

Abstract
Small molecule inhibitors of HER2 are clinically active in women with advanced HER2 positive breast cancer who have progressed on trastuzumab treatment. However, the effectiveness of this class of agents is limited by either primary resistance or acquired resistance. Using an unbiased genetic approach we performed a genome wide loss-of-function shRNA screen to identify novel modulators of resistance to lapatinib, a recently approved anti-HER2 tyrosine kinase inhibitor. Here, we have identified the tumour suppressor PTEN as a modulator of lapatinib sensitivity in vitro and in vivo. In addition, we demonstrate that two dominant activating mutations in PIK3CA (E545K and H1047R), which are prevalent in breast cancer, also confer resistance to lapatinib. Furthermore, we show that PI3K induced lapatinib resistance can be abrogated through the use of NVP-BEZ235, a dual inhibitor of PI3K/mTOR. Our data show that deregulation of the PI3K pathway, either through loss-of-function mutations in PTEN or dominant activating mutations in PIK3CA, leads to lapatinib resistance which can be effectively reversed by NVP-BEZ235.
Keywords: Breast cancer, lapatinib, barcode screen, PI3K pathway, PI3K inhibitors
Introduction
The HER2 (ErbB2/neu) gene is amplified/overexpressed in 20-30% of invasive breast carcinomas with its overexpression being associated with increased metastatic potential and poor clinical outcome(1, 2). As a result HER2 is an attractive target for therapeutic drug development. A myriad of inhibitors targeting HER2 have been developed, most notably, the humanised monoclonal antibody trastuzumab (Herceptin ®), which targets the extracellular domain of HER2. The mechanisms underlying trastuzumab activity include downregulation of HER2 expression via endocytosis (3), deregulation of the PI3K-AKT pathway, either through disruption of HER2 signalling or by increased PTEN membrane localisation(4), or the induction of a G1 growth arrest through the stabilisation of the cyclin dependent kinase inhibitor p27(5). Interestingly, trastuzumab has also been shown to induce apoptosis in multiple breast cancer cell lines via antibody-dependent cell-mediated cytotoxity (ADCC)(6). Clinical studies have established that trastuzumab provides substantial clinical benefits in patients with HER2-overexpressing metastatic breast cancers. However, the objective response rate to single agent trastuzumab is low with only 12-34% of patients responding to monotherapy (7, 8).
A number of mechanisms have been identified which consequently limit the effect of trastuzumab-based therapy in patients including hyperactivation of HER2 family members or the dimerization of HER2 with the insulin-like growth factor I receptor (IGFR1)(9, 10). Furthermore, the recent identification of a truncated form of the HER2 receptor that lacks the extracellular trastuzumab-binding domain (p95 CTF) has been reported to affect trastuzumab sensitivity (11).
Mutations in PIK3CA have been reported to occur at high frequency in a number of human cancers (12). Increasing evidence indicates that a functional PI3K-AKT pathway is also critical for trastuzumab sensitivity. Hyperactivation of PI3K signalling, downstream from HER2, either through loss-of-function PTEN mutations or dominant activating mutations in the catalytic subunit of PI3K, PIK3CAα, appear to decrease trastuzumab activity in breast cancer (4, 13). Interestingly, in primary breast cancer, a significant correlation between HER2 overexpression and the presence of PI3K mutations has been described insinuating that multiple oncogenic inputs are required to overcome the strong tumour suppressor capability of wild-type PTEN (14).
Lapatinib is an orally active small molecule inhibitor of the EGFR and HER2 tyrosine kinase domains. Treatment with lapatinib has been shown to deregulate baseline and ligand stimulated HER2 activity resulting in the inhibition of downstream effector pathways(15). Initial experiments have shown that lapatinib potently inhibits cell survival in trastuzumab resistant breast cancer cells through the induction of apoptosis(16, 17). Furthermore, in contrast to trastuzumab, lapatinib effectively inhibits the transactivation of EGFR and HER2 by IGF-1 signalling (16). Recent data has also described the ability of lapatinib to potently inhibit the tumour forming potential of p95 CTF derived breast cancer cell lines in mouse xenograft models (11).
A series of clinical trials have shown that lapatinib is active in patients with HER2 overexpressing breast cancer and a pivotal phase III study in patients with advanced disease has shown that lapatinib in combination with capecitabine prolongs the progression free survival in patients who have progressed on trastuzumab (18, 19). However, as with trastuzumab, patients with advanced disease who initially respond to this TKI almost invariably develop resistance. Therefore a clear understanding of the mechanisms underlying lapatinib secondary or acquired resistance will be advantageous on deciding which patients may benefit the most. Moreover, prior identification of patients who are unlikely to respond to lapatinib therapy due to upfront or primary resistance may lead to the development of rational drug combinations that are likely to circumvent resistance. Here using an unbiased functional genetic approach we have identified that dominant activating mutations in the PI3K pathway lead to lapatinib resistance in vitro and in vivo. Furthermore, we demonstrate that combination therapy with lapatinib plus the dual PI3K/mTOR inhibitor NVP-BEZ235 leads to complete growth arrest in PI3K pathway induced lapatinib resistance.
Materials and Methods
shRNA Barcode Screen
The pooled NKI library representing 23,742 vectors was retrovirally infected into BT474 cells and selected with puromycin (2.0 μg/ml) for 3 days. After selection cells were trypsinized and plated into two populations at a density of 2 × 105 cells in a 15-cm dish. A total of 2 × 106 cells were plated for each population. One population remained untreated, while the other population was cultured in 27nM lapatinib. Media was refreshed every 3 days. After 2 weeks cells were trypsinized and replated out at 2 × 105 cells in a 15-cm dish. After a total of 4 weeks in culture the treated and untreated populations were collected and genomic DNA was isolated using DNAzol (Life Technologies). The shRNA inserts were amplified from genomic DNA by PCR. Primers used for PCR are as follows; Forward: GGC CAG TGA ATT GTA ATA CGA CTC ACT ATA GGG AGG CGG CCC TTG AAC CTC CTC GTT CGA CC, and Reverse: TAA AGC GCA TGC TCC AGA CT. Purified PCR products were used for linear RNA amplification and purified products were labelled with cyanine-3 (Cy3) or cyanine-5 (Cy5) fluorescent isotopes.(Kreatech). Labelled RNA probes from both untreated and Lapatinib treated cells were combined and hybridized to microarrays. Quantification of the microarray images was performed with Imagene 5.6 (Biodiscovery). Microarray data was normalised and 2log transformed. Barcode protocols can be accessed at http://www.screeninc.nki.nl/.
Plasmids and Antibodies
pJP1520, pJP1520-PIK3CAα, pJP1520-E545K, pJP1520-H1047R were kind gifts from Joan Brugge. The second PTEN hairpin was a kind gift from Roderik Kortlever. Antibodies anti-p-AKT (S473), anti-p-AKT (S308), anti-p-ERK, anti p-S6 (240/244), anti-S6, IRS1 and PTEN were from Cell Signaling; anti-AKT, anti-ERK were purchased from Santa Cruz. Anti-tubulin was purchased from Sigma Aldrich. Anti-pTyr was purchased from Upstate.
Cell Culture and Transient Tranfections
The HER2 positive cell lines BT474 (PTEN +, PI3K (K111N), KRAS wt, HRAS wt, NRAS wt), and SkBR3 (PTEN +, PI3K WT, p53 mut, KRAS wt, HRAS wt, NRAS wt). cells were cultured in Dulbecc’s modified Eagle medium (DMEM-F12 + Glutamax), while Phoenix cells were cultured in Dulbecc’s modified Eagle medium (DMEM). Both media were supplemented with 10% fetal calf serum and Penicillin/Streptomycin. Phoenix cells were divided in 10cm dishes 1 day prior to transfection. Subconfluent cells were tranfected with 25 g of pRetroSuper DNA using the calcium phosphate transfection method (20). Cells were incubated overnight and washed twice in PBS. 48 hours after transfection the viral supernatant was collected, purified with a 45 μm filter and supplemented with polybrene (0.8 μg/ml). Infection of desired cells was repeated 3-5 times. Infected cells were selected with puromycin (2 μg/ml) for 3 days. When desired, stable cell lines were treated with Trastuzumab (5 μg/ml) (Herceptin; kindly provided by Genetech Inc, South San Francisco, CA), Lapatinib (27nM) (Tykerb, kindly provided by GlaxoSmithKline, Research Triangle Park, NJ), or NVP-BEZ235 (15nM) (kindly provided by Novartis, Bern, Switzerland), or in combination overnight unless otherwise indicated. PI-103 was purchased from Echelon Biosciences.
Commassie Staining
BT474 or SkBR3 cells were cultured in the presence of trastuzumab (5 mg/ml), lapatinib (27nM) or both for 3-4 weeks. Cells were washed twice in PBS and fixed with methanol and acetic acid (3:1). After 30 minutes cells were washed once in water and 10 ml commassie stain (0.2% commassie, 50% methanol, and 10% acetic acid) was added. After 30 minutes cells were washed 3 times in H2O and air-dried.
Western Blotting
Cells were lysed in solubilizing buffer (50mM Tris pH 8.0, 150mM NaCl, 1 % NP-40, 0.5% deoxycholic acid, 0.1% SDS, 1mM Sodium Vanadate, 1mM pyrophosphate, 50 mM sodium fluoride, 100 mM β-glycerol phosphate), supplemented with protease inhibitors (Complete; Roche). Whole cell extracts were then separated on 7%-12% SDS-Page gels and transferred to polyvinylidene difluoride membranes (Millipore). Membranes were blocked with bovine serum albumin and probed with specific antibodies. Blots were then incubated with an HRP-linked second antibody and resolved with chemiluminescence (Pierce).
Growth Curves
BT474 cells were retrovirally infected, selected, and polyclonal cell lines were seeded in 12-well plates (2×104). 24 hours later cells were treated with either 27nM lapatinib, 5 g/ml trastuzumab, or 15nM NVP-BEZ235 where appropriate. Cell numbers were quantified at the indicated time points by fixing cells with 4% glutaraldehyde, washing the cells twice in H2O and staining the cells with crystal violet (0.1% Sigma). The dye was subsequently extracted with 10% acetic acid and its optical density determined (570nm). Growth curves were performed in triplicate.
Tumour Xenografts in Nude Mice
Mice were maintained under the institutional guidelines set by the Vall d’Hebron University Hospital Care and Use Committee. Six to eight week old female BALB/c athymic (nu+/nu+, n=32) mice were acquired from Charles Rivers Laboratories (Paris, France). Mice were housed in air-filtered laminar flow cabinets with a 12 -hour light cycle and food and water ad libitum. Mice were acclimatized for 2 weeks. A 17 β-estradiol pellet (Innovative Research of America, Sarasota, Fl) was inserted subcutaneously to each mouse 1 day prior to injection with BT474 VH2 (pRS-GFP) or BT474 VH2 (pRS-PTEN-B) (21). For BT474 VH2 clones 2 × 107 cells were injected subcutaneously and treatment was initiated when the tumours achieved a mean size of 400 mm3. Lapatinib was administered daily by oral gavage in 0.5% hydroxypropylmethycellulose, 0.1 % Tween 80. Tumour xenografts were measured with callipers every 2-3 days, and tumour volume was determined using the formula: (length × width2) × (π/6). When appropriate mice were anesthetized with 1.5 % isofluorane-air mixture and killed by cervical dislocation. Tumours were homogenized in solubilizing buffer (see above).
Results
Loss of PTEN expression confers resistance to Lapatinib
To identify genes whose suppression by shRNA cause resistance to lapatinib we infected BT474 HER2 overexpressing breast cancer cells with a retroviral library that comprises 23,742 shRNA vectors targeting 7914 genes (22). After selection with puromycin, cells were plated out at low density and treated with 27nM lapatinib. The IC50 value of BT474 cells was predetermined to be approximately 25nM (data not shown and (17, 23)). To rapidly identify shRNAs that are capable of circumventing the proliferation arrest induced by lapatinib we employed shRNA Barcode technology(24). After 4 weeks DNA was harvested from the surviving lapatinib treated cells and, as control, from untreated cells (Sup. Fig. 1A). shRNA cassettes were recovered by PCR and RNA probes were generated by linear amplification and fluorescent labelling. The relative representation of each shRNA in the population was measured using a microarray. To minimize experimental variation we combined the data from two individual experiments. Sup. Fig. 1B shows the relative abundance of the shRNA vectors in the lapatinib treated population as compared to untreated controls. Interestingly, we identified 8 shRNA vectors (C20ORF44, DNMT3A, GRAP2, PPP1R14B, PTEN, TK1, ZAP70, and ZIC3) for which the same shRNA vector was identified in both individual barcode screens (Table S1 and S2). However, when tested in second round selection of the 8 shRNA vectors tested, only the hairpin targeting PTEN conferred resistance to lapatinib (Fig 1A and data not shown). As expected, loss of PTEN expression also abrogated trastuzumab sensitivity (Fig. 1A). Critically, a second non-overlapping shRNA capable of inhibiting PTEN expression (Fig. 1B), also conferred resistance to lapatinib and trastuzumab therefore arguing against an off target effect (Fig. 1A) (25). An shRNA targeting GFP was used as a negative control in all experiments. Interestingly, treatment with both trastuzumab and lapatinib conferred an enhanced response to the proliferation potential of HER2 positive cells compared to either treatment alone, confirming the results of others which have indicated that combining lapatinib with trastuzumab enhances their biological effect (Fig. 1A) (26). However, while combination treatment with lapatinib and trastuzumab limited cellular proliferation in PTEN knockdown cells, viable cells remained. (Fig 1A).
Fig.1.

PTEN silencing decreases sensitivity to trastuzumab and lapatinib. When required cells were either treated with trastuzumab (5 ug/ml), or lapatinib (27 nM) unless otherwise stated. A) Colony formation assay in BT474 cells infected with either NKI library vector PTENkdA, a second independent shRNA vector PTENkdB, or relevant controls. Infected cells were left untreated or treated with trastuzumab, or lapatinib, or both. After 4 weeks cells were photographed and stained with crystal violet. B) Western Blot analysis monitoring PTEN expression in stably infected PTEN knockdown cells. C). Growth curves of stably infected PTEN knockdown BT474 cells treated for 3 weeks with trastuzumab, or lapatinib, or both. Cell numbers were quantified as described in Experimental Procedures. Growth curves were performed in triplicate with the error bars depicting the mean ± s.d. D) Western blot analysis of stably infected BT474 cells with shRNA PTENkdB vector treated overnight with trastuzumab, or lapatinib, or both in 10% serum. Whole cell extracts were analyzed with indicated antibodies.
To investigate the sensitivity of the PTEN knockdown cell lines to the different HER2 targeted therapies we analysed the proliferation potential of PTEN knockdown cells when treated with trastuzumab (5ug/ml), lapatinib (27nM) or both for 4 weeks. Treatment with HER2 directed therapies completely inhibited the proliferation potential of control cells. However, the ablation of PTEN expression in BT474 cells decreased the growth inhibitory properties of both trastuzumab and lapatinib (Fig. 1C). Collectively these data suggest that PTEN expression is required for both trastuzumab and lapatinib sensitivity in BT474 cells.
As has previously been reported lapatinib growth inhibition correlates with downregulation of HER2 dependent PI3K signalling (17, 26). Therefore, in order to study the effects of lapatinib on PI3K signalling in cells which lack PTEN activity, we treated BT474 cells or BT474 PTEN depleted cells with lapatinib (Fig. 1D). Indeed, similar to trastuzumab, there was a significant downregulation in AKT473 phosphorylation in lapatinib treated control cells compared to untreated control cells. In contrast downregulation of AKT phosphorylation was attenuated in lapatinib treated PTEN knockdown cells compared to lapatinib treated controls. However, unlike trastuzumab, no change was observed in MAPK phosphorylation upon treatment with lapatinib. In addition, treatment of cells with both lapatinib and trastuzumab resulted in an additive inhibitory effect on AKT activity suggesting that trastuzumab and lapatinib may function through partially non-overlapping mechanisms to disrupt HER2 dependent PI3K signalling.
The approved dose in patients of lapatinib when used in combination with capecitabine is a daily dose of 1250mg (18). This dosage results in a minimal plasma drug concentration of approximately 500 nM (27). Therefore to test if PTEN loss can overcome lapatinib sensitivity at clinically relevant concentrations we performed a colony formation assay. As shown in figure 2A, loss of PTEN expression significantly enhanced the growth potential of BT474 cells when treated at clinically relevant doses of lapatinib, which correlates with an increase in AKT activity (Fig. 2B).
Fig. 2.
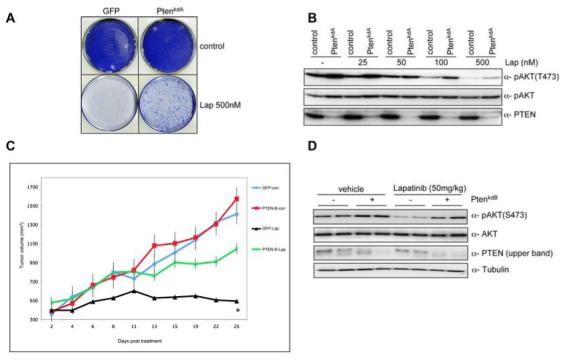
PTEN silencing decreases sensitivity to lapatinib at clinically relevant concentrations A) Colony formation assay in BT474 cells infected with vector PTENkdA. Infected cells were left untreated or treated with lapatinib (500 nM). After 4 weeks (controls) or after 12 weeks (lapatinib treated) cells were photographed and stained with crystal violet. B) pAKT in relation to the total level of unphosphorylated protein in lapatinib treated samples (0, 25, 50, 100, 500 nM) in the presence or absence of PTENkdA vector. C) Mouse xenograft assay with BT474 VH2 cells stably infected with vector PTENkdB, or relevant controls. Mice were treated daily with lapatinib (50 mg/kg) or vehicle. The results shown are the mean tumour volume ± SE. A two-tailed student T test compares the two treated populations *p< 0.02 D) Western blot analysis of mouse xenograft BT474 VH2 tumours stably infected with vector PTENkdB or relevant controls. Whole cell extracts were analyzed with indicated antibodies.
To investigate if PTEN deficiency leads to lapatinib resistance in vivo, we retrovirally infected BT474 cells with a shRNA targeting PTEN or a relevant control and injected athymic nude mice subcutaneously. When tumour xenografts reached a mean size of 400 mm3 (approximately 14 days) we treated the mice with lapatinib (50 mg/kg) or vehicle daily. BT474 PTEN depleted cells exhibited similar growth rates to controls in vehicle treated mice (Fig 2C). However, loss of PTEN significantly inhibited the anti-tumorigenic effects of lapatinib compared to controls (Fig 2C). Furthermore, western blot analysis of tumours clearly demonstrates a decrease in AKT dephosphorylation in PTEN knockdown tumours compared to controls (Fig. 2D). Together these data demonstrate that loss of PTEN expression attenuates lapatinib sensitivity in vitro and in vivo possibly by maintaining the activation of the AKT signalling pathway.
Breast Cancer relevant PI3K mutations confer resistance to Lapatinib
The PI3K pathway is frequently mutated in cancer. Loss-of-function mutations in PTEN have been described in a variety of cancers resulting in hyperactivation of the PI3K pathway (28). In addition a number of recent reports have indicated that activating mutations in PI3K subunit PIK3CAα occur in 18% to 40% of primary breast cancers (29). The majority of these mutations reside within two hotspot regions leading to single amino acid substitutions within the helical domain (E545K) and kinase domain (H1047R) resulting in enhanced PI3K signalling (30). Importantly, deregulation of the PI3K pathway appears to be poor prognostic indicator towards trastuzumab sensitivity (13).
To investigate whether cancer associated PI3K mutations result in lapatinib resistance, we retrovirally transduced BT474 cells with hemaggllutinin (HA)-tagged PIK3CAα, or the breast cancer relevant isoforms, HA-E545K, or HA-H1047R. Both PI3K dominant activating mutations rendered BT474 cells nearly completely refractory to the growth inhibitory effects of lapatinib and trastuzumab (Fig 3A). However, unlike trastuzumab, lapatinib appears to limit the growth potential of PIK3CAα overexpressing BT474 cells (Fig 3A). Interestingly, expression of PIK3CA(E545K) and PIK3CA(H1047R) also conferred resistance to the growth arrest conferred by the combined treatment of lapatinib and trastuzumab (Fig 3A). Similar results were observed in the HER2 overexpressing cell line SKBR3 (Supplementary figure 2).
Fig 3.
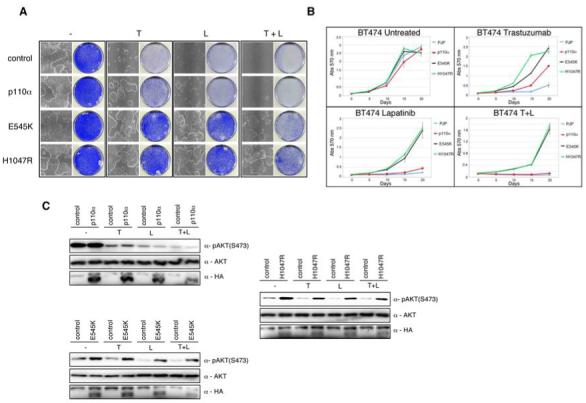
Constitutive activation of the PI3K pathway decreases sensitivity to trastuzumab and lapatinib. When required cells were either treated with trastuzumab (5 ug/ml), or lapatinib (27 nM) unless otherwise stated. A) Colony formation assay in BT474 cells infected with either WT PIK3CAα or PIK3CAα mutants E545K and H1047R, or relevant controls. Infected cells were treated with trastuzumab, or lapatinib, or both. After 4 weeks cells were photographed and stained with crystal violet. B) Growth curves of stably infected PI3K wildtype or mutant PIK3CAα BT474 cells treated for 3 weeks with trastuzumab, or lapatinib, or both. Cell numbers were quantified as described in Experimental Procedures. Growth curves were performed in triplicate with the error bars depicting the mean ± s.d. C) Western blot analysis of BT474 cells infected with HA-tagged PI3K PIK3CAα (upper left panel), PI3K(E545K) PIK3CAα (lower left panel), PI3K(H1047R) PIK3CAα (right panel), or controls were treated overnight with trastuzumab, or lapatinib, or both. Whole cell extracts were analyzed with indicated antibodies.
Next we analyzed the proliferation potential of BT474 cells retrovirally infected with the different PI3K alleles when treated with trastuzumab (5ug/ml), lapatinib (27nM), or both for 3 weeks. As expected, expression of activated PI3K mutants abrogated the growth inhibitory effects of these anti-HER2 therapies when used as either as treatment alone or in combination (Fig 3B). In contrast, in PIK3CAα overexpressing cells, both trastuzumab and lapatinib were active although lapatinib was superior at the concentrations tested (Fig 3B). In cells harbouring mutant PI3K, there was no difference in proliferation relative to WT expressing cells in non-treated samples. Together these data suggest that PI3K breast cancer prevalent mutations can counteract lapatinib and trastuzumab sensitivity in HER2 positive cells.
Since both PTEN loss-of-function mutations and oncogenic mutations in PI3K leads to constitutive AKT signalling we reasoned that AKT inhibition by lapatinib might be attenuated in the presence of dominant activating mutations in PI3K (29, 31). Indeed both E545K and H1047R mutant alleles bypassed the inhibitory effects of lapatinib and trastuzumab on AKT activity as measured by AKT473 phosphorylation (Fig. 3C). Consistent with this, both E545K and H1047R mutants decreased the sensitivity of lapatinib towards AKT activity at clinically relevant concentrations (Fig. 4C and D) resulting in a marked increase in cellular survival (Fig. 4A). In contrast, no difference was observed in phosphorylated AKT levels in PIK3CAα overexpressing cells compared to controls in lapatinib treated samples (Fig. 3C). Collectively this data suggests that hyperactivation of the PI3K-AKT pathway by hot spot mutations is a critical regulator of the anti-HER2 therapies; trastuzumab and lapatinib. Interestingly, while similar effects were observed in PIK3CAα overexpressing cells treated with trastuzumab, only a minor degree of resistance was noted in lapatinib treated samples.
Fig.4.

Constitutive activation of the PI3K pathway decreases sensitivity to lapatinib at clinically relevant concentrations A) Colony formation assay in BT474 cells infected with either WT PIK3CAα or PIK3CAα mutants E545K and H1047R, or relevant controls. Infected cells were left untreated or treated with lapatinib (500 nM). After 5 weeks cells were photographed and stained with crystal violet. B) Western blot analysis showing pAKT in relation to the total level of unphosphorylated protein in lapatinib treated samples (0, 25, 50, 100, 500 nM) in the presence or absence of WT PIK3CAα vector. C) Western blot analysis showing pAKT in relation to the total level of unphosphorylated protein in lapatinib treated samples (0, 25, 50, 100, 500 nM) in the presence or absence of PIK3CAα mutant E545K. D) Western blot analysis showing pAKT in relation to the total level of unphosphorylated protein in lapatinib treated samples (0, 25, 50, 100, 500 nM) in the presence or absence of PIK3CAα mutant H10147R.
Lapatinib and the PI3K inhibitor NVP-BEZ235 collaborate to suppress the PI3K-AKT-mTOR axis driven by loss-of-function PTEN mutations
The above data clearly demonstrates that hyperactivation of the PI3K pathway confers lapatinib resistance. Therefore we reasoned that the use of PI3K antagonists would restore the sensitivity of HER2 directed therapies. To do this we made use of the dual PI3K/mTOR inhibitor NVP-BEZ235. NVP-BEZ235 is an imidazo[4,5-c]quinoline derivative that binds equivalently to the ATP binding cleft of these enzymes and is presently undergoing Phase I clinical trials (32). Of note, we have recently reported that the IC50 for Ser473-P-Akt was 6.4 fold higher than that of P-S6 (77±29 nM compared to 12±10 nM) in NVP-BEZ235 treated samples (33).
Stably infected BT474 PTEN knockdown cells were treated with either trastuzumab (5 mg/ml), lapatinib (27nM), NVP-BEZ235 (15nM), or in combination. The IC50 value for NVP-BEZ235 in BT474 cells is approximately 15nM (data not shown). As shown in figure 5A, BT474 cells are exquisitely sensitive to NVP-BEZ235 treatment alone, which is only slightly improved by the addition of trastuzumab or lapatinib. In contrast, and in line with previous observations, BT474 PTEN knockdown cells inhibited trastuzumab, lapatinib, or NVP-BEZ235 mediated growth inhibition compared to control cells. However, combination treatment in BT474 PTEN knockdown cells with either trastuzumab and NVP-BEZ235 or lapatinib and NVP-BEZ235 was additive (Fig. 5A). Similar observations were noted when we analysed the proliferation potential of BT474 cells expressing hairpins targeting PTEN exposed to either lapatinib, NVP-BEZ235, or the combination (Fig. 5B).
Fig. 5.

Lapatinib and NVP-BEZ235 suppress the PI3K-AKT-mTOR axis driven by loss-of-function PTEN mutations. When required cells were either treated with trastuzumab (5 ug/ml), or lapatinib (27 nM), or NVP-BEZ235 (15nM) unless otherwise stated. A) Colony formation assay in BT474 cells infected with either PTENkdA or relevant controls. Infected cells were treated with trastuzumab, or lapatinib, or NVP-BEZ235, or in combination. After 4 weeks cells were photographed and stained with crystal violet. B).Growth curves of stably infected PTEN knockdown BT474 cells (PTENkdA or PTENkdB) treated for 3 weeks with lapatinib or NVP-BEZ235, or both. Cell numbers were quantified as described in Experimental Procedures. Growth curves were performed in triplicate with the error bars depicting the mean ± s.d. C) Western blot analysis of stably infected BT474 cells with shRNA PTENkdA vector treated overnight with lapatinib (27 nM), or NVP-BEZ235 (100nM) or both. Whole cell extracts were analyzed with the indicated antibodies. D) Western blot analysis of stably infected BT474 cells with shRNA PTENkdA vector treated overnight with lapatinib (27 nM), or NVP-BEZ235 (500nM) or both. Whole cell extracts were analyzed with the indicated antibodies.
To elucidate the mechanisms behind the additive effect observed between lapatinib and NVP-BEZ235 we compared the intercellular responses of BT474 or BT474 PTEN depleted cells treated with lapatinib or NVP-BEZ235 alone or in combination (Fig. 5C). In wild type cells, as expected, HER2 inhibition by lapatinib reduced phosphorylation of AKT473 and downstream mTOR signalling exhibited by reduced S6240/244 phosphorylation. Similarly, NVP-BEZ235 (100nM) treatment reduced phosphorylation of both AKT473 and S6240/244, which was accompanied by an increase in the phosphorylation of ERK in control cells, but not in PTEN knockdown cells (Fig.5C). Similar observations were seen with another dual PI3K/mTOR inhibitor, PI-103, albeit at higher concentrations (Sup. Fig. 3A, B). Recent data demonstrates that mTOR inhibition results in a mobility shift of IRS1 due to decreased serine phosphorylation (34). The loss of IRS1 serine phosphorylation inhibits degradation of the protein. Consequently, IRS1 is phosphorylated on tyrosine residues nullifying the inhibitory feedback loop and permitting the downstream activation of AKT (35). In agreement with this, BT474 cells treated with NVP-BEZ235 exhibited a decreased mobility shift, stabilization of IRS1, and increased IRS1 tyrosine phosphorylation (Sup. Fig.3 C). Surprisingly, NVP-BEZ235 did not augment IRS1 tyrosine phosphorylation in PTEN knockdown cells. IRS-1 is the major substrate of IGFR1 signalling promoting the activation of downstream effector pathways (36). Recent observations have demonstrated that treatment with the mTOR inhibitor everolimus (RAD001) induces MAPK activation through a negative feedback loop that relies on a S6K-PI3K-Ras-Raf-MEK1/2 dependent mechanism (37). The observed increase in ERK phosphorylation in NVP-BEZ235 treated samples is likely to be a consequence of mTOR inhibition resulting in the suppression of this negative feedback loop.
In contrast, loss of PTEN attenuated AKT dephosphorylation but not S6 dephosphorylation in NVP-BEZ235 treated cells. This suggests that at the concentration tested the inhibitory properties of NVP-BEZ235 are insufficient to completely abrogate the kinase activity of PI3K. In line with these results, treatment of cells with a higher concentration of NVP-BEZ235 (500 nM) reduced phosphorylation of AKT473 to levels comparable with those seen in control cell lines (Fig. 5D). This data indicates that only a limited degree of PI3K activity is sufficient to maintain activated AKT in the absence of PTEN phosphatase activity. More importantly, however, the combination treatment of BT474 PTEN knockdown cells with lapatinib and NVP-BEZ235 caused a marked decrease in AKT473 phosphorylation similar to that observed with either lapatinib or NVP-BEZ235 treatment alone in control cells. Collectively these data demonstrate an additive effect with lapatinib and NVP-BEZ235 in cell lines with decreased PTEN expression through the inhibition of both upstream and downstream signalling in the HER2/PI3K/AKT/mTOR axis, accounting for the lethal collaboration exhibited between these two drugs.
NVP-BEZ235 suppresses the PI3K-mTOR axis driven by activating mutations in the PI3K pathway in trastuzumab and lapatinib resistant cells
Next we wanted to examine if NVP-BEZ235 would circumvent the observed resistance of breast cancer relevant mutations towards trastuzumab and lapatinib. Importantly, recent observations have demonstrated that NVP-BEZ235 works equally well at repressing the activity of both WT PIK3CA or the two mutant forms E545K and H1047R (IC50: 4 nM, 4.6 nM and 5.7 nM, respectively) (38). Retrovirally transduced BT474 cells expressing either wild type PIK3CAα or the breast cancer associated PI3K isoforms were treated with either trastuzumab (5 mg/ml), lapatinib (27nM), NVP-BEZ235 (15nM) or in combination (Fig. 6A). Unsurprisingly, treatment with NVP-BEZ235 alone completely inhibited cellular outgrowth of the PI3K mutant containing cells. These results are in line with previous observations which demonstrate that PI3K mutant cell lines are highly sensitive to mTOR inhibition by rapamycin analogs (29, 39). Similar observations were later confirmed when we quantified the proliferation rates of the PI3K mutant BT474 cell lines (Fig. 6B).
Fig. 6.
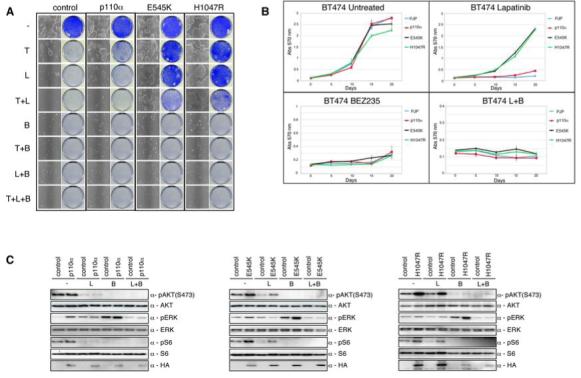
Lapatinib and NVP-BEZ235 suppress the PI3K-AKT-mTOR axis driven by gain of function PIK3CA mutations. When required cells were either treated with trastuzumab (5 ug/ml), or lapatinib (27 nM), or NVP-BEZ235 (15nM) unless otherwise stated. A) Colony formation assay in BT474 cells infected with either WT PIK3CAα or PIK3CAα mutants E545K and H1047R, or relevant controls. Infected cells were treated with trastuzumab, or lapatinib, or NVP-BEZ235, or in combination. After 4 weeks cells were photographed and stained with crystal violet. B) Growth curves of stably infected PI3K wildtype or mutant PIK3CAα BT474 cells treated for 3 weeks with lapatinib, or NVP-BEZ235 or both. Cell numbers were quantified as described in Experimental Procedures. Growth curves were performed in triplicate with the error bars depicting the mean ± s.d. C) Western blot analysis of BT474 cells infected with HA-tagged PI3K PIK3CAα (left panel), PI3K(E545K) PIK3CAα (middle panel), PI3K(H1047R) PIK3CAα (right panel), or controls were treated overnight with lapatinib or NVP-BEZ235, or both. Whole cell extracts were analyzed with indicated antibodies.
Next we wanted to determine if treatment with NVP-BEZ235 would alleviate the enhanced downstream signalling exhibited in PI3K mutant cell lines. Indeed NVP-BEZ235 treatment alone was sufficient to completely prevent phosphorylation of AKT473 and S6240/244, to levels comparable with those seen in control cell lines (Fig. 6C). Furthermore, this data demonstrates that treatment with NVP-BEZ235 overcomes PI3K dependent lapatinib resistance in BT474 cells.
Discussion
Lapatinib is approved for the therapy of patients with HER2 positive breast cancer who have progressed on trastuzumab. However, the effectiveness of this compound is limited by both primary and acquired resistance. In order to identify novel mechanisms of resistance to lapatinib we have performed a genome wide loss-of-function shRNA screen. Here we have identified the tumour suppressor PTEN as a mediator of lapatinib sensitivity in vitro and in vivo. Previous reports have shown that lapatinib activity is not dependent upon PTEN (19, 40). However, using an unbiased approach, we clearly demonstrate that loss of PTEN, and the resulting activation of the PI3K pathway, leads to deregulation of lapatinib sensitivity in our model. Consistent with this, we have identified that the two most prevalent breast cancer mutations in PIK3CA (E545K and H1047R) also confer resistance to lapatinib. Therefore, hyperactivation of the PI3K pathway by either loss of PTEN function or by activating mutations of PI3K result in resistance to lapatinib. In addition, our findings are consistent with recently reported observations utilising the anti-HER2 monoclonal antibody trastuzumab (4, 13). However it must be noted that while overexpression of wt PIK3CA diminished the effectiveness of trastuzumab in BT474 cells it was unable to circumvent the growth inhibitory properties of lapatinib, suggesting that lapatinib may function as a single agent in patients overexpressing wt PIK3CA.
A number of possibilities might explain the differing effect of PTEN loss and lapatinib resistance observed between our group and others, including the efficiency of PTEN knockdown in targeted cell lines, the use of stably infected cell lines to determine the long term effects of PTEN knockdown and lapatinib treatment, and that a 20-fold lower dose of lapatinib was used in the initial screen, reducing the chance of non-specific effects. Be that as it may, a number of studies have identified that PTEN loss does not predict for lapatinib response in patients (19, 40). Similar results have been observed in trastuzumab resistance whereby no significant correlation has been observed in PTEN loss and time to progression in trastuzumab treated patients (13). This data indicates that a larger cohort of patients may be needed in order to observe differences in response in PTEN deficient tumours. An additional explanation is the lack of a validated test to determine PTEN loss in human tumours. Until a validated test becomes available it will be difficult to try to establish reliable clinical correlations between PTEN loss and response to lapatinib and other agents. However, subsequent analysis combining both PTEN status and PI3K status has clearly demonstrated the potential of PI3K pathway hyperactivation as a biomarker for trastuzumab efficacy. As such, it will be of critical importance to equally assess PI3K pathway hyperactivation as a predictor to lapatinib response.
Abnormal activation of the PI3K pathway is frequent in breast cancer. Loss-of-function PTEN or PIK3CA mutations have been observed in approximately 20%-25% and 18%-40% of primary breast cancers, respectively (13, 14, 29). Taking into consideration the near mutual exclusivity between loss-of-function PTEN mutations and PI3K mutations (14), it is not surprising that deregulation of the PI3K pathway likely occurs in over 50% of breast cancers (29). In addition, a significant correlation between HER2 overexpression and the presence of PI3K mutations has been described (14).
There are several potential implications of these observations. One such implication is that PTEN status and the presence of PI3K activating mutations should be taken into account in clinical studies with anti-HER2 agents since they could predict for resistance. A second consequence of our findings is that hyperactivation of the PI3K pathway may be pharmacologically targeted which could in turn result in reversal of lapatinib resistance. This has been a focus of our study. We have demonstrated a near complete loss of PI3K downstream signalling in BT474 cells harbouring a deregulated PI3K pathway upon treatment with the dual PI3K/mTOR inhibitor NVP-BEZ235 and lapatinib. Interestingly, treatment of NVP-BEZ235 alone in PI3K mutant cell lines was sufficient to inhibit AKT phosphorylation. This is in contrast to cells with PTEN loss where the same NVP-BEZ235 dose fails to completely abrogate AKT activity. Considering PI3K mutant cell lines retain PTEN, this result highlights a collaboration between mechanisms to downregulate signalling through the cascade- NVP-BEZ235 inhibiting PIK3CA and PTEN dephosphorylating its downstream target PIP3. Ultimately, this could impact clinical decision making, where lower doses of NVP-BEZ235 may be selected for patients harbouring activating mutations of PI3K, with higher doses for those individuals with PTEN loss.
Recent data has highlighted the use of the PI3K inhibitors LY294002 and wortmanin in the restoration of trastuzumab sensitivity in PTEN-deficient cells (4). However, the use of these compounds in the clinic has been limited by their poor pharmacokinetics and excessive toxicity (reviewed in Marone et al. (41)). Similarly, the use of rapamycin in patients with an activated PI3K pathway has shown promising results in clinical trials(42). Again, however, patients who rapidly progressed on rapamycin treatment exhibited enhanced PRAS40 phosphorylation, a downstream target of AKT. Although highly promising, this data suggests that rapamycin efficacy in patients is limited due to the inhibition of the negative feedback loop.
Here our data suggests that combination therapy with NVP-BEZ235, which is in early-stage clinical trials, and lapatinib should be considered in patients whose tumours have a defined deregulated PI3K pathway.
Deciphering the molecular basis of response to lapatinib and other HER2 directed therapies is of great importance to maximizing the clinical efficacy of these compounds. In this present study we demonstrate the power of genome wide loss of function screens to identify critical components of lapatinib sensitivity. Furthermore our data justifies the need for future clinical trails to validate the PI3K pathway as a biomarker for lapatinib sensitivity and to explore a combined blockade with anti-PI3K inhibitors and lapatinib in a selected patient population with tumors with HER2 amplification and hyperactivation of the PI3K pathway by PTEN deletion or activating PI3K mutations.
Supplementary Material
Sup. Fig.1 shRNA Barcode screen identifies PTEN as a regulator of lapatinib sensitivity. A) Schematic representation of lapatinib Barcode screen. Library infected BT474 cells were left untreated (control) or were treated with 27nM lapatinib. After 2 weeks or 4 weeks for controls and lapatinib treated samples, respectively, shRNA cassettes were recovered by PCR, fluorescently labelled, and their abundance measured by microarray. B) Dot Plot representing the relative abundance of the recovered shRNA cassettes (M) versus relative signal intensity (A). Red dots represent shRNAs that are significantly enriched in the population compared to controls. Green dots represent shRNAs that are significantly decreased in the population compared to controls. Black dots represent no change. Data were normalized and 2log transformed. Figure represents combined results of two independent experiments. All hybridizations were performed with reverse colour swap.
Sup. Fig. 2 Constitutive activation of the PI3K pathway decreases sensitivity of trastuzumab and lapatinib. A) Colony formation assay in SkBR3 cells infected with either WT PIK3CAα or PIK3CAα mutants E545K and H1047R, or relevant controls. Infected cells were treated with trastuzumab (5 ug/ml), or lapatinib (27nM), or both. After 4 weeks cells were photographed and stained with crystal violet.
Sup. Fig. 3 Lapatinib and PI-103 suppress the PI3K-AKT-mTOR axis driven by loss-of-function PTEN mutations. A) Western blot analysis of stably infected BT474 cells with shRNA PTENkdA vector treated overnight with lapatinib (27 nM), or PI-103 (100 nM) or both. Whole cell extracts were analyzed with the indicated antibodies. B) Western blot analysis of stably infected BT474 cells with shRNA PTENkdA vector treated overnight with lapatinib (27 nM), or PI-103 (500 nM) or both. Whole cell extracts were analyzed with the indicated antibodies. C) Western blot analysis of stably infected BT474 cells with shRNA PTENkdA vector treated overnight with NVP-BEZ235 (100 nM). Equal amounts of cell lysate (500 μg) were immunprecipitated with-IRS1 and analyzed by western blot for either tyrosine phosphorylation (top panel) or total IRS1 (bottom panel).
Acknowledgments
The authors would like to thank Ben Markman for critical reading of this manuscript. This grant was supported by the Breast Cancer Research Foundation and the NIH. Dr. Baselga is currently involved in conducting clinical trials sponsored by Hoffman-La Roche, GlaxoSmithKline and Novartis. The sponsors are not responsible for the study design, data collection and analysis, interpretation of the data, or the preparation of the manuscript.
This work was supported by the Breast Cancer Research Foundation and the NIH.
References
- 1.Slamon DJ. Studies of the HER-2/neu proto-oncogene in human breast cancer. Cancer Invest. 1990;8(2):253. doi: 10.3109/07357909009017573. [DOI] [PubMed] [Google Scholar]
- 2.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 3.Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004;23(11):2057–70. doi: 10.1038/sj.onc.1207390. [DOI] [PubMed] [Google Scholar]
- 4.Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 5.Shin I, Yakes FM, Rojo F, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8(10):1145–52. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 6.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6(4):443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 7.Baselga J, Tripathy D, Mendelsohn J, et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14(3):737–44. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- 8.Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639–48. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 9.Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin) J Natl Cancer Inst. 2001;93(24):1852–7. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- 10.Sergina NV, Rausch M, Wang D, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445(7126):437–41. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaltriti M, Rojo F, Ocana A, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99(8):628–38. doi: 10.1093/jnci/djk134. [DOI] [PubMed] [Google Scholar]
- 12.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 13.Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 14.Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65(7):2554–9. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- 15.Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94. [PubMed] [Google Scholar]
- 16.Nahta R, Yuan LX, Du Y, Esteva FJ. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther. 2007;6(2):667–74. doi: 10.1158/1535-7163.MCT-06-0423. [DOI] [PubMed] [Google Scholar]
- 17.Konecny GE, Pegram MD, Venkatesan N, et al. Activity of the dual kinase inhibitor lapatinib ( GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–9. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 18.Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–43. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 19.Johnston S, Trudeau M, Kaufman B, et al. Phase II Study of Predictive Biomarker Profiles for Response Targeting Human Epidermal Growth Factor Receptor 2 (HER-2) in Advanced Inflammatory Breast Cancer With Lapatinib Monotherapy. J Clin Oncol. 2008 doi: 10.1200/JCO.2007.13.9949. [DOI] [PubMed] [Google Scholar]
- 20.van der Eb AJ, Graham FL. Assay of transforming activity of tumor virus DNA. Methods Enzymol. 1980;65(1):826–39. doi: 10.1016/s0076-6879(80)65077-0. [DOI] [PubMed] [Google Scholar]
- 21.Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998;58(13):2825–31. [PubMed] [Google Scholar]
- 22.Berns K, Hijmans EM, Mullenders J, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428(6981):431–7. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 23.Rusnak DW, Alligood KJ, Mullin RJ, et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007;40(4):580–94. doi: 10.1111/j.1365-2184.2007.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brummelkamp TR, Fabius AW, Mullenders J, et al. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol. 2006;2(4):202–6. doi: 10.1038/nchembio774. [DOI] [PubMed] [Google Scholar]
- 25.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877–84. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia W, Gerard CM, Liu L, Baudson NM, Ory TL, Spector NL. Combining lapatinib ( GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene. 2005;24(41):6213–21. doi: 10.1038/sj.onc.1208774. [DOI] [PubMed] [Google Scholar]
- 27.Burris HA, 3rd, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib ( GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23(23):5305–13. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- 28.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96(8):4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isakoff SJ, Engelman JA, Irie HY, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65(23):10992–1000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 30.Miled N, Yan Y, Hon WC, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317(5835):239–42. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 31.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102(51):18443–8. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 33.Serra V, Markman B, Scaltriti M, et al. NVP-BEZ-235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits growth of cancer cells with activating PI3K mutations; Proceedings of the 99th Annual Meeting of the American Association for Cancer Research; 2008; [DOI] [PubMed] [Google Scholar]
- 34.Pederson TM, Kramer DL, Rondinone CM. Serine/Threonine Phosphorylation of IRS-1 Triggers Its Degradation. Diabetes. 2001:5024–31. doi: 10.2337/diabetes.50.1.24. [DOI] [PubMed] [Google Scholar]
- 35.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4(5):335–48. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 37.Carracedo A, Ma L, Teruya-Feldstein J, et al. A novel feedback loop leads toa ctivation of MAPK pathway by mTORC1 inhibitors. JCI. 2008 doi: 10.1172/JCI34739. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maira S, Stauffer F, Brueggen J, et al. Identification and chracterization of NVP-BEZ235 a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 39.Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103(5):1475–9. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia W, Husain I, Liu L, et al. Lapatinib antitumor activity is not dependent upon phosphatase and tensin homologue deleted on chromosome 10 in ErbB2-overexpressing breast cancers. Cancer Res. 2007;67(3):1170–5. doi: 10.1158/0008-5472.CAN-06-2101. [DOI] [PubMed] [Google Scholar]
- 41.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase-Moving towards therapy. Biochim Biophys Acta. 2008;1784(1):159–85. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 42.Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5(1):e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sup. Fig.1 shRNA Barcode screen identifies PTEN as a regulator of lapatinib sensitivity. A) Schematic representation of lapatinib Barcode screen. Library infected BT474 cells were left untreated (control) or were treated with 27nM lapatinib. After 2 weeks or 4 weeks for controls and lapatinib treated samples, respectively, shRNA cassettes were recovered by PCR, fluorescently labelled, and their abundance measured by microarray. B) Dot Plot representing the relative abundance of the recovered shRNA cassettes (M) versus relative signal intensity (A). Red dots represent shRNAs that are significantly enriched in the population compared to controls. Green dots represent shRNAs that are significantly decreased in the population compared to controls. Black dots represent no change. Data were normalized and 2log transformed. Figure represents combined results of two independent experiments. All hybridizations were performed with reverse colour swap.
Sup. Fig. 2 Constitutive activation of the PI3K pathway decreases sensitivity of trastuzumab and lapatinib. A) Colony formation assay in SkBR3 cells infected with either WT PIK3CAα or PIK3CAα mutants E545K and H1047R, or relevant controls. Infected cells were treated with trastuzumab (5 ug/ml), or lapatinib (27nM), or both. After 4 weeks cells were photographed and stained with crystal violet.
Sup. Fig. 3 Lapatinib and PI-103 suppress the PI3K-AKT-mTOR axis driven by loss-of-function PTEN mutations. A) Western blot analysis of stably infected BT474 cells with shRNA PTENkdA vector treated overnight with lapatinib (27 nM), or PI-103 (100 nM) or both. Whole cell extracts were analyzed with the indicated antibodies. B) Western blot analysis of stably infected BT474 cells with shRNA PTENkdA vector treated overnight with lapatinib (27 nM), or PI-103 (500 nM) or both. Whole cell extracts were analyzed with the indicated antibodies. C) Western blot analysis of stably infected BT474 cells with shRNA PTENkdA vector treated overnight with NVP-BEZ235 (100 nM). Equal amounts of cell lysate (500 μg) were immunprecipitated with-IRS1 and analyzed by western blot for either tyrosine phosphorylation (top panel) or total IRS1 (bottom panel).