

Abstract
A transition in hemoglobin behavior at close to body temperature has been discovered recently by micropipette aspiration experiments on single red blood cells (RBCs) and circular dichroism spectroscopy on hemoglobin solutions. The transition temperature was directly correlated to the body temperatures of a variety of species. In an exploration of the molecular basis for the transition, we present neutron scattering measurements of the temperature dependence of hemoglobin dynamics in whole human RBCs in vivo. The data reveal a change in the geometry of internal protein motions at 36.9°C, at human body temperature. Above that temperature, amino acid side-chain motions occupy larger volumes than expected from normal temperature dependence, indicating partial unfolding of the protein. Global protein diffusion in RBCs was also measured and the findings compared favorably with theoretical predictions for short-time self-diffusion of noncharged hard-sphere colloids. The results demonstrated that changes in molecular dynamics in the picosecond time range and angstrom length scale might well be connected to a macroscopic effect on whole RBCs that occurs at body temperature.
INTRODUCTION
The function of biological macromolecules depends not only on their structure but also on their dynamics. Many functional properties of proteins cannot be completely understood from the average protein structure as determined by x-ray crystallography or NMR. Reaction rates, for example, are closely related to protein dynamics (1). Protein dynamics covers a very large range of timescales, from fast electronic rearrangements in the femtosecond scale to slow protein folding events in the order of seconds to minutes (2). Motions in the pico- and nanosecond timescales are believed to act as a lubricant for much slower protein dynamics in the millisecond time range (3).
Below a dynamical transition temperature between 180 and 240 K, atomic motions are predominantly harmonic (4,5). In this state, the atoms vibrate around their structural equilibrium positions. Above the dynamical transition temperature at sufficient hydration, the mean-square displacements increase significantly because of the contribution of internal anharmonic diffusive motions. These diffusive motions may contribute to the sampling of different so-called conformational substates on a complex energy landscape (4,5). The forces that maintain biological molecular structure and allow atomic motions are “weak”, because they are similar to thermal energy at physiological temperatures (3). The amplitudes of atomic motions are a measure of the internal macromolecular flexibility, because they correspond to the width of the potential well in which atoms move (6). In this picture, equilibrium protein stabilization would correspond to the depth of the well (7). The dependence of mean-square displacements on temperature has been interpreted in terms of an effective force constant, called “resilience” by Zaccai (6), which is related to the shape of the well. Protein thermal stability was supposed to be inversely correlated to protein flexibility (8,9), although this point is still largely debated (7,10). The dynamics of lysozyme has been studied with quasielastic neutron scattering during the unfolding process (11), and it was concluded that compared to the folded state, the unfolded state is characterized by significantly larger amplitudes of diffusive motion of amino acid side chains. Protein unfolding was therefore related to a loss of macromolecular resilience.
There is considerable evidence that protein dynamics is strongly influenced by the local environment and the level of hydration (8,12–14). Solvent molecules can act either as plasticizers or stabilizers by allowing or preventing the protein groups to jump between conformational substates (15). Water, which is the natural solvent of biomolecules, is a well known plasticizer. Pacciaroni et al. (13) could show that internal lysozyme dynamics gets activated when the environment is altered by pure glycerol, which is a stabilizer, toward a plasticizer by increasing the level of hydration.
The main natural environment of proteins is within the cell, and protein function necessarily is adapted to these conditions. In bacteria and eukaryotic cells, macromolecular interactions are likely to be influenced by the high cellular protein concentration (16–18). This effect, called “crowding”, results from high-volume occupancy and steric hindrance of the protein molecules between each other. The free distance between macromolecules in the cell is in the order of some angstroms (19) and intercellular water is in close vicinity to the protein surfaces. There is much interest and discussion about similarities and differences in structure and dynamics of cytoplasmic and bulk water (20). There is still little known about how protein dynamics is influenced by the crowded cytoplasmic environment and the special properties of cellular water. Doster and Longeville (21) have measured the global diffusion of hemoglobin in red blood cells with neutron spin-echo spectroscopy. They found a reduced diffusion coefficient of hemoglobin compared to dilute solution and attributed the reduction to direct interactions between protein molecules and to hydrodynamic interactions with the solvent. The authors deduced that hydrodynamic effects dominate macromolecular transport at high protein concentration. In recent work, Jasnin et al. have investigated macromolecular and water dynamics in Escherichia coli bacteria with quasielastic neutron scattering in a wide range of timescales (22,23). An appreciable increase in internal molecular flexibility compared to fully hydrated protein powders was revealed. The authors concluded that intracellular complexity influences protein dynamics, which is necessary for biological activity. The study of global and internal protein diffusive motions is of importance for a fundamental understanding of macromolecular transport and functional protein internal flexibility in cells.
The fast trajectories in the pico- and nanosecond timescales can be directly accessed by molecular dynamics simulations (24) and incoherent neutron scattering (25). Incoherent neutron scattering has the advantage that the proteins under study are not limited to a maximum molecular weight, samples do not have to be crystalline or even monodisperse. Neutrons penetrate deeply into the sample without causing radiation damage, which makes them an ideal tool to probe radiation-sensitive biological matter. The incoherent scattering cross section of hydrogen atoms is one magnitude larger than all other elements that occur in biological material. As hydrogen atoms are uniformly distributed in proteins, neutron scattering probes average protein dynamics. Incoherent neutron scattering experiments have been performed on hydrated protein powders (25–27), protein solutions (14,28,29), and even in vivo on whole cells (21,22,30).
Hemoglobin is the main protein constituent of red blood cells (92% of dry weight). Its biological function is to carry oxygen from the lungs to the tissues. The discovery of the structure of hemoglobin by Max Perutz is one of the major breakthroughs in the history of molecular biology (31). The protein is a tetramer with a molecular weight of ∼64 kDa. It consists of two α-chains and two β-chains, each having 141 and 146 amino acid residues, respectively. The α-chains contain seven and the β-chains eight helices (32). Every chain carries one heme group in a pocket, to which oxygen and several other small molecules can bind reversibly. The concentration of hemoglobin in the red blood cells is ∼330 mg/ml (19), which corresponds to a volume fraction of 0.25. It has become clear recently that hemoglobin at higher concentration shows a variety of interesting effects. Micropipette experiments with aspirated single human red blood cells showed a sudden passage phenomenon of the cells that is very close to human body temperature. A drop in viscosity of concentrated hemoglobin solutions at temperatures higher than the transition temperature was found and it was hypothesized that protein aggregation is the cause for the cellular passage effect and the drop in viscosity (33). In addition, it was found that hemoglobin shows a pronounced loss of its α-helical content at body temperature. To our amazement, the transition temperatures were directly correlated to the body temperature of a big variety of species (34,35). It was speculated that this reflects partial unfolding of the helical structure and goes hand in hand with an increase in surface hydrophobicity, which promotes protein aggregation (34).
Here, we present an incoherent quasielastic neutron scattering study on the temperature dependence of hemoglobin dynamics in whole red blood cells. The experiment was performed at temperatures between 16.9°C and 45.9°C. Global and internal protein motions could be separated. A change in the amplitudes of protein side-chain diffusion was found close to human body temperature, which was attributed to partial unfolding of hemoglobin.
MATERIALS AND METHODS
Sample preparation
For neutron scattering experiments, samples of human venous blood from healthy adults were drawn with tubes containing heparin to prevent blood coagulation. The blood samples were suspended in HEPES buffer solution at pH 7.4 and 290 mOsm (137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 0.8 mM Na2HPO4, 0.2 mM NaH2PO4, 0.7 mM MgSO4, 8.4 mM HEPES, and 4 mM NaOH). The cells were washed twice and collected by centrifugation at 560 relative centrifugal force (RCF) for 10 min. The supernatant was removed together with the “buffy coat” on top of the cells. The washed cells and the successively used buffer solutions were then gassed with CO to increase the stability of hemoglobin. The cells were resuspended in TRIS buffer (20 mM TRIS, 145 mM NaCl) at pH 5.5 and treated with neuraminidase (from Clostridium perfringens Type VI, purchased from Sigma-Aldrich, St. Louis, MO) to remove the glycocalyx matrix, as described elsewhere (36). Afterward, the cells were washed in H2O HEPES buffer. To reduce the neutron scattering contribution of the buffer, the cells were washed with D2O HEPES buffer (pD 7.4, 290 mOsm), incubated for ∼30 min, and centrifuged at 560 rcf. The washing steps were repeated until the level of H2O was estimated to be <0.1 vol %, assuming that the H2O-D2O exchange through the cell membrane during this time reaches a constant value. No cell lysis was detected during the preparation, and the shape of the cells was checked with optical microscopy at the end. After a final centrifugation step, the cell pellet was sealed in a flat aluminum sample holder of 0.2 mm thickness for the neutron scattering experiment. The scattering from the aluminum screws was blocked using a cadmium mask. It was checked by weighting that no loss of sample material occurred during the experiment.
Hemoglobin samples for dynamic light scattering experiments were prepared from ∼75 μl of human red blood cells taken from the finger tip with a heparinized glass capillary. We tried to perform the light scattering measurements in conditions that resembled the saline environment in the red blood cell. Although the extracellular concentration of sodium is ∼145 mM, the intercellular concentration is only between 5 and 15 mM. Inversely, the intracellular concentration of potassium is ∼140 mM and the extracellular concentration is ∼5 mM (37). Therefore, we used potassium chloride instead of sodium chloride for the light scattering experiments. The cell sample was washed with buffer solution, as described above (0.1 M KCl, 61.3 mM K2HPO4, 5.33 mM KH2PO4, pH 7.4, 290–300 mOsm). The cells were then hemolyzed in 200 μl distilled H2O and diluted with 800 μl buffer. The solution was centrifuged twice at 20,000 RCF to remove cytoskeleton and membrane parts. Oxyhemoglobin solutions of 4.0 mg/ml, 1.2 mg/ml, and 0.3 mg/ml were prepared and filtered twice, using 0.25-μm nitrocellulose filters, into dust-free glass scintillation vials used for dynamic light scattering experiments. The hemoglobin concentration was determined spectrophotometrically using extinction coefficients of 13.8 at 541 nm and 128 at 405 nm for oxyhemoglobin (38).
Neutron scattering experiments
The experiment was performed on the cold time-of-flight spectrometer FOCUS at the neutron spallation source SINQ (Paul Scherrer Institut, Villigen, Switzerland). A detailed description of the instrument can be found in Janssen et al. (39). The incident wavelength was set to 6 Å. The q-dependent elastic energy resolution ranges from 41 μeV (full width at half-maximum) at q = 0.5 Å−1 to 61 μeV at q = 1.6 Å−1, as determined from a vanadium measurement. During the experiment, we could largely profit from the high neutron flux delivered by the liquid metal target of the MEGAPIE project (40). Samples were measured in the temperature range 16.9–45.9°C, in which no hemolysis occurs. All samples, including the 1-mm-thick vanadium slab and empty sample holder, were oriented at 135° with respect to the incident neutron beam direction. The measured time-of-flight spectra were corrected for empty cell scattering, normalized to vanadium, transformed into energy transfer and scattering vector space, and binned into 12 groups with 0.5 Å−1 ≤ q ≤ 1.6 Å−1. The spectra were corrected with a detailed balance factor. Data treatment was done using the DAVE package (41). Multiple scattering was neglected as the transmission of all samples was between 0.9 and 0.95.
Data analysis
An exhaustive description of quasielastic neutron scattering can be found in Bée (42). The application to protein dynamics has been reviewed in Gabel et al. (43) and Smith (44). In concentrated protein solutions, both internal and global diffusive motions of the proteins contribute to the measured signal. In the case where internal and global motions are uncorrelated, the theoretical incoherent quasielastic scattering function can be written as (42)
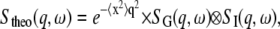 |
(1) |
where the scattering function  corresponds to global protein motions, the scattering function
corresponds to global protein motions, the scattering function 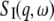 to internal motions, and
to internal motions, and 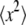 stands for the mean-square vibrational displacements. The scattering function for internal motions can be separated into an elastic and a Lorentzian part
stands for the mean-square vibrational displacements. The scattering function for internal motions can be separated into an elastic and a Lorentzian part
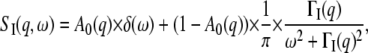 |
(2) |
with 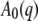 the elastic incoherent structure factor (EISF) that contains information about the geometry of internal motions. Global protein motions are a combination of translational and rotational diffusion. A recent work by Perez et al. (14) showed that the global scattering function for proteins in solution can be approximated by a single Lorentzian
the elastic incoherent structure factor (EISF) that contains information about the geometry of internal motions. Global protein motions are a combination of translational and rotational diffusion. A recent work by Perez et al. (14) showed that the global scattering function for proteins in solution can be approximated by a single Lorentzian
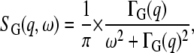 |
(3) |
with the apparent diffusion coefficient 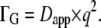 This study found that in the case of the small protein, lysozyme, the value of Dapp is 1.27 times higher than free Brownian diffusion due to the contribution of rotational motion. We followed the calculations given in Perez et al. (14) for the bigger protein hemoglobin and found that the apparent diffusion coefficient is also 1.27 times higher than an assumed free translational diffusion coefficient of D0 = 8.61 × 10−7 cm2/s with a radius of R = 31 Å and a rotational diffusion coefficient given by
This study found that in the case of the small protein, lysozyme, the value of Dapp is 1.27 times higher than free Brownian diffusion due to the contribution of rotational motion. We followed the calculations given in Perez et al. (14) for the bigger protein hemoglobin and found that the apparent diffusion coefficient is also 1.27 times higher than an assumed free translational diffusion coefficient of D0 = 8.61 × 10−7 cm2/s with a radius of R = 31 Å and a rotational diffusion coefficient given by 
The theoretical scattering function then reads
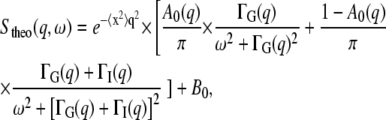 |
(4) |
where B0 is an inelastic background due to vibrational modes of lowest energy (phonons) (42).
The measured data were fitted with the PAN routine of the DAVE software package (41) using the relation
 |
(5) |
in which 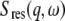 is the instrumental resolution determined by vanadium. The fits were performed over the energy transfer range from −0.75 meV to +0.75 meV.
is the instrumental resolution determined by vanadium. The fits were performed over the energy transfer range from −0.75 meV to +0.75 meV.
Here, we outline the essential steps of the calculation of the global scattering function for proteins in solution: The scattering function of free translational diffusion takes the form of
 |
(6) |
with the diffusion coefficient 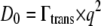 (42). Rotational diffusion on the surface of a sphere is described by the formalism developed by Sears (45). We assume that the protein has a spherical shape of radius R, and that hydrogen atoms are homogeneously distributed within this sphere. The Sears model then needs to be integrated over the protein volume to correctly account for the distribution of hydrogen atoms (14). The scattering function for rotational protein diffusion is then (14)
(42). Rotational diffusion on the surface of a sphere is described by the formalism developed by Sears (45). We assume that the protein has a spherical shape of radius R, and that hydrogen atoms are homogeneously distributed within this sphere. The Sears model then needs to be integrated over the protein volume to correctly account for the distribution of hydrogen atoms (14). The scattering function for rotational protein diffusion is then (14)
 |
(7) |
with 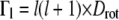 and the rotational diffusion coefficient Drot as defined above. The integrals in the terms
and the rotational diffusion coefficient Drot as defined above. The integrals in the terms  and
and 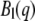 are an extension of the Sears model to describe the distribution of hydrogen atoms within the protein,
are an extension of the Sears model to describe the distribution of hydrogen atoms within the protein,
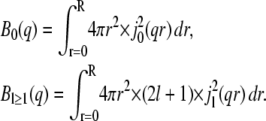 |
(8) |
The term jl is the lth-order spherical Bessel function of the first kind. Rotational and translational diffusion are assumed to be uncorrelated. Therefore, the global scattering function 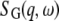 is then the convolution of both terms, and reads
is then the convolution of both terms, and reads
 |
(9) |
The terms 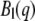 were integrated numerically using the mathematical software Maple (Waterloo Maple, Waterloo, Canada) for q-values in the range 0.5–2.0 Å−1. The Lorentzians in Eq. 9 were summed over the experimentally covered energy range. Depending on the radius of the protein and the q-value, different numbers of terms are needed to reach convergence in the infinite sum of Eq. 9. At the scattering vector q = 2.0 Å−1, it was necessary to include 39 terms for the small protein lysozyme, with a radius of R = 19 Å (14), and 70 terms for the bigger protein, hemoglobin, with radius R = 31 Å. The obtained global scattering function
were integrated numerically using the mathematical software Maple (Waterloo Maple, Waterloo, Canada) for q-values in the range 0.5–2.0 Å−1. The Lorentzians in Eq. 9 were summed over the experimentally covered energy range. Depending on the radius of the protein and the q-value, different numbers of terms are needed to reach convergence in the infinite sum of Eq. 9. At the scattering vector q = 2.0 Å−1, it was necessary to include 39 terms for the small protein lysozyme, with a radius of R = 19 Å (14), and 70 terms for the bigger protein, hemoglobin, with radius R = 31 Å. The obtained global scattering function  could be perfectly approximated by a single Lorentzian, as stated in Eq. 3 (data not shown). The apparent diffusion coefficient, Dapp, was calculated from the half-widths using
could be perfectly approximated by a single Lorentzian, as stated in Eq. 3 (data not shown). The apparent diffusion coefficient, Dapp, was calculated from the half-widths using 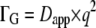 and compared to the assumed free translational diffusion coefficient D0, which gave the relation of
and compared to the assumed free translational diffusion coefficient D0, which gave the relation of 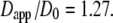
Although incoherent scattering is predominantly due to nonexchangeable hydrogen atoms of the protein, scattering from D2O solvent contributes partially to the signal. The incoherent scattering cross section of human carbonmonoxy hemoglobin was estimated from the amino acid sequence taken from pdb file 2DN3 (46), assuming that 13% of the protons exchange with deuterons (47). The fraction of D2O in the sample was determined by drying and weighting of an aliquot. These theoretical estimations give a D2O solvent incoherent scattering cross section of ∼9% of the total incoherent scattering cross section. Even so, it still could be that D2O dynamics contribute strongly to the measured signal as the characteristic relaxation times of D2O are mostly in the picosecond time range, whereas protein dynamics might be slower and appear more dominant at slower timescales only, which could be out of the accessible time-space window of the neutron spectrometer. The coherent structure factor of D2O begins to increase above 0.8 Å−1 and reaches a maximum at ∼2 Å−1 (48); a stronger contribution of D2O to the measured signal is expected at higher q-values.
To estimate the incoherent and coherent contributions of D2O to the measured signal, we compared the measured elastic intensity of equal amounts of D2O buffer and red blood cell sample (data not shown). The elastic intensity of D2O is ∼6% at low q-values and reaches ∼10% above 1.3 Å−1 of the elastic intensity of the red blood cell sample. When we correct for the fraction of D2O in the red blood cell sample, we obtain D2O contributions of 4.5% at low scattering vectors and 7.5% above 1.3 Å−1. This demonstrates that protein dynamics gives a strong signal compared to D2O buffer at the energy resolution of 50 μeV.
Dynamic light scattering measurement
Dynamic light scattering was measured with a DAWN-EOS instrument equipped with a quasielastic light scattering module (Wyatt Technology, Santa Barbara, CA) in the temperature range 16.9–45.9°C. Temperature variations never exceeded ±0.2°C. The ASTRA 5 software package from the manufacturer was used for data acquisition and calculation of the diffusion coefficients. The instrument was used in batch mode and a∼5 ml of sample was measured in glass scintillation cells.
RESULTS
Neutron scattering
Typical quasielastic neutron spectra at 16.9°C, 31.9°C, and 45.9°C at q = 1.6 Å−1 together with the results from the fits using Eq. 5, are shown in Fig. 1. The narrow Lorentzian corresponds to global protein motions and the broad Lorentzian to internal protein dynamics.
FIGURE 1.

(A–C) Quasielastic neutron scattering spectra of hemoglobin in human red blood cells at the different temperatures 16.9°C (A), 31.9°C (B), and 45.9°C (C) at the scattering vector q = 1.6 Å−1. Circles show measured data and the solid line presents the fit over the energy transfer range from −0.75 meV to +0.75 meV. The components correspond to the narrow Lorentzian (dashed line) and the broad Lorentzian (dotted line). The instrumental energy resolution determined by vanadium is indicated in A (dash-dotted line).
Global motions
The line widths ΓG(q) of the narrow Lorentzian at the temperatures 16.9°C and 45.9°C as a function of q2 are shown in Fig. 2 A. ΓG(q) does not intercept q2 = 0, as would be expected for free translational diffusion. Above ∼1.0 Å−2, the ΓG values increase with q2, and below this q2 value the half-widths, ΓG(q), converge to a plateau Γ0 at q2 = 0. We interpret this as resulting from global hemoglobin diffusion within a restricted volume formed by the neighboring protein molecules. All protein hydrogen atoms are involved in the global motions, and the plateau in the line widths at small q2 therefore represents the center of mass diffusion. The model of Volino and Dianoux was developed for free diffusion in a restricted spherical volume of radius r (49). It is characterized by a plateau of the half-widths 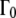 until
until 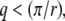 and when
and when  by a limiting behavior of the half-widths at large q2 with
by a limiting behavior of the half-widths at large q2 with 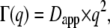 where Dapp is the apparent translational diffusion coefficient. In a preliminary, simplified, and speculative approach we use the model of free diffusion to describe the measured data. Especially at the lowest temperature, deviations from linear behavior are visible. The linear fits appear more justified at higher temperatures. An alternative interpretation with a jump-diffusion model is presented further below.
where Dapp is the apparent translational diffusion coefficient. In a preliminary, simplified, and speculative approach we use the model of free diffusion to describe the measured data. Especially at the lowest temperature, deviations from linear behavior are visible. The linear fits appear more justified at higher temperatures. An alternative interpretation with a jump-diffusion model is presented further below.
FIGURE 2.

(A) Half-widths at half-maximum ΓG of the narrow Lorentzian as a function of q2 at T = 16.9°C (squares) and 45.9°C (circles). The line widths reach a plateau at small q2, which is interpreted as global diffusion within a confined space. The solid lines are linear fits in the q2-range 1.0−2.56 Å−2 that are extended to intersect the constant plateaus at smaller q2-values. The linear fits pass through zero (dotted lines). The dashed lines are fits according to a jump-diffusion model in the q2-range 0.49–2.56 Å−2. (B) Temperature behavior of the apparent translational diffusion coefficients of hemoglobin in red blood cells. Error bars are within the symbols. The dashed line indicates expected normal thermal behavior according to the Stokes-Einstein equation, with  where
where  is the viscosity of pure D2O and Rh = 31.3 Å is the hydrodynamic radius of hemoglobin.
is the viscosity of pure D2O and Rh = 31.3 Å is the hydrodynamic radius of hemoglobin.
The apparent translational diffusion coefficients, Dapp, were calculated according to 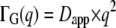 in the q2 range of 1.0–2.56 Å−2. The obtained values of Dapp as a function of temperature are presented in Fig. 2 B. According to the model of Volino and Dianoux (49) and the corrected translational diffusion coefficient Dtrans = Dapp/1.27, the radius, r, of the spherical volume can be estimated by
in the q2 range of 1.0–2.56 Å−2. The obtained values of Dapp as a function of temperature are presented in Fig. 2 B. According to the model of Volino and Dianoux (49) and the corrected translational diffusion coefficient Dtrans = Dapp/1.27, the radius, r, of the spherical volume can be estimated by  The obtained radii lie between r = 2.1 ± 0.2 Å at 16.9°C and r = 2.3 ± 0.2 Å at 45.9°C, which are constant within the errors.
The obtained radii lie between r = 2.1 ± 0.2 Å at 16.9°C and r = 2.3 ± 0.2 Å at 45.9°C, which are constant within the errors.
Normal-temperature behavior of the diffusion coefficient follows the Stokes-Einstein equation 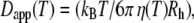 with solvent viscosities
with solvent viscosities 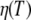 and hydrodynamic radius of the protein Rh. The Stokes-Einstein equation was fitted to the diffusion coefficients Dapp by taking literature values of the viscosities
and hydrodynamic radius of the protein Rh. The Stokes-Einstein equation was fitted to the diffusion coefficients Dapp by taking literature values of the viscosities 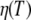 of pure D2O (50) and the hydrodynamic radius Rh as a free parameter (Fig. 2 B). We obtain a value of Rh = 31.3 Å, which is nearly identical to the published hydrodynamic radius of human hemoglobin of 31.7 Å (34).
of pure D2O (50) and the hydrodynamic radius Rh as a free parameter (Fig. 2 B). We obtain a value of Rh = 31.3 Å, which is nearly identical to the published hydrodynamic radius of human hemoglobin of 31.7 Å (34).
To check the validity of the Hall and Ross model (42) for restricted jump-diffusion, we examined the behavior of the plateau  at small q. The sphere radius, r, is then given by the upper limit
at small q. The sphere radius, r, is then given by the upper limit 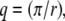 at which the plateau
at which the plateau 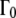 ends. This yields radii between r = 3.4 Å at 16.9°C and 4.0 Å at 45.9°C, which are on average 1.7 times bigger than the values from the approach of free diffusion at large q2. Diffusion coefficients, Dlocal, were estimated according to
ends. This yields radii between r = 3.4 Å at 16.9°C and 4.0 Å at 45.9°C, which are on average 1.7 times bigger than the values from the approach of free diffusion at large q2. Diffusion coefficients, Dlocal, were estimated according to 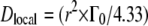 (49). We get values ranging between 13.7 × 10−7 cm2/s at 16.9°C and 22.0 × 10−7 cm2/s at 45.9°C. The reasons for the discrepancy are considered in the Discussion section.
(49). We get values ranging between 13.7 × 10−7 cm2/s at 16.9°C and 22.0 × 10−7 cm2/s at 45.9°C. The reasons for the discrepancy are considered in the Discussion section.
At larger q2 values, the half-widths exhibit some deviation from linear behavior. There might be signs for saturation at 16.9°C and 45.9°C, which is less visible at high temperatures. In the Volino and Dianoux model for restricted free diffusion, the elementary steps of motion are assumed to be infinitely small, and therefore the half-widths show linear behavior at large q2 values. In the case of elementary steps of motion with a finite size, these steps become observable at high q2. A plateau at high q2 values is then a sign for a possible jump-diffusion mechanism. The line widths were approximated with a jump-diffusion model 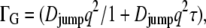 with diffusion coefficient Djump and mean residence time
with diffusion coefficient Djump and mean residence time 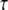 (42). The fits are presented in Fig. 2 A. The residence time on one site between jumps is
(42). The fits are presented in Fig. 2 A. The residence time on one site between jumps is 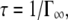 where
where 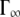 is obtained from the asymptotic behavior at high q, whereas Γ approaches a constant value. The Γ value at high q is still increasing over the q-range examined, and has not yet reached a constant value. Therefore, the constant value was estimated using the presented equation of the jump-diffusion model and then extrapolated to higher q. The behavior of the residence times,
is obtained from the asymptotic behavior at high q, whereas Γ approaches a constant value. The Γ value at high q is still increasing over the q-range examined, and has not yet reached a constant value. Therefore, the constant value was estimated using the presented equation of the jump-diffusion model and then extrapolated to higher q. The behavior of the residence times,  can be approximated by an Arrhenius relation with
can be approximated by an Arrhenius relation with 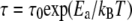 in the investigated temperature range. The residence times
in the investigated temperature range. The residence times 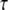 are presented in an Arrhenius plot in Fig. 3. We obtained a value of Ea = 6.6 ± 1.8 kcal/mol for the activation energy. All values Djump are constant within the errors and have the average value of 12.6 ± 0.2 × 10−7 cm2/s.
are presented in an Arrhenius plot in Fig. 3. We obtained a value of Ea = 6.6 ± 1.8 kcal/mol for the activation energy. All values Djump are constant within the errors and have the average value of 12.6 ± 0.2 × 10−7 cm2/s.
FIGURE 3.

Arrhenius plot of the residence time, τ, of the jump-diffusion model for global hemoglobin motion.
The measured line widths at large q2 can be approximated with either a jump-diffusion model or the Volino-Dianoux model for free diffusion in confined space. However, the errors are too big to decide clearly which model is appropriate. A jump-diffusion mechanism seems to be more favorable at low temperatures than at high temperatures. How far a whole globular protein with a molecular weight of 64 kDa can perform jump-diffusive motion is still not understood. The diffusion coefficient is mostly determined by the behavior of the line widths at small q2. The diffusion coefficients were estimated from the half-widths at small q in the Volino-Dianoux model, and with an approach of free confined diffusion at large q2. The level of the resolution ranges between 20 μeV at q = 0.5 Å−1 and 30 μeV at q = 1.6 Å−1 (half-width at half-maximum). All measured line-widths are at the lower limit of energy resolution. If limited energy resolution should play a role, then this would influence the low temperature data stronger as the line-widths are generally smaller than at high temperature. At this point, we cannot decide clearly if the plateau at high q2-values is an artifact or a real feature. The obtained diffusion coefficients for free diffusion should be considered rather as average values, as the plateau at high q2 influences the data. The validity of these average diffusion coefficients is checked by the comparison to expected normal temperature behavior.
Internal motions
Amplitudes
Conclusions about protein internal motions can be drawn from the amplitudes A0(q) and the half-widths ΓI(q) of the broad Lorentzian. Information about the average geometry of internal protein motion and the fraction of hydrogen atoms participating in this motion is contained in the EISF, which appears in front of the delta-function in Eq. 2. In the case of a protein solution the delta-function for internal protein motion is convoluted with the Lorentzian for global protein motion (14,28). We used a pseudo-EISF, which appears as the prefactor A0 of the narrow Lorentzian in Eq. 4. The values of A0 were fitted using a model for “diffusion in a sphere” proposed by Volino and Dianoux (49) with
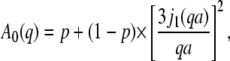 |
(10) |
where j1 is the first-order spherical Bessel function of the first kind and a is the sphere radius. The populations of hydrogen atoms that appear as immobile and mobile are represented by the fractions p and (1 − p), respectively.
Molecular dynamics simulations showed that the polydispersity of protein internal motions and geometries are best represented by a free discrete distribution of sphere radii (51). As the measured experimental results cover only a limited q-region of A0(q), a fit with a completely free discrete distribution of sphere radii was not feasible. A simplified model assuming a Gaussian distribution f(a) of the sphere radii with 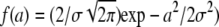 and the standard deviation σ as free parameter (14) gave good results. The mean value of the sphere radius is then given by
and the standard deviation σ as free parameter (14) gave good results. The mean value of the sphere radius is then given by 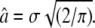 The fits to the EISF, together with the Gaussian distributions, are shown in Fig. 4, A and B. Because the model we used is a Gaussian distribution of spheres, the EISF decays slowly with q and reaches the constant value of the immobile hydrogen fraction only at very high q-values. The inset in Fig. 4 A illustrates this behavior of the EISF. With increasing temperature, the Gaussian distributions change from narrow to broad above 36.9°C. The mean radii, â, as a function of temperature are given in Fig. 5 A. The values between 16.9°C and 31.9°C show a slight linear increase with temperature, whereas a kink appears at 36.9°C, with a much steeper increase at higher temperatures. The fraction of immobile hydrogen atoms is shown in Fig. 5 B. They are roughly constant within the errors between 16.9°C and 39.9°C and have an average value of 0.35 ± 0.01. The values at 42.9°C and 45.9°C are slightly above the average.
The fits to the EISF, together with the Gaussian distributions, are shown in Fig. 4, A and B. Because the model we used is a Gaussian distribution of spheres, the EISF decays slowly with q and reaches the constant value of the immobile hydrogen fraction only at very high q-values. The inset in Fig. 4 A illustrates this behavior of the EISF. With increasing temperature, the Gaussian distributions change from narrow to broad above 36.9°C. The mean radii, â, as a function of temperature are given in Fig. 5 A. The values between 16.9°C and 31.9°C show a slight linear increase with temperature, whereas a kink appears at 36.9°C, with a much steeper increase at higher temperatures. The fraction of immobile hydrogen atoms is shown in Fig. 5 B. They are roughly constant within the errors between 16.9°C and 39.9°C and have an average value of 0.35 ± 0.01. The values at 42.9°C and 45.9°C are slightly above the average.
FIGURE 4.

(A) Variation of the EISF as a function of q at 16.9°C (squares) and 45.9°C (circles).The solid and dashed lines are fits with a model for diffusion in a sphere with a Gaussian distribution of radii. The inset shows the slow decay of the model, which reaches the limiting value of the immobile hydrogen fraction only at high q-values. (B) Gaussian distribution of the sphere radius f(a) at different temperatures.
FIGURE 5.

(A) Mean value, â, of the Gaussian distribution as a function of temperature. The dashed line is a linear fit to the values between 16.9°C and 31.9°C and serves as a guide for the eye. (B) Fraction of hydrogen atoms that appear immobile within the instrumental energy resolution. The average value between 16.9°C and 39.9°C is indicated by the dashed line.
In a recent study on the dynamics of apocalmodulin, Gibrat et al. used a lognormal distribution to describe the polydispersity of protein motions and to fit the measured EISF (52). The lognormal distribution is defined by 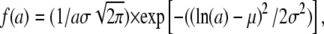 where
where 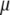 and
and 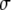 are the mean and the standard deviation of the variable's natural logarithm. A lognormal distribution is appropriate if the variable is the product, whereas a Gaussian distribution is valid if the variable is the sum of a large number of independent, identically distributed variables.
are the mean and the standard deviation of the variable's natural logarithm. A lognormal distribution is appropriate if the variable is the product, whereas a Gaussian distribution is valid if the variable is the sum of a large number of independent, identically distributed variables.
It was shown that the radius of the sphere of diffusion of a hydrogen atom along an aliphatic chain fixed at one end increases linearly with distance from the fixed end (53). It was argued by Gibrat et al. that the motions of the hydrogen atoms that are bound to a carbon atom of the aliphatic chain are then the “product” of the motions of the previous hydrogen atoms in the chain (52). In this sense, it was concluded that a lognormal distribution would be more appropriate than a Gaussian distribution (52) to describe the polydispersity of hydrogen motion in the amino acid side-chains of a protein.
In fact, we found that both a Gaussian and a lognormal distribution for the Volino and Dianoux model could fit the measured EISF equally well. The obtained average sphere radii of the lognormal and Gaussian distributions were identical within the errors and showed similar temperature behavior, but the standard deviations of the lognormal distributions were much bigger than those of the Gaussian distributions. Therefore, we use the results of the fits with the Gaussian distributions in the following. Fits to the EISF using a single sphere radius gave only poor results.
Widths
The half-widths ΓI(q) of the broad Lorentzian are shown in Fig. 6. The half-widths tend toward a constant value of ∼100 μeV for 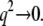 At higher q2-values, the line-widths follow the behavior of a jump-diffusion model (42). At highest q2-values, a plateau
At higher q2-values, the line-widths follow the behavior of a jump-diffusion model (42). At highest q2-values, a plateau 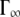 is approached that ranges approximately between 170 μeV at 16.9°C and 240 μeV at 45.9°C.
is approached that ranges approximately between 170 μeV at 16.9°C and 240 μeV at 45.9°C. 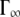 corresponds to a correlation time of
corresponds to a correlation time of 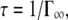 which is roughly between 3.9 ps at 16.9°C and 2.7 ps at 45.9°C. However, the determination of ΓI is inaccurate and doesn't allow a more precise analysis.
which is roughly between 3.9 ps at 16.9°C and 2.7 ps at 45.9°C. However, the determination of ΓI is inaccurate and doesn't allow a more precise analysis.
FIGURE 6.

Half-width at half-maximum 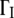 values of the broad Lorentzian as a function of q2 at T = 16.9°C (squares) and 45.9°C (circles).
values of the broad Lorentzian as a function of q2 at T = 16.9°C (squares) and 45.9°C (circles).
Dynamic light scattering
The diffusion coefficients of hemoglobin in H2O buffer at the concentrations 0.3 mg/ml, 1.2 mg/ml, and 4.0 mg/ml were measured with dynamic light scattering in the temperature range 16.9–45.9°C. The diffusion coefficients at infinite dilution were obtained by linearly extrapolating the measured values to zero concentration. The measurements in D2O buffer showed pronounced hemoglobin aggregation. Therefore, the values in H2O buffer were corrected for pure D2O and H2O viscosities (50) to estimate the diffusion coefficients of hemoglobin in D2O buffer.
DISCUSSION
Global motions
Hemoglobin molecules are densely packed in the cytoplasm with intermolecular distances in the order of angstroms (19). Suspensions of colloidal particles are well studied model systems for such a highly crowded protein solution (54). Colloidal particles are small enough that their motions are governed by thermal energy and big enough that the solvent molecules participate in the interactions only in an averaged way. The diffusion of these colloidal particles at infinite dilution D0 is described by the Stokes-Einstein relation. At higher concentrations, the interactions between individual colloidal particles become important. In this case, direct van der Waals and electrostatic forces determine particle diffusion, but hydrodynamic interactions mediated by the solvent play an important role (55). These hydrodynamic interactions describe how a colloidal particle moves in the flow-field, which is determined by its neighboring particles. If a colloidal particle has collided and interacted directly with other particles, the diffusion coefficient approaches a constant value that is now termed long-time self-diffusion coefficient 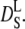 The time that a particle needs to cross a typical interparticle distance d is referred to as structural relaxation time τD. At timescales shorter than this structural relaxation time, the particle moves in an approximately constant configuration of the surrounding particles. This stage is governed by hydrodynamic interactions and the displacements of the colloidal particle are characterized by the so-called short-time self-diffusion coefficient
The time that a particle needs to cross a typical interparticle distance d is referred to as structural relaxation time τD. At timescales shorter than this structural relaxation time, the particle moves in an approximately constant configuration of the surrounding particles. This stage is governed by hydrodynamic interactions and the displacements of the colloidal particle are characterized by the so-called short-time self-diffusion coefficient 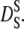 The relaxation time is then given by
The relaxation time is then given by 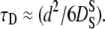 Beenakker and Mazur (56) first evaluated the coefficient
Beenakker and Mazur (56) first evaluated the coefficient 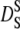 over a large range of volume fractions. Due to configurational relaxation at long timescales, the long-time self-diffusion coefficient
over a large range of volume fractions. Due to configurational relaxation at long timescales, the long-time self-diffusion coefficient 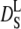 is always slower than the short-time self-diffusion coefficient
is always slower than the short-time self-diffusion coefficient 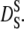
The values of 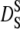 and
and  as a function of the volume fraction ϕ for noncharged hard-sphere particles were calculated by Tokoyama and Oppenheim (57), taking into account both short- and long-range hydrodynamic interactions. Their theory predicts experimental data of uncharged colloids at intermediate and high volume fraction quite well. The volume fraction of hemoglobin in red blood cells is ϕ = 0.25 (19) and their calculations yield values of
as a function of the volume fraction ϕ for noncharged hard-sphere particles were calculated by Tokoyama and Oppenheim (57), taking into account both short- and long-range hydrodynamic interactions. Their theory predicts experimental data of uncharged colloids at intermediate and high volume fraction quite well. The volume fraction of hemoglobin in red blood cells is ϕ = 0.25 (19) and their calculations yield values of 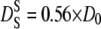 and
and 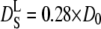 with the diffusion coefficient at infinite dilution D0.
with the diffusion coefficient at infinite dilution D0.
Doster and Longeville measured the long-time self-diffusion coefficient of hemoglobin in red blood cells with neutron spin-echo spectroscopy (21) and applied for the first time in neutron spectroscopy the abovementioned concepts of hydrodynamic theory to interpret their data. They found that the long-time self-diffusion coefficient of hemoglobin was slightly reduced compared to the predicted theoretical value. This reduction was attributed to the influence from the protein hydration shell, which was supposed to stick to the surface of the protein and move in a joint way.
In our work, we used the behavior of the line widths ΓG(q) of the narrow Lorentzian to gain information about the short-time self-diffusion coefficient, the interparticle distance d, and the residence time of hemoglobin diffusion in red blood cells. Neutron scattering results show the dependency of the length scale. The form of the half-widths is in agreement with a model for confined diffusion within a restricted spherical volume (49). At small q2-values, longer real-space length scales become visible and the effects of the boundaries dominate; the line widths tend toward a constant value Γ0 instead of a zero intercept, and a Dq2 behavior for q2→0. At large q2-values, the dynamic behavior at small real-space length scales dominates. In the case of free diffusion, the elementary steps of motions are supposed to be infinitely small and the half-widths follow a limiting behavior of 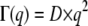 at large q2. If the elementary steps of motions have a finite size, then the half-widths tend toward an asymptotic value,
at large q2. If the elementary steps of motions have a finite size, then the half-widths tend toward an asymptotic value,  (42).
(42).
In our study, we calculated apparent average diffusion coefficients for assumed free diffusion from the behavior of the half-widths at large q2 according to 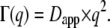 The temperature behavior of these apparent diffusion coefficients is well approximated with the Stokes-Einstein equation and a hydrodynamic radius of Rh = 31.3 Å, which is nearly identical to the published hydrodynamic radius of human hemoglobin of 31.7 Å (34). The sphere radius r of global confined motion was estimated from the value of constant
The temperature behavior of these apparent diffusion coefficients is well approximated with the Stokes-Einstein equation and a hydrodynamic radius of Rh = 31.3 Å, which is nearly identical to the published hydrodynamic radius of human hemoglobin of 31.7 Å (34). The sphere radius r of global confined motion was estimated from the value of constant 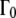 at small q2, and the diffusion coefficients according to the model of Dianoux and Volino, with
at small q2, and the diffusion coefficients according to the model of Dianoux and Volino, with  If the assumptions are valid, then the sphere radius, r, should be given equally by the value of
If the assumptions are valid, then the sphere radius, r, should be given equally by the value of  at which the plateau
at which the plateau 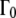 ends. These sphere radii are on average 1.7 times larger than the radii calculated using the diffusion coefficients of free diffusion and the value of
ends. These sphere radii are on average 1.7 times larger than the radii calculated using the diffusion coefficients of free diffusion and the value of 
Dellerue et al. performed molecular dynamics simulations of a globular protein. They were able to analyze amino acid side-chain and backbone dynamics separately with a model for diffusion within a sphere with a distribution of radii (51). The authors calculated scattering functions from the simulations that were comparable to neutron scattering results. They demonstrated that only in an ideal case of diffusion in a single-sphere with radius r does the plateau  end promptly at
end promptly at  In the case of a distribution of sphere radii, the plateau exhibits a more gradual change and ends at an apparent smaller q′ value than in the single-sphere case. Consequently, if a sphere size is determined from the position of q′, the obtained apparent sphere radius is bigger than the real average value of the sphere radii distribution (51). For the side chains and the backbone, the apparent sphere radii were between 1.7 and 1.8 times bigger than the average value of the distribution.
In the case of a distribution of sphere radii, the plateau exhibits a more gradual change and ends at an apparent smaller q′ value than in the single-sphere case. Consequently, if a sphere size is determined from the position of q′, the obtained apparent sphere radius is bigger than the real average value of the sphere radii distribution (51). For the side chains and the backbone, the apparent sphere radii were between 1.7 and 1.8 times bigger than the average value of the distribution.
In our study, we found a factor of 1.7 between the sphere radii obtained using the two methods, which agrees nicely with the work of Dellerue et al. In this sense, the validity of our approach is confirmed. Obviously, this also demonstrates that confined global diffusion of hemoglobin in the red blood cells is best described by a distribution of sphere radii. In fact, recent small-angle neutron scattering work points to the existence of a higher concentration of hemoglobin close to the internal membrane surface than on average in the cell (58). Of necessity, the factor between apparent and average sphere radius will depend on the precise nature of the sphere distribution. Here, we limit our discussion to the sphere radii obtained from the value of constant  at small q2 and the average diffusion coefficients obtained from linear approximation at higher q2-values.
at small q2 and the average diffusion coefficients obtained from linear approximation at higher q2-values.
We estimated a temperature-independent radius r = 2.2 ± 0.2 Å of confined global hemoglobin diffusion. This has to be understood as the radius of the sphere in which the center of mass of the protein moves. This is in agreement with high-resolution neutron-backscattering measurements of protein dynamics in whole Escherichia coli bacteria, with average jump lengths of the protein population of 2.2 ± 0.3 Å at 10.9°C and 1.9 ± 0.2 Å at 29.9°C (22). Another neutron-backscattering study with concentrated myoglobin solution at ϕ = 0.26 found a protein jump-diffusion length of 1.4 Å (59). In that experiment, the pH was not controlled, and this might have influenced the packing density of the protein, leading to a slightly reduced jump length.
From our measurements, we conclude that the hemoglobin molecules diffuse in a cage of 2.2-Å radius before they interact directly with the neighbor proteins. The diffusion coefficient D0 at infinite dilution in D2O buffer was obtained from dynamic light scattering experiments. The theoretical prediction yields 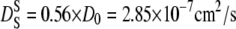 at 16.9°C and
at 16.9°C and  at 45.9°C (57). From these values, we obtain structural relaxation times ranging from 280 to 140 ps. Even if the real short-time self-diffusion coefficient might deviate from the theoretical predictions, the calculations show that the structural relaxation time is in the order of several hundred picoseconds.
at 45.9°C (57). From these values, we obtain structural relaxation times ranging from 280 to 140 ps. Even if the real short-time self-diffusion coefficient might deviate from the theoretical predictions, the calculations show that the structural relaxation time is in the order of several hundred picoseconds.
We deduced the global apparent diffusion coefficient from the line-widths of the narrow Lorentz function. The correlation times of these line-widths range from 47 ps at 1.0 Å−2 to 150 ps at 2.56 Å−2. This is smaller than the structural relaxation time, and therefore the experimentally measured apparent diffusion coefficients reflect short-time self-diffusion behavior of hemoglobin.
The measured apparent global diffusion coefficients are bigger than the true translational diffusion coefficients, as they contain the contribution of rotational motion (14). Calculations for free diffusion of hemoglobin at infinite dilution give a correction factor of 1.27 between the apparent and true translational diffusion coefficients, similar to myoglobin and lysozyme solutions (14). However, the translational diffusion coefficient of hemoglobin in red blood cells is strongly reduced compared to free diffusion (21). This is due to the high concentration of hemoglobin and the resulting crowding effects; the correction factor can therefore serve only as an estimate.
The apparent global diffusion coefficients of hemoglobin were divided by 1.27 to obtain the translational diffusion coefficients Dtrans. The diffusion coefficients at infinite dilution were scaled by a factor of 0.56 (57) to yield the theoretical predictions for short-time self-diffusion. The calculated quantities as a function of temperature are compared in Fig. 7. The lines in the graph represent the expected normal thermal behavior. In the temperature range investigated, this is nearly linear in temperature. The values for translational diffusion coefficients in red blood cells are ∼1.5 times larger than those for predicted short-time self-diffusion. An explanation could be that the concepts for noncharged hard-sphere colloids are not fully applicable to the much smaller protein molecules. Future studies may determine whether the solvent can be treated as a continuum equivalently for large colloid particles and smaller protein molecules.
FIGURE 7.

Apparent global diffusion coefficients of hemoglobin were divided by 1.27 to obtain the translational diffusion coefficients Dtrans (circles). The diffusion coefficients at infinite dilution were scaled by a factor 0.56 to yield the theoretical predictions for short-time self-diffusion  (squares). The dashed and dotted lines show expected thermal behavior of the respective diffusion coefficients.
(squares). The dashed and dotted lines show expected thermal behavior of the respective diffusion coefficients.
Hemoglobin carries only a small total charge at the pH values used (19) but it has a distribution of positively and negatively charged residues on the protein surface. The interactions between proteins in concentrated solution are not expected to be described only by hard-sphere interactions. Recent investigations using small-angle scattering and molecular dynamics simulations have shown the importance of both attractive and repulsive interactions between proteins in concentrated solution (60–63). How these repulsive and attractive protein-protein interactions influence hemoglobin self-diffusion in red blood cells requires further study. Doster and Longeville argue that the hydration shell moves with the protein and needs to be included in the volume fraction to correctly describe hemoglobin diffusion in red blood cells measured with neutron spin-echo spectroscopy (21). In this way, they obtain effective volume fractions between 0.32 and 0.36. Taking into account these effective volume fractions, the discrepancy between our measured translational diffusion coefficients and the predicted short-time self-diffusion coefficients would then be even bigger. This interesting fact could be investigated further, in detail, by using neutron spectrometers with higher energy resolution to observe the changeover from hemoglobin short-time to long-time self-diffusion.
As discussed above, the line widths might show signs of saturation at high q2-values. We calculated the residence times between jumps by analysis with a jump-diffusion model. Busch et al. (59) found a residence time of 80 ps at 20°C in concentrated myoglobin solution, and Jasnin et al. (22) obtained a residence time of 590 ps at 30°C in whole bacteria with high-resolution neutron backscattering. In our work, we obtained residence times of 51 ps at 21.9°C and 31 ps at 31.9°C, which is close to the values for concentrated myoglobin solution, but differs strongly from those for whole bacteria. We obtained an activation energy of Ea = 6.6 ± 1.8 kcal/mol from the residence times. The residence time of jump-diffusion of pure water cannot be described in a general way by an Arrhenius law over a broad temperature range (64). We applied an Arrhenius law to literature values for the purpose of comparison for a restricted temperature range (64). In this way, the activation energy of pure water residence times in the range between 5°C to 20°C was found to be 6.7 ± 0.5 kcal/mol. This is very close to our obtained activation energy of hemoglobin residence times, and could point out that hydrogen bond fluctuations are involved in hemoglobin global diffusion. The measured line widths at large q2 can be approximated using either a jump-diffusion model or assumed free diffusion in a confined space. A jump-diffusion model might be more favorable to describe the measured data. However, the errors are too big to decide clearly which model is appropriate. How far a whole globular protein with a molecular weight of 64 kDa can perform jump-diffusive motion is still not understood.
Global protein diffusion was estimated from the broadening of the narrow Lorentzian. The experimentally measured line widths reach down to 3 μeV, which corresponds to correlation times of 200 ps. Usually, any motions that result in line widths smaller than the Gaussian resolution function—in our case, 25 μeV on average (half-width at half-maximum)—are considered to produce elastic scattering. It was shown by Perez et al. (14), and verified by Russo et al. (28), that for protein solution this is not the case. Global protein diffusion results in detectable line widths that are even smaller than the instrumental resolution itself. However, it should be noted that a Lorentzian has a significantly wider tailing than the Gaussian resolution function, which facilitates detection of small line widths.
Internal motions
Internal protein dynamics covers a very broad distribution of timescales, with motions ranging from the pico- to nanosecond scale up to the millisecond range. Fast internal motions in the picosecond time range are contained within the measured broad Lorentzian, whereas slow motions that cannot be resolved by the instrument contribute to the elastic part in the internal scattering function. Therefore, the extracted quantities yield only phenomenological information about average internal dynamics. Nevertheless, the obtained information is valid for studying changes in the average internal dynamics as a function of temperature. We gained information about the geometries from the analysis of the EISF.
Micropipette experiments with aspirated single human red blood cells revealed a sudden change in the behavior of the cells from blockage of the pipette below TPipette = 36.4 ± 0.4°C to easy passage above. The transition temperature TPipette of cell passage was surprisingly very close to human body temperature. The effect was interpreted as a pronounced reduction in the viscosity of concentrated hemoglobin solution at TPipette (33). Further studies with circular dichroism (CD) investigated changes in the protein secondary structure around the transition temperature. A partial loss of α-helical content was found at temperature TCD = 37.2 ± 0.6°C (65). The loss of hemoglobin α-helical structure at a specific TCD was also observed for hemoglobin molecules of a large variety of different species (34,35). To our surprise, the transition temperatures TCD were directly correlated to the body temperature of the animals, ranging from 34°C for the duck-billed platypus to 42°C for a bird, the spotted nutcracker. It was excluded that the partial loss of protein structure at TCD results from irreversible protein denaturation that occurs at distinctly higher temperatures. Independent of these experiments, two-dimensional infrared correlation spectroscopy suggested a structural perturbation stage of bovine hemoglobin between 30°C and 44°C (66). The observed perturbations were assigned to hydrogen-bonded extended chains that connect the helices. It was concluded that the passage transition of red blood cells is caused by hemoglobin molecules and that the observed small structural changes of hemoglobin at TCD might be the cause of the drop in viscosity at TPipette (67).
In our work, we were able to determine the mean radius, â, of the distribution of spheres in which the mobile protein protons—in the model used, mostly hydrogen atoms in side chains—diffuse. The mean radii increase linearly between 16.9°C and 31.9°C due to higher thermal amplitudes. At 36.9°C and higher temperatures, the mean radii deviate from the low-temperature linear behavior and increase with a significantly steeper slope. This pronounced increase in average sphere size is interpreted as partial unfolding of hemoglobin, which begins at human body temperature.
In the partially unfolded state, the side chains can move in a larger space compared to the more compact low-temperature state. At 45.9°C, the accessible volume is around three times bigger than at 31.9°C. The fraction of immobile protons, p, remains roughly constant within the errors; only at 42.9°C and 45.9°C is p slightly above the average. Hydrogen atoms that move so slowly that they contribute to the immobile fraction p are mainly located in the protein interior and in the protein backbone; amino acids toward the exterior and on the surface of the protein contribute mostly to the mobile fraction (51).
It was proposed that aggregation of hemoglobin in red blood cells occurs above body temperature and that an increase in surface hydrophobicity could be the reason for this (34). At first sight, the aggregation effect at 36.9°C seems to be in contradiction to the observed larger volume of side-chain diffusion above this temperature. The observed change in side-chain mobility might reflect structural rearrangements that cause the increase in surface hydrophobicity. In this way, the observed dynamical and structural changes in the picosecond time range and angstrom length scale could influence protein-protein interactions in the crowded cell environment. The cooperative effects of the protein interactions would then cause the measured passage effect of whole red blood cells seen with micropipette experiments. This might be an intriguing example of how atomic effects influence macroscopic properties of whole red blood cells.
The onset of protein aggregation also might be the cause for the small increase in the fraction of immobile hydrogen atoms at 42.9°C and 45.9°C. Although the majority of external protein side chains can access a larger space at these temperatures, a small side-chain fraction might get slowed down due to strong protein-protein interactions. The hydrogen atoms of these slowed-down side chains could then contribute to the fraction of immobile protons. Removing the protons from the average would also lead to a larger sphere radius in the Volino-Dianoux model.
Because the immobile proton fraction p changes only slightly as the mean radii increase strongly above 36.9°C, we conclude that the observed changes in mobility occur mostly in amino acid side chains on the hemoglobin surface. If hydrogen atoms in the protein interior were involved during partial unfolding, then we would expect more pronounced changes in p. Large changes in p go hand in hand with protein unfolding and denaturation (28,52,68) when internal side-chain and backbone hydrogen atoms become more mobile. In this case, the immobile fraction approaches zero in the unfolded state. Our results confirm that the internal structure of hemoglobin unfolds only partially and does not denature completely in the investigated temperature range.
It is not yet known whether the observed effects of hemoglobin around body temperature are related to hemoglobin function in the human body. The temperature close to the core of the human body is some degrees Celsius higher then in the extremities. Whether and how hemoglobin function and the properties of red blood cells are influenced by this temperature difference is still an open question.
CONCLUSIONS
We have used incoherent quasielastic neutron scattering to measure the dynamics of hemoglobin in whole human red blood cells around body temperature. In our approach, we were able to separate global protein diffusion from internal hemoglobin motions. We complemented our neutron experiments with dynamic light scattering measurements to obtain the diffusion coefficient of hemoglobin at infinite dilution. Theoretical predictions for concentrated suspensions of noncharged hard-sphere colloids were used to compare the global diffusion coefficients of hemoglobin in red blood cells with hemoglobin diffusion coefficients at infinite dilution. The values of the hemoglobin diffusion coefficients in the cells are in the order expected for short-time self-diffusion. The hemoglobin molecules are trapped in cages formed by neighboring proteins. The cage radius is in agreement with results of neutron backscattering studies on the average protein population in Escherichia coli cells and in concentrated myoglobin solutions (22,59). Internal protein motion was analyzed with a model for diffusion in a sphere with a distribution of radii. At temperatures >36.9°C, amino acid side-chain motions occupy larger volumes than expected from normal temperature dependence. These results indicate partial unfolding of hemoglobin at around human body temperature, which is in agreement with the results of CD experiments (1,3,47). It was deduced that these changes are caused mostly by amino acid side chains toward the exterior and on the surface of the proteins. Micropipette experiments with single red blood cells found a passage phenomenon from complete blockage of the pipette below body temperature to sudden passage at higher temperatures. It was suggested that intracellular hemoglobin aggregation at temperatures above human body temperature is responsible for this passage effect (33). The observed changes in the geometry of amino acid side-chain motion at body temperature could reflect an increase in surface hydrophobicity, which might cause an increase in protein-protein attraction and thus lead to protein aggregation. Further studies on hemoglobin interactions in red blood cells would help to shed light on this issue. It is still unknown whether the changes in the behavior of hemoglobin at body temperature are necessary for hemoglobin function in the human body. Protein dynamics and structural changes in the picosecond time range and angstrom length scale might be connected to a macroscopic effect of whole red blood cells.
Acknowledgments
We thank Prof. Aysegül Temiz Artmann and Dipl. Ing. Dariusz Porst for taking blood and help during the sample preparation. This work is based on experiments performed at the Swiss spallation neutron source SINQ, Paul Scherrer Institute, Villigen, Switzerland.
This research project was supported by the European Commission under the 6th Framework Programme through Key Action: Strengthening the European Research Area, Research Infrastructures, contract No. RII3-CT-2003-505925.
Editor: Jill Trewhella.
References
- 1.Austin, R. H., K. W. Beeson, L. Eisenstein, H. Frauenfelder, and I. C. Gunsalus. 1975. Dynamics of ligand binding to myoglobin. Biochemistry. 14:5355–5373. [DOI] [PubMed] [Google Scholar]
- 2.Creighton, T. E. 1992. Proteins: Structures and Molecular Properties. W.H. Freeman, New York.
- 3.Brooks III, C. L., M. Karplus, and B. M. Pettitt. 1988. Proteins: A Theoretical Perspective of Dynamics, Structure, and Thermodynamics. John Wiley & Sons, New York.
- 4.Frauenfelder, H., F. Parak, and R. D. Young. 1988. Conformational substates in proteins. Annu. Rev. Biophys. Biophys. Chem. 17:451–479. [DOI] [PubMed] [Google Scholar]
- 5.Fraunfelder, H., S. G. Sligar, and P. G. Wolynes. 1991. The energy landscapes and motions of proteins. Science. 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- 6.Zaccai, G. 2000. How soft is a protein? A protein dynamics force constant measured by neutron scattering. Science. 288:1604–1607. [DOI] [PubMed] [Google Scholar]
- 7.Tehei, M., D. Madern, C. Pfister, and G. Zaccai. 2001. Fast dynamics of halophilic malate dehydrogenase and BSA measured by neutron scattering under various solvent conditions influencing protein stability. Proc. Natl. Acad. Sci. USA. 98:14356–14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai, A. M., T. J. Udovic, and D. A. Neumann. 2001. The inverse relationship between protein dynamics and thermal stability. Biophys. J. 81:2339–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang, K. E. S., and K. Dill. 1998. Native protein fluctuations: the conformational-motion temperature and the inverse correlation of protein flexibility with protein stability. J. Biomol. Struct. Dyn. 16:397–411. [DOI] [PubMed] [Google Scholar]
- 10.Fitter, J., R. Herrmann, N. A. Dencher, A. Blume, and T. Hauss. 2001. Activity and stability of a thermostable α-amylase compared to its mesophilic homologue: mechanisms of thermal adaptation. Biochemistry. 40:10723–10731. [DOI] [PubMed] [Google Scholar]
- 11.De Francesco, A., M. Marconi, S. Cinelli, G. Onori, and A. Paciaroni. 2004. Picosecond internal dynamics of lysozyme as affected by thermal unfolding in nonaqueous environment. Biophys. J. 86:480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehnert, U., V. Reat, M. Weik, G. Zaccai, and C. Pfister. 1998. Thermal motions in bacteriorhodopsin at different hydration levels studied by neutron scattering: correlation with kinetics and light-induced conformational changes. Biophys. J. 75:1945–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paciaroni, A., S. Cinelli, and G. Onori. 2002. Effect of the environment on the protein dynamical transition: a neutron scattering study. Biophys. J. 83:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez, J., J. M. Zanotti, and D. Durand. 1999. Evolution of the internal dynamics of two globular proteins from dry powder to solution. Biophys. J. 77:454–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gregory, R. B. 1995. Protein-Solvent Interactions. Marcel Dekker, New York.
- 16.Hall, D., and A. P. Minton. 2003. Macromolecular crowding: qualitative and semiquantitive successes, quantitative challenges. Biochim. Biophys. Acta. 1649:127–139. [DOI] [PubMed] [Google Scholar]
- 17.Minton, A. P. 2001. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 276:10577–10580. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerman, S. B., and A. P. Minton. 1993. Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 22:27–65. [DOI] [PubMed] [Google Scholar]
- 19.Krueger, S., and R. Nossal. 1988. SANS studies of interacting hemoglobin in intact erythrocytes. Biophys. J. 53:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ball, P. 2008. Water as an active constitutent in cell biology. Chem. Rev. 108:74–108. [DOI] [PubMed] [Google Scholar]
- 21.Doster, W., and S. Longeville. 2007. Microscopic diffusion and hydrodynamic interactions of hemoglobin in red blood cells. Biophys. J. 93:1360–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jasnin, M., M. Moulin, M. Haertlein, G. Zaccai, and M. Tehei. 2008. In vivo measurement of internal and global macromolecular motions in E. coli. Biophys. J. 95:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jasnin, M., M. Moulin, M. Haertlein, G. Zaccai, and M. Tehei. 2008. Down to atomic-scale intracellular water dynamics. EMBO Rep. 9:543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCammon, J. A., and S. C. Harvey. 1987. Dynamics of Proteins and Nucleic Acids. Cambridge University Press, Cambridge, UK.
- 25.Doster, W., S. Cusack, and W. Petry. 1989. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature. 337:754–756. [DOI] [PubMed] [Google Scholar]
- 26.Reat, V., H. Patzelt, M. Ferrand, C. Pfister, D. Oesterhelt, and G. Zaccai. 1998. Dynamics of different functional parts of bacteriorhodopsin: H-2H labeling and neutron scattering. Proc. Natl. Acad. Sci. USA. 95:4970–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zanotti, J. M., M. C. Bellissent-Funel, and J. Parello. 1999. Hydration-coupled dynamics in proteins studied by neutron scattering and NMR: The case of the typical EF-hand calcium-binding parvalbumin. Biophys. J. 76:2390–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russo, D., J. Perez, J. M. Zanotti, M. Desmadril, and D. Durand. 2002. Dynamic transition associated with the thermal denaturation of a small β protein. Biophys. J. 83:2792–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Longeville, S., W. Doster, and G. Kali. 2003. Myoglobin in crowded solutions: structure and diffusion. Chem. Phys. 292:413–424. [Google Scholar]
- 30.Tehei, M., B. Franzetti, D. Madern, M. Ginzburg, B. Z. Ginzburg, M. T. Giudici-Orticoni, M. Bruschi, and G. Zaccai. 2004. Adaptation to extreme environments: macromolecular dynamics in bacteria compared in vivo by neutron scattering. EMBO Rep. 5:66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perutz, M. F., M. G. Rossmann, A. F. Cullis, H. Muirhead, G. Will, and A. C. T. North. 1960. Structure of haemoglobin: a three-dimensional Fourier synthesis at 5.5-Å resolution, obtained by x-ray analysis. Nature. 185:416–422. [DOI] [PubMed] [Google Scholar]
- 32.Perutz, M. F., G. Fermi, and B. Luisi. 1987. Stereochemistry of cooperative mechanisms in hemoglobin. Acc. Chem. Res. 20:309–321. [DOI] [PubMed] [Google Scholar]
- 33.Artmann, G. M., C. Kelemen, D. Porst, G. Büldt, and S. Chien. 1998. Temperature transitions of protein properties in human red blood cells. Biophys. J. 75:3179–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Digel, I., C. Maggakis-Kelemen, K. F. Zerlin, P. Linder, N. Kasischke, P. Kayser, D. Porst, A. Temiz Artmann, and G. M. Artmann. 2006. Body temperature-related structural transitions of monotremal and human hemoglobin. Biophys. J. 91:3014–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zerlin, K. F., N. Kasischke, I. Digel, C. Maggakis-Kelemen, A. Temiz Artmann, D. Porst, P. Kayser, P. Linder, and G. M. Artmann. 2007. Structural transition temperature of hemoglobins correlates with species' body temperature. Eur. Biophys. J. 37:1–10. [DOI] [PubMed] [Google Scholar]
- 36.Elgsaeter, A., and D. Branton. 1974. Intramembrane particle aggregation in erythrocyte ghosts. I. The effects of protein removal. J. Cell Biol. 63:1018–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alberts, B., A. Johnson, J. Lewis, M. Raff, K. Roberts, and P. Walter. 2002. Molecular Biology of the Cell. Garland, New York.
- 38.Antonini, E., and M. Brunori. 1970. Hemoglobin. Annu. Rev. Biochem. 39:977–1042. [DOI] [PubMed] [Google Scholar]
- 39.Janssen, S., J. Mesot, L. Holitzner, A. Furrer, and R. Hempelmann. 1997. FOCUS: a hybrid TOF-spectrometer at SINQ. Physica B (Amsterdam). 234:1174–1176. [Google Scholar]
- 40.Bauer, G. S., M. Salvatores, and G. Heusener. 2001. MEGAPIE, a 1 MW pilot experiment for a liquid metal spallation target. J. Nucl. Mater. 296:17–33. [Google Scholar]
- 41.http://www.ncnr.nist.gov/dave.
- 42.Bee, M. 1988. Quasielastic neutron scattering. Principles and Applications in Solid State Chemistry, Biology and Materials Science. Adam Hilger, Philadelphia.
- 43.Gabel, F., D. Bicout, U. Lehnert, M. Tehei, M. Weik, and G. Zaccai. 2002. Protein dynamics studied by neutron scattering. Q. Rev. Biophys. 35:327–367. [DOI] [PubMed] [Google Scholar]
- 44.Smith, J. C. 1991. Protein dynamics: comparison of simulations with inelastic neutron-scattering experiments. Q. Rev. Biophys. 24:227–291. [DOI] [PubMed] [Google Scholar]
- 45.Sears, V. F. 1966. Theory of cold neutron scattering by homonuclear diatomic liquids. 2. Hindered rotation. Can. J. Phys. 44:1299–1311. [Google Scholar]
- 46.Park, S. Y., T. Yokoyama, N. Shibayama, Y. Shiro, and J. R. H. Tame. 2006. 1.25 Å resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J. Mol. Biol. 360:690–701. [DOI] [PubMed] [Google Scholar]
- 47.Elantri, S., O. Sire, and B. Alpert. 1990. Relationship between protein solvent proton-exchange and progressive conformation and fluctuation changes in hemoglobin. Eur. J. Biochem. 191:163–168. [DOI] [PubMed] [Google Scholar]
- 48.Bosio, L., J. Teixeira, and M. C. Bellissent-Funel. 1989. Enhanced density fluctuations in water analyzed by neutron scattering. Phys. Rev. A. 39:6612–6613. [DOI] [PubMed] [Google Scholar]
- 49.Volino, F., and A. J. Dianoux. 1980. Neutron incoherent-scattering law for diffusion in a potential of spherical symmetry: general formalism and application to diffusion inside a sphere. Mol. Phys. 41:271–279. [Google Scholar]
- 50.Cho, C. H., J. Urquidi, S. Singh, and G. Wilse Robinson. 1999. Thermal offset viscosities of liquid H2O, D2O, and T2O. J. Phys. Chem. B. 103:1991–1994. [Google Scholar]
- 51.Dellerue, S., A. J. Petrescu, J. C. Smith, and M. C. Bellissent-Funel. 2001. Radially softening diffusive motions in a globular protein. Biophys. J. 81:1666–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibrat, G., L. Assairi, Y. Blouquit, C. T. Craescu, and M. C. Bellissent-Funel. 2008. Biophysical study of thermal denaturation of apo-calmodulin: II. Dynamics of native and unfolded states. Biophys. J. 10.1529/biophysj:107.120147. [DOI] [PMC free article] [PubMed]
- 53.Carpentier, L., M. Bee, A. M. Giroud-Godquin, P. Maldivi, and J. C. Marchon. 1989. Alkyl chain motions in columnar mesophases: a quasielastic neutron scattering study of dicopper tetrapalminat. Mol. Phys. 68:1367–1378. [Google Scholar]
- 54.Hunter, R. J. 2001. Foundations of Colloid Science. Oxford University Press, Oxford.
- 55.Dhont, J. K. G. 1996. An Introduction to Dynamics of Colloids. Elsevier, Amsterdam.
- 56.Beenakker, C. W. J., and P. Mazur. 1984. Diffusion of spheres in a concentrated suspension II. Physica A. 126:349–370. [Google Scholar]
- 57.Tokuyama, M., and I. Oppenheim. 1994. Dynamics of hard-sphere suspensions. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 50:R16–R19. [DOI] [PubMed] [Google Scholar]
- 58.Garvey, C. J., R. B. Knott, E. Drabarek, and P. W. Kuchel. 2004. Shear-induced alignment of self-associated hemoglobin in human erythrocytes: small angle neutron scattering studies. Eur. Biophys. J. 2004:589–595. [DOI] [PubMed] [Google Scholar]
- 59.Busch, S., W. Doster, S. Longeville, V. Garcia Sakai, and T. Unruh. 2007. Microscopic protein diffusion at high concentration. In QENS2006. P. E. Sokol, H. Kaiser, D. Baxter, R. Pynn, D. Bossev, M. Leuschner, editors. Materials Research Society, Bloomington, IN. 107–114.
- 60.Cardinaux, F., A. Stradner, P. Schurtenberger, F. Sciortino, and E. Zaccarelli. 2007. Modeling equilibrium clusters in lysozyme solutions. Europhys. Lett. 77:1–5. [Google Scholar]
- 61.Stradner, A., F. Cardinaux, and P. Schurtenberger. 2006. A small-angle scattering study on equilibrium clusters in lysozyme solutions. J. Phys. Chem. B. 110:21222–21231. [DOI] [PubMed] [Google Scholar]
- 62.Stradner, A., G. Foffi, N. Dorsaz, G. Thurston, and P. Schurtenberger. 2007. New insight into cataract formation: enhanced stability through mutual attraction. Phys. Rev. Lett. 99:1–4. [DOI] [PubMed] [Google Scholar]
- 63.Stradner, A., H. Sedgwick, F. Cardinaux, W. C. Poon, S. U. Egelhaaf, and P. Schurtenberger. 2004. Equilibrium cluster formation in concentrated protein solutions and colloids. Nature. 432:492–495. [DOI] [PubMed] [Google Scholar]
- 64.Teixeira, J., M. C. Bellissentfunel, S. H. Chen, and A. J. Dianoux. 1985. Experimental determination of the nature of diffusive motions of water molecules at low-temperatures. Phys. Rev. A. 31:1913–1917. [DOI] [PubMed] [Google Scholar]
- 65.Artmann, G. M., L. Burns, J. M. Canaves, A. Temiz-Artmann, G. W. Schmid-Schonbein, S. Chien, and C. Maggakis-Kelemen. 2004. Circular dichroism spectra of human hemoglobin reveal a reversible structural transition at body temperature. Eur. Biophys. J. 33:490–496. [DOI] [PubMed] [Google Scholar]
- 66.Yan, Y. B., Q. Wang, H. W. He, and H. M. Zhou. 2004. Protein thermal aggregation involves distinct regions: sequential events in the heat-induced unfolding and aggregation of hemoglobin. Biophys. J. 86:1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelemen, C., S. Chien, and G. M. Artmann. 2001. Temperature transition of human hemoglobin at body temperature: effects of calcium. Biophys. J. 80:2622–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fitter, J. 2001. Dynamical properties of a-amylase in the folded and unfolded state: the role of thermal equilibirum fluctuations for conformational entropy and protein stabilisation. Physica B (Amsterdam). 301:1–7. [Google Scholar]