyl substrates, see xviia.
- xix.Protonolysis of the precursor Pt-Me with [Ph2NH2][BF4] to give (EtPPPEt)Pt2+ is rapid (<10 min), see: Feducia JA, Campbell AN, Anthis JW, Gagné MR. Organometallics. 2006;25:3114–3117.
- xx.Chemical shifts are given for the central P of the triphosphine ligand. This signal is the most diagnostic as the chemical shifts for the terminal phosphines in the Pt(η2-alkenyl) and the Pt(alkyl) overlap.
- xxi.Over time in CH2Cl2, the 31P NMR spectrum became more complex as chloride abstraction from the solvent generated (EtPPPEt)PtCl (JPt-P = 3030 Hz), among other decomposition species, see: Liaw B, Lobana TS, Lin Y, Wang J, Liu CW. Inorg Chem. 2005;44:9921–9929. doi: 10.1021/ic051166+.Angulo IM, Bouwman E, Lok SM, Lutz M, Mul WP, Spek AL. Eur J Inorg Chem. 2001:1465–1473. doi: 10.1021/ic0006955.Oster SS, Lachicotte RJ, Jones WD. Inorg Chim Acta. 2002;330:118–124.Wang Q, Marr AC, Blake AJ, Wilson C, Schröder M. Chem Commun. 2003:2776–2777. doi: 10.1039/b309523a.
- xxii.The Pt(η2-alkene) is observed in a 2:1 ratio which correlates to the E:Z ratio of diene 1 (Figure S1). The complex upfield splitting of the Pt(η2-alkene) is presumably a result of hindered rotation of the bound olefin by the terminal phosphine substituents. The same splitting pattern is observed in [(EtPPPEt)Pt(1-hexene)][BF4]2 (Figure S2).
- xxiii.The equilibrium constant was calculated from the following equation where [Pt-alkyl] and [Pt(η2-alkene)] were obtained from 31P NMR:
- xxiv.Equilibrium constant determinations were performed using a C6D6 solution of PPh3 as an external standard (sealed capillary). No net loss of Pt(η2-alkene) and Pt-alkyl was detected (other than to Pt-Cl in CH2Cl2). Additional experimental details are presented in the Supporting Information.
- xxv.While using 1 as a trapping ligand during protonolysis studies, there was no observation of a Pt(η2-alkene) when using PPP or EtPPP as the supporting ligand; see footnote xvi.
- xxvi.31P NMR data for these compounds is given in Table S1 of the Supporting Information.
- xxvii.DFT calculations on a 1,6-dienyl phenol indicated that in the presence of base, cyclization was semi-concerted. Nowroozi-Isfahani T, Musaev DG, Morokuma K, Gagné MR. Organometallics. 2007;26:2540–2549.
- xxviii.These results are presented in Table S2 of the Supporting Information.
- xxix.We presume an equatorially disposed Pt-C bond in each case.
- xxx.(a) Wiberg KB. Angew Chem Int Ed. 1986;25:312–322. [Google Scholar]; (b) Allinger NL, Tribble MT, Miller MA, Wertz DH. J Am Chem Soc. 1971;93:1637–1648. [Google Scholar]
- xxxi.This structure displays the largest C-P-C angle deviations for PPP at the central phosphorus (117.0°). For a discussion on the importance of C-P-C bond angles, see footnote xvi.
- xxxii.Mullen CA, Gagné MR. J Am Chem Soc. 2007;129:11880–11881. doi: 10.1021/ja073573l. Also see footnote x and xii. [DOI] [PubMed] [Google Scholar]
- xxxiii.Chan J, Jamison TF. J Am Chem Soc. 2004;126:10682–10691. doi: 10.1021/ja0470968. [DOI] [PubMed] [Google Scholar]
- xxxiv.No detectable quantities of a second isomer was observed either at very short or very long reaction times.
- xxxv.Benzofuran naming convention.
- xxxvi.For clarity, the same diastereoface is used with the opposite C-2 stereocenter in Scheme 4, BB.
- xxxvii.The C-P-C bond angle at the central phosphorus is reduced in 7 (110.8°) compared to 2 (117.0°), which we have previously taken to indicate less strain at the central phosphorus for this square planar complex (footnote xix).
- xxxviii.Kocienski P, Wadman S, Cooper K. J Org Chem. 1989;54:1215–1217. [Google Scholar]
- xxxix.31P NMR analysis indicated no decomposition, and so the stoppage is difficult to explain.
- xl.Molecular mechanic calculations (AM1, MacSpartan 04) on the minor isomer indicated that the THF β-methyl adopts a position to minimize unfavorable Me--Me interactions, which consequently reduces the key H-C-C-H dihedral angle to 35°. The calculated JHH in this orientation is 6.7 Hz, which helps to explain the usually large observed coupling constant (9.0 Hz). See: Kemp W. Organic Spectroscopy. W. H. Freeman and Company; New York: 1991. pp. 154–155.
- xli.Molecular mechanics indicate that the chair conformer of cyclohexane is more stable than the boat conformer by 6.4 kcal/mol, see: Allinger NL, Miller MA, VanCatledge FA, Hirsch JA. J Am Chem Soc. 1967;89:4345–4357.Allinger NL. J Am Chem Soc. 1977;99:8127–8134.
- xlii.Footnote xii also describes a similar situation with a pyridyl-bisphosphine (PNP) pincer ligand on Pd2+, which gives an initial diastereoselectivity of 93:7 and >99:1 after cleavage. Since both diastereoisomers converge to the same trans product, the initial dr was interpreted as reflecting the boat-chair vs. chair-chair preference.
- xliii.By molecular mechanics the chair conformer of cyclohexane is more stable than the boat conformer by 6.4 kcal/mol, see: Allinger NL, Miller MA, VanCatledge FA, Hirsch JA. J Am Chem Soc. 1967;89:4345–4357.Allinger NL. J Am Chem Soc. 1977;99:8127–8134.
- xliv.Since the forward rate is too slow for EtPPPEt under standard conditions, and additional stronger base is poorly behaved, we have not been successful at rigorously proving that the tricyclization is irreversible. However, the expected larger driving force for tri- versus bicyclization, the significant difference in the ground state structures of model structures (footnote 45), and the modest dr all suggest otherwise.
-
xlv.Hartree-Fock calculations (6–31G*) indicate the following ground state relative energies (kcal/mol). Calculations were carried out using MacSpartan 04. The trans-anti-cis structures were not considered viable since a strong nOe between the angular CH3 and the CH3 alpha to the THF oxygen was observed in both diastereomers.
- xlvi.Such non-classical carbocations form the basis of the early experimental rationalizations for the non-concerted, but stereospecific, cascade cyclizations. See footnote 1a, 2, and 3.
- xlvii.For a classic example of this phenomenon in acid promoted bicyclization of farnesic acid derivatives, see: Stadler PA, Eschenmoser A, Schinz H, Stork G. Helv Chim Acta. 1957;40:2191–2198. See also footnote 1a.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables and figures for equilibrium studies including sample 31P NMR spectra, full experimental procedures, and X-ray structural data for 4, 8 and 9. This material is free of charge via the Internet at http://pubs.acs.org.