

Abstract
Matrix metalloproteinase-2 (MMP-2) expression is often upregulated in advanced cancers and known to play important role in tumor angiogenesis. We previously showed that adenoviral-mediated delivery of siRNA for MMP-2 (Ad-MMP-2-Si) inhibited lung cancer growth, angiogenesis and metastasis. In this study, we investigated the signaling mechanisms involved in Ad-MMP-2-Si-mediated inhibition of angiogenesis. Ad-MMP-2-Si treatment inhibited neo-vascularization in vivo as determined by mouse dorsal air sac model, and conditioned medium from Ad-MMP-2-Si-infected A549 lung cancer cells (Ad-MMP-2-Si-CM) inhibited endothelial tube formation in vitro. Ad-MMP-2-Si-CM decreased proliferation as determined by Ki-67 immunofluorescence and induced apoptosis in endothelial cells as determined by TUNEL assay. Furthermore, Ad-MMP-2-Si-CM inhibited AKT phosphorylation and induced phosphorylation of ERK-MAPKs in endothelial cells. Overexpression of constitutively active-AKT reversed the Ad-MMP-2-Si-CM-mediated inhibition of tube formation and induction of ERK phosphorylation. Conversely, Ad-MMP-2-Si-CM induced TIMP-3 expression, and the interaction of VEGFR2 and TIMP-3 was determined by co-immunoprecipitation experiments. TIMP-3 induction was mediated by ERK activation. In addition, electrophoretic mobility shift and chromatin immunoprecipitation assays demonstrate that Sp1 transcription factor mediated Ad-MMP-2-Si-CM-stimulated increase of TIMP-3. Vasculature destruction was confirmed with co-localization studies with TUNEL and an endothelial marker, CD31, in tumor sections of Ad-MMP-2-Si-treated mice. Our data collectively suggest MMP-2 inhibition induces endothelial apoptosis in vivo and inhibits endothelial tube formation. These experiments provide the first evidence that inhibition of p-AKT and induction of p-ERK1/2 are crucial events in the induction of TIMP-3-mediated endothelial apoptosis in MMP-2 inhibited lung tumors.
Keywords: ERK, TIMP-3, MMP-2, siRNA, angiogenesis, apoptosis, lung cancer
INTRODUCTION
The initiation and development of adequate neo-vascularization from existing blood vessels to furnish nutrients and oxygen to tumor cells is crucial for tumor growth. Angiogenesis, a process of the origination of functional microcapillaries from existing vasculature, plays a pivotal role in tumor survival, growth, and metastasis (1). The angiogenic switch, a discrete step in tumor development, implies a shift in equilibrium between angiogenesis inducers and countervailing inhibitors in the local environment of the tumor (2). A common line of attack for shifting the balance involves altered gene transcription of positive and negative regulators of angiogenesis. Among these, vascular endothelial growth factor (VEGF) is the most potent and specific of the endothelial cell mitogens, and acts as both an endothelial cell survival factor and a key factor in mobilizing circulating endothelial cell precursors to nascent blood vessels (3–6). Not only does VEGF promote the vascularization and growth of the primary tumor, but it also seems to play a key role in the early stages of establishing new metastatic foci (5). Malignant pleural effusions from lung cancer have high levels of VEGF, and VEGF secretion has also been associated with the development of ascites (7). Moreover, many tumors including lung tumors evidence increased expression of VEGF compared to their normal tissue counterparts (8, 9). VEGF receptors are also expressed on lung cancer cells and mediate anti-apoptotic and metastatic signals (10).
Matrix metalloproteinases (MMPs), a family of extracellular proteolytic enzymes, and their tissue inhibitors (TIMPs) are known to play significant roles in tumor invasion, angiogenesis and metastasis (11). The possible roles of MMP-2 and MMP-9 (also known as gelatinases) in tumor angiogenesis have been explored using different in vivo and in vitro experimental models. Human umbilical vein endothelial cells (HUVECs) undergoing tubulogenesis within a 3-dimensional fibrin matrix have exhibited increased expression of several MMPs (MMP-2, MT1-MMP, MT2-MMP and MT3-MMP) in comparison to HUVECs growing in monolayer cultures (12). MMP-2 was expressed mostly by the microvascular cells of the blood vessels within and surrounding the tumor and by fibroblasts adjacent to the tumor stroma, while MMP-9 was expressed in the tissue macrophages in the vicinity of tumor nodules (13, 14). MMP inhibition studies support the notion that angiogenesis is dependent at least in part on the actions of MMPs since both TIMPs and synthetic MMP inhibitors, such as BB94 (Batimastat) and BMS-275291, display anti-angiogenic properties in vitro and in vivo (15–17). The anti-angiogenic signaling capacity of TIMPs was shown to be distinct from their MMP inhibitory activity, and more specifically, the ability of TIMP-3 to interact with VEGFR2, competed with VEGF for binding to its receptor (18). MMP-2-deficient mice displayed reduced tumor-induced angiogenesis as measured by the dorsal air sac assay (19). In a Swarm rat chondrosarcoma model, in which MMP-2 activation was associated with the angiogenic phenotype, suppression of MMP-2 activity by antisense oligonucleotides inhibited angiogenic potential and tumor growth (20). However, the mechanisms underlying decreased angiogenesis via MMP-2 inhibition remain largely unknown.
We have previously shown that downregulation of MMP-2 by adenovirus-mediated delivery of MMP-2 siRNA (Ad-MMP-2-Si) reduced spheroid invasion and angiogenesis in vitro and metastasis and tumor growth in vivo (21). Hence, we hypothesized that MMP-2 inhibition in the tumor cells causes a shift in the balance between positive and negative regulators of neovascularization, thereby resulting in the inhibition of tumor-induced angiogenesis and subsequent inhibition of tumor growth. These studies, as detailed below, document that MMP-2 inhibition in lung cancer cells resulted in decreased induction of VEGF in endothelial cells and the subsequent cascade of signaling mechanisms that culminated in TIMP-3 induction leading to inhibition of angiogenesis and endothelial apoptosis.
MATERIALS AND METHODS
Cells and reagents
A549 cells were cultured on RPMI-1640 (ATCC, Manassas, VA) supplemented with 10% fetal bovine serum (FBS), 50 units/mL penicillin, and 50 μg/mL streptomycin (Life Technologies Inc., Frederick, MD). In addition, glutamine, EGF (San Jose, CA) and hydrocortisone (Stem Cell Technologies, British Columbia, Canada) were added to advanced MEM medium (Invitrogen, Carlsbad, CA) for human dermal microvascular endothelial cells (HMECs). Cells were incubated in a humidified 5% CO2 atmosphere at 37°C. We used antibodies specific for VEGF, VEGFR2, phospho-VEGFR2, ERK, phospho-ERK, Ki-67, Sp1 (Santa Cruz Biotechnology, Santa Cruz, CA), PI3K, AKT, phospho-AKT (Cell Signal, Boston, MA), GAPDH (Novus Biologicals, Inc., Littleton, CO), TIMP-3, CD31 and HRP-conjugated secondary antibodies (Biomeda, Foster City, CA). Chromatin Immunoprecipitation (ChIP) Assay Kit (Upstate Biotechnology, Lake Placid, NY), diffusion chambers (Fisher Scientific, Pittsburgh, PA), constitutively expressed AKT (myr-AKT) (Addgene, Plasmid 10841), Protein A/G PLUS-Agarose, TIMP-3 siRNA and control siRNA oligos (Santa Cruz Biotechnology, Santa Cruz, CA) were used in this study.
Adenoviral siRNA constructs and infection
Adenoviral siRNA for MMP-2 (Ad-MMP-2-Si) and scrambled vector (Ad-SV) were constructed and amplified as described by us previously (21). Viral titers were quantified as pfu/ml following infection of 293 cells. Titers were obtained for the viruses used in this work are Ad-SV (7.6 × 1011 pfu/ml) and Ad-MMP-2 (5.0 × 1011). The specificity of this virus to inhibit MMP-2 was documented in our previous studies (21, 22). The amount of infective adenoviral vector per cell (pfu/cell) in culture media was expressed as multiplicity of infection (MOI). Virus constructs were diluted in serum-free culture media to the desired concentration, added to cells, and incubated at 37°C for 1 h. Then, the necessary amount of complete medium was added and cells were incubated.
Preparation of tumor conditioned media (TCM)
1.5 × 106 A549 cells were seeded in 100mm Petri dishes and left overnight. Cells were infected with mock (PBS), 100 MOI of either Ad-SV (adenovirus carrying a scrambled sequence) or Ad-MMP-2-Si (adenovirus carrying siRNA against MMP-2) and incubated for a further 24 h. The medium was replaced with serum-free DMEM/F-12 50/50 medium and incubated for 16 h. This conditioned medium was removed, centrifuged for 5 min, and used to grow endothelial cells. Conditioned media collected from mock, Ad-SV and Ad-MMP-2-Si-infected A549 cells were designated as mock-CM, Ad-SV-CM and Ad-MMP-2-Si-CM, respectively. All the experiments were performed in the presence of serum-free endothelial culture media to see if conditioned medium by itself had any effect on endothelial cells.
In vitro angiogenic assay
Angiogenesis was performed as described earlier (23) with little modification. HMECs (2×104 cells/well) were grown in the presence of TCM in 96-well plates coated with matrigel and incubated for 16 h at 37°C. The formation of capillary-like structures was captured. The degree of angiogenesis was measured as the number of branch points per view and tube length of branches was counted.
Mouse dorsal air sac model
Athymic nude mice were maintained within a specific-pathogen, germ-free environment. The implantation technique of the dorsal skin-fold chamber model has been described previously (24). Briefly, diffusion chambers with mock or Ad-SV or Ad-MMP-2-Si-infected A549 cells (2×106) were placed underneath the skin into the superficial incision made horizontally along the edge of the dorsal air sac. After 10 days, the mice were carefully skinned around the implanted chambers and the skin fold covering the chambers was photographed under a visible light microscope. The number of blood vessels within the chamber in the area of the air sac fascia was counted and quantitated.
Immunocytochemical analysis for Ki-67 index
HMECs (5×103 cells/well) seeded in 8-well chamber slides were allowed to grow for 72 h on A549-CM. The effects of the TCM on HMEC cellular proliferation were measured by analysis for Ki-67 immunoreactivity. Cells were fixed in cold methanol and permeabilized in 0.1% Triton X-100 in PBS. After blocking with 1% BSA in PBS for 1 h at room temperature (RT), cells were incubated overnight anti-Ki-67 (1:100 dilution). Mouse IgG was used as a negative control. After incubation with HRP-conjugated secondary antibody (1:200 dilution) for 1 h, 3,3-diaminobenzidine solution (Sigma, St. Louis, MO) was used for developing chromogen and counterstained with hematoxylin and mounted. The bright field images were captured with an Olympus BX-60 research fluorescence microscope attached to a CCD camera.
Terminal deoxy nucleotidyl transferase-mediated nick labeling (TUNEL) assay and immunohistochemistry
We detected apoptosis in HMECs cultured in TCM as well as the subcutaneous tumor tissue sections of Ad-MMP-2-Si-treated mice using TUNEL enzyme reagent as per the manufacturer’s instructions. Briefly, 5 × 103 HMECs were cultured in TCM for the indicated time periods (fresh TCM was added every 24 h), fixed in 4% paraformaldehyde in PBS for 1 h at RT, and permeabilized in 0.1% Triton-X100 in 0.1% sodium citrate in PBS for 2 min (for cells) or 10 min (for tissue sections) on ice. The samples were incubated in TUNEL reaction mixture in a humidified atmosphere at 37°C for 1 h in the dark. For tissue sections, after TUNEL, immunohistochemical analysis was performed using anti-CD31 antibody as described previously (22). Images were captured with an Olympus BX 60 research fluorescence microscope attached to a CCD camera, and cells were counted. The apoptotic index was defined as follows: apoptotic index (%) = 100 × (apoptotic cells/total cells).
Extraction of extracellular matrix protein
After 16 h in the presence of TCM, HMEC cells were washed and ECM extracted as described previously (22). The extracts were subjected to immunoblotting.
Immunoprecipitation and immunoblotting
HMECs were grown in TCM for 16 h. Then, cells were collected, and whole cell lysates were prepared by lysing cells in RIPA lysis buffer containing 1 mM sodium orthovanadate, 0.5 mM PMSF, 10 μg/mL aprotinin, and 10 μg/mL leupeptin. Equal amounts of protein fractions or immunoprecipitates of lysates with the indicated antibodies, were resolved by SDS-PAGE and transferred to PVDF membrane. Proteins were detected with primary antibodies followed by HRP-conjugated secondary antibodies. Comparable loading of proteins on the gel was verified by re-probing the blots with an antibody specific for the housekeeping gene product GAPDH.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from HMECs grown in TCM for 16 h as described previously (25). The protein was quantified and 5 μg nuclear extract were incubated in 20 μL of buffer (20mM HEPES pH 7.9, 0.4 mM EDTA pH 8.4, 0.4 mM DTT, 10% glycerol, 100 μg/mL poli-dI/dC, 1% NP-40) with P32-labeled, double-stranded oligonucleotides containing the SP1 binding site with 80,000 cpm for 15 min at RT. For super shift, Sp1 antibodies were used. The reaction mixture was electrophoresed through a 5% polyacrylamide gel, and the gel was dried and subjected to autoradiography using phosphor screens at −70°C.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed as per the manufacturer's instructions. Briefly, HMECs were growm in TCM for 16 h, fixed with formaldehyde (final concentration of 1%), and incubated for 10 min at 37°C. The cells were washed twice with ice-cold PBS containing protease inhibitors (1 mM PMSF, 1 μg/mL aprotinin and 1 μg/mL pepstatin-A), harvested, and lysed with SDS lysis buffer for 10 min on ice. The lysates were sonicated to shear the DNA to fragment lengths ~1000 bp (amplitude 60%, 4 bursts of 10 sec, Fisher Sonic Dismembrator 60, Pittsburgh, PA). From each sonicated sample, 5% was used as the input control for immunoprecipitated fragments. The complexes were immunoprecipitated with antibodies specific for Sp1. Complexes were collected using salmon sperm DNA/protein-A agarose slurry and washed as per the manufacturer's instructions. The immune complexes were eluted with 1% SDS and 0.1 M NaHCO3, and the crosslinks were reversed by incubation at 65°C for 4 h in the presence of 200 nM NaCl. After treatment with proteinase-K at 45°C for 1 h, the DNA was purified by phenol/chloroform extraction, ethanol precipitation, and resuspended in 3 μL of nuclease water. PCR was carried out for 30 cycles with primers specific for the TIMP-3 promoter. PCR products were resolved on 2% agarose gels and analyzed using ethidium bromide staining.
Xenograft animal models
SCID mice were maintained and experiments were performed as described previously (21). Subcutaneous tumors were fixed in 10% phosphate-buffered formaldehyde.
Statistical analysis
All data are presented as means ± standard error (SE) of at least three independent experiments, each performed at least in triplicate. One way analysis of variance (ANOVA) combined with the Tukey post-hoc test of means was used for multiple comparisons in cell culture experiments. Statistical differences are presented at probability levels of p < 0.05, p < 0.01 and p < 0.001.
RESULTS
Ad-MMP-2-Si inhibits angiogenesis
We have shown previously that the downregulation of MMP-2 with Ad-MMP-2-Si inhibited tumor growth and lung metastasis in a mouse model (21). In the present study, tumor conditioned medium from A549 cells infected with mock (mock-CM) or 100 MOI of either Ad-SV (Ad-SV-CM) or Ad-MMP-2-Si (Ad-MMP-2-Si-CM) was used to induce human dermal microvascular endothelial cells (HMECs) to form a capillary network in an in vitro angiogenic assay. Fig. 1A show that MMP-2 activity is inhibited by more than 75% in the conditioned medium of A549 cells infected with 100 MOI of Ad-MMP-2 Si compared to mock and Ad-SV controls. Mock-CM and Ad-SV-CM elicited a strong angiogenic response and induced HMECs to differentiate into capillary-like structures within 16 h. In contrast, Ad-MMP-2-Si-CM inhibited micro vessel morphogenesis Cells grown the presence with endothelial culture media without serum (no serum) were just beginning to differentiate into capillaries at 16h time point. (Fig. 1B). Quantification indicated a ~85% decrease in branch points and a ~90% decrease in vessel length in HMECs cultured with Ad-MMP-2-Si-CM as compared to mock-CM and Ad-SV-CM (Fig. 1B).
Figure 1. Ad-MMP-2-Si inhibits tumor-induced angiogenesis.
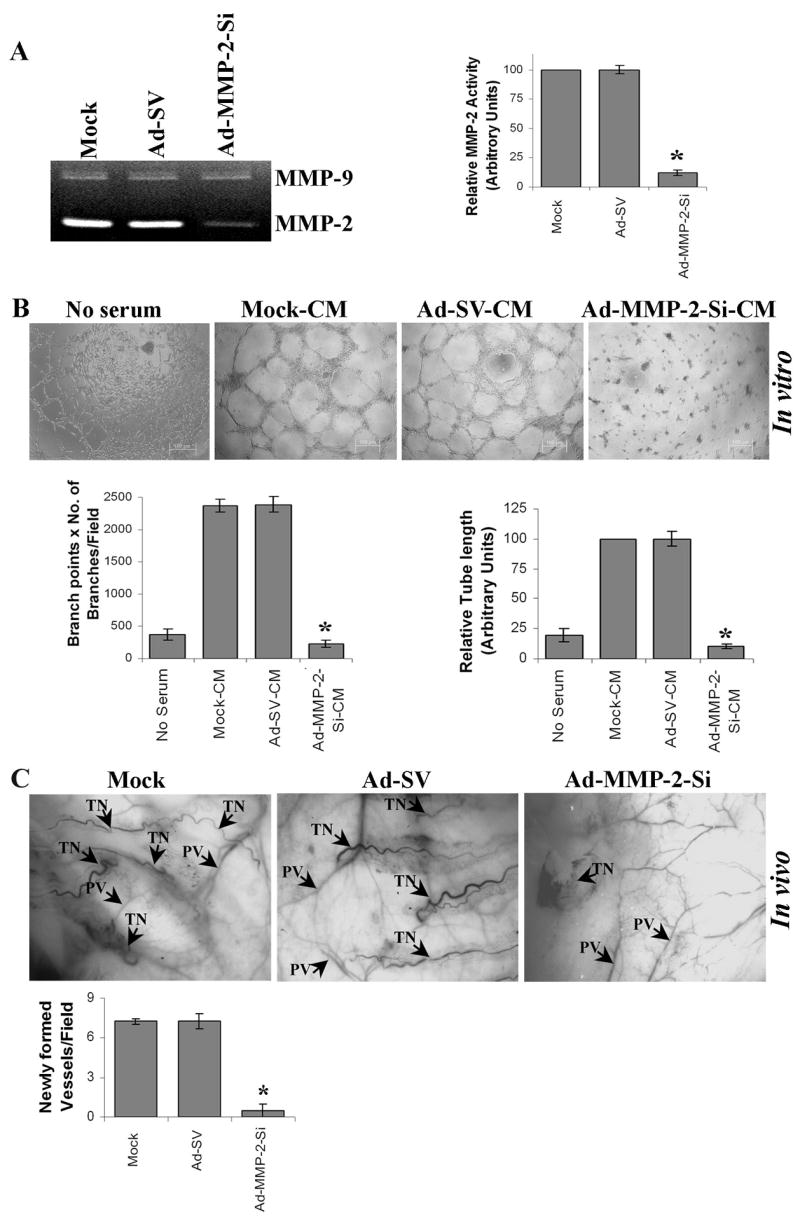
(A & B) A549 cells were infected as described in Materials and Methods. Briefly, A549 cells were infected with 100 MOI of either Ad-SV or Ad-MMP-2-Si, medium was aspirated after 36 h of incubation and 3 mL of serum-free medium was added, and cells incubated overnight. (A) Gelatin zymographic analysis of tumor conditioned medium. The band intensities of MMP-2 activity were quantified by densitometry and normalized with the intensity of the mock-conditioned medium band. Columns: mean of triplicate experiments; bars: SE; *p<0.01, significant difference from mock-CM. (B) Upper panel: In vitro angiogenesis: The tumor conditioned medium (TCM) was added into 96-well plates, which were coated with matrigel and pre-seeded with human dermal microvascular endothelial cells (HMECs) (2 × 104 cells/well). After overnight incubation at 37°C, cells were observed under the bright field microscope for formation of capillary-like structures. Bottom panel: The degree of angiogenic induction by Mock-CM, Ad-SV-CM and Ad-MMP-2-Si-CM was quantified for the numerical value of the product of the relative capillary length and number of branch points per field. The capillary length and number of branch points were reduced drastically in HMECs grown in conditioned medium from Ad-MMP-2-Si-infected A549 cells. Columns: mean of quadruplicate experiments; bars: SE; *p<0.01, significant difference from mock-CM.
(C) In vivo angiogenic assay using the dorsal skin fold model as described in Materials and Methods. Briefly, the animals were implanted with diffusion chambers containing mock, 100 MOI of either Ad-SV or Ad-MMP-2-Si-infected cells in a dorsal cavity. Ten days after implantation, the animals were sacrificed and the skin fold covering the diffusion chamber was observed under bright field light microscope for the presence of tumor-induced neovasculature (TN) and preexisting vasculature (PV) and photographed (upper panel). The mean number of neovasculature (>20μm) per field was quantified (Bottom panel). Columns: mean of quadruplicate experiments; bars: SE; *p<0.01, significant difference from mock.
We also examined whether Ad-MMP-2-Si could inhibit tumor angiogenesis in vivo as assessed by the dorsal window model. Implantation of a chamber containing mock or Ad-SV-infected A549 cells in the dorsal skinfold chamber resulted in the development of microvessels (as indicated by arrows) with curved thin structures and many tiny bleeding spots. In contrast, implantation of A549 cells infected with Ad-MMP-2-Si resulted in the development of a few additional microvessels (Fig. 1C).
Ad-MMP-2-Si-CM inhibits endothelial cell proliferation and survival
To elucidate the cellular mechanisms underlying the inhibitory effect of tumor conditioned medium (TCM) from tumor cells infected with Ad-MMP-2-Si on angiogenesis, we investigated the effect of TCM on the proliferation and survival of endothelial cells. We examined cell proliferation indices in endothelial cells grown in Ad-MMP-2-Si-CM. We assayed for the presence of the Ki-67 molecular marker (expressed in the G1-S-G2 phases of the cell cycle), and following immunocytochemical analysis, we quantified the number of positive cells in all treatment groups at 400X magnification. Figure 2A indicates that Ad-MMP-2-Si-CM caused a decrease in the fraction of cells expressing the proliferation marker Ki-67 at 72 h. The controls had Ki-67 labeling indices of 45.01±4.81% in mock-CM and 44.59±5.26% in Ad-SV-CM, whereas Ad-MMP-2-Si-CM had a significantly lower index at 15.7±2.11%. Having observed a marked reduction in the Ki-67 index in endothelial cells grown in Ad-MMP-2-Si-CM, we determined the presence of apoptosis by TUNEL assay. Figure 2B indicates a time-dependent increase in apoptotic index in endothelial cells grown in Ad-MMP-2-Si-CM. The number of cells staining positive for apoptosis were counted in 5 randomly selected fields at 400X magnification. TUNEL labeling indices in endothelial cells grown in the presence of mock-CM and Ad-SV-CM showed hardly any apoptosis for up to 72 h (Fig. 2B). However, endothelial cells grown in the presence of Ad-MMP-2-Si-CM displayed a time-dependent increase in apoptosis. At the 24 h time point, 7–8% of the cells were TUNEL-positive; at the 48 h time point, 31.1±2.83% at the 72 h time point, 62.1±5.15% of the cells were TUNEL-positive.
Figure 2. Ad-MMP-2-Si-CM inhibits endothelial cell survival and proliferation.
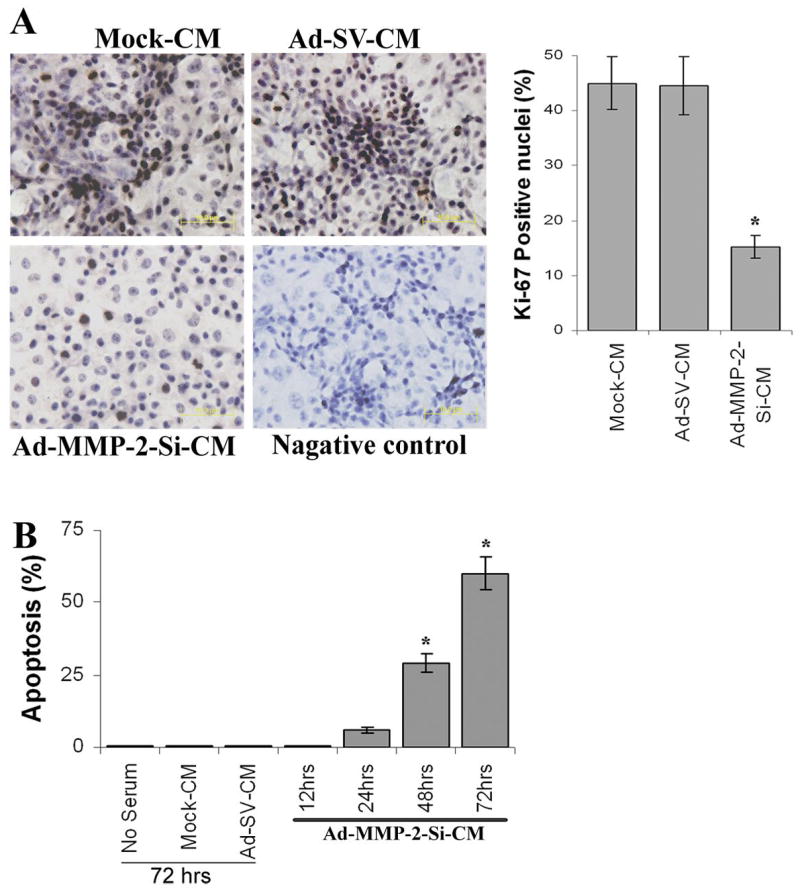
(A) Proliferating HMECs were identified by Ki-67 indirect immunofluorescence. Values were calculated as the number of Ki67-positive cells/number of cells with intact nuclei (hematoxylin staining) at 400X magnification. Columns: mean of quadruplicate experiments; bars: SE; *p<0.05, significant difference from mock-CM.
(B) Determination of apoptosis in endothelial cells grown in the presence of Ad-MMP-2-Si-CM by TUNEL staining up to 72 h. The % apoptotic cells were shown over time. Columns: mean of quadruplicate experiments; bars: SE; *p<0.001, significant difference from mock-CM.
Ad-MMP-2-Si-CM reduces VEGF expression and VEGFR2 phosphorylation in endothelial cells
To determine the effect of Ad-MMP-2-Si-CM on the expression of angiogenesis-inducing molecules, we used western blotting and immunoprecipitation to assess the VEGF protein content and VEGFR2 phosphorylation of endothelial cells grown in TCM for 16 h. Figure 3A indicates VEGF protein levels were decreased in HMECs grown in Ad-MMP-2-Si-CM as compared to HMECs grown in mock-CM and Ad-SV-CM. The phosphorylation state of VEGFR2 was assessed by immunoprecipitation of the receptor followed by phosphotyrosine immunodetection in endothelial cells. Decreased tyrosine phosphorylation of VEGFR2 was observed in endothelial cells grown in the presence Ad-MMP-2-Si-CM (Fig. 3B). Densitometric normalization of VEGFR2 phosphorylation against VEGFR2 band density indicated that there was a 55% decrease in VEGFR phosphorylation.
Figure 3. Ad-MMP-2-Si-CM inhibits expression of VEGF and phosphorylation of VEGFR2 in endothelial cells.
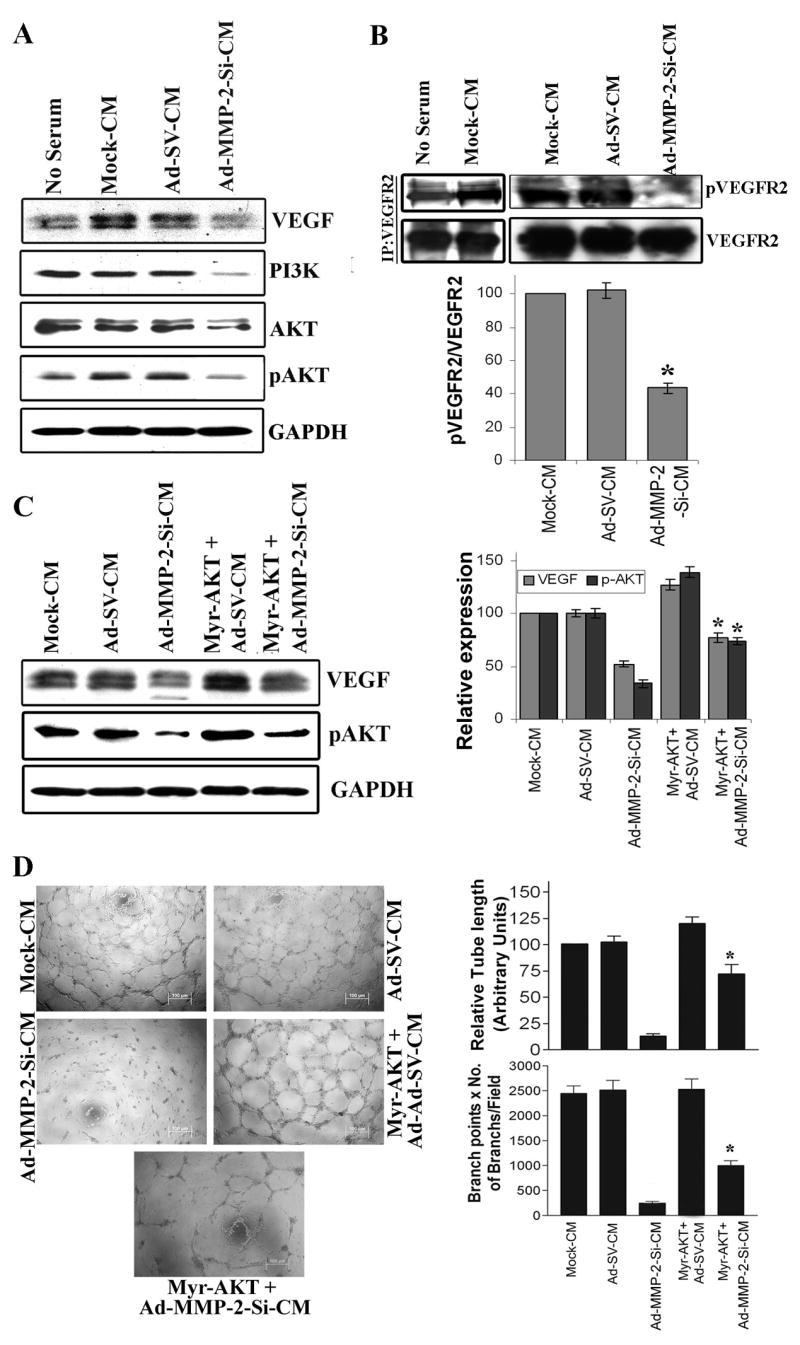
(A) HMECs grown on the Mock-CM, Ad-SV-CM or Ad-MMP-2-Si-CM for 16 h, cells were collected and lysed and immunoblotted for VEGF, PI3K, AKT and pAKT using specific antibodies. The experiments were carried out three times and representative immunoblot was shown. The blots were stripped and re-probed with GAPDH antibody to detect total amounts of the respective proteins.
(B) VEGFR2 was immunoprecipitated from solubilized cell extracts with a polyclonal anti VEGFR2 antibody, and VEGFR2 phosphorylation was determined by immunoblotting using an anti-phosphotyrosine antibody. The experiments were carried out three times, and representative autoradiograms are presented. Densitometric normalization of VEGFR2 phosphorylation against VEGFR2 band density is shown. Columns: mean of triplicate experiments; bars: SE; *p<0.01, significant difference from mock-CM.
(C) Endothelial cells were transfected with a plasmid expressing constitutively active AKT (myr-AKT) and were grown in presence of conditioned medium as described above. Cell lysates were immunoblotted for pAKT and VEGF. Densitometric normalization of VEGF and phospho-AKT is shown. Columns: mean of quadruplicate experiments; bars: SE; *p<0.05, significant difference from Ad-MMP-2-Si-CM treatment alone.
(D) An in vitro angiogenic assay was performed under the same conditions as described in Figure 1. The experiments were carried out three times, and representative pictures are shown. The degree of angiogenic induction was quantified for the numerical value of the product of the relative capillary length and number of branch points per field. Columns: mean of quadruplicate experiments; bars: SE; *p<0.05, significant difference from Ad-MMP-2-Si-CM treatment alone.
Ad-MMP-2-Si-CM suppresses AKT activation and enhances ERK phosphorylation
Because Ad-MMP-2-Si-CM inhibited VEGFR2 phosphorylation, we sought to determine whether the downstream mediators of VEGFR2 signaling were potential targets of Ad-MMP-2-Si-CM. VEGFR2 is characterized by tyrosine kinase activity and phosphorylation of secondary messengers. These secondary messengers appear to regulate endothelial cell proliferation via activation of extracellular signal-regulated kinase (ERK) 1/2 mitogen-activated protein kinases (26) as well as endothelial cell survival via AKT activation (27). To assess the effect of Ad-MMP-2-Si-CM on these pathways, endothelial cells grown in the presence of the TCM for 16 h and cell lysates were subjected to immunodetection using antibodies against PI3K, AKT, phosphorylated AKT (Ser473), ERK and phosphorylated ERK1/2. There is an increase in AKT phosphorylation in endothelial cells grown in the presence of tumor conditioned medium from mock treated cells compared to no serum medium. However, Ad-MMP-2-Si-CM significantly decreased PI3K expression and phosphorylation of AKT in endothelial cells as compared to the controls (Fig. 3A). The serine/threonine protein kinase AKT is an important component in migratory and pro-survival signaling pathways and Akt-mediated signaling is stimulated by several angiogenic factors and plays a pivotal role for endothelial cell survival, migration, and proliferation, all of which are intimately involved in angiogenesis and vascular repair (28).
To assess this hypothesis directly, we transfected cells with constitutively active AKT, myr-AKT (29), and examined the implications for Ad-MMP-2-Si-CM-mediated inhibition on VEGF expression and endothelial cord formation. As shown in Figure 3C, myr-AKT transfection reversed the inhibitory effect of Ad-MMP-2-Si-CM on phosphor-AKT and VEGF expression in endothelial cells. Moreover, overexpression of AKT in the presence of Ad-MMP-2-Si-CM reversed the inhibitory effect of Ad-MMP-2-Si-CM on endothelial tube formation (Fig. 3D). Quantification of tube length and branch points indicated that there was a ~5-fold increase in tube length and a ~2-fold increase in the number of branch points in myr-AKT-overexpressed cells grown on Ad-MMP-2-Si-CM compare to cell grown on Ad-MMP-2-Si-CM alone.
Conversely, an unexpected increase in ERK1/2 phosphorylation was observed in endothelial cells grown in Ad-MMP-2-Si-CM as compared to mock-CM and Ad-SV-CM (Fig. 4A). The ratio between phosphorylated ERK and total ERK indicated a 2-fold increase in pERK levels in Ad-MMP-2-Si-CM-treated endothelial cells as compared to the controls. Moreover, overexpression of AKT inhibited Ad-MMP-2-Si-CM-induced phosphorylation of ERK, thereby confirming that this effect was a result of AKT (Fig. 4B). Taken together, these results suggest that inhibition of AKT phosphorylation by Ad-MMP-2-Si-CM plays a key role the inhibition of angiogenesis. In addition, Ad-MMP-2-Si-CM not only inhibited the downstream effects of the AKT survival pathway, but also allowed overactivation of the ERK pathway via AKT blockade.
Figure 4. Ad-MMP-2-Si-CM induces ERK-mediated expression of TIMP-3.

Endothelial cells were grown in the presence of mock-CM, Ad-SV-CM or Ad-MMP-2-Si-CM for 16 h.
(A) Cellular extracts were subjected to immunoblotting with antibodies for phosphorylation-state-specific ERK1 and total ERK1 followed by chemiluminescence. Densitometric normalization of ERK phosphorylation against total protein band density is shown. *p<0.01, significant difference from mock-CM.
(B) Endothelial cells were transfected with a plasmid expressing constitutively active AKT (myr-AKT) and were grown in presence of conditioned medium as described above. Cell lysates were immunoblotted for pERK, total ERK and TIMP-3. Densitometry normalization of ERK phosphorylation against total protein band density is shown. Columns: mean of quadruplicate experiments; bars: SE; *p<0.01, significant difference from control; **p<0.01, significant difference from Ad-MMP-2-Si-CM treatment.
(C) Endothelial cells were cultured as described above and extracellular protein were analyzed by immunoblotting for Ad-MMP-2-Si-CM-induced TIMP-3 in endothelial cells. Densitometric analysis of TIMP-3 band density is shown in columns. Columns: mean of quadruplicate experiments; bars: SE; *p<0.001, significant difference from mock-CM.
(D) Impact of ERK pathway on Ad-MMP-2-Si-CM-stimulated expression of TIMP-3 in endothelial cells. Endothelial cells were transfected with Dn-ERK followed by culturing on the tumor-conditioned medium from A549 cells infected with mock, Ad-SV or Ad-MMP-2-Si. Cell lysates were immunoblotted with antibodies for phosphorylation-state-specific ERK1/2 and total ERK. ECM protein was analyzed for TIMP-3 immunoblotting. Densitometric normalization of ERK phosphorylation against total protein band density is shown. The quantification of TIMP-3 bands is shown in arbitrary units. Columns: mean of quadruplicate experiments; bars: SE; *p<0.01, significant difference from mock-CM; **p<0.01, significant difference from Ad-MMP-2-Si-CM treatment.
Ad-MMP-2-Si-CM-mediated ERK activation leads to TIMP-3 expression in endothelial cells
TIMP-3 has been reported to inhibit capillary morphogenesis of endothelial cells in vitro and bFGF-induced angiogenesis in vivo (15). Therefore, we examined whether Ad-MMP-2-Si-CM induced TIMP-3 expression in endothelial cells. Figure 4C indicates a 4-fold increase in the 24-kDa-band equivalent to non-glycosylated TIMP-3 in endothelial cells grown in Ad-MMP-2-Si-CM as compared to the controls. To explore the requirement of ERK pathway stimulation for Ad-MMP-2-Si-CM-induced TIMP-3 expression, endothelial cells were transiently transfected with a dominant negative mutant of ERK (Dn-ERK) to inhibit ERK signaling and then grown in the presence of Ad-MMP-2-Si-CM for 16 h. To ascertain if Dn-ERK affected ERK phosphorylation, we assessed ERK phosphorylation under these conditions. As shown in Figure 4D, Dn-ERK transfection inhibited ERK phosphorylation and inhibited Ad-MMP-2-Si-CM-induced TIMP-3 expression. Similarly there was decrease in Ad-MMP-2-Si-CM-induced TIMP-3 expression in Myr Akt transfected cells (Fig. 4B). Since p42/p44 (Erk1/2) MAPKs can phosphorylate Sp1 in vitro and in vivo (30) and the promoter region of the human TIMP-3 gene contains four Sp1 transcription factor-binding sites (31), we determined Sp1 binding activity in endothelial cells grown in the presence of Ad-MMP-2-Si-CM. As shown in Figure 5A, Sp1 activity increased in endothelial cells grown in Ad-MMP-2-Si-CM as compared to cells grown in conditioned media from mock and Ad-SV-transfected cells. Pre-incubation of nuclear extracts with Sp1 antibody indicated a shift, thereby confirming the specificity of the band.
Figure 5. Sp1 is a transcription factor for TIMP-3.

(A) Endothelial cells were grown as described above and nuclear extracts subjected to EMSA for Sp1 binding to its consensus sequence.
(B) HMECs were grown, fixed in formaldehyde and immunoprecipitated with Sp1 antibody as described in Materials and Methods. Purified DNA was subjected to PCR with TIMP-3 primers. Densitometric analysis of the intensity of the bands is shown. Columns: mean of triplicate experiments; bars: SE; *p<0.01, significant difference from mock-CM.
(C) Immunoprecipitation (IP) and immunoblot (IB) analyses from HMECs cultured as described as in Materials and Methods. Immunoprecipitation of cell lysates with antibodies specific for VEGFR2 followed by immunoblotting with TIMP-3 and VEGF. For loading controls, bolts were reprobed with VEGFR2 antibody.
(D) After 24 h of treatment with either TIMP-3 siRNA or control siRNA transfection, HMECs were grown in conditioned media for 72 h, and the apoptosis in endothelial cells grown in the presence of Ad-MMP-2-Si-CM was detected using the TUNEL assay. Columns: mean of triplicate experiments; bars: SE; *p<0.001, versus cells grown on Mock-CM **p<0.001, versus cells grown on Ad-MMP-2-Si-CM.
To assess whether Sp1 interacts with the TIMP-3 promoter, we performed ChIP assays. Using an antibody specific for Sp1, protein chromatin complexes were immunoprecipitated, and the amount of TIMP-3-specific DNA that was released from the immunoprecipitates was analysed by PCR. As shown in Figure 5B, ChIP assay results indicated that Sp1 is strongly associated with TIMP-3 in HMECs grown in Ad-MMP-2-Si-CM as compared to the controls. Quantitative analysis indicated that there was a 2-fold increase in Sp1 binding to the TIMP-3 promoter in endothelail cells grown in the repsense of AD-MMP-2-SI-CM compared to Mock-CM and Ad-SV-CM..
TIMP-3 expression contributes to decreased angiogenesis and endothelial apoptosis
TIMP-3 was shown to block the binding of VEGF to VEGFR2, thereby inhibiting downstream signaling and angiogenesis (18). We performed immunoprecipitation experiments to analyze VEGFR2 interactions with TIMP-3 and VEGF in endothelial cells. Figure 5C indicates that binding of VEGF to VEGFR2 was reduced in endothelial cells grown in Ad-MMP-2-Si-CM as compared to mock-CM and Ad-SV-CM. We also observed increased binding of TIMP-3 to VEGFR2 in endothelial cells cultured with Ad-MMP-2-Si-CM as compared to mock-CM and Ad-SV-CM (Fig. 5C). Since TIMP-3 induces apoptosis in various types of cells (32–35), we further evaluated whether the elevated TIMP-3 levels were responsible for the apoptosis in HMECs grown in Ad-MMP-2-Si-CM. Endothelial cells were transfected with TIMP-3 siRNA and grown in the TCM for 72 h. Ad-MMP-2-Si-CM-induced apoptosis induction was partially reversed by TIMP-3 siRNA, thereby confirming that the apoptosis was mediated via upregulation of TIMP-3 (Fig. 5D).
Ad-MMP-2-Si treatment suppresses angiogenesis in vivo
Having shown that Ad-MMP-2-Si infection in lung cancer cells inhibited angiogenesis using capillary formation assay and an in vivo dorsal air sac assay, we next sought to determine whether the tumor growth inhibition observed in a spontaneous metastasis model after Ad-MMP-2-Si treatment (21) was due to an extension of the anti-angiogenic activity. The tumor cores of the mock, Ad-SV and Ad-MMP-2-Si-treated SCID mice were analyzed immunohistologically with anti-CD31. The immunoreactivity of CD31 in the tissue sections from the tumors derived from mice that received each treatment provided some measure of vascularity as affected by Ad-MMP-2-Si treatment (Fig. 6). Compared to mock and Ad-SV-treated mice, tumor sections from Ad-MMP-2-Si-treated mice showed decreased CD31. To confirm endothelial cell apoptosis, we performed TUNEL staining on the same sections. Endothelial cell apoptosis as determined by TUNEL and CD31 double staining was significantly higher in the tumor sections from mice treated with Ad-MMP-2-Si as compared to the mock and Ad-SV-treated mice (18±2.1 versus 0.5±1.5 and respectively). These results suggest that inhibition of MMP-2 inhibited angiogenesis through stimulation of apoptosis in endothelial cells.
Figure 6. MMP-2 inhibition using Ad-MMP-2-Si decreased angiogenesis and induced endothelial cell apoptosis in lung tumors.
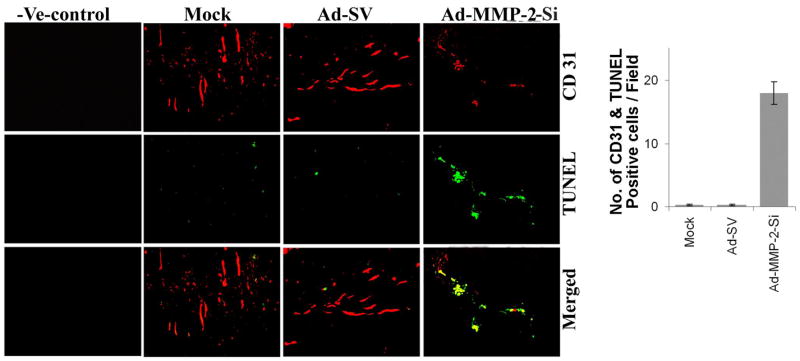
Representative sections (400X magnification) from mock and Ad-SV treated tumors and those treated with Ad-MMP-2-Si. Ad-MMP-2-Si-treated mice showed a decrease in angiogenesis as determined by CD31 staining. Also shown is a negative staining control for CD31 where the antibody was replaced by non-immune serum (negative control). Dual staining for TUNEL positive and CD31 positive cells indicating endothelial apoptosis in Ad-MMP-2-Si-treated mice compared to mock and Ad-SV-treated mice.
DISCUSSION
Treatment for cancer is focused on developing treatments based on inhibiting tumor angiogenesis, as tumors require a blood supply for nutrition and growth. Such therapy, termed “anti-angiogenic” therapy, has now been documented to prolong the survival of patients with lung cancer in large randomized clinical trials. In the present study, we demonstrate the role of MMP-2 inhibition in tumor cells by targeting angiogenic components of tumor growth and identifying particular molecules and/or signaling pathways involved in the angiogenic process. We provide evidence that MMP-2 inhibition in tumor cells triggers signaling pathways that ultimately lead to endothelial cell death.
As an initial step of our study, we observed that tumor conditioned medium from lung cancer cells infected with Ad-MMP-2-Si inhibited capillary tube formation in endothelial cells as compared to the controls. We also observed decreased proliferative index Ki-67 and increased apoptosis in endothelial cells grown in the presence of tumor-conditioned medium from lung cancer cells infected with Ad-MMP-2-Si (Ad-MMP-2-Si-CM). We next carried out experiments designed to identify signaling molecules secreted by endothelial cells that might be responsible for the observed effect on endothelial cells. Endothelial cell differentiation leading to tubule formation is primarily established by activation of VEGF and its receptor. VEGFR2 is the main human receptor responsible for both physiological and pathological vascular development, and the VEGF-VEGFR2 signaling pathway has become an important target for the development of anti-angiogenic agents (36). In a study of the significance of the various VEGF isoforms in NSCLC, Yuan et al. (37) reported that a high VEGF-189 level correlated with high intratumoral microvessel density, short survival, and early postoperative relapse, while VEGF-121 was correlated with short survival and relapse. Another group also reported a similar correlation between poor prognosis and localized elevated microvessel density at the advancing front of NSCLC tumors accompanied by increased localized staining of VEGF and its receptor (38). In this study, we demonstrate that a mechanism in which the paracrine effect of MMP-2-downregulated lung tumor cells inhibited VEGF expression in endothelial cells, thereby leading to inhibition of the PI3K/AKT pathway. Activated PI3K and AKT are strong inducers of neovascularization and endothelial cell proliferation (39). We also show that TIMP-3 induction occurred under the same conditions and binds to VEGFR2. Previous studies have also shown that TIMP-3 binding to VEGFR2 inhibited downstream signaling and angiogenesis (18).
To determine the functional consequences of the upregulation of ERK pathway, we manipulated Ad-MMP-2-Si-CM-induced ERK pathway upregulation and determined the implications for endothelial cell apoptosis. The inhibitory effect of Dn-ERK on TIMP-3 expression implies that the ERK1/2 signal pathway is critical for TCM-induced TIMP-3 expression. To begin to understand the underlying mechanisms of the correlation between levels of ERK and TIMP-3, we evaluated the activity of Sp1, a known transcriptional regulator of TIMP-3 (40). Our results show that Sp1 was induced in endothelial cells grown in the presence of Ad-MMP-2-Si-CM. However, we cannot rule out the possible involvement of other transcription factors in TIMP-3 expression. Nevertheless, it is certain that the Sp1 transcription factor is dominantly involved in the induction of TIMP-3 as determined by ChIP assay. Our studies indicate that tumor conditioned medium from lung cancer cells infected with Ad-MMP-2-Si activates the ERK-MAPK pathway, which in turn, could increase Sp1 phosphorylation and its DNA binding activity leading to TIMP-3 induction. Our results support previous findings that Sp1 is an important link between increased activation of the ERK pathway and TIMP-3 expression (40). We further demonstrate the significance of TIMP-3 expression in the induction of endothelial apoptosis using siRNA. These results suggest that the induction of TIMP-3 in Ad-MMP-2-Si-treated mice inhibited in vivo angiogenesis by inhibition of VEGF/VEGFR2 signaling and induction of endothelial apoptosis. Induction of apoptotic cell death by TIMP-3 overexpression is well documented and has been shown in vascular cells and cancer cells in vitro and in vivo (41–44). Intratumoral injection with TIMP-3-expressing adenovirus inhibits growth of melanomas and squamous cell carcinomas by inducing apoptosis and inhibiting tumor angiogenesis (45). Further, Mino et al. observed that TIMP-3 expression status is significantly correlated with pathologic stage and nodal involvement in resected NSCLC, and that the overall survival rate of patients with elevated TIMP-3 expression is high (46).
In summary, we show that MMP-2 inhibition in lung tumor cells inhibits VEGF-mediated AKT activation in endothelial cells, leading to increased ERK activation that contributes to TIMP-3 induction. We have also established the importance of these pathways in the induction of endothelial cell apoptosis. These data provide insights about the mechanisms of MMP-2 inhibition in tumor cells, which led to decreased angiogenesis and resulted in tumor growth inhibition.
Supplementary Material
Acknowledgments
We thank Noorjehan Ali for technical assistance, Shellee Abraham for assistance in manuscript preparation, and Diana Meister and Sushma Jasti for review of this paper. We thank Professor Richard A. Roth for the construct encoding the constitutively active myristoylated AKT (myr-AKT delta 4-129).
This research was supported by National Cancer Institute Grant CA75557, CA92393, CA95058, CA116708 and N.I.N.D.S. NS47699, NS57529, NS61835 and Caterpillar, Inc., OSF Saint Francis, Inc. Peoria, IL (to J.S.R.). and American Cancer Society Grant #06-03 (S.S.L.)
Abbreviations used
- ERK
extracellular signal-regulated kinase
- TIMP
tissue inhibitor of metalloproteinases
- MMP
matrix metalloproteinase
- siRNA
short interfering RNA
- HMEC
human dermal microvascular endothelial cells
- TCM
tumor conditioned medium
- TUNEL
terminal deoxynucleotidyltransferase-mediated nick end labeling
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- EMSA
electrophoretic mobility shift assay
- ChIP
chromatic immunoprecipitation
Reference List
- 1.Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757–63. doi: 10.1056/NEJM199512283332608. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Asahara T, Takahashi T, Masuda H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18:3964–72. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrara N. Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int. 1999;56:794–814. doi: 10.1046/j.1523-1755.1999.00610.x. [DOI] [PubMed] [Google Scholar]
- 5.Li CY, Shan S, Huang Q, et al. Initial stages of tumor cell-induced angiogenesis: evaluation via skin window chambers in rodent models. J Natl Cancer Inst. 2000;92:143–7. doi: 10.1093/jnci/92.2.143. [DOI] [PubMed] [Google Scholar]
- 6.Lyden D, Hattori K, Dias S, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 7.Yano S, Shinohara H, Herbst RS, et al. Production of experimental malignant pleural effusions is dependent on invasion of the pleura and expression of vascular endothelial growth factor/vascular permeability factor by human lung cancer cells. Am J Pathol. 2000;157:1893–903. doi: 10.1016/S0002-9440(10)64828-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bremnes RM, Camps C, Sirera R. Angiogenesis in non-small cell lung cancer: the prognostic impact of neoangiogenesis and the cytokines VEGF and bFGF in tumours and blood. Lung Cancer. 2006;51:143–58. doi: 10.1016/j.lungcan.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Volpert OV, Dameron KM, Bouck N. Sequential development of an angiogenic phenotype by human fibroblasts progressing to tumorigenicity. Oncogene. 1997;14:1495–502. doi: 10.1038/sj.onc.1200977. [DOI] [PubMed] [Google Scholar]
- 10.Dy GK, Adjei AA. Angiogenesis inhibitors in lung cancer: a promise fulfilled. Clin Lung Cancer. 2006;7(Suppl 4):S145–S149. doi: 10.3816/clc.2006.s.006. [DOI] [PubMed] [Google Scholar]
- 11.Bodey B, Bodey B, Jr, Siegel SE, Kaiser HE. Immunocytochemical detection of the expression of members of the matrix metalloproteinase family in adenocarcinomas of the pancreas. In Vivo. 2001;15:71–6. [PubMed] [Google Scholar]
- 12.Lafleur MA, Handsley MM, Knauper V, Murphy G, Edwards DR. Endothelial tubulogenesis within fibrin gels specifically requires the activity of membrane-type-matrix metalloproteinases (MT-MMPs) J Cell Sci. 2002;115:3427–38. doi: 10.1242/jcs.115.17.3427. [DOI] [PubMed] [Google Scholar]
- 13.John A, Tuszynski G. The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol Oncol Res. 2001;7:14–23. doi: 10.1007/BF03032599. [DOI] [PubMed] [Google Scholar]
- 14.Vacca A, Moretti S, Ribatti D, et al. Progression of mycosis fungoides is associated with changes in angiogenesis and expression of the matrix metalloproteinases 2 and 9. Eur J Cancer. 1997;33:1685–92. doi: 10.1016/s0959-8049(97)00186-x. [DOI] [PubMed] [Google Scholar]
- 15.Anand-Apte B, Pepper MS, Voest E, et al. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase-3. Invest Ophthalmol Vis Sci. 1997;38:817–23. [PubMed] [Google Scholar]
- 16.Fisher C, Gilbertson-Beadling S, Powers EA, Petzold G, Poorman R, Mitchell MA. Interstitial collagenase is required for angiogenesis in vitro. Dev Biol. 1994;162:499–510. doi: 10.1006/dbio.1994.1104. [DOI] [PubMed] [Google Scholar]
- 17.Naglich JG, Jure-Kunkel M, Gupta E, et al. Inhibition of angiogenesis and metastasis in two murine models by the matrix metalloproteinase inhibitor, BMS-275291. Cancer Res. 2001;61:8480–5. [PubMed] [Google Scholar]
- 18.Qi JH, Ebrahem Q, Moore N, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9:407–15. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 19.Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H, Itohara S. Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res. 1998;58:1048–51. [PubMed] [Google Scholar]
- 20.Fang J, Shing Y, Wiederschain D, et al. Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proc Natl Acad Sci USA. 2000;97:3884–9. doi: 10.1073/pnas.97.8.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chetty C, Bhoopathi P, Joseph P, Chittivelu S, Rao JS, Lakka SS. Adenovirus-mediated siRNA against MMP-2 suppresses tumor growth and lung metastasis in mice. Mol Cancer Ther. 2006;5:2289–99. doi: 10.1158/1535-7163.MCT-06-0169. [DOI] [PubMed] [Google Scholar]
- 22.Chetty C, Bhoopathi P, Lakka SS, Rao JS. MMP-2 siRNA induced Fas/CD95-mediated extrinsic II apoptotic pathway in the A549 lung adenocarcinoma cell line. Oncogene. 2007;26:7675–83. doi: 10.1038/sj.onc.1210584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gondi CS, Lakka SS, Dinh D, Olivero W, Gujrati M, Rao JS. Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biology. 2004;1:165–76. doi: 10.1017/s1740925x04000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leunig M, Yuan F, Menger MD, et al. Angiogenesis, microvascular architecture, microhemodynamics, and interstitial fluid pressure during early growth of human adenocarcinoma LS174T in SCID mice. Cancer Res. 1992;52:6553–60. [PubMed] [Google Scholar]
- 25.Chaturvedi MM, Mukhopadhyay A, Aggarwal BB. Assay for redox-sensitive transcription factor. Methods Enzymol. 2000;319:585–602. doi: 10.1016/s0076-6879(00)19055-x. [DOI] [PubMed] [Google Scholar]
- 26.Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res. 2001;49:568–81. doi: 10.1016/s0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 27.Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3'-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–43. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 28.Dimmeler S, Zeiher AM. Akt takes center stage in angiogenesis signaling. Circ Res. 2000;86:4–5. doi: 10.1161/01.res.86.1.4. [DOI] [PubMed] [Google Scholar]
- 29.Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996;271:21920–6. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- 30.Milanini-Mongiat J, Pouyssegur J, Pages G. Identification of two Sp1 phosphorylation sites for p42/p44 mitogen-activated protein kinases: their implication in vascular endothelial growth factor gene transcription. J Biol Chem. 2002;277:20631–9. doi: 10.1074/jbc.M201753200. [DOI] [PubMed] [Google Scholar]
- 31.Wick M, Haronen R, Mumberg D, et al. Structure of the human TIMP-3 gene and its cell cycle-regulated promoter. Biochem J. 1995;311(Pt 2):549–54. doi: 10.1042/bj3110549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bond M, Murphy G, Bennett MR, Newby AC, Baker AH. Tissue inhibitor of metalloproteinase-3 induces a Fas-associated death domain-dependent type II apoptotic pathway. J Biol Chem. 2002;277:13787–95. doi: 10.1074/jbc.M111507200. [DOI] [PubMed] [Google Scholar]
- 33.Drynda A, Quax PH, Neumann M, et al. Gene transfer of tissue inhibitor of metalloproteinases-3 reverses the inhibitory effects of TNF-alpha on Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. J Immunol. 2005;174:6524–31. doi: 10.4049/jimmunol.174.10.6524. [DOI] [PubMed] [Google Scholar]
- 34.Majid MA, Smith VA, Easty DL, Baker AH, Newby AC. Adenovirus mediated gene delivery of tissue inhibitor of metalloproteinases-3 induces death in retinal pigment epithelial cells. Br J Ophthalmol. 2002;86:97–101. doi: 10.1136/bjo.86.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mannello F, Gazzanelli G. Tissue inhibitors of metalloproteinases and programmed cell death: conundrums, controversies and potential implications. Apoptosis. 2001;6:479–82. doi: 10.1023/a:1012493808790. [DOI] [PubMed] [Google Scholar]
- 36.Shinkaruk S, Bayle M, Lain G, Deleris G. Vascular endothelial cell growth factor (VEGF), an emerging target for cancer chemotherapy. Curr Med Chem Anticancer Agents. 2003;3:95–117. doi: 10.2174/1568011033353452. [DOI] [PubMed] [Google Scholar]
- 37.Yuan A, Yu CJ, Kuo SH, et al. Vascular endothelial growth factor 189 mRNA isoform expression specifically correlates with tumor angiogenesis, patient survival, and postoperative relapse in non-small-cell lung cancer. J Clin Oncol. 2001;19:432–41. doi: 10.1200/JCO.2001.19.2.432. [DOI] [PubMed] [Google Scholar]
- 38.Koukourakis MI, Giatromanolaki A, Thorpe PE, et al. Vascular endothelial growth factor/KDR activated microvessel density versus CD31 standard microvessel density in non-small cell lung cancer. Cancer Res. 2000;60:3088–95. [PubMed] [Google Scholar]
- 39.Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001;12:363–9. [PubMed] [Google Scholar]
- 40.Qureshi HY, Ahmad R, Sylvester J, Zafarullah M. Requirement of phosphatidylinositol 3-kinase/Akt signaling pathway for regulation of tissue inhibitor of metalloproteinases-3 gene expression by TGF-beta in human chondrocytes. Cell Signal. 2007;19:1643–51. doi: 10.1016/j.cellsig.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Ahonen M, Baker AH, Kahari VM. Adenovirus-mediated gene delivery of tissue inhibitor of metalloproteinases-3 inhibits invasion and induces apoptosis in melanoma cells. Cancer Res. 1998;58:2310–5. [PubMed] [Google Scholar]
- 42.Baker AH, Zaltsman AB, George SJ, Newby AC. Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J Clin Invest. 1998;101:1478–87. doi: 10.1172/JCI1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker AH, George SJ, Zaltsman AB, Murphy G, Newby AC. Inhibition of invasion and induction of apoptotic cell death of cancer cell lines by overexpression of TIMP-3. Br J Cancer. 1999;79:1347–55. doi: 10.1038/sj.bjc.6690217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith MR, Kung H, Durum SK, Colburn NH, Sun Y. TIMP-3 induces cell death by stabilizing TNF-alpha receptors on the surface of human colon carcinoma cells. Cytokine. 1997;9:770–80. doi: 10.1006/cyto.1997.0233. [DOI] [PubMed] [Google Scholar]
- 45.Ahonen M, la-Aho R, Baker AH, et al. Antitumor activity and bystander effect of adenovirally delivered tissue inhibitor of metalloproteinases-3. Mol Ther. 2002;5:705–15. doi: 10.1006/mthe.2002.0606. [DOI] [PubMed] [Google Scholar]
- 46.Mino N, Takenaka K, Sonobe M, et al. Expression of tissue inhibitor of metalloproteinase-3 (TIMP-3) and its prognostic significance in resected non-small cell lung cancer. J Surg Oncol. 2007;95:250–7. doi: 10.1002/jso.20663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.