

Abstract
2,3-Disubstituted benzo[b]furans are readily prepared under very mild reaction conditions by the palladium/copper-catalyzed cross-coupling of various o-iodoanisoles and terminal alkynes, followed by electrophilic cyclization using I2, PhSeCl or p-O2NC6H4SCl. Aryl- and vinylic-substituted alkynes undergo electrophilic cyclization in excellent yields. Biologically important furopyridines can be prepared by this approach in high yields.
Introduction
The benzo[b]furan nucleus is prevalent in a wide variety of biologically active natural and unnatural compounds.1 Many 2-arylbenzofuran derivatives are well known to exhibit a broad range of biological activities, including anticancer,2 antiproliferative,3 antiviral,4 antifungal,5 immunosuppressive,6 antiplatelet,7 antioxidative,8 insecticidal,9 anti-inflammatory,10 antifeedant,11 and cancer preventative activity.12 These compounds are also important calcium blockers13 and phytoestrogens.14 For instance, XH-1415 was the first reported potent nonnucleoside adenosine A1 agonist16 and obovaten is known as an active antitumor agent.17

There has been growing interest in developing a general and versatile synthesis of benzo[b]furan derivatives. A number of synthetic approaches to this class of compounds have been introduced in recent years.18 One common approach to heterocycles that has been utilized for the synthesis of benzo[b]furans,19 benzo[b]thiophenes,20 indoles21 and isoquinolines22 has been electrophilic cyclization of the corresponding 2-(1-alkynyl)-phenols, -thioanisoles, -anilines and -imines respectively (Scheme 1).
Scheme 1.
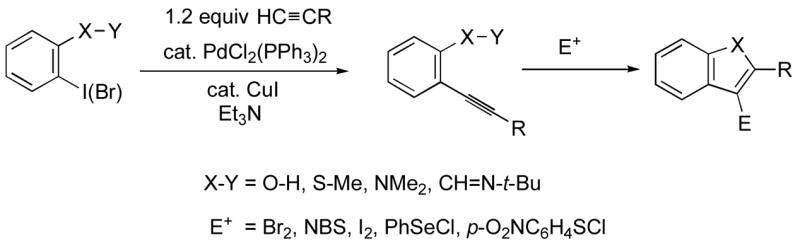
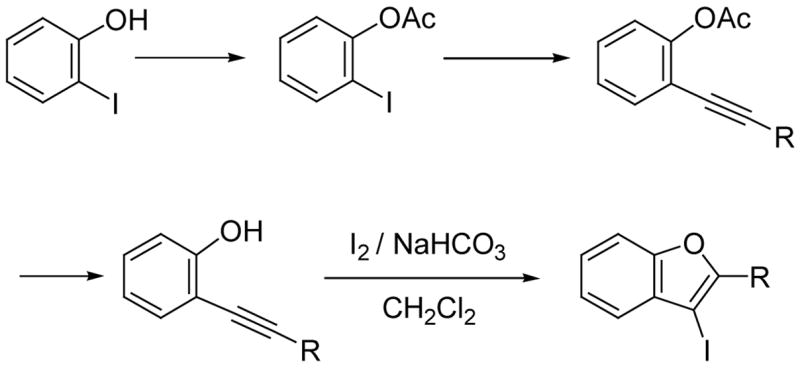
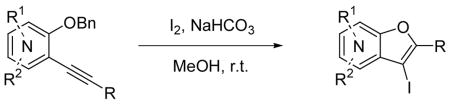
Our and other’s recent success in this area encouraged us to examine the possibility of preparing benzo[b]furans by the same strategy involving a palladium/copper-catalyzed alkyne coupling, followed by electrophilic cyclization. Cacchi and co-workers have previously reported an approach to the synthesis of 3-iodobenzo[b]furans by a related process involving iodocyclization (Scheme 2).19 Unfortunately, the protecting and deprotecting steps required to synthesize the alkynylphenol are not particularly attractive synthetically. Some of the alkynylphenols are also relatively unstable. In another paper, Cacchi has demonstrated an analogous cyclization of benzylic ethers to generate furopyridines and reported that the o-hydroxyalkynylpyridines are not stable and cyclize spontaneously to give furopyridines (eq 1).23 We have attempted to make this overall approach more attractive synthetically by examining the preparation and cyclization of the corresponding methyl ethers using a variety of commercially available electrophiles. Herein, we wish to report an efficient approach to 2,3-disubstituted benzo[b]furans and furopyridines involving the palladium/copper-catalyzed coupling of various iodoanisoles and an iodomethoxypyridine and terminal alkynes, followed by electrophilic cyclization.
Scheme 2.
 |
(1) |
Results and Discussion
The arylalkynes required for our approach to benzo[b]furans are readily prepared by the Sonogashira coupling24 of commercially available o-iodoanisole (5.0 mmol) and terminal alkynes (6.0 mmol) using a catalyst consisting of 2 mol % PdCl2(PPh3)2 and 1 mol % CuI in the presence of Et3N (12.5 mL) as the solvent at room temperature. The yields of this process range from 70 to 94% and this procedure should readily accommodate considerable functionality.
We first examined the reaction of our methoxy-substituted aryl alkynes (0.25 mmol in 3 mL of CH2Cl2) with I2 (2.0 equiv in 2 mL of CH2Cl2) under our well established reaction conditions for the synthesis of benzo[b]thiophenes20 and indoles21 (Scheme 1). We were pleased to see that 2-(phenylethynyl)anisole reacted in less than 3 h at room temperature to afford 3-iodo-2-phenylbenzo[b]furan in an 87% yield (Table 1, entry 1). In order to extend this approach to other benzo[b]furans, we have also looked at a range of other readily available electrophiles. So far p-O2NC6H4SCl and PhSeCl have been successfully employed in this electrophilic cyclization, providing excellent yields of the desired cyclization products (Table 1, entries 2–4).
Table 1.
Synthesis of benzo[b]furans by electrophilic cyclization.a
| entry | alkyne | electrophile | product | % yield |
|---|---|---|---|---|
| 1 |
1 |
I2 |
2 |
87 |
| 2 | 1 | ICl | 2 | 98 |
| 3 | 1 | PhSeCl |
3 |
100 |
| 4 | 1 | p-O2NC6H4SCl |
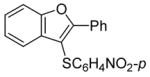
4 |
85 |
| 5 |
5 |
I2 |
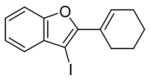
6 |
80 |
| 6 | 5 | PhSeCl |
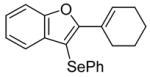
7 |
88 |
| 7 |
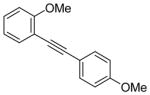
8 |
I2 |
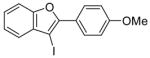
9 |
100 |
| 8 | 8 | PhSeCl |
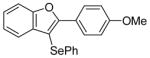
10 |
96 |
| 9 |
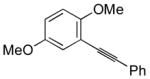
11 |
I2 |
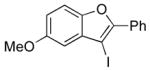
12 |
74 |
| 10 | 11 | PhSeCl |
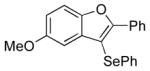
13 |
90 |
| 11 |
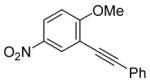
14 |
I2 |
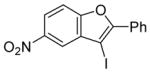
15 |
80b |
| 12 |
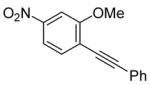
16 |
I2 |
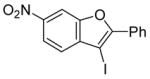
17 |
67b |
| 13 |
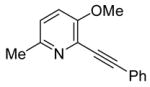
18 |
I2 |
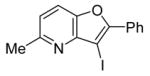
19 |
67b |
| 14 |
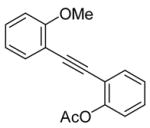
20 |
I2 |
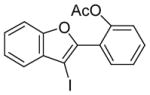
21 |
95 |
| 15 |
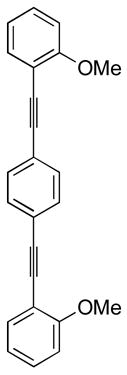
22 |
I2 |
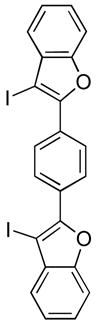
23 |
97 |
| 16 |
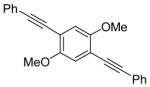
24 |
I2 |
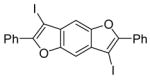
25 |
60 |
| 17 |
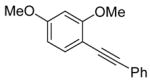
26 |
I2 |
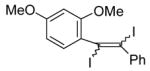
|
66c |
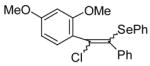
| 18 | 26 | PhSeCl |
|
80c |
| 19 |
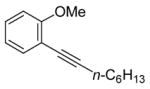
27 |
I2 |
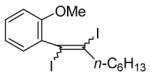
|
98c |
| 20 | 27 | PhSeCl | -c | |
| 21 |
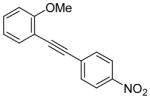
28 |
I2 |
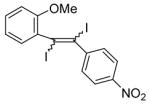
|
-d |
| 22 | 28 | PhSeCl | -d | |
| 23 |
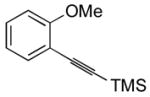
29 |
I2 | -d | |
| 24 |
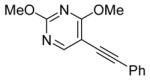
30 |
I2 |
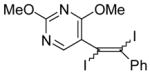
31 |
68 |
| 25 |
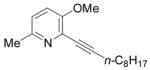
32 |
I2 |
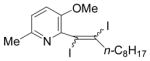
33 |
66 |
All reactions were run with 0.25 mmol of the alkyne, 2 equiv of electrophile in 5 mL of CH2Cl2 at 25 °C for 3 h.
The reaction took 12 h.
None of the desired cyclization product was observed.
An inseparable mixture was obtained.
The nature of the substituents attached to the triple bond and the arene have a major impact on the success of the reaction. Virtually no difference in the rates of reaction or the overall yields have been observed using a vinylic alkyne and arylalkynes bearing certain types of functionality on the aromatic ring (entries 1–16). However, alkynes bearing an alkyl group (entries 19, 20 and 25) fail to undergo electrophilic cyclization. Instead, an almost quantitative yield of the product of simple addition of the electrophile to the alkyne triple bond was obtained (entries 19, 20 and 25). In order to form a furan moiety, the oxygen of the methoxy group has to undergo a five endo-dig attack on the carbon-carbon triple bond. The failure to undergo the desired cyclization in entries 19, 20, 21, 22 and 25 is possibly due to more favorable formation of a “vinylic” cation on the carbon of the triple bond next to the aryl group bearing the methoxy group, instead of on the more remote carbon (see the later mechanistic discussion). Similarly, a methoxy group para to the triple bond increases the electron density on the distal end of the triple bond (Figure 1), which favors electrophilic attack at that position and disfavors a five endo-dig cyclization. This, in turn, leads to addition of the electrophile to the triple bond, rather than cyclization (entries 17, 18 and 24).
Figure 1.
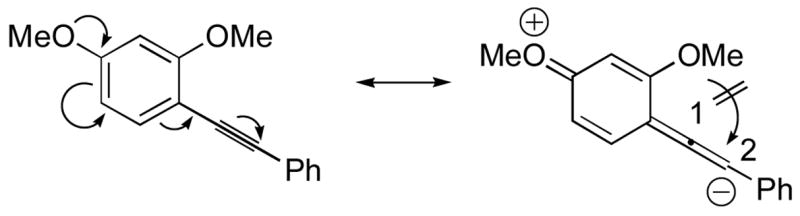
Methoxy electron-donating effect on the triple bond
The presence of a nitro group in compound 28 decreases the electron density on C1 (Figure 2), and again favors simple addition of the electrophile to the alkyne triple bond (entries 21 and 22). On the other hand, the presence of an electron-withdrawing group, such as a nitro group on the methoxy-substituted arene favors electrophilic cyclization, although a longer reaction time (12 h) is generally required (entries 11 and 12). The more electron deficient C2 position is more likely to undergo attack by the nucleophilic oxygen of the methoxy group than the C1 position, because of either the resonance effect of the nitro group para to the carbon-carbon triple bond (Figure 3) or the inductive effect of the electron-poor arene. The trimethylsilyl-substituted alkyne 29 also failed to undergo electrophilic cyclization, providing instead an inseparable mixture of unidentifiable compounds, which is consistent with Cacchi’s earlier results (entry 23).19
Figure 2.
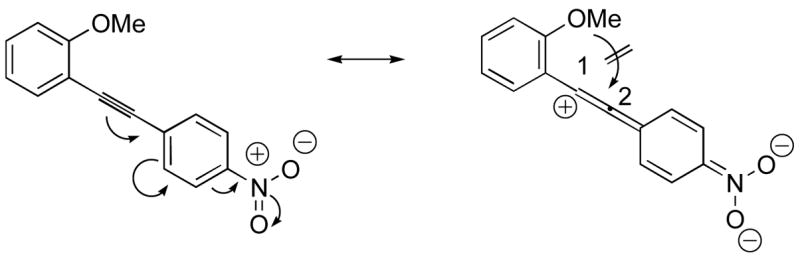
Nitro electron-withdrawing effect on the triple bond
Figure 3.
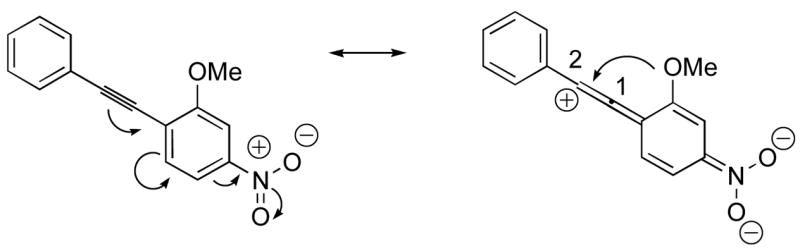
Nitro electron-withdrawing effect on the triple bond
Compound 20 with both a methoxy and an acetoxy group in positions ortho to the triple bond undergoes exclusive electrophilic cyclization onto the methoxy group to produce compound 21 in a 95% yield (entry 14). This should be quite useful for the regioselective synthesis of benzofurans. Thus, the more nucleophilic methoxy group more readily attacks the triple bond, affording the corresponding cyclization product.
We have also investigated the possibility of carrying out double iodocyclizations, which might be quite useful for the quick assembly of systems with extended conjugation. Compounds 22 and 24 undergo iodocyclization to afford double cyclization products in 97% and 60% yields respectively (entries 15 and 16).
While Cacchi reported the successful synthesis of several furopyridines by electrophilic cyclization of o-(benzyloxy)alkynylpyridines, we have examined the cyclization of an o-methoxyalkynylpyridine and found that a methyl group can also be a good leaving group in this reaction. Pyridine derivative 18 was treated with I2 under our standard electrophilic cyclization conditions to afford the desired furopyridine in a 67 % yield. This provides a convenient alternative route to the synthesis of furopyridines, since methoxypyridines are more readily available than (benzyoxy)pyridines in many cases.
Mechanistically, we believe that these cyclizations proceed by anti attack of the electrophile and the oxygen of the methoxy group on the alkyne to produce an intermediate A, which undergoes methyl group removal via SN2 displacement by nucleophiles present in the reaction mixture (Scheme 3). In most cases, the nucleophile is presumably the halide remaining in solution.
Scheme 3.
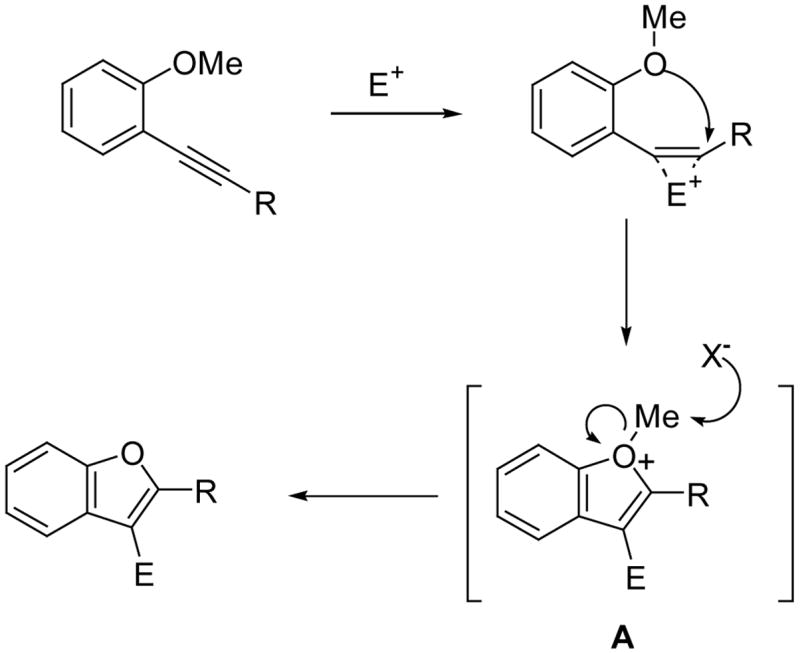
Conclusions
We believe that this approach to 3-iodobenzo[b]furans and furopyridines should prove quite useful in synthesis, particularly when one considers that there are many ways to transform the resulting iodide functionality into other substituents. For example, the resulting heterocyclic iodides should be particularly useful as intermediates in many palladium-catalyzed processes, like Sonogashira,24 Suzuki,25 and Heck26 cross-coupling processes.
Experimental Section
General
1H and 13C NMR spectra were recorded at 300 and 75.5 MHz or 400 and 100 MHz respectively. Thin layer chromatography was performed using commercially prepared 60-mesh silica gel plates (Whatman K6F), and visualization was effected with short wavelength UV light (254 nm) or a basic KMnO4 solution [3 g of KMnO4 + 20 g of K2CO3 + 5 mL of NaOH (5 %) + 300 mL of H2O]. All melting points are uncorrected. Low resolution mass spectra were recorded on a Finnigan TSQ700 triple quadrupole mass spectrometer (Finnigan MAT, San Jose, CA). High resolution mass spectra were recorded on a Kratos MS50TC double focusing magnetic sector mass spectrometer using EI at 70 eV.
Reagents
All reagents were used directly as obtained commercially unless otherwise noted. Anhydrous forms of ethyl ether, hexanes, ethyl acetate, and CH2Cl2 were purchased from Fisher Scientific Co. 2-Iodoanisole, 2-bromo-1,4-dimethoxybenzene, 1-bromo-2,4-dimethoxybenzene, 2-iodo-4-nitroanisole, 2-iodo-5-nitroanisole, resorcinol, 4-iodoanisole, 1-iodo-4-nitrobenzene, phenylacetylene, 1-cyclohexenyl acetylene, 1-octyne, trimethylsilylacetylene, and Et3N were purchased from Aldrich Chemical Co., Inc. The palladium salts were donated by Johnson Matthey Inc. and Kawaken Fine Chemicals Co. Ltd.
General procedure for the palladium/copper-catalyzed formation of o-(1-alkynyl)anisoles
To a solution of Et3N (12.5 mL), PdCl2(PPh3)2 (2 mol %), 5.0 mmol of o-iodoanisole and 6.0 mol of terminal acetylene (stirring for 5 min beforehand), CuI (1 mol %) was added and stirring was continued for another 2 min before flushing with Ar. The flask was then sealed. The mixture was allowed to stir at room temperature for 3–6 h and the resulting solution was filtered, washed with satd aq NaCl and extracted with diethyl ether (2 × 15 mL). The combined ether fractions were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product. The crude product was purified by flash chromatography on silica gel using ethyl acetate/hexane as the eluent.
2-(Phenylethynyl)anisole (1)
The product was obtained as a yellow oil: 1H NMR (CDCl3) δ 3.90 (s, 3H), 6.89 (d, J = 8.4 Hz, 1H), 6.93 (t, J = 7.6 Hz, 1H), 7.29–7.33 (m, 4H), 7.50 (d, J = 7.2 Hz, 1H), 7.55–7.57 (m, 2H); 13C NMR (CDCl3) δ 56.0, 85.9, 93.6, 110.9, 112.6, 120.7, 123.7, 128.2, 128.3, 129.9, 131.8, 133.7, 160.1; IR (neat, cm−1) 3058, 2926, 2855, 2226; HRMS calcd for C15H12O 208.0888, found 208.0894.
General procedure for the iodocyclizations
To a solution of 0.25 mmol of the alkyne and 3 mL of CH2Cl2, 2 equiv of I2 dissolved in 2 mL of CH2Cl2 was added gradually. The reaction mixture was flushed with Ar and allowed to stir at room temperature for the desired time. The excess I2 was removed by washing with satd aq Na2S2O3. The mixture was then extracted by diethyl ether (2 × 10 mL). The combined ether layers were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product, which was purified by flash chromatography on silica gel using ethyl acetate/hexanes as the eluent.
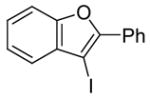
3-Iodo-2-phenylbenzo[b]furan (2)
The product was obtained as a yellow oil: 1H NMR (CDCl3) δ 7.28–7.51 (m, 7H), 8.16–8.19 (m, 2H); 13C NMR (CDCl3) δ 61.3, 111.4, 122.1, 123.7, 125.9, 127.7, 128.7, 129.4, 130.2, 132.7, 153.2, 154.1; IR (neat, cm−1) 3058, 1450; HRMS calcd for C14H9IO 319.9698, found 319.9700.
General procedure for the p-O2NC6H4SCl and PhSeCl cyclizations
To a solution of 0.25 mmol of the alkyne and CH2Cl2 (5 mL), 0.375 mmol of p-O2NC6H4SCl or PhSeCl was added. The mixture was flushed with Ar and allowed to stir at 25 °C for 2–6 h. The reaction mixture was washed with 20 mL of water and extracted with diethyl ether. The combined ether layers were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product, which was further purified by flash chromatography on silica gel using ethyl acetate/hexanes as the eluent.
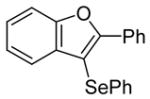
2-Phenyl-3-(phenylselenyl)benzo[b]furan (3)
The product was obtained as a yellow oil: 1H NMR (CDCl3) δ 7.13–7.17 (m, 3H), 7.22 (t, J = 4.0 Hz, 1H), 7.27–7.32 (m, 4H), 7.43 (t, J = 7.6 Hz, 2H), 7.51 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 8.19–8.21 (m, 2H); 13C NMR (CDCl3) δ 99.9, 111.4, 121.4, 123.6, 125.4, 126.4, 128.0, 128.7, 129.4, 129.5, 130.3, 131.6, 132.1, 154.3, 157.4; IR (neat, cm−1) 3058, 2926; HRMS calcd for C20H14OSe 350.0211, found 350.0220.
Supplementary Material
Acknowledgments
We thank the National Institute of General Medical Sciences (GM070620) for support of this research and Johnson Matthey, Inc., and Kawaken Fine Chemicals Co., Ltd., for donations of palladium salts.
References
- 1.(a) Dean FM. In: The Total Synthesis of Natural Products. ApSimon J, editor. Vol. 1. Wiley; New York: 1973. pp. 467–562. [Google Scholar]; (b) Cagniant P, Cagniant D. In: Advances in Heterocyclic Chemistry. Katritzky AR, Boulton AJ, editors. Vol. 18. Academic Press; New York: 1975. pp. 337–482. [Google Scholar]; (c) Dean FM, Sargent MV. In: Comprehensive Heterocyclic Chemistry. Katritzky AR, Rees CW, editors. Vol. 4. Pergamon Press; England: 1984. pp. 531–596. [Google Scholar]; (d) Katritzky AR, Rees CW, Scriven EFV, editors. Comprehensive Heterocyclic Chemistry II. Vol. 2. Pergamon Press; Oxford: 1996. pp. 259–321. [Google Scholar]
- 2.(a) Navarro E, Alonso SJ, Trujillo J, Jorge E, Perez C. J Nat Prod. 2001;64:134. doi: 10.1021/np9904861. [DOI] [PubMed] [Google Scholar]; b Lambert JD, Meyers RO, Timmermann BN, Dorr RT. Cancer Lett. 2001;171:47. doi: 10.1016/s0304-3835(01)00560-2. [DOI] [PubMed] [Google Scholar]; c Takasaki MT, Komatsu K, Tokuda H, Nishino H. Cancer Lett. 2000;158:53. doi: 10.1016/s0304-3835(00)00499-7. [DOI] [PubMed] [Google Scholar]; d Banskota A, Tezuka Y, Midorikawa K, Matsushige K, Kadota S. J Nat Prod. 2000;63:1277. doi: 10.1021/np000143z. [DOI] [PubMed] [Google Scholar]; e Lee SK, Cui B, Mehta RR, Kinghorn AD, Pezzuto JM. Chem-Bio Interact. 1998;115:215. doi: 10.1016/s0009-2797(98)00073-8. [DOI] [PubMed] [Google Scholar]; f Thompson LU, Rickard SE, Orcheson LJ, Seidl MM. Carcinogenesis. 1996;17:1373. doi: 10.1093/carcin/17.6.1373. [DOI] [PubMed] [Google Scholar]; g Thompson LU, Seidl MM, Rickard SE, Orcheson LJ, Fong HH. Nutr Cancer. 1996;26:159. doi: 10.1080/01635589609514472. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda R, Nagao T, Okabe H, Nakano Y, Matsunaga H, Katano M, Mori M. Chem Pharm Bull. 1998;46:871. doi: 10.1248/cpb.46.871. [DOI] [PubMed] [Google Scholar]
- 4.(a) Craigo J, Callahan M, Huang RCC, DeLucia AL. Antiviral Res. 2000;47:19. doi: 10.1016/s0166-3542(00)00089-9. [DOI] [PubMed] [Google Scholar]; b Leung C, Charlton JL, Cow C. Can J Chem. 2000;78:553. [Google Scholar]; c Charlton JL. J Nat Prod. 1998;61:1447. doi: 10.1021/np980136z. [DOI] [PubMed] [Google Scholar]
- 5.(a) Carter GA, Chamberlain K, Wain RL. Ann Appl Biol. 1978;88:57. doi: 10.1111/j.1744-7348.1975.tb01621.x. [DOI] [PubMed] [Google Scholar]; b Zacchino S, Rodriguez G, Pezzenati G, Orellana G, Enriz R, Gonzalez SM. J Nat Prod. 1997;60:659. doi: 10.1021/np9605504. [DOI] [PubMed] [Google Scholar]
- 6.(a) Gordaliza M, Castro M, del Corral JM, Lopez-Vazquez M, Feliciano AS, Faircloth GT. Bioorg Med Chem Lett. 1997;7:2781. [Google Scholar]; b Cho JY, Kim AR, Yoo ES, Baik KU, Park MH. J Pharm Pharmacol. 1999;51:1267. doi: 10.1211/0022357991777001. [DOI] [PubMed] [Google Scholar]
- 7.Chen CC, Hsin WC, Ko FN, Huang YL, Ou JC, Teng CM. J Nat Prod. 1996;59:1149. doi: 10.1021/np960443+. [DOI] [PubMed] [Google Scholar]
- 8.(a) Masuda S, Hasuda H, Tokoroyama T. Chem Pharm Bull. 1994;42:2500. doi: 10.1248/cpb.42.2500. [DOI] [PubMed] [Google Scholar]; b Lu H, Liu GT. Planta Med. 1992;58:311. doi: 10.1055/s-2006-961473. [DOI] [PubMed] [Google Scholar]; c Silva DHS, Pereira FC, Zanoni MVB, Yoshida M. Phytochemistry. 2001;57:437. doi: 10.1016/s0031-9422(00)00477-5. [DOI] [PubMed] [Google Scholar]
- 9.(a) Findlay JA, Buthelezi S, Li G, Seveck M, Miller JD. J Nat Prod. 1997;60:1214. [Google Scholar]; b Brader G, Greger H, Bacher M, Kalchhauser H, Hofer O, Vajrodaya S. J Nat Prod. 1998;61:1482. doi: 10.1021/np9801965. [DOI] [PubMed] [Google Scholar]
- 10.(a) Day SH, Chiu NY, Tsao LT, Wang JP, Lin CN. J Nat Prod. 2000;63:1560. doi: 10.1021/np000191j. [DOI] [PubMed] [Google Scholar]; b Borsato MLC, Grael CFF, Souza GEP, Lopes NP. Phytochemistry. 2000;55:809. doi: 10.1016/s0031-9422(00)00388-5. [DOI] [PubMed] [Google Scholar]
- 11.Ward RS. Nat Prod Rep. 1995;12:183. doi: 10.1039/np9951200183. [DOI] [PubMed] [Google Scholar]
- 12.(a) den Tonkelaar I, Keinan-Boker L, Van’t Veer P, Arts CJM, Adlercreutz H, Thijssen JHH, Peeters PHM. Cancer Epidemiol Biomark Prev. 2001;10:223. [PubMed] [Google Scholar]; b Knekt P, Adlercreutz H, Rissanen H, Aromaa A, Teppo L, Heliovaara M. Br J Cancer. 2000;82:1107. doi: 10.1054/bjoc.1999.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hirose M, Lin C, Kimoto N, Futakuchi M, Kono T, Nishibe S, Shirai T, Yamaguchi T. Cancer Lett. 2000;155:79. doi: 10.1016/s0304-3835(00)00411-0. [DOI] [PubMed] [Google Scholar]; d Oikannen SI, Pajari AM, Mutanen M. Cancer Lett. 2000;159:183. doi: 10.1016/s0304-3835(00)00543-7. [DOI] [PubMed] [Google Scholar]
- 13.Gubin J, de Vogelaer H, Inion H, Houben C, Lucchetti J, Mahaux J, Rosseels G, Peiren M, Polstre P, Chatelain P, Clinet M. J Med Chem. 1993;36:1425. doi: 10.1021/jm00062a015. [DOI] [PubMed] [Google Scholar]
- 14.Pietinen P, Stumpf K, Mannisto S, Kataja V, Uusitupa M, Adlercreutz H. Cancer Epidemiol Biomark Prev. 2001;10:339. [PubMed] [Google Scholar]
- 15.Chang HM, Cheng KP, Choang TF, Chow HF, Chui KY, Hon PM, Tan FWL, Yang Y, Zhong ZP, Lee CM, Sham HL, Chan CF, Cui YX, Wong HNC. J Org Chem. 1990;55:3537. [Google Scholar]
- 16.Yang Z, Hon PM, Chui KY, Xu ZL, Chang HM, Lee CM, Cui YX, Wong HNC, Poon CD, Fung BM. Tetrahedron Lett. 1991;32:2061. [Google Scholar]
- 17.Tsai IL, Hsieh CF, Duh CY. Phytochemistry. 1998;48:1371. doi: 10.1016/s0031-9422(97)00948-5. [DOI] [PubMed] [Google Scholar]
- 18.(a) Horaguchi T, Iwanami H, Tanaka T, Hasegawa E, Shimizu T, Suzuki T, Tanemura K. J Chem Soc, Chem Commun. 1991:44. [Google Scholar]; b Horaguchi T, Kobayashi H, Miyazawa K, Hasegawa E, Shimizu T. J Heterocycl Chem. 1990;27:935. [Google Scholar]; c Boehm TL, Showalter HDH. J Org Chem. 1996;61:6498. doi: 10.1021/jo961242d. [DOI] [PubMed] [Google Scholar]; d Nicolaou KC, Snyder SA, Bigot A, Pfefferkorn JA. Angew Chem, Int Ed Engl. 2000;39:1093. [PubMed] [Google Scholar]; e Kondo Y, Shiga F, Murata N, Sakamoto T, Yamanaka H. Tetrahedron. 1994;50:11803. [Google Scholar]; f Arcadi A, Cacchi S, Rosario MD, Fabrizi G, Marinelli F. J Org Chem. 1996;61:9280. [Google Scholar]; g Cacchi S, Fabrizi G, Moro L. Tetrahedron Lett. 1998;39:5101. [Google Scholar]; h Monteiro N, Arnold A, Balme G. Synlett. 1998:1111. [Google Scholar]; i Kraus GA, Zhang N, Verkade JG, Nagarajan M, Kisanga PB. Org Lett. 2000;2:2409. doi: 10.1021/ol0000758. [DOI] [PubMed] [Google Scholar]; j Katritzky AR, Ji Y, Fang Y, Prakash I. J Org Chem. 2001;66:5613. doi: 10.1021/jo010278p. [DOI] [PubMed] [Google Scholar]; k Kao CL, Chern JW. J Org Chem. 2002;67:6772. doi: 10.1021/jo0258960. [DOI] [PubMed] [Google Scholar]
- 19.Arcadi A, Cacchi S, Fabrizi G, Marinelli F, Moro L. Synlett. 1999:1432. [Google Scholar]
- 20.(a) Yue D, Larock RC. J Org Chem. 2002;67:1905. doi: 10.1021/jo011016q. [DOI] [PubMed] [Google Scholar]; b Flynn BL, Verdier-Pinard P, Hamel E. Org Lett. 2001;3:651. doi: 10.1021/ol0067179. [DOI] [PubMed] [Google Scholar]
- 21.(a) Barluenga J, Trincado M, Rubio E, Gonzalez JM. Angew Chem, Int Ed. 2003;42:2406. doi: 10.1002/anie.200351303. [DOI] [PubMed] [Google Scholar]; b Yue D, Larock RC. Org Lett. 2004;6:1037. doi: 10.1021/ol0498996. [DOI] [PubMed] [Google Scholar]; c Amjad M, Knight DW. Tetrahedron Lett. 2004;45:539. [Google Scholar]
- 22.Huang Q, Hunter JA, Larock RC. J Org Chem. 2002;67:3437. doi: 10.1021/jo020020e. [DOI] [PubMed] [Google Scholar]
- 23.Arcadi A, Cacchi S, di Giuseppe S, Fabrizi G, Marinelli F. Org Lett. 2002;4:2409. doi: 10.1021/ol0261581. [DOI] [PubMed] [Google Scholar]
- 24.(a) Sonogashira K. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Wiley-VCH; Weinheim: 1998. pp. 203–229. [Google Scholar]; (b) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467. [Google Scholar]
- 25.(a) Suzuki A. J Organomet Chem. 1999;576:147. [Google Scholar]; b Lloyd-Williams P, Giralt E. Chem Soc Rev. 2001;30:145. [Google Scholar]; (c) Suzuki A. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Wiley-VCH; Weinheim: 1998. pp. 49–97. [Google Scholar]; (d) Suzuki S, Miyaura N. Chem Rev. 1995;95:2457. [Google Scholar]
- 26.(a) Crisp GT. Chem Soc Rev. 1998;27:427. [Google Scholar]; (b) Brase S, de Meijere A. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Wiley-VCH; Weinheim: 1998. pp. 99–166. [Google Scholar]; (c) Heck RF. Palladium Reagents in Organic Synthesis. Academic Press; San Diego: 1985. pp. 276–287. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.