

Abstract
We studied allosteric potentiation of rat α3β2 neuronal nicotinic acetylcholine receptors (nAChRs) by the anthelmintic compound morantel. Macroscopic currents evoked by acetylcholine (ACh) from nAChRs expressed in Xenopus laevis oocytes increase up to 8-fold in the presence of low concentrations of morantel (≤10 μM); the magnitude of the potentiation depends on both agonist and modulator concentrations. It is noteworthy that the potentiated currents exceed the maximum currents achieved by saturating (millimolar) concentrations of agonist. Studies of macroscopic currents elicited by prolonged drug applications (100–300 s) indicate that morantel does not increase α3β2 receptor activity by reducing slow (≥1 s) desensitization. Instead, using outside-out patch-clamp recordings, we demonstrate that morantel increases the frequency of single-channel openings and alters the bursting characteristics of the openings in a manner consistent with enhanced channel gating; these results quantitatively explain the macroscopic current potentiation. Morantel is a very weak agonist alone, but we show that the classic competitive antagonist dihydro-β-erythroidine inhibits morantel-evoked currents noncompetitively, indicating that morantel does not bind to the canonical ACh binding sites.
nAChRs mediate rapid synaptic transmission throughout the central and peripheral nervous systems and are implicated in a wide range of important pathologic conditions, the most pervasive of which is nicotine addiction (e.g., Berrettini and Lerman, 2005). With recent advances in high-resolution structure determination (Brejc et al., 2001; Unwin, 2005), and a clearer understanding of the agonist binding sites (for review, see Sine, 2002) and agonist-mediated conformational changes (e.g., Lyford et al., 2003; Mitra et al., 2004; Lee and Sine, 2005), the prospect of rational drug design applied to nAChRs is becoming a distinct possibility.
The anthelmintic compounds levamisole, pyrantel, and morantel are full agonists of nAChRs in lower species such as nematodes, where they are used widely to clear parasitic infections in livestock (Martin, 1997). However, for mammalian nAChRs, levamisole and pyrantel are generally poor agonists alone (e.g., Rayes et al., 2004) but allosterically potentiate responses when coapplied with agonist (Levandoski et al., 2003).
A small number of compounds have been shown to potentiate neuronal nicotinic receptors from mammalian species. In addition to the aforementioned anthelmintics, these include curare (Cachelin and Rust, 1994), choline (Zwart and Vijverberg, 2000), and novel compounds such as PNU-120596 (Hurst et al., 2005) and various (2-amino-5-keto) thiazoles (Broad et al., 2006). More extensive work characterizing potentiation has focused on anticholinesterases, primarily physostigmine and galantamine (Pereira et al., 1994; Schrattenholz et al., 1996; Zwart et al., 2000). The diversity of modulator structures and nAChR subtypes represented in these studies suggest allosteric potentiation is a general phenomenon of nicotinic receptors.
However, mechanisms underlying allosteric potentiation and the binding sites involved remain poorly understood. Instead, studies have focused largely on finding new modulatory compounds with subtype specificity (Hurst et al., 2005; Broad et al., 2006). In fact, in the most-studied case of the anticholinesterases, fundamental disagreements remain: Vijverberg and coworkers suggest that these compounds potentiate competitively [i.e., by binding at the canonical acetylcholine (ACh) binding site] (Zwart et al., 2000; Smulders et al., 2005). In contrast, other groups suggest that these compounds are noncompetitive because physostigmine- and galantamine-evoked single-channel currents are not blocked by the competitive antagonists dihydro-β-erythroidine (DHβE), methyllycaconitine, or α-bungarotoxin (Pereira et al., 1993, 1994; Akk and Steinbach, 2005). Given the use of anticholinesterases to improve cognitive function (e.g., Ferreri et al., 2006), and the potential for developing allosteric modulators as a new class of therapeutic drugs, more work is needed to understand the mechanisms of action and binding sites of this important class of compounds.
We report here studies of the potentiation mechanism of the anthelmintic morantel, elucidated through combined macroscopic and single-channel current recordings. Morantel alone is a very weak agonist of rat nAChRs but substantially potentiates agonist-evoked responses in the low micromolar concentration range. We initially postulated that macroscopic potentiation could be explained by changes in either desensitization kinetics or channel gating. Our results distinguish between these two possibilities and show that morantel potentiates by enhancing channel gating through a binding site distinct from the canonical agonist binding site.
Materials and Methods
Chemicals and Rat Subunit Constructs
All reagents used, unless otherwise noted, were reagent grade and obtained from Sigma (St. Louis, MO). Morantel (Mor), shown as the inset of Fig. 1, is 1,4,5,6-tetrahydro-1-methyl-2-(2-[3-methyl-2-thienyl]ethenyl)pyrimidine, tartrate salt. pGEMHE-based vectors bearing the cDNA for rat α3 and β2 subunits were obtained from C. W. Luetje (University of Miami, Miami, FL) and were prepared using standard procedures (Levandoski et al., 2003). cRNA transcripts were made using the T7 mMESSAGE mMACHINE kit from Ambion (Austin, TX).
Fig. 1.
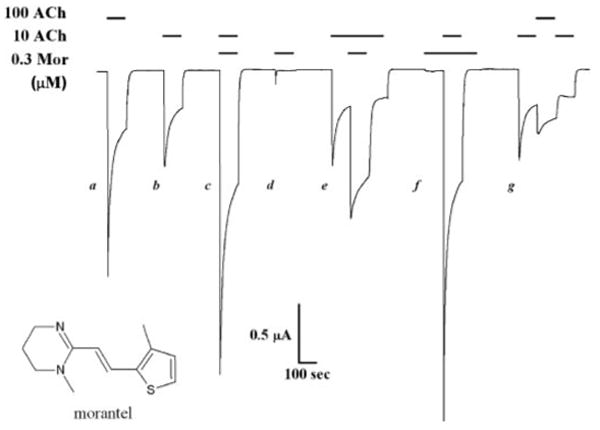
Morantel does not affect desensitization. A sample continuous current trace for an oocyte expressing α3β2 receptors is shown. Drug applications were for 100 or 300 s when either ACh or morantel was used to “presaturate,” in which case the coapplication was begun after 100 s. The holding potential was −60 mV.
Oocyte Preparation and Injections
Whole ovaries from Xenopus laevis frogs were obtained from Nasco (Ft. Atkinson, WI). Oocytes were prepared from the ovaries as described in Levandoski et al. (2003). After collagenase treatment to remove the follicular cell layer, healthy stage V to VI oocytes were manually selected and maintained at 16°C in Barth medium (88 mM NaCl, 1.0 mM KCl, 2.5 mM NaHCO3, 0.3 mM Ca(NO3)2, 0.41 mM CaCl2, 0.82 mM MgSO4, 15 mM HEPES, and 2.5 mM sodium pyruvate, pH 7.6) supplemented with 100 units/ml penicillin/streptomycin (Invitrogen, Carlsbad, CA), and 50 μg/ml gentamicin (Lonza Walkersville, Inc., Walkersville, MD). Oocytes were injected with 23 ng of total cRNA in 46 nl of solution consisting of an equimolar ratio of both subunits using a Drummond Nanoject and maintained in the Barth antibiotic medium for 2 to 4 days before recording. For most of the single-channel experiments, 6 to 8 ng of cRNA was injected to achieve a lower receptor density. Throughout the text, the number of “donors” refers to the number of oocyte batches prepared from separate frogs.
Electrophysiological Recordings and Analysis
Macroscopic Currents
Drug-evoked currents were measured from injected oocytes using the two-electrode voltage clamp method with a GeneClamp 500B amplifier (Axon Instruments, Foster City, CA). Unless otherwise noted, the membrane potential was held at −60 mV. Oocytes were perfused with oocyte Ringer's medium (OR2; 115 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, and 10 mM HEPES, pH 7.3). Concentrated stocks of the various drugs (in water) were diluted directly in OR2. Recordings were performed in a RC-1Z chamber (Warner Instruments, Hamden, CT) with an incubation volume of ∼300 μl and a gravity perfusion flow of 5 to 10 ml/min. The flow of the various drug solutions was controlled using solenoid valves driven by Warner VC-6 valve controllers. Electrodes of resistance 0.5 to 4.0 MΩ were filled with 3 M KCl. Data were acquired on a personal computer using pClamp 9 software, through an Digidata 1322A (Axon Instruments) analog/digital converter.
Measurements of current response for each drug concentration were made one to three times on each oocyte. Applications were typically 5 s, but 10-, 30-, and 100-s applications were also used. After each application, 100- or 200-s washout by continuous perfusion ensured baseline current for at least 30 s before the next application. Most experiments included applications of 100 μM ACh and 100 μM ACh + 10 μM Mor for standardization purposes. No appreciable rundown of the response was observed. Repeat measurements from the same oocyte were averaged; the normalization used depended on the experiment and is described in the figure legends. Data are reported as means ± S.E.M.
Single-Channel Currents
Cell-attached and outside-out patch recordings from oocytes were made generally according to the methods of Cooper et al. (1996). The vitelline membrane was removed with fine forceps after incubation for ∼10 min in the standard hypertonic solution. Oocytes were bathed in OR2 for recording. For cell-attached patches, the pipette contained drugs dissolved in the bath solution from concentrated stocks made up in water. For outside-out patch recordings, the pipette was filled with internal solution 80 mM potassium fluoride, 20 mM KCl, 10 mM HEPES, and 10 mM EGTA, adjusted to pH 7.2. Oocytes were placed in 35-mm dishes containing 3.0 ml of bath solution. After forming stable outside-out patches and establishing a membrane potential of −60 mV, recordings for 3 to 4 min in the absence of agonist served as a negative control. An aliquot of a concentrated stock of ACh was then added to the bath, and agonist-evoked currents were recorded only after 5 min had elapsed to allow for diffusion of agonist. This procedure was then used to expose the same patch to morantel in the presence of ACh, again waiting 5 min for diffusion of the drug. We found empirically that this was sufficient time for diffusion by observing a dye in control experiments, as well as by noting steady channel activity after this period. All told, experiments on individual patches lasted 30 – 45 min, with no detectable rundown. This is in contrast to some reports of nAChR single-channel recordings (e.g., Papke and Heinemann, 1991; Hsiao et al., 2008) and may be due to the low ACh concentrations used and the low inherent activity of α3β2 receptors.
Data were sampled at 50 kHz and recorded via Acquire software. To determine unitary conductances, a subset of events (100–300 per record) for each voltage was analyzed using TAC (Bruxton Corp., Seattle, WA) in the track events mode. The resulting amplitude distribution was then fitted to a single Gaussian function using TACFit. Subsequent detailed analysis of patches used the half-amplitude threshold criterion and cubic spline interpolation of the digital signal (Colquhoun and Sigworth, 1985). Open- and closed-time distributions were fitted by maximum likelihood using TACFit. Control experiments included recording from uninjected or sham-injected oocytes. In addition, in 5 outside-out experiments on oocytes expressing receptors, morantel was added before ACh; for morantel alone, events were very infrequent (<0.08 s−1) but increased upon adding ACh to levels observed in the other ACh + Mor experiments. Popen values were stable over 5 to 10 min of recording with ACh or ACh + Mor.
In the various recordings for a given experiment, single-channel currents were identified by their conductance and kinetic properties. In most patches, several unitary current amplitudes were observed; we analyzed the predominant form, for which the effect of morantel addition was obvious on inspection. Unitary conductances, determined as the slope of the current-voltage plot for three or four voltages in the range −20 to −100 mV, were 19.3 ± 1.6 pS (range, 11.3 – 30.2; n = 14 from five donors) and are in good agreement with those reported for rat α3β2 (Papke and Heinemann, 1991). Closed time distributions were fitted to four exponential components, except for two patches (at 10 μM ACh and 10 μM Mor) that required five components. Open time distributions were fitted to two components. Following previous work (see Mathie et al., 1991; Papke and Heinemann, 1991; Lee and Sine, 2004), burst durations were defined by the point of intersection of the second and third briefest closed-time components (τc2 and τc3; Fig. 4A, arrows). This time is dependent on agonist and modulator concentrations, and the closed times shorter than this value are assumed to represent closures of the fully liganded receptor. Critical times ranged from 2 to 8 ms. However, other critical closed times were tested, and the effect of morantel was clear regardless of this choice.
Fig. 4.
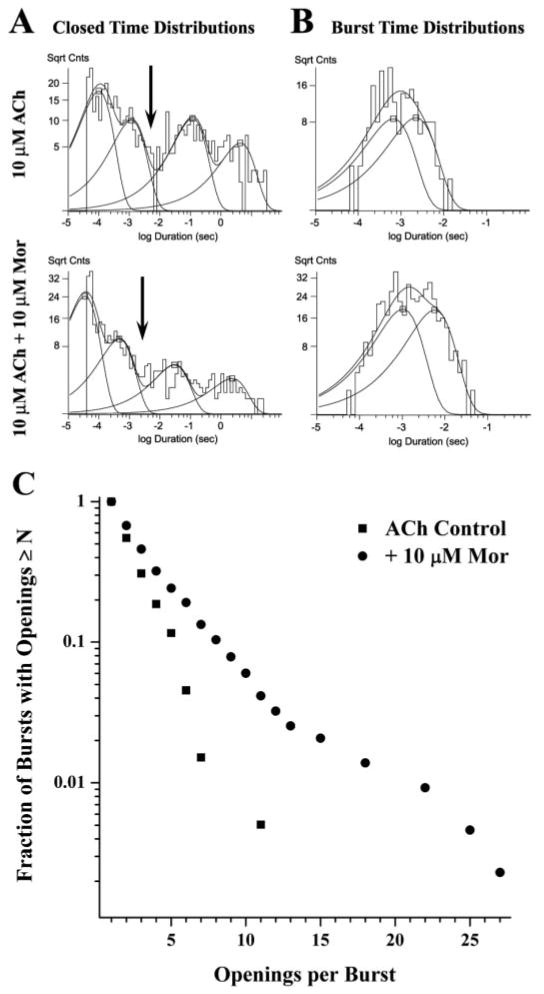
Morantel increases the bursting behavior of single-channel openings. A, the closed time histograms for the experiment shown in Fig. 3 are given for ACh alone (top) and with morantel (bottom). The four-component fits were as follows (relative area, τ in ms), for 10 μM ACh alone: 0.385, 0.112; 0.234, 1.12; 0.247, 110; 0.135, 4230; and with 10 μM Mor: 0.543, 0.052; 0.248, 0.664; 0.128, 47.6; 0.08, 1610. B, the burst time histograms for this same patch are given for ACh alone (top) and with morantel (bottom). The two-component fits were as follows (relative area, τ in ms), for 10 μM ACh alone: 0.48, 0.669; 0.52, 2.17; and with 10 μM Mor: 0.57, 1.23; 0.43, 6.37. The arrows in A indicate tcritical = 3.0 ms, defining the burst (see Materials and Methods). The ordinate in A and B is the square root of counts per bin. C, the distributions of the number of openings per burst for the two conditions in the experiment are plotted to indicate the difference in bursting character.
Results
Morantel Greatly Enhances Macroscopic Currents
Morantel, shown in the inset of Fig. 1, substantially increases currents when coapplied with ACh but is a very weak agonist alone. Figure 1 shows a continuous trace for current responses evoked from an oocyte expressing α3β2 receptors. In this recording, a low concentration of morantel (0.3 μM) enhances the peak response evoked by 10 μM ACh by 3-fold. Morantel applied alone elicits a negligible response (Fig. 1, peak d and first 100 s off). We can appreciate the efficacy of morantel by noting that adding 0.3 μM morantel to 10 μM ACh (Fig. 1, response c versus b) elicits 45% more current than a 10-fold increase in ACh concentration (Fig. 1, compare responses a versus b).
After a screen of agonists and anthelmintics coapplied to rat neuronal nAChRs, we focused on Mor because of its considerably larger potentiation compared with that of levamisole (Levandoski et al., 2003). For the four receptor types formed by the pair-wise combinations of α3, α4, β2, and β4 subunits, potentiation by morantel was the largest for α3β2 receptors. For example, in the presence of an EC50 concentration of ACh, 10 μM Mor potentiates responses by 1.5-fold for α3β4 (n = 5), compared with 3.2-fold for α3β2 (at 30 μM ACh; Fig. 2A). We therefore chose to study in detail the action of morantel on α3β2 receptors. We studied rat receptors for consistency with other ongoing studies, but we found that morantel potentiates human α3β2 nAChRs equally well (not shown).
Fig. 2.
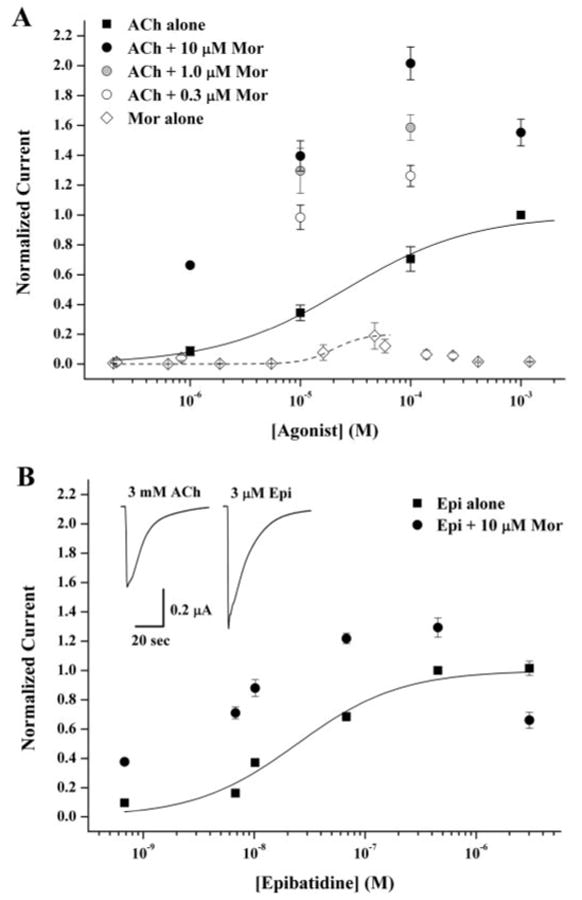
Morantel potentiates currents at saturating ACh. A, oocytes expressing rat α3β2 receptors were challenged with 5- or 10-s applications of ACh or morantel alone as agonist, or as coapplication of ACh and Mor at various concentrations. In one experiment, the peak current responses to increasing ACh concentration alone (■) or with 10 μM morantel (●) were all normalized to that evoked by 1.0 mM ACh; n = 4, two donors. The best fit to the Hill equation is shown as the solid curve through the ACh data, with parameters EC50 = 30 ± 5 μM, nH = 0.75 ± 0.11. In a separate experiment, oocytes were challenged by coapplication with 0.3 μM (○) or 1.0 μM morantel (
 ), and the response was normalized to the respective 10 or 100 μM ACh evoked response. Because in this case 1.0 mM ACh was not used, the current ratios (I+Mor/I−Mor) were corrected for the fractional response elicited by ACh alone, using 0.34 for 10 μM ACh and 0.71 for 100 μM ACh; n = 5, one donor. In a third experiment, currents elicited by 5-s application of morantel alone at the indicated concentrations were normalized to the response evoked by 100 μM ACh alone and the data corrected for the fractional response at 100 μM ACh of 0.71; the best fit to the Hill equation is shown as the dashed curve with EC50 = 20 ± 2 μM, nH = 3.1 ± 1.6, and fixed Emax = 0.20. n = 4 to 5, two donors. B, coapplication experiments, as described in A, using epibatidine as the agonist are shown. ■ are epibatidine responses alone, and ● are plus 10 μM morantel, each normalized to the 0.4 μM epibatidine response; n = 5 to 13, two donors. The best fit to the Hill equation is shown as the solid curve through the epibatidine data, with parameters EC50 = 25 ± 5 nM, nH = 0.94 ± 0.17. The inset shows sample current traces from the same oocyte for maximum responses evoked by a saturating concentration of ACh (left) or epibatidine (right). Values are presented as mean ± S.E.M.
), and the response was normalized to the respective 10 or 100 μM ACh evoked response. Because in this case 1.0 mM ACh was not used, the current ratios (I+Mor/I−Mor) were corrected for the fractional response elicited by ACh alone, using 0.34 for 10 μM ACh and 0.71 for 100 μM ACh; n = 5, one donor. In a third experiment, currents elicited by 5-s application of morantel alone at the indicated concentrations were normalized to the response evoked by 100 μM ACh alone and the data corrected for the fractional response at 100 μM ACh of 0.71; the best fit to the Hill equation is shown as the dashed curve with EC50 = 20 ± 2 μM, nH = 3.1 ± 1.6, and fixed Emax = 0.20. n = 4 to 5, two donors. B, coapplication experiments, as described in A, using epibatidine as the agonist are shown. ■ are epibatidine responses alone, and ● are plus 10 μM morantel, each normalized to the 0.4 μM epibatidine response; n = 5 to 13, two donors. The best fit to the Hill equation is shown as the solid curve through the epibatidine data, with parameters EC50 = 25 ± 5 nM, nH = 0.94 ± 0.17. The inset shows sample current traces from the same oocyte for maximum responses evoked by a saturating concentration of ACh (left) or epibatidine (right). Values are presented as mean ± S.E.M.
Altered Desensitization Does Not Account for Potentiation
Nicotinic receptors undergo desensitization, a process in which the agonist-evoked current decreases upon continued exposure to agonist (Giniatullin et al., 2005). We therefore asked whether morantel potentiates by inhibiting desensitization of α3β2 nAChRs. As seen in the multiple responses from the same oocyte (Fig. 1), the current response decays during continued exposure to agonist for 100 to 300 s, and this decay occurs even in the presence of morantel. The time course of desensitization is best fitted with two exponential terms. As indicated in Table 1 for experiments using 10 μM ACh and 0.3 μM Mor (as in Fig. 1), the fitted relative areas and time constants are unchanged in the presence of morantel (p > 0.2 for each paired comparison). In addition, the extent of desensitization (Iplateau/Ipeak) is the same in both cases: 0.32 ± 0.02 for the ACh control and 0.34 ± 0.03 with morantel (n = 7). The data in Fig. 1 and Table 1 are from a comprehensive study of desensitization that included four pairs of ACh and morantel concentrations (10 and 100 μM ACh, 0.3 and 1.0 μM Mor) and the saturating concentration of 1 mM ACh alone. Across all these conditions, the mean time constants for desensitization ranged from 5 to 10 s and 30 to 100 s for the two exponential terms and are in general agreement with other studies of desensitization in neuronal nAChRs (e.g., Fenster et al., 1997). Although we cannot exclude a minor contribution of enhanced slow desensitization to the potentiation of α3β2 receptors by morantel, this clearly does not account for the manyfold enhancement of macroscopic currents.
TABLE 1. Sample current decay parameters.
Values represent means ± S.E.M.; n = 7 for each measurement. In this experiment, the control was 10 μM ACh and the coapplication was 10 μM ACh + 0.3 μM Mor, delivered in the standard manner (see response c in Fig. 1). Both challenges were for 100 s.
| A1 | τ1 | A2 | τ2 | |
|---|---|---|---|---|
| s | s | |||
| Control | 0.36 ± 0.06 | 7 ± 2 | 0.64 ± 0.06 | 37 ± 4 |
| Coapplication | 0.39 ± 0.03 | 9 ± 2 | 0.61 ± 0.03 | 43 ± 5 |
The design of the prolonged agonist exposure experiments (Fig. 1) also reveals additional features of morantel potentiation of α3β2 receptors. First, as indicated in the fifth response (Fig. 1, e), morantel can potentiate receptors even after significant decay of the initial peak response. Second, exposure to morantel for 100 s before applying ACh (sixth response; Fig. 1, f) results in a peak response that is larger than for the standard simultaneous coapplication (Fig. 1, response c). For example, in experiments with 10 μM ACh and 0.3 μM Mor as in Fig. 1, the “Mor presaturation” response (Fig. 1, i.e., response f) was 25% larger than that elicited by the standard coapplication (p < 0.01; n = 5). This result suggests that even the macroscopic recordings harbor a kinetic component of morantel potentiation, a full understanding of which requires further study. Most importantly, the experiment in Fig. 1 demonstrates that the responses are robust and that desensitization is readily reversible upon washout. In more than 50 min of recording, the peak current elicited by 10 μM ACh remains the same (Fig. 1, responses b, e, and g), even after repeated cycles of desensitization and recovery. These observations held true for all experiments of this study using the following combinations of ligand concentrations: 0.3 μM Mor, 10 μM ACh [n = 10, two donors]; 0.3 μM Mor, 100 μM ACh [n = 11, two donors]; 1.0 μM Mor, 10 μM ACh [n = 11, two donors]; 1.0 μM Mor, 100 μM ACh [n = 9, three donors].
Morantel Enhances Responses Even at Saturating Concentrations of Ach
To understand morantel potentiation quantitatively, we further explored this enhancement as a function of agonist and modulator concentrations. Figure 2A shows that coapplication of 10 μM morantel enhances the response to a wide range of ACh concentrations, including the saturating concentration of 1.0 mM, to which all these responses are normalized. Thus, a saturating concentration of ACh alone elicits only a nominal maximum response of α3β2 receptors. A shift in potency also accompanies the morantel effect; in the presence of 10 μM morantel, the EC50 is 5 μM, a 6-fold decrease relative to ACh alone. Potentiation also depends on the concentration of morantel. For example, in the series 0.3, 1.0, and 10 μM morantel coapplied with 100 μM ACh; the currents are 1.3, 1.6, and 2.0 relative to the response evoked by 1.0 mM ACh alone (Fig. 2A).
Morantel, like levamisole and pyrantel, is a potent agonist at nematode muscle-type nAChRs (Martin, 1997). We therefore studied currents from rat neuronal α3β2 receptors elicited by morantel alone. Representative traces for morantel-evoked currents are shown in Fig. 1 (response d and the first 100 s of f) and again in Fig. 5A. The full concentration-response relationship is shown in Fig. 2A (◇). The mean maximum response is only ∼0.2 relative to the ACh maximum. Note that the response decreases at morantel concentrations above 50 μM; such currents (not shown) display the “rebound” upon washout of the drug that is characteristic of open channel block (e.g., Levandoski et al., 2003). Excluding responses to morantel concentrations above 50 μM, the data can be fitted by the Hill equation, giving EC50 = 20 ± 2 μM, nH = 3.1 ± 1.6 (for a fixed Emax = 0.20). These measurements show that morantel is a weak partial agonist of α3β2 receptors.
Fig. 5.
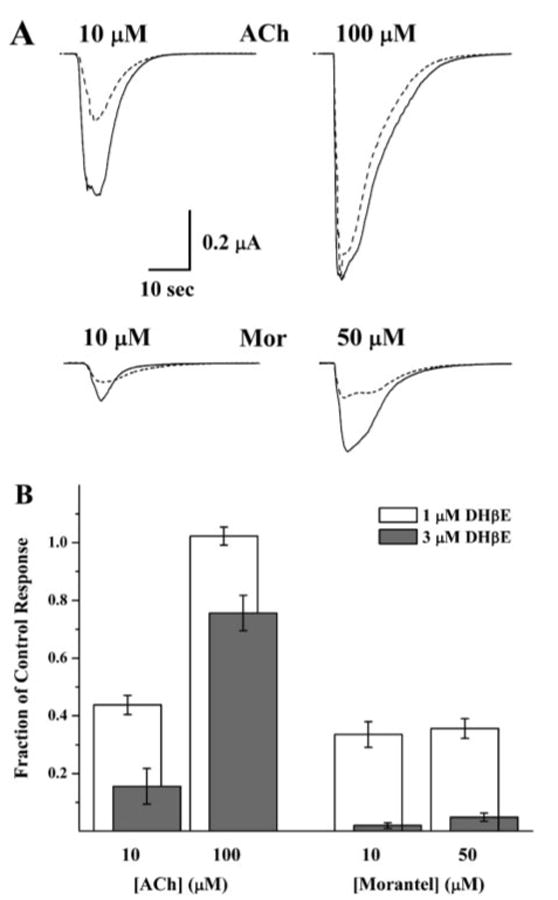
DHβE inhibits morantel-evoked currents noncompetitively. A, currents, all from the same α3β2-expressing oocyte, evoked by ACh or morantel (solid traces) are overlaid with their pairs, the currents evoked by coapplication of the same concentration and 1.0 μM dihydro-β-erythroidine (DHβE; dashed traces). All applications were 5 s. B, collated data of the type shown in A are plotted to indicate the degree of inhibition by DHβE [i.e., the fraction of control current remaining upon coapplication with DHβE (I+DHβE/Iagonist alone)]. The open bars are inhibition by 1.0 μM DHβE (n = 5, one donor) and the solid bars are inhibition by 3.0 μM DHβE (n = 8, one donor, different from the 1 μM experiment). For both experiments, all 8 challenges were given to the entire set of oocytes. The choices of 10 and 50 μM morantel were constrained by the need to have measurable responses both in the absence and presence of DHβE and to avoid the self-inhibition at concentrations >50 μM (see Figure 2A). This 5-fold range constitutes the same relative change in evoked response as the 10-fold range for ACh.
Morantel also potentiates responses evoked by epibatidine, as shown in Fig. 2B. As observed with ACh, the enhancement occurs even at saturating concentrations of epibatidine. However, the relative degree of potentiation is smaller; this dependence of potentiation on the choice of agonist was also observed with levamisole (Levandoski et al., 2003). In side-by-side measurements (e.g., inset of Fig. 2B), we determined that epibatidine is 50 ± 5% more efficacious than ACh on these receptors (n = 21 total, three donors), in agreement with the reduced potentiation by morantel. That ACh and epibatidine responses are both potentiated by morantel at saturating concentrations but to differing degrees suggests an effect on channel gating.
Single-Channel Behavior Reveals the Potentiation Mechanism
Because reduced desensitization does not explain the potentiating effect of morantel on α3β2 receptors, we recorded single-channel currents from cell-free, outside-out patches containing these receptors. After forming the patch and setting the membrane potential to −60 mV, we recorded current for 3 to 4 min in the absence of agonist and morantel and observed no unitary currents (not shown). Upon adding 10 μM ACh to the bath, unitary currents appeared as mainly brief, single-current pulses or occasionally as groups of closely spaced pulses, flanked by long (>10 ms) closed times (Fig. 3A, top). Addition of morantel to the patch greatly increased the frequency of channel openings, and many of the openings occurred in bursts of several openings in quick succession (Fig. 3A, bottom).
Fig. 3.

Morantel increases the frequency of single-channel openings. A, representative single-channel current traces are shown from an experiment using 10 μM ACh alone (top) and with 10 μM morantel added (bottom) to the same outside-out patch held at −60 mV. Traces are not continuous; filtering was at 5 kHz. Downward deflections are the brief openings. The trace marked with * in each set is shown at the higher time resolution as the bottom trace in each set. B, the frequency of channel openings, calculated as averages in 10-s bins to approximate an “instantaneous” measure, is plotted for each entire 300-s recording for the experiment shown in A. The dashed lines indicate the overall average frequencies of 1.5 s−1 for ACh alone and 5.1 s−1 in the presence of morantel.
The substantial increase in opening frequency is clear from the display of the entire experiment, shown in Fig. 3B, which plots the activity in 10-s intervals against recording time. Both the ACh-alone and ACh-plus-Mor recordings are 300 s long (exclusive of the first and last closed times); in the presence of ACh alone, there are 445 openings (total duration, 180 ms), whereas after morantel addition, there are 1518 openings (total duration 1123 ms). It is noteworthy that in agreement with the small macroscopic currents evoked by morantel alone, the increased frequency of single-channel openings is not due to a new population of events evoked solely by morantel; in control experiments with 10 μM morantel in the cell-attached configuration (n = 5) or applied to outside-out patches in the absence of ACh (n = 5), opening frequency was less than 5 per minute.
The increase in single-channel activity upon addition of morantel for all experiments in this study is summarized in Table 2. We quantified potentiation at the single-channel level by taking the ratio [(ACh+Mor)/(ACh control)] of the total open-state time for a given recording, normalized to the total recording time. The resulting potentiation of single-channel currents is consistent with potentiation of both peak and integrated macroscopic currents (Figs. 1 and 2). For example, 10 μM morantel potentiates single-channel currents elicited by 1.0 μM ACh by a factor of 4, compared with the 6-fold potentiation of peak macroscopic currents. Similar potentiation of ACh-evoked single-channel currents is observed when the ratio of the number of openings per unit time is calculated (not shown). The ratio of macroscopic currents after 100 s of coapplication of 10 μM ACh and 1.0 μM morantel, a condition perhaps more similar to the single-channel experiment is 3.1 ± 0.2 (n = 11). The analysis presented here and below is based on 14 patches total. We also observed increased single-channel activity upon addition of morantel to 16 other patches over the four combinations of ACh and morantel concentrations, but we did not analyze these experiments further because of the low frequency of ACh-evoked events (fewer than ∼100 openings in 5 min of recording).
TABLE 2. Morantel potentiation of single-channel currents.
Fold potentiation was calculated as the total open time (normalized to total recording time) in the presence of morantel divided by the ACh-alone control recording for outside-out patch experiments. Values represent means ± S.E.M. for n replicates indicated in parentheses.
| Macroscopic | ||||||
|---|---|---|---|---|---|---|
| Single-Channel Currents | Peak Currents | Integrated Areas | ||||
| 1 μM Mor | 10 μM Mor | 1 μM Mor | 10 μM Mor | 1 μM Mor | 10 μM Mor | |
| 1 μM ACh | 3.1 ± 0.6 (3) | 4.1 ± 1.2 (4) | N.D. | 6.0 ± 1.3 (5) | N.D. | 8.2 ± 1.1 (5) |
| 10 μM ACh | 7.9 ± 2.4 (3) | 9.2 ± 2.2 (4) | 3.0 ± 0.3 (16) | 3.8 ± 0.6 (5) | 3.4 ± 0.3 (5) | 3.6 ± 0.3 (5) |
N.D., not determined.
For the patch in Fig. 3, the unitary current amplitude was 2.06 ± 0.33 pA at −60 mV, and the slope conductance was 12 pS both in the absence and presence of morantel. This observation, true of all our experiments, immediately excludes the possibility that morantel potentiates by increasing the unitary channel conductance.
As noted above, the bursting character of these channels changes upon addition of morantel. Closed time distributions for the representative experiment in Fig. 3 are shown in Fig. 4A. Not surprisingly, given the greater frequency of openings per unit time (Fig. 3B), the closed time distribution shifts toward shorter times in the presence of morantel. Moreover, the relative weights of the fitted exponential components also change, resulting in a greater proportion of the two briefest components of closings, indicating a greater number of openings within bursts (successive openings flanked by closings briefer than a specified critical time) when morantel is present. Relative areas and time constants for the brief closed-time components for all outside-out patch experiments are summarized in Table 3. Although means are tabulated for convenience, in all cases our statistical analysis is a paired comparison t test. The time constant of the second-briefest component (τ2) decreases approximately 5-fold with either 1.0 or 10 μM morantel, dropping from ∼10 ms with ACh alone to ∼2 ms (p = 0.001). However, the increase in the summed areas of the two brief components is only significant with 10 μM morantel added (p = 0.05).
TABLE 3. Characteristics of brief closed states.
Values represent means ± S.E.M. for n replicates indicated in parentheses. The quantity A1 + A2 (unitless) is the combined fractional area of the two briefest closed components.
| Control | + 1 μM Mor | + 10 μM Mor | |
|---|---|---|---|
| A1 + A2 | |||
| 1 μM ACh | 0.49 ± 0.04 (8) | 0.45 ± 0.11 (4) | 0.60 ± 0.10 (4)* |
| 10 μM ACh | 0.47 ± 0.05 (7) | 0.36 ± 0.04 (3) | 0.59 ± 0.09 (4)* |
| τ1 (ms) | |||
| 1 μM ACh | 0.19 ± 0.04 (8) | 0.10 ± 0.02 (4)* | 0.19 ± 0.04 (4) |
| 10 μM ACh | 0.18 + 0.04 (7) | 0.09 + 0.02 (3)* | 0.13 + 0.03 (4) |
| τ2 (ms) | |||
| 1 μM ACh | 10.0 ± 4.0 (8) | 2.1 ± 0.2 (4)* | 2.5 ± 0.5 (4)* |
| 10 μM ACh | 14.0 ± 3.8 (7) | 1.5 ± 0.5 (3)* | 1.4 ± 0.5 (4)* |
Significant difference from control in paired t test (P < 0.05).
Changes in the bursts of channel openings mediated by morantel are even more pronounced (Fig. 4B). Bursts are defined to include all openings separated by closures shorter than a specified critical time, which is taken as the point of intersection of the two intermediate closed time components (e.g., tcritical = 3 ms for the ACh + Mor case in Fig. 4A, arrow). Most importantly, as indicated in Table 4, morantel increases the time constant for the longer component of bursts (τ2) almost 3-fold (p = 0.004). On average for ACh alone, the relative areas for the two burst components are 75% for the briefer component and 25% for the longer component (Table 4). Upon addition of morantel, the relative area of the long component of bursts (A2) increases significantly, nearly doubling for the 10 μM ACh and 10 μM Mor case (p = 0.01). It is noteworthy that the mean duration of openings within the long burst component does not increase in the presence of morantel (e.g., 2.3 ± 0.6 ms for 10 μM ACh + 10 μM Mor, n = 4, versus 2.4 ± 0.8 ms for 10 μM ACh alone, n = 7), suggesting that the major effect of morantel is to increase the number of openings per burst. Although the 1.0 μM Mor data also followed all the trends described above, the changes in these parameters were only significant with 10 μM morantel. No appreciable change in the brief burst component (τ1) occurs with morantel present (p = 0.10); these values are 0.27 ± 0.04 ms (n = 16) at 1.0 μM ACh and 0.54 ± 0.09 ms (n = 14) at 10 μM ACh.
TABLE 4. Burst characteristics.
Values represent means ± S.E.M. for n replicates indicated in parentheses.
| Control | + 1 μM Mor | + 10 μM Mor | |
|---|---|---|---|
| A2 | |||
| 1 μM ACh | 0.28 ± 0.03 (8) | 0.35 ± 0.11 (4) | 0.36 ± 0.05 (4)* |
| 10 μM ACh | 0.23 ± 0.06 (7) | 0.23 ± 0.01 (3) | 0.44 ± 0.03 (4)* |
| τ2 (ms) | |||
| 1 μM ACh | 3.9 ± 1.3 (8) | 3.6 ± 1.2 (4) | 9.6 ± 4.4 (4)* |
| 10 μM ACh | 2.9 ± 0.9 (6) | 2.1 ± 0.3 (3) | 8.1 ± 1.5 (3)* |
| Openings per burst | |||
| 1 μM ACh | 1.7 ± 0.2 (8) | 2.1 ± 0.6 (4) | 3.1 ± 0.9 (4)* |
| 10 μM ACh | 1.4 ± 0.1 (7) | 1.5 ± 0.1 (3) | 2.6 ± 0.4 (4)* |
Significant difference from control in paired t test (P < 0.05).
As just suggested, Fig. 4C demonstrates that morantel increases the number of openings per burst; the fraction of bursts with any given number of openings is greater in the presence of morantel than for the ACh control, as expected for the statistical distribution containing longer bursts. In addition for this patch, morantel seems to activate a second type of burst characterized by long trains of openings (see also Fig. 3A), represented by the region of the plot with 15 to 27 openings per burst. The average number of openings per burst for this experiment is 2.2 for ACh alone and 3.5 with morantel added. When considered separately for the morantel-added case, the two classes of burst average 3.2 (98% of bursts) and 19.7 (2%) openings per burst, respectively. Across all patches, for both 1.0 and 10 μM ACh, 10 μM morantel nearly doubles the number of openings per burst (p = 0.002; Table 4).
The analysis of the second, long component of bursts (A2, τ2; Fig. 4B and Table 4) indicates that morantel increases the total ACh-evoked current by 5- to 6-fold relative to the ACh control. All told, our single-channel results—more frequent openings of longer and greater burst-like character—account for the macroscopic observations in a quantitative manner and establish the underlying mechanism of morantel potentiation.
Morantel Is a Noncompetitive Ligand
Morantel dramatically enhances agonist-evoked currents, even at concentrations that should yield low saturation of morantel binding. However, the site of morantel binding to the α3β2 receptor remains unknown. To address the question of where morantel binds, we studied inhibition of morantel-evoked currents by DHβE, a competitive antagonist of the ACh binding site. Sample macroscopic current traces for one of these experiments, using 1.0 μM DHβE, are shown in Fig. 5A. DHβE inhibits the current evoked by 10 μM ACh by some 50%, but it has no effect on current evoked by 100 μM ACh (upper traces). However, although DHβE also inhibits responses evoked by 10 and 50 μM morantel (lower traces), each response is inhibited by approximately 50%; in other words, the higher concentration of morantel does not relieve the DHβE block, unlike ACh. Figure 5B shows data from multiple oocytes in which DHβE was applied at concentrations of 1.0 and 3.0 μM, and the fraction of current remaining in the presence of DHβE is plotted. Increasing the ACh concentration from 10 to 100 μM relieves the block by both 1.0 and 3.0 μM DHβE, equivalent to a rightward shift in the concentration-response curve in the presence of DHβE. This is the established demonstration of competitive antagonism by a functional assay (e.g., Rang, 1981; Harvey and Luetje, 1996). In contrast, although DHβE inhibits morantel-evoked currents, it does so noncompetitively, because increasing the morantel concentration at a fixed DHβE concentration does not relieve the block.
Discussion
We have shown that morantel alone is a weak agonist but strongly potentiates ACh-evoked currents, including up to a 2-fold enhancement at saturating agonist concentrations. Single-channel recordings reveal that morantel increases the frequency and bursting character of unitary currents. In addition, morantel is noncompetitive with ACh and DHβE. Below we consider these results in terms of a mechanism for morantel potentiation and their implications for a morantel binding site.
On the Mechanism of Morantel Potentiation
Macroscopic current measurements show that morantel increases receptor activity even at saturating ACh concentrations, and it increases agonist potency (Fig. 2). We first investigated whether morantel alters the desensitization processes of α3β2 nAChRs (Fig. 1). Upon prolonged exposure to agonist, the time course and extent of desensitization are unchanged (Table 1) by low morantel concentrations that nonetheless enhance peak currents 2- to 4-fold.
That morantel prevents entry into a very rapidly desensitizing state is, however, a formal possibility. A desensitization step with a half-time for onset as long as 0.2 s would be nearly complete, and therefore undetectable, during the 1- to 2-s rise times of our macroscopic currents. The novel compound PNU-120596 (Hurst et al., 2005), which potentiates α7 receptors by decreasing the rate of entry into a desensitized state, provides a precedent for such a mechanism. However, given our observation of enhanced single-channel activity, appealing to fast desensitization to explain the morantel effect is unnecessary.
Morantel potentiation might be explained by enhancement of ligand binding, channel gating, or both. Unlike the mechanistic understanding possible for muscle-type nAChRs (e.g., Sine et al., 1990; Zhang et al., 1995; Ohno et al., 1996), detailed kinetic modeling of binding and gating steps has proven difficult for neuronal nAChRs (e.g., Mathie et al., 1991; Papke and Heinemann, 1991). Figure 4A demonstrates that our recordings display at least four closed states and two open states. This kinetic complexity does not allow for analysis according to the classic linear binding-gating scheme (Colquhoun and Sakmann, 1985; Colquhoun and Ogden, 1988), for which, in the limit of low agonist concentration, only two closed states and one open state are expected. Such kinetic complexity, previously observed with neuronal nAChRs (e.g., Mathie et al., 1991), might arise from exceeding the limit of low agonist concentration, especially when morantel is added, or from channels of different subunit stoichiometry in the same patch. Nonetheless, we can use this simple model as a starting point to understand changes in kinetics caused by morantel.
The simplest interpretation of our data is that channel gating improves, direct evidence for which is seen in the number of openings per burst increasing in the presence of morantel (Figs. 3 and 4, Table 4). For a simple linear mechanism, the number of openings per burst is defined by 1 + β/k−2, where β is the channel opening rate and k−2, is the rate of dissociation of one molecule of agonist (Colquhoun and Sakmann, 1985). An increase in channel opening rate, a decrease in agonist dissociation rate, or both would increase the number of openings per burst. Consideration of our single-channel and macroscopic current measurements together allows us to distinguish between morantel effects on binding versus gating. We find that morantel increases macroscopic currents evoked by a saturating concentration of ACh by approximately 2-fold (Fig. 2A). In the limit of high ACh concentration, k−2 becomes kinetically insignificant, because any agonist dissociation event is immediately followed by reassociation, and only the channel gating equilibrium constant, β/α, determines the peak current. The decrease in channel closing rate constant (α = 1/τopen) in the presence of morantel is modest (70% average over all experiments). Therefore, the potentiation of macroscopic currents seen at saturating ACh concentrations most likely arises from an increased channel opening rate constant.
The probability of channel opening at saturating agonist concentration is given by Popen, max = β/(α + β) (Colquhoun and Ogden, 1988). Because morantel enhances macroscopic currents even at a saturating concentration of ACh, our data suggest that α3β2 receptors have inherently low gating efficacy, as suggested by Papke and Heinemann (1991). Our observation that morantel enhances epibatidine-evoked currents to a lesser degree than ACh suggests epibatidine is more efficacious than ACh, in agreement with our finding (Fig. 2B).
Morantel also decreases the EC50 for ACh-evoked macroscopic currents by 6-fold (Fig. 2A). This increased potency can be explained by enhanced channel gating (Colquhoun, 1998), which at the single-channel level manifests as an increased frequency of openings and increased bursting character (Fig. 3, Table 2). Our observations do not exclude the possibility that the rate of agonist association increases, or dissociation decreases, in the presence of morantel.
Physostigmine and galantamine are noncompetitive potentiators of several nAChR subtypes (e.g., Pereira et al., 1993; Akk and Steinbach, 2005), but single-channel studies have not examined enhanced activation by both agonist and modulator, probably because these compounds evoke appreciable single-channel currents on their own. The benzodiazepine diazepam increases the frequency of GABAA receptor single-channel openings with agonist present, but does not increase the duration of any of three observed classes of openings (Rogers et al., 1994). Although benzodiazepines enhance agonist binding and shift the dose-response curves leftward, they do not increase the maximum response at saturating concentrations of GABA, so whether they enhance agonist binding or channel gating remains unresolved (Hevers and Lüddens, 1998). Downing et al. (2005) showed that benzodiazepines enhance GABAA receptor responses to saturating concentrations of partial agonists, accompanied by decreased EC50 and increased cooperativity. These findings can be reconciled if GABA is a very efficacious agonist alone, with Popen, max approaching unity, which would mask enhancement of channel gating by benzodiazepines at the single-channel level. Instead, improved channel gating would cause a shift to higher potency (Colquhoun, 1998). In this light, morantel, which increases the frequency of openings and the degree of bursting, shifts the agonist potency and increases macroscopic currents at saturating agonist concentrations, because in α3β2 receptors, ACh and epibatidine are not fully efficacious. Thus, benzodiazepines and positive allosteric modulators of nAChRs may operate by a common mechanism.
On the Nature of the Morantel Binding Site
Our understanding of potentiation mechanisms is incomplete without knowing binding site locations. Two main observations from this study suggest morantel does not bind to the canonical ACh binding sites. First, low concentrations of morantel (≤10 μM, which elicit less than 30% of the maximum response when morantel is applied alone) increase currents evoked by otherwise saturating concentrations of ACh (Fig. 2A). If morantel potentiated by occupying one of the two canonical binding sites while the other was bound with ACh (see Smulders et al., 2005), potentiation would be eliminated at saturating ACh concentrations, contrary to our finding.
More importantly, we tested directly whether morantel competed with DHβE binding, and found that it does not (Fig. 5). In these experiments, ACh and DHβE are competitive at α3β2 receptors, in agreement with previous work (e.g., Harvey and Luetje, 1996), because increasing the ACh concentration surmounts the DHβE inhibition, but increasing the morantel concentration does not.
Our results indicate that morantel is a noncompetitive allosteric modulator. Given the diversity of structures of subunit interfaces and modulators, competitive or noncompetitive binding sites for nAChR modulators are formal possibilities. However, noncompetitive modulation has been demonstrated in many systems (e.g., Maelicke and Albuquerque, 2000; Levandoski et al., 2003; Broad et al., 2006). Likewise, SEP-174559 inhibits rat α3β4 noncompetitively, but without the voltage dependence expected for a channel blocker (Fleck, 2002), which was also observed for clozapine block of α7 receptors (Singhal et al., 2007). We are intrigued by the possibility that disparate ligands act through a common type of binding site. We previously hypothesized that levamisole potentiates nAChRs by binding to alternate, noncanonical subunit interfaces based on the conclusion that it does not compete with agonist and on the logical parallel to the benzodiazepine modulators of GABAA receptors (Levandoski et al., 2003). This idea is further supported by the demonstration that zinc potentiates nAChRs by binding to alternate subunit interfaces and increasing burst duration (Hsiao et al., 2006, 2008), and indirectly by the demonstration that noncompetitive inhibitors bind to subunit interfaces in ACh-binding protein (Hansen and Taylor, 2007). We are pursuing the hypothesis that morantel binds to the noncanonical α/β; interface of α3β2 receptors using chemical modification of cysteines substituted in these regions.
Implications of this Work
Morantel potentiation at saturating ACh or epibatidine concentrations clearly demonstrates that these are partial agonists on α3β2 receptors, a fact not fully appreciated previously. More work is needed to understand mechanisms of nAChR allosteric potentiators and their binding sites, as well as their partial agonist behavior. If morantel or positive modulators generally bind at alternate interfaces, we must re-examine how such compounds activate nAChRs on their own (e.g., Hogg and Bertrand, 2007), because this challenges the concept that channel gating is activated solely by agonist binding to canonical sites. Given the great promise for drug discovery (Maelicke and Albuquerque, 2000), more low-efficacy nicotinic ligands that potentiate agonist responses are needed, and alternate subunit interfaces offer new possibilities for the design of subtype-specific drugs.
Acknowledgments
M.M.L thanks the members of the Sine lab for their generosity and support, especially Dr. Won Yong Lee for advice and sharing the rig.
This work was supported by a Mellon 8 Sabbatical Award, National Institutes of Health AREA R15-NS043163-01A1 and Research Corporation Cottrell Award CC5765 (to M.M.L.) and National Institutes of Health R37-NS31744 (to S.M.S.).
Abbreviations
- nAChR
nicotinic acetylcholine receptor
- PNU-120596
1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)urea
- ACh
acetylcholine
- DHβE
dihydro-β-erythroidine
- Mor
Morantel (1,4,5,6-tetrahydro-1-methyl-2-(2-[3-methyl-2-thienyl]ethenyl)pyrimidine, tartrate salt)
- OR2
oocyte Ringer's medium
- SEP-174559
(S)-desmethylzopiclone
References
- Akk G, Steinbach JH. Galantamine activates muscle-type nicotinic acetylcholine receptors without binding to the acetylcholine-binding site. J Neurosci. 2005;25:1992–2001. doi: 10.1523/JNEUROSCI.4985-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrettini WH, Lerman CE. Pharmacotherapy and pharmacogenetics of nicotine dependence. Am J Psychiatry. 2005;162:1441–1451. doi: 10.1176/appi.ajp.162.8.1441. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Broad LM, Zwart R, Pearson KH, Lee M, Wallace L, McPhie GI, Emkey R, Hollinshead SP, Dell CP, Baker SR, et al. Identification and pharmacological profile of a new class of selective nicotinic acetylcholine receptor potentiators. J Pharmacol Exp Ther. 2006;318:1108–1117. doi: 10.1124/jpet.106.104505. [DOI] [PubMed] [Google Scholar]
- Cachelin AB, Rust G. Unusual pharmacology of (+)-tubocurarine with rat neuronal nicotinic acetylcholine receptors containing β4 subunits. Mol Pharmacol. 1994;46:1168–1174. [PubMed] [Google Scholar]
- Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Ogden DC. Activation of ion channel in the frog end-plate by high concentrations of acetylcholine. J Physiol Lond. 1988;395:131–159. doi: 10.1113/jphysiol.1988.sp016912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol Lond. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth B. Fitting and statistical analysis of single channel records. In: Sakmann B, Neher E, editors. Single Channel Recording. Plenum Publishing; New York: 1985. pp. 191–264. [Google Scholar]
- Cooper JC, Gutbrod O, Witzemann V, Methfessel C. Pharmacology of the nicotinic acetylcholine receptor from fetal rat muscle expressed in Xenopus oocytes. Eur J Pharmacol. 1996;309:287–298. doi: 10.1016/0014-2999(96)00294-4. [DOI] [PubMed] [Google Scholar]
- Downing SS, Lee YT, Farb DH, Gibbs TT. Benzodiazepine modulation of partial agonist efficacy and spontaneously active GABAA receptors supports an allosteric model of modulation. Br J Pharmacol. 2005;145:894–906. doi: 10.1038/sj.bjp.0706251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–5759. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreri F, Agbokou C, Gauthier S. Cognitive dysfunctions in schizophrenia: potential benefits of cholinesterase inhibitor adjunctive therapy. J Psychiatry Neurosci. 2006;31:369–376. [PMC free article] [PubMed] [Google Scholar]
- Fleck MW. Molecular actions of (S)-desmethylzopiclone (SEP-174559), an anxiolytic metabolite of zopiclone. J Pharmacol Exp Ther. 2002;302:612–618. doi: 10.1124/jpet.102.033886. [DOI] [PubMed] [Google Scholar]
- Giniatullin R, Nistri A, Yakel JL. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. 2005;28:371–378. doi: 10.1016/j.tins.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Taylor P. Galanthamine and non-competitive inhibitor binding to ACh-binding protein: Evidence for a binding site on non-a-subunit interfaces of heteromeric neuronal nicotinic receptors. J Mol Biol. 2007;369:895–901. doi: 10.1016/j.jmb.2007.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Luetje CW. Determinants of competitive antagonist sensitivity on neuronal nicotinic receptor beta subunits. J Neurosci. 1996;16:3798–3806. doi: 10.1523/JNEUROSCI.16-12-03798.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevers W, Luüddens H. The diversity of GABAA receptors. Mol Neurobiol. 1998;18:35–86. doi: 10.1007/BF02741459. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Bertrand D. Partial agonists as therapeutic agents at neuronal nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;73:459–468. doi: 10.1016/j.bcp.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Hsiao B, Mihalak KB, Repicky SE, Everhart D, Mederos AH, Malhotra A, Luetje CW. Determinants of zinc potentiation on alpha4 neuronal nicotinic receptors. Mol Pharmacol. 2006;69:27–36. doi: 10.1124/mol.105.015164. [DOI] [PubMed] [Google Scholar]
- Hsiao B, Mihalak KB, Magleby KL, Luetje CW. Zinc potentiates neuronal nicotinic receptors by increasing burst duration. J Neurophysiol. 2008;99:999–1007. doi: 10.1152/jn.01040.2007. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Hajoós M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, Rutherford-Root KL, Berkenpas MB, Hoffman WE, Piotrowski DW, et al. A novel allosteric modulator of the α7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Sine SM. Invariant aspartic Acid in muscle nicotinic receptor contributes selectively to the kinetics of agonist binding. J Gen Physiol. 2004;124:555–567. doi: 10.1085/jgp.200409077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- Levandoski MM, Piket B, Chang J. The anthelmintic levamisole is an allosteric modulator of human neuronal nicotinic acetylcholine receptors. Eur J Pharmacol. 2003;471:9–20. doi: 10.1016/s0014-2999(03)01796-5. [DOI] [PubMed] [Google Scholar]
- Lyford LK, Sproul AD, Eddins D, McLaughlin JT, Rosenberg RL. Agonist-induced conformational changes in the extracellular domain of α7 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;64:650–658. doi: 10.1124/mol.64.3.650. [DOI] [PubMed] [Google Scholar]
- Maelicke A, Albuquerque EX. Allosteric modulation of nicotinic acetylcholine receptors as a treatment strategy for Alzheimer's disease. Eur J Pharmacol. 2000;393:165–170. doi: 10.1016/s0014-2999(00)00093-5. [DOI] [PubMed] [Google Scholar]
- Martin RJ. Modes of action of anthelmintic drugs. Vet J. 1997;154:11–34. doi: 10.1016/s1090-0233(05)80005-x. [DOI] [PubMed] [Google Scholar]
- Mathie A, Cull-Candy SG, Colquhoun D. Conductance and kinetic properties of single nicotinic acetylcholine receptor channels in rat sympathetic neurones. J Physiol. 1991;439:717–750. doi: 10.1113/jphysiol.1991.sp018690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Bailey TD, Auerbach AL. Structural dynamics of the M4 transmembrane segment of during acetylcholine receptor gating. Structure. 2004;12:1909–1918. doi: 10.1016/j.str.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S, Quiram P, Pruitt JN, Sine SM, Engel AG. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- Papke RL, Heinemann SF. The role of the β4-subunit in determining the kinetic properties of rat neuronal nicotinic acetylcholine α3-receptors. J Physiol. 1991;440:95–112. doi: 10.1113/jphysiol.1991.sp018698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira EF, Reinhardt-Maelicke S, Schrattenholz A, Maelicke A, Albuquerque EX. Identification and functional characterization of a new agonist site on nicotinic acetylcholine receptors of cultured hippocampal neurons. J Pharmacol Exp Ther. 1993;265:1474–1491. [PubMed] [Google Scholar]
- Pereira EF, Alkondon M, Reinhardt S, Maelicke A, Peng X, Lindstrom J, Whiting P, Albuquerque EX. Physostigmine and galanthamine: probes for a novel binding site on the α4β2 subtype of neuronal nicotinic acetylcholine receptors stably expressed in fibroblast cells. J Pharmacol Exp Ther. 1994;270:768–778. [PubMed] [Google Scholar]
- Rang HP. Drugs and ionic channels: mechanisms and implications. Postgrad Med J. 1981;57(Suppl 1):89–97. [PubMed] [Google Scholar]
- Rayes D, De Rosa MJ, Bartos M, Bouzat C. Molecular basis of the differential sensitivity of nematode and mammalian muscle to the anthelmintic agent levamisole. J Biol Chem. 2004;279:36372–36381. doi: 10.1074/jbc.M403096200. [DOI] [PubMed] [Google Scholar]
- Rogers CJ, Twyman RE, Macdonald RL. Benzodiazepine and β-carboline regulation of single GABAA receptor channels of mouse spinal neurones in culture. J Physiol. 1994;475:69–82. doi: 10.1113/jphysiol.1994.sp020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrattenholz A, Pereira EF, Roth U, Weber KH, Albuquerque EX, Maelicke A. Agonist responses of neuronal nicotinic acetylcholine receptors are potentiated by a novel class of allosterically acting ligands. Mol Pharmacol. 1996;49:1–6. [PubMed] [Google Scholar]
- Sine SM. The nicotinic receptor ligand binding domain. J Neurobiol. 2002;53:431–446. doi: 10.1002/neu.10139. [DOI] [PubMed] [Google Scholar]
- Sine SM, Claudio T, Sigworth FJ. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts: single-channel current kinetics reveal distinct agonist binding affinities. J Gen Physiol. 1990;96:395–437. doi: 10.1085/jgp.96.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal SK, Zhang L, Morales M, Oz M. Antipsychotic clozapine inhibits the function of alpha7-nicotinic acetylcholine receptors. Neuropharmacology. 2007;52:387–394. doi: 10.1016/j.neuropharm.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Smulders CJ, Zwart R, Bermudez I, van Kleef RG, Groot-Kormelink PJ, Vijverberg HP. Cholinergic drugs potentiate human nicotinic alpha4beta2 acetylcholine receptors by a competitive mechanism. Eur J Pharmacol. 2005;509:97–108. doi: 10.1016/j.ejphar.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen J, Auerbach A. Activation of recombinant mouse acetylcholine receptors by acetylcholine, carbamylcholine and tetraethylammonium. J Physiol. 1995;486:189–206. doi: 10.1113/jphysiol.1995.sp020802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, van Kleef RG, Gotti C, Smulders CJ, Vijverberg HP. Competitive potentiation of acetylcholine effects on neuronal nicotinic receptors by acetylcho-linesterase-inhibiting drugs. J Neurochem. 2000;75:2492–2500. doi: 10.1046/j.1471-4159.2000.0752492.x. [DOI] [PubMed] [Google Scholar]
- Zwart R, Vijverberg HP. Potentiation and inhibition of neuronal α3β4 nicotinic acetylcholine receptors by choline. Eur JBiochem. 2000;393:209–214. doi: 10.1016/s0014-2999(00)00002-9. [DOI] [PubMed] [Google Scholar]