

Abstract
p38 mitogen-activated protein kinase (MAPK) mediates cellular responses to injurious stress and immune signaling. Among the multiple p38 isoforms, p38α is the most widely expressed in adult tissues and can be targeted by various pharmacological inhibitors. Here we investigated how p38α activation is linked to cell type-specific outputs using mouse models of cutaneous inflammation. We showed that both myeloid and epithelial p38α can signal to evoke inflammatory responses, yet the mode of skin irritation determines the cell type in which p38α serves the function. In addition, myeloid p38α induces self-limitation of acute inflammation via activation of MSK-dependent anti-inflammatory gene expression. These suggest a dual role of p38α in regulation of inflammation, and reveal a mixed potential for its inhibition as a therapeutic strategy.
INTRODUCTION
The inflammatory response is initiated upon infection and tissue injury, and mobilizes a variety of effector mechanisms that contain and eliminate the causative injurious agent. Cellular and molecular mediators of inflammation also promote tissue repair and regeneration, thus restoring the disturbed tissue organization to a functional state. Although inflammation protects and heals against pathogenic stimuli, it may cause more damage than the inciting event if its magnitude and duration are not strictly controlled by intrinsic negative regulators. Unbalanced and unrestrained inflammatory responses underlie diverse forms of chronic inflammatory diseases regardless of the pathogenic mechanisms involved.
Central to inflammatory signaling is reversible phosphorylation of protein regulators and effectors by protein kinases and phosphatases. In particular, the distinct mitogen-activated protein kinase (MAPK) pathways mediated by ERK, JNK and p38 MAPK family members play a pivotal role in linking inflammatory stimuli to cellular responses. Mammalian p38 MAPK was originally discovered as an evolutionarily conserved protein kinase whose activity is induced by lipopolysaccharide (LPS) and interleukin (IL)-1, and also as a protein that binds with high affinity to a group of anti-inflammatory compounds such as SB2021901–4. Therefore, its functional relevance to inflammation was predicted at the very outset. In addition to the first identified p38 MAPK protein, now referred to as p38α, three additional paralogs--p38β, p38γ, and p38δ--exist in mammals5,6. Although the four p38 isoforms share a certain degree of structural and enzymatic properties, only p38α and p38β are sensitive to inhibition by SB202190 and its derivatives7,8. p38α is the most ubiquitously expressed in human and mouse tissues9 and, in particular, the most abundant in inflammatory cells of myeloid origin10.
p38 MAPK mediates inflammatory responses partly through activating gene expression. Proteins phosphorylated by a mechanism dependent on p38 MAPK activity include sequence-specific transcription factors, transcriptional coregulators, nucleosomal proteins, and regulators of mRNA stability and translation11. p38 MAPK either directly phosphorylates these proteins or induces their phosphorylation by activating other protein kinases termed MAPK-activated protein kinases (MKs). The MKs that are phosphorylated by and functionally subordinate to p38 MAPK include MK2 and MK3, mitogen- and stress-activated kinase 1 (MSK1) and MSK2, MAPK-interacting kinase 1 (MNK1) and MNK2, and p38 regulated/activated kinase (PRAK)11,12. MK2 and MK3 have recently been shown to phosphorylate and activate another class of MKs, the p90 ribosomal S6 kinases (RSKs), albeit specifically in dendritic cells, illustrating the multilayered configuration of the protein kinase cascades downstream of p38 MAPK13. Phosphorylation by p38 MAPK and its subordinate kinases induces changes in the activity, turnover, and subcellular location of substrate proteins, and consequently the expression of their target genes.
Attempts to determine how p38α contributes to immunity and inflammatory disease have been hampered by limited target specificity of p38 MAPK inhibitors14 and early lethality of p38α-null mouse embryos due to placental and vascular defects15–18. Gene disruption methods that ablate p38α alleles in embryonic but not placental tissues19 or at postnatal stages in a drug-inducible fashion20 permitted survival of the mutant mice. However, those p38α-null animals were found to develop spontaneous anomalies in homeostasis of pulmonary epithelial and fetal hematopoietic tissues, thus precluding further characterization of their response in experimentally induced diseases. Mice with p38β deficiency were also generated, but they manifested no discernible phenotypes in the inflammation models tested21.
In this study, we generated two mouse mutants lacking p38α in different types of cells--myeloid and epithelial--wherein p38 MAPK likely plays distinct roles in inflammation. These cell type-specific p38α knockout mice, which did not exhibit overt tissue disturbances similar to those observed in systemic p38α-null mice, were subjected to different mechanisms of tissue injury, and their inflammatory responses examined. We found that, in both myeloid and epithelial cells, p38α contributes to the onset of inflammation by activating production of leukocyte chemoattractants and other pro-inflammatory mediators. p38α also induced anti-inflammatory gene expression, particularly in myeloid cells such as macrophages, thereby preventing pathology resulting from unrestrained inflammatory responses.
RESULTS
Targeted deletion of p38α
The initiation and progression of inflammation depends on coordinated molecular signaling in both hematopoietic and parenchymal cells. Myeloid cells, such as macrophages, are amongst the principal hematopoietic subpopulations serving the inflammatory role. To investigate cell type-specific functions of p38α using cutaneous inflammation models, we generated two different mutant mice in which deletion of loxP site-flanked (floxed; fl) alleles of the p38α gene22, Mapk14, is mediated by Cre recombinase expressed under the control of the myeloid-specific lysozyme M (LysM) or skin epithelial-specific keratin 14 (K14) promoters23,24. Both of the resulting mutant mice, Mapk14fl/fl;LysMCre and Mapk14fl/fl;K14Cre (hereafter called p38αΔM and p38αΔK, respectively; Supplementary Fig. 1a, online), were born alive and grew to adulthood without exhibiting discernible anomalies or developing spontaneous disease under the specific pathogen-free condition (data not shown).
We observed highly efficient Mapk14 deletion (Supplementary Fig. 1b, online) and corresponding removal of p38α protein (Fig. 1a) in bone marrow-derived macrophages (BMDMs) prepared from p38αΔM mice. Peritoneal exudate macrophages (PEMs) from thioglycollate-injected p38αΔM mice showed a similar degree of p38α ablation (data not shown). The yield of p38αΔM PEMs was comparable to that of p38α wild-type (WT; Mapk14fl/fl) PEMs. WT and p38αΔM mice also exhibited similar numbers of skin- and spleen-resident cell populations expressing F4/80, a macrophage marker (Fig. 1b). These indicate that the recruitment and maintenance of tissue macrophages occur independently of macrophage-autonomous p38α function.
Figure 1. Disruption of Mapk14 results in efficient removal of its protein product, p38α, in myeloid and epithelial cells.
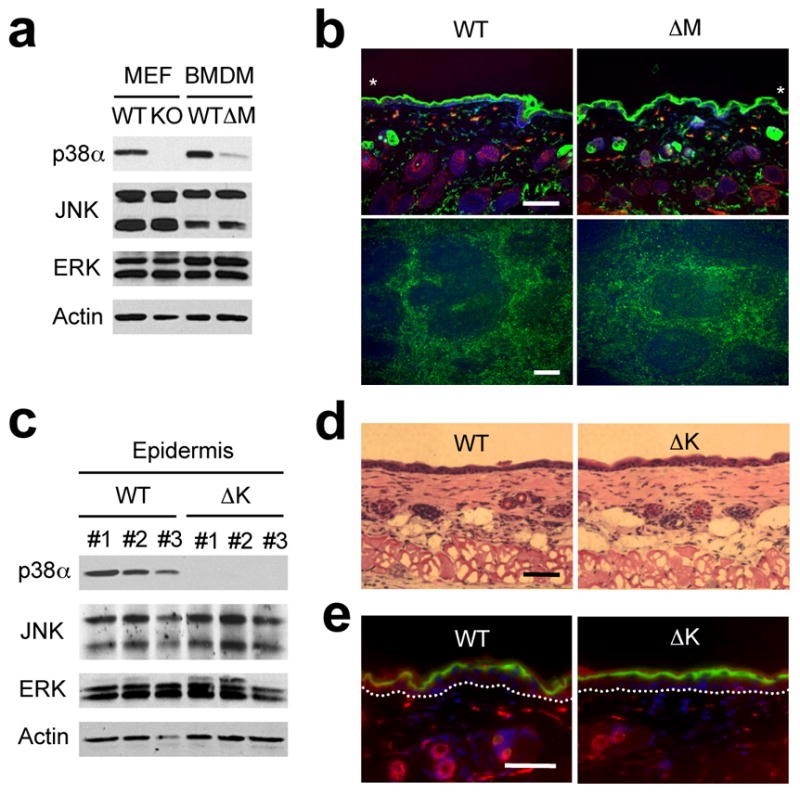
(a) Whole cell lysates from Mapk14+/+ and Mapk14−/−MEFs (WT and KO, respectively), and WT and p38αΔM BMDMs were analyzed by immunoblotting with antibodies against the proteins indicated on the left. Data are representative of at least five independent experiments.
(b) Skin (top) and spleen (bottom) tissue sections from WT and p38αΔM mice were analyzed by immunostaining with an antibody against the macrophage marker F4/80 (green). The counter staining of F-actin (red; skin) and DNA (blue; skin and spleen) is shown together. Asterisks indicate nonspecific staining of stratum corneum. Scale bar, 100 μm. Data are representative of two independent experiments.
(c) Whole cell lysates from WT and p38αΔK epidermis were analyzed by immunoblotting as in (a). The numbers indicate individual animals. Data are representative of analysis of at least ten litters.
(d, e) Skin tissue sections from WT and p38αΔK mice were analyzed by H&E staining (d) and immunostaining (e) with an antibody against loricrin (green). The counter staining is shown as in (b). Scale bar, 100 μm. Data are representative of at least ten (d) or two (e) independent experiments.
p38αΔK mice exhibited Mapk14 deletion in the epidermis, as well as cultured keratinocytes derived thereof, but not in other tissues (Supplementary Fig. 1c, online and data not shown). Consistently, p38αΔK epidermis was devoid of detectable p38α protein (Fig. 1c). The deficiency of p38α in keratinocytes did not interfere with the differentiation of the stratified skin epithelium or epidermal-dermal organization (Fig. 1d); p38αΔK skin expressed WT amounts of loricrin, a marker for terminal epidermal differentiation (Fig. 1e).
Ablation of p38α did not perturb the expression of the other MAPK proteins, JNK and ERK (Fig. 1a,c), or other p38 isoforms (Supplementary Fig. 2, online). We also confirmed that Cre expression as such was without effect on the viability of and inflammatory gene expression in macrophages and keratinocytes (Supplementary Fig. 3, online and data not shown).
Myeloid p38α in chronic skin inflammation
The absence of spontaneous skin disturbances in p38αΔM and p38αΔK mice enabled analysis of skin pathology that ensues from experimentally induced inflammation. We first employed a method that induces chronic skin inflammation by repeated topical treatment with sodium dodecyl sulfate (SDS). Detergent-mediated disruption of epidermal barrier function effects cytokine production, leukocyte infiltration, acanthosis (epidermal hyperplasia) and keratosis25,26. We painted shaved mouse back skin daily with an SDS solution. WT mice began to exhibit skin irritation, as indicated by erythema, in less than 5 days of SDS treatment, which then continued to increase in severity. The inflamed skin expressed high amounts of tumor necrosis factor (TNF) of macrophage origin; the TNF-expressing cells were localized in the dermis, and most stained positive for F4/80 (Fig. 2a).
Figure 2.
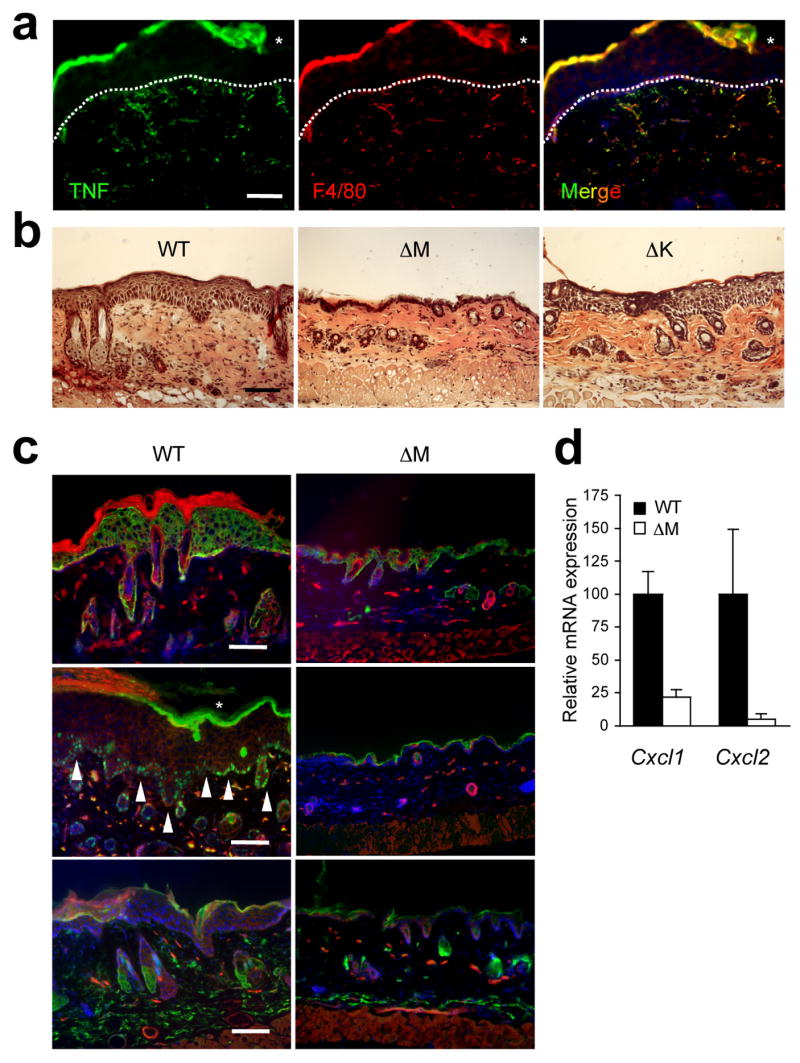
Myeloid p38α signaling is essential for chronic inflammation and acanthosis. The shaved back skin of mice was treated daily with 5% SDS for 7 days to induce chronic inflammation, and analyzed. Asterisks (a, c) indicate nonspecific staining of stratum corneum. Scale bar, 100 μm.
(a) Skin section from a WT mouse was immunostained with antibodies against TNF (green; left) and F4/80 (red; middle). Merged image (right), also includes the counter stain of DNA (blue). Dotted lines denote the epidermal-dermal boundaries. Data are representative of at least three independent experiments.
(b, c) Skin tissue sections from the indicated mice were analyzed by H&E staining (b), and immunostaining (c) with antibodies against K14, Ki67, and Gr-1 (green; top, middle, bottom, respectively). The counter staining of DNA (blue) and F-actin (red) is shown together. Arrowheads (white) indicate Ki67-positive nuclei. Data are representative of six independent experiments.
(d) Relative mRNA expression in skin tissues (n=3) from the indicated mice was determined by qPCR.
Next we compared the response of WT and the p38α mutant mice to SDS irritation. Histological analysis of skin tissues in SDS-treated WT mice indicated a massive epidermal hyperproliferation and a dermal inflammatory infiltration (Fig. 2b). These changes were detectable as early as day 4 of treatment and reached their maximal levels by day 7. The inflammatory response in p38αΔK skin was comparable in severity and kinetics to that in WT skin, as determined by macroscopic and histological examination (Fig. 2b and data not shown), suggesting the dispensability of epithelial p38α in this inflammation model. By contrast, p38αΔM mice were highly refractory to topical SDS treatment. No clear signs of irritation such as erythema or scaliness occurred in their skin throughout the course of the treatment. In particular, the p38αΔM skin tissues were completely devoid of epidermal thickening (Fig. 2b). Immunofluorescence analysis of SDS-treated skin areas confirmed that the epidermal layer expressing the basal keratinocyte marker K14 was greatly expanded, and proliferating Ki67-positive cells were detected in WT but not p38αΔM skin (Fig. 2c). Infiltration of Gr-1+ neutrophils was also evident in WT but scarce and mild in p38αΔM skin (Fig. 2c). In line with these observations, p38αΔM mice expressed in SDS-challenged skin tissues greatly diminished amounts of transcripts encoding the neutrophilic chemokines KC and MIP-2 (Cxcl1 and Cxcl2, respectively) (Fig. 2d). Removal of myeloid p38α with the resultant dampening of skin inflammation prevented the development of keratotic skin lesions at the end of 7 days of SDS treatment (Supplementary Fig. 4, online). These results indicate an essential role for myeloid p38α in evoking cutaneous inflammatory responses and concomitant tissue alterations upon SDS-mediated skin barrier disruption.
p38α in acute epidermal injury and irritation
To examine whether myeloid and epithelial p38α serve different functions depending on the mode of inflammatory insult, we subjected WT and the p38α mutant mice to epidermal injury caused by ultraviolet B radiation (hereafter called UVB). UVB can only penetrate down to the epidermis and thus inflict primary damage on the skin surface layer. In our test, a single UVB irradiation of shaved back skin resulted in apoptotic death of keratinocytes in similar degrees in WT, p38αΔM, and p38αΔK mice (Fig. 3a). The acute UVB damage was followed by acanthosis and dermal neutrophil infiltration as early as 48 h post-irradiation in WT mice (Fig. 3b). Deficiency of myeloid p38α did not interfere with the onset of either of these UVB-induced responses (Fig. 3b and data not shown), indicating distinct signaling requirements for UVB- and SDS-induced inflammatory responses.
Figure 3.
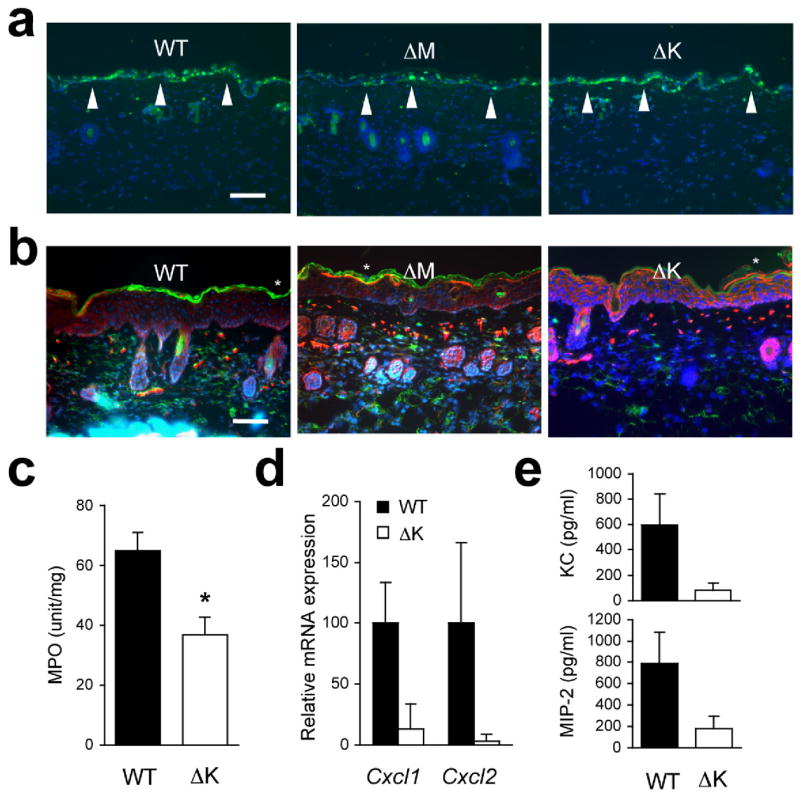
UVB-induced inflammatory infiltration and injury depend on epithelial p38α signaling. The shaved back skin of mice was irradiated with UVB (160 mJ/cm2) to induce epidermal injury (a–d).
(a, b) Skin tissue sections from the indicated mice were analyzed by TUNEL staining 24 h post-irradiation (a) and immunostaining with Gr-1-specific antibody 96 h post-irradiation (b). Scale bar, 100 μm. Apoptotic nuclei and neutrophils (green) are shown together with the counter staining of DNA (blue) and/or F-actin (red). Arrowheads (white) indicate TUNEL-positive nuclei. Data are representative of at least three independent experiments.
(c, d) Skin tissues from the indicated mice were analyzed for MPO activity (n=4; *P = 0.015) (c) and gene expression (n=3) (d) 96 h after irradiation. Relative mRNA expression was determined by qPCR.
(e) Culture supernatants of the indicated keratinocytes were collected 6 h after UVB irradiation, and KC and MIP-2 concentrations were measured. Data are representative of two independent experiments.
UVB-irradiated p38αΔK skin displayed unabated epidermal hyperproliferation but a substantially lower Gr-1+ neutrophil influx up to 4 days post-irradiation (Fig. 3b). This finding was verified by measuring the activity of myeloperoxidase, a neutrophil marker enzyme, in the skin tissues (Fig. 3c). p38αΔK mice showed markedly reduced amounts of KC and MIP-2 mRNA in irradiated skin compared with WT mice (Fig. 3d); this findingn may reflect a defect of chemokine gene expression that is inherent in the keratinocytes. In support of this notion, cultured keratinocytes isolated from p38αΔK mice produced much lower amounts of KC and MIP-2 than WT cells following in vitro UVB exposure (Fig. 3e). Macroscopically, keratinocyte-specific p38α ablation protected UVB-irradiated skin from ‘sunburn’ injury (focal blistering and erosions), which WT and p38αΔM skin incurred in less than 2 days after UVB exposure (Supplementary Fig. 5, online). However, p38αΔK mice developed a delayed skin pathology starting on the 5th day post-irradiation (data not shown). Therefore, unlike in the SDS model where myeloid p38α played a dominant pro-inflammatory role, it is in the keratinocyte that p38α signaling appears essential for initiation of inflammation in response to UVB. In addition, UVB-induced acanthosis appears to occur independently of p38α function in either cell type.
Intriguingly, p38αΔM skin manifested a more severe swelling, indicative of an increased edema formation, than WT and p38αΔK skin upon UVB exposure (Fig. 4a, b). Hence, in UVB-damaged skin, ablation of myeloid p38α not only spared acanthotic and leukocyte chemotactic responses but could even intensify certain aspects of inflammatory pathology. To address whether myeloid p38α negatively regulates vascular permeability and edema formation in other inflammatory settings, we compared acute swelling in the auricle skin of WT and p38αΔM mice after a single topical application of 12-O-tetradecanoylphorbol-13-acetate (TPA). Similarly to UVB-induced damage, TPA irritation induced edema to a greater extent in 38αΔM skin (Fig. 4c, d). To directly assess the role of myeloid p38α in regulating vascular permeability, we intravenously injected Evans Blue, a dye highly adsorbent to serum albumin, into WT and p38αΔM mice after TPA irritation. p38αΔM mice exhibited a greater extravascular leakage of injected dye in TPA-treated but not control skin (Fig. 4e). TPA-treated p38αΔM skin also expressed higher amounts of cyclooxygenase-2 (COX-2), a prostaglandin-synthesizing enzyme responsible for triggering various inflammatory symptoms including vascular hyperpermeability (Fig. 4f). Therefore, the appreciable role of myeloid p38α in UVB-and TPA-induced skin responses was confined to attenuating, rather than augmenting, inflammatory pathology.
Figure 4. Myeloid p38α functions to limit edema formation in epidermal injury and irritation.
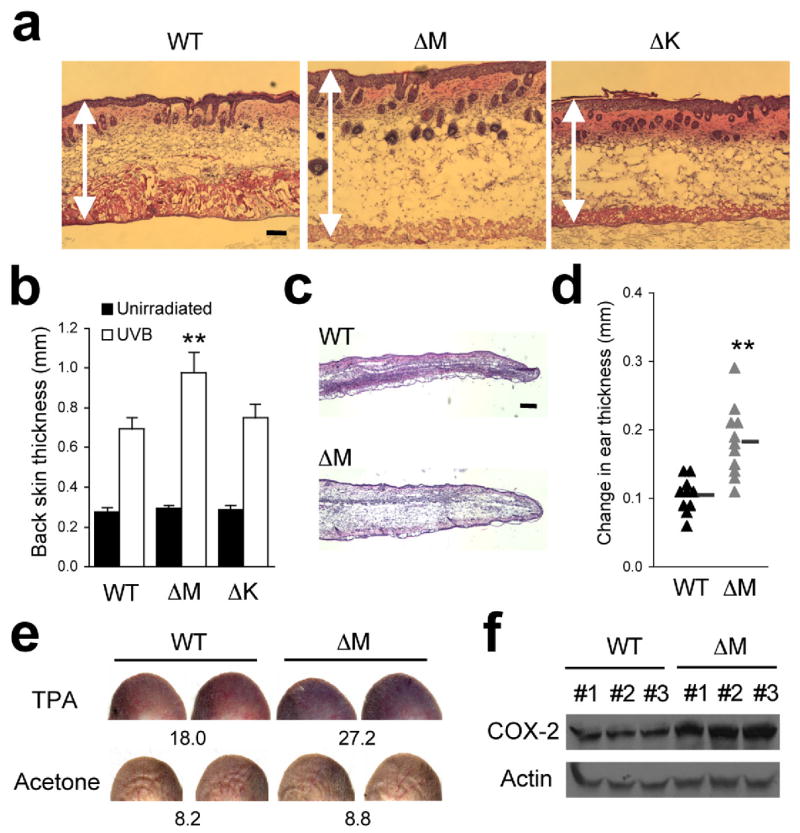
(a) The shaved back skin of mice was irradiated with UVB as in Fig. 3. Skin tissue sections were prepared from the indicated mice 48 h after irradiation and analyzed by H&E staining. Double-head arrows indicate skin thickness. Scale bar, 100 μm. Data are representative of at least three independent experiments.
(b) The thickness of back skin without or with UVB irradiation is measured. Data represent mean ± standard error (n=5; **P = 0.0015 relative to WT-UVB).
(c) Skin tissue sections from the auricles of the indicated mice were prepared 24 h after TPA treatment and analyzed by H&E staining. Scale bar, 100 μm. Data are representative of analysis of five animals of each genotype.
(d) Changes in ear thickness of individual mice before and after TPA treatment were determined (triangle). The horizontal bars denote the mean values in the two groups (**P=0.00024).
(e) Evans Blue was injected intravenously into mice 6 h after treatment with TPA (right auricle) or acetone (left auricle). The auricles were photographed 30 min after dye injection. The values at the bottom of auricle images indicate relative dye leakage. Data are representative of two independent experiments.
(f) Auricle skin extracts were prepared 6 h after TPA treatment and analyzed by immunoblotting with anti-COX-2 and anti-actin. Data are from one experiment involving three animals of each genotype. The numbers indicate individual animals.
p38α-dependent gene expression in macrophages
The data described thus far show that both myeloid- and epithelial p38α serve to initiate inflammatory responses depending on the mode of tissue insult. In addition, myeloid p38α exerted negative regulatory effects on certain inflammatory processes. The dual role of myeloid p38α in the coordination of inflammation may be partly attributed to a functional diversity of genes whose expression in macrophages is regulated by p38α. We therefore focused our subsequent analyses on identifying the role of p38α in effecting inflammatory gene expression in macrophages.
In an inflammatory milieu, tissue-resident macrophages are exposed to complex p38 MAPK-activating stimuli that include cytokines, oxidants, and products of necrotic cells and invading microbes. For our gene expression analysis, lipopolysaccharide (LPS) was used to activate p38 signaling in cultured macrophages, as its receptor, Toll-like receptor 4 (TLR4), serves as a sensor for the detection of both septic and aseptic injury and is a potent inducer of inflammatory gene transcription. We performed quantitative real-time PCR (qPCR) to measure the expression of a panel of genes whose fold-induction by LPS is at least 5-fold and ranges up to several thousand-fold (data not shown). Of 48 LPS-induced genes analyzed, expression of 7 genes was greatly diminished (to 36% or lower relative to WT) in p38αΔM BMDMs (Fig. 5a). Those genes exhibited a similar p38α dependence in PEMs (Supplementary Fig. 6, online). We also examined in parallel LPS-induced gene expression in IκB kinase β (IKKβ)-null fetal liver-derived macrophages, which are defective in NF-κB-dependent transcription, and found that 77% of the tested genes were IKKβ-dependent by the same criteria for target gene assignment (Fig. 5a). Therefore, in contrast to a very broad target spectrum of IKKβ-NF-κB signaling, p38α is required for the induction of a relatively restricted number of genes in macrophages.
Figure 5. p38α is required for transcriptional induction of specific genes in LPS-treated macrophages.
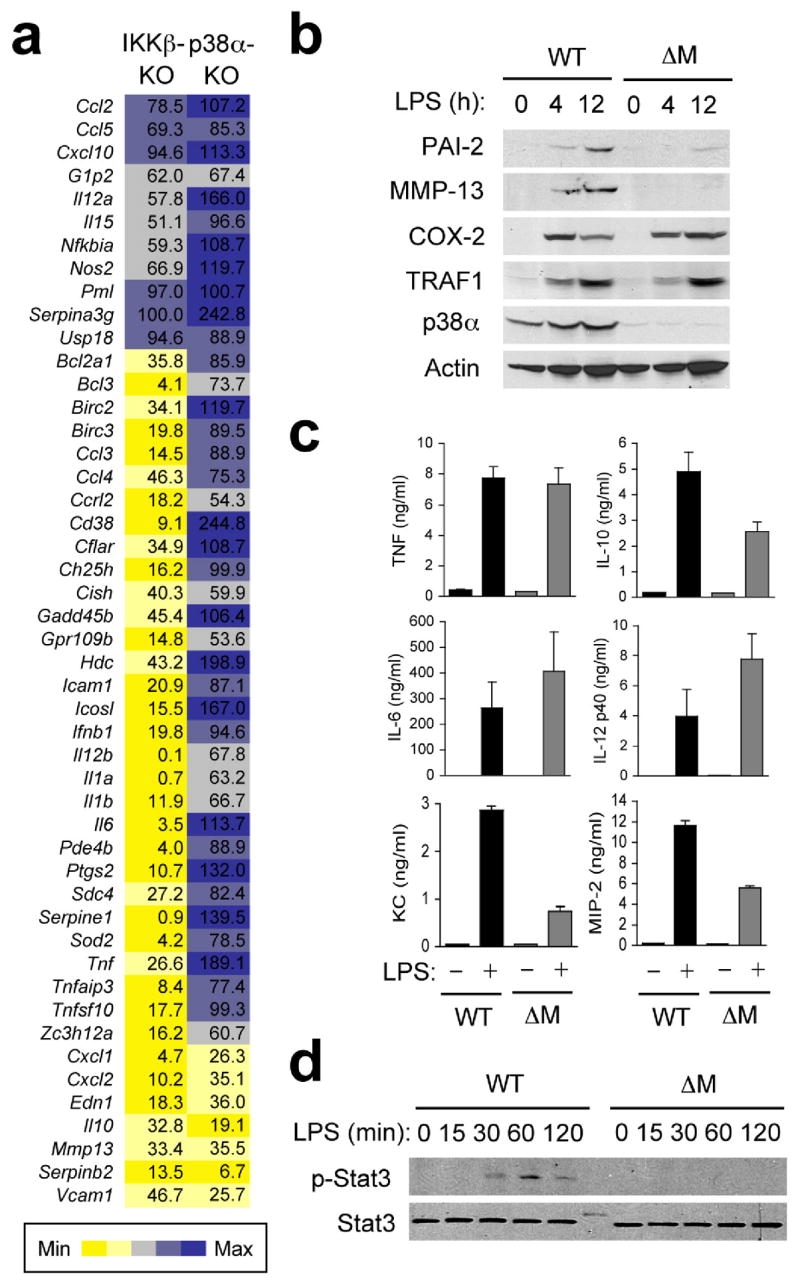
(a) Gene expression in Ikbkb−/−FLDMs (IKKβ-KO), and p38αΔM BMDMs (p38α KO) 4 h after treatment with LPS (100 ng/ml) was analyzed by qPCR. The values represent percentage of mRNA in these samples relative to WT samples.
(b) WT and p38αΔM BMDMs were treated with LPS. At the indicated time points, whole cell lysates were prepared and analyzed by immunoblotting with antibodies against the proteins indicated on the left. PAI-2, MMP-13, COX-2, and TRAF1 are encoded by Serpinb2, Mmp13, Ptgs2, and Traf1, respectively. Data are representative of two independent experiments.
(c) Culture supernatants of WT and p38αΔM BMDMs (n=3) were collected 12 h after LPS treatment and cytokine and chemokine concentrations were measured.
(d) WT and p38αΔM BMDMs were treated with LPS and analyzed by immunoblotting as in (b). Data are representative of three independent experiments.
The p38α-dependent genes identified in this screen include Cxcl1 and Cxcl2, the chemokine genes underexpressed in SDS-treated p38αΔM skin (Fig. 2d), Serpinb2, a previously reported p38 target gene27, Edn1, Il10, Mmp13, and Vcam1. The role of p38α in mediating LPS-induced expression of these genes was confirmed by decreased production of their protein products in LPS-treated p38αΔM macrophages (Fig. 5b, c). In particular, reduced IL-10 release from p38α-deficient macrophages was accompanied by abrogation of Stat3 phosphorylation in the cells (Fig. 5d), an event effected by autocrine IL-10 action28.
Inflammatory phenotypes of p38αΔM mice may arise from a reduction of p38α target gene expression in macrophages. In particular, the impaired neutrophil recruitment in SDS-treated p38αΔM skin (Fig. 2c) is likely attributable to the defect of macrophages to express KC and MIP-2. We considered the alternative explanation that ablation of myeloid p38α might disturb the chemotactic motility of neutrophils, which express the LysMCre transgene. This possibility was, however, ruled out, as neutrophils isolated from WT and p38αΔM mice showed similar migration in gradients of KC and MIP-2 (Supplementary Fig. 7, online). Production of the chemokines in UVB-irradiated keratinocytes also occurred via a p38α-dependent mechanism (Fig. 3e) and was associated with UVB-induced neutrophil infiltration (Fig. 3b). Therefore, although distinct inflammatory responses may involve different cellular sources of the chemokines, infiltration of neutrophils in the inflamed tissue appears to rely on p38α-dependent production of their chemoattractants.
The excessive edema in the UVB- and TPA-challenged skin of p38αΔM mice could conceivably be due to failure of a p38α-mediated anti-inflammatory mechanism. We have identified IL-10, a cytokine with strong anti-inflammatory properties, as a target gene of myeloid p38α signaling. Of note, previous studies demonstrated a crucial role for IL-10 in moderating edema and other inflammatory reactions in chemically irritated, and UVB-damaged skin29,30. It was also shown that removal of myeloid-derived but not T cell-derived IL-10 exacerbated the skin pathology resulting from subcutaneous LPS injection31. Therefore, skin-resident macrophages appear to impose restraint on vascular permeability at least partly through p38α-dependent IL-10 induction.
Feedback regulation of p38 by Dusp1
Most inflammatory stimuli that induce p38 MAPK activity also trigger other intracellular signaling cascades. We examined whether p38α deficiency affects NF-κB, ERK, and JNK activation in response to LPS and UVB. In both macrophages and keratinocytes challenged by the respective stimuli, rapid degradation of IκBα and nuclear translocation of the NF-κB proteins RelA and c-Rel occurred independently of p38α (Supplementary Fig. 8, online and data not shown). By contrast, p38α-deficient macrophages exhibited an enhanced and prolonged phosphorylation of ERK and JNK in response to LPS (Fig. 6a). Similarly, removal of p38α resulted in constitutive ERK phosphorylation in cultured keratinocytes, and increased the magnitude of their JNK activation upon UVB exposure (Fig. 6b). These observations are consistent with the role of p38 MAPK in antagonizing ERK and JNK signaling in a variety of cell types examined by others19,32–35.
Figure 6. p38α signaling limits the activation of other MAP kinase signaling pathways.
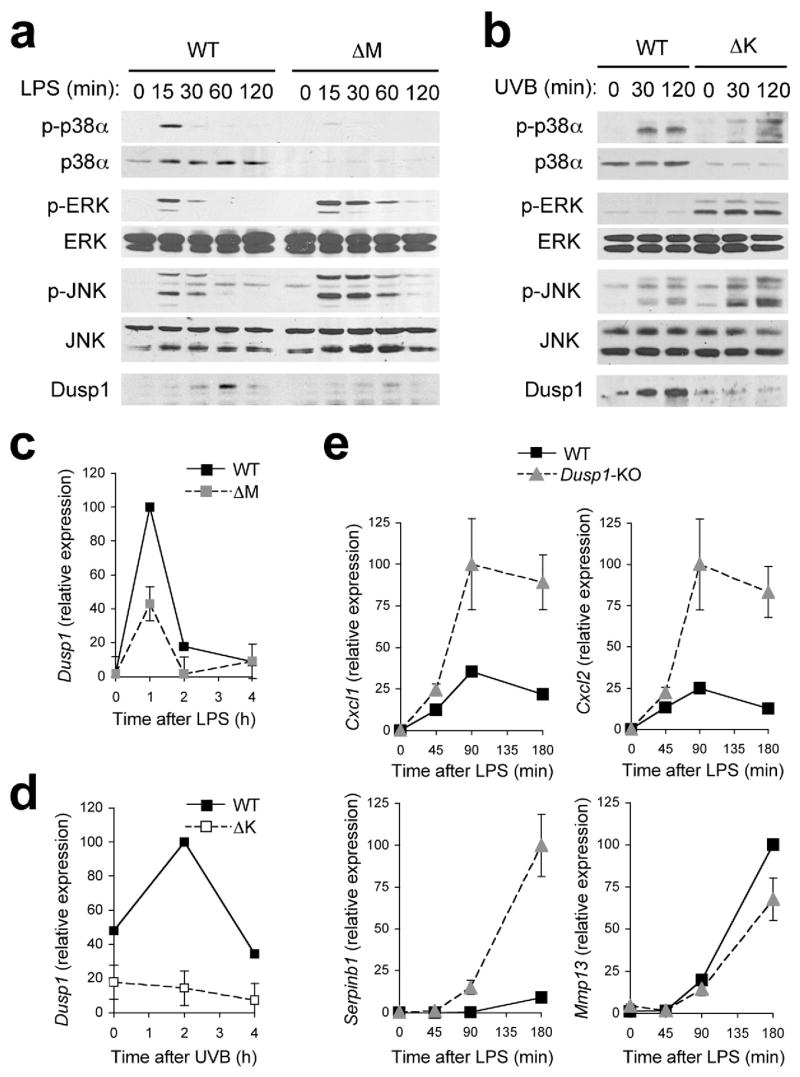
(a, b) Whole cell lysates from BMDMs treated with LPS (a), and keratinocytes irradiated with UVB (b) were prepared after the indicated durations of stimulation and analyzed by immunoblotting with antibodies against the proteins indicated on the left. Data are representative of at least five independent experiments.
(c, d) Dusp1 mRNA expression in BMDMs (c) and keratinocytes (d) at the indicated time points following LPS treatment (c) and UVB irradiation (d) was analyzed by qPCR. Data are representative of two independent experiments.
(e) Expression of genes in WT and Dusp1-KO BMDMs at the indicated time points following LPS treatment was analyzed by qPCR. Data are representative of two independent experiments.
Possible mechanisms linking p38α deficiency and JNK hyperactivation include inadequate expression or activity of endogenous JNK inhibitors such as MAPK phosphatases (MKPs). It was previously reported that MKPs are sensitive to oxidative inactivation by reactive oxygen species (ROS)36, and that p38α acts to counter ROS accumulation in cells transformed by certain oncogenes37. In cultured macrophages, however, p38α deficiency did not lead to an elevation in either basal or LPS-induced ROS expression (Supplementary Fig. 9, online). However, p38α-deficient macrophages showed greatly diminished induction of Dusp1--an MKP that deactivates JNK and p38 MAPK38–41--protein and mRNA (Fig. 6a, c). Dusp1 was induced only transiently in LPS-stimulated macrophages and thus escaped detection in our screen for p38α target genes whose induction is sustained up to 4 h post-LPS treatment (Fig. 5a). Dusp1 protein and mRNA induction in UVB-irradiated keratinocytes was also highly dependent on p38α (Fig. 6b, d). Rapid induction of Dusp1 in LPS- and UVB-exposed cells likely contributes to the cross-inhibitory effect of p38α signaling on JNK activation although there may exist other mechanisms that function in parallel. Furthermore, Dusp1 induction by p38α signaling represents a regulatory loop in which activation of an inflammatory signaling module is restrained by the action of its own output. Failure of this self-limiting mechanism may contribute to hyperinflammatory phenotypes resulting from p38α removal, especially in p38αΔM mice. Consistent with this notion, loss of Dusp1 was sufficient to accentuate the LPS-induced expression of several but not all p38α target genes in macrophages (Fig. 6e).
Transcriptional regulation downstream of p38α
p38 MAPK signaling influences multiple tiers of gene expression. We first explored whether the decreased expression of specific LPS-inducible genes in p38α-deficient macrophages was due to reduced mRNA stability. To this end, LPS-stimulated WT and p38αΔM macrophages were incubated with the RNA synthesis inhibitor actinomycin D, and changes in the abundance of mRNAs analyzed over time. None of the p38α target gene transcripts examined showed a more rapid decay in p38αΔM macrophages (Supplementary Fig. 10, online), suggesting that the effect of p38α deficiency on target mRNA expression might instead be attributable to transcriptional mechanisms. In this regard, we noted that MSK1 and MSK2 have been shown to relay transcription activation signals from p38 MAPK to various components of the transcriptional machinery. We therefore tested the role of the two MSKs in p38α target gene expression by using BMDMs prepared from Msk1, Msk2-DKO (double knockout) mice. A subset of the p38α target genes tested was strictly dependent on MSKs for LPS-induced mRNA expression; expression of Il10, Dusp1, Serpinb2, and Edn1 was almost abolished or significantly reduced in the double knockout cells while that of the other target genes, Cxcl1, Cxcl2, Mmp13, and Vcam1, was unaffected (Fig. 7a). This observation indicates that p38α delivers transcriptional activation signals to at least two distinct downstream regulatory modules: MSK-dependent and -independent.
Figure 7. Transcriptional induction of a subset of p38α target genes depends on MSKs.
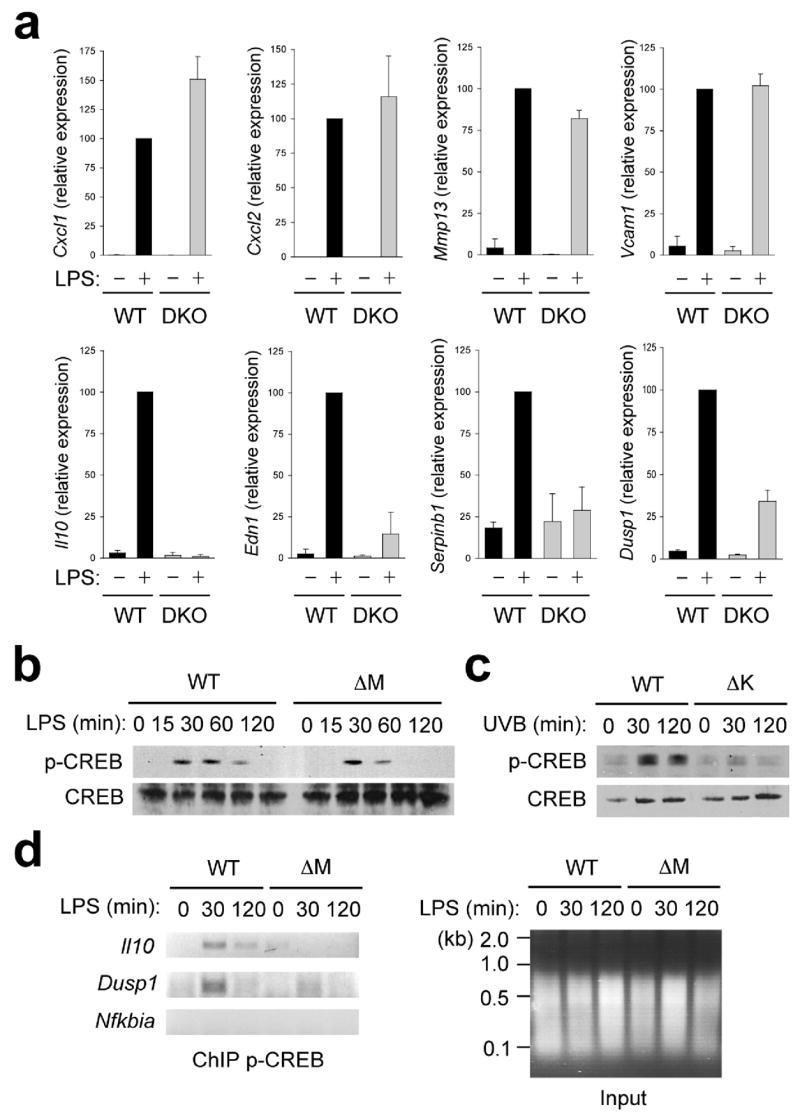
(a) Expression of genes in WT and Msk1, Msk2-DKO BMDMs at 0 and 4 h after LPS treatment was analyzed by qPCR. Data are representative of three independent experiments.
(b, c) Whole cell lysates from BMDMs treated with LPS (b), and keratinocytes irradiated with UVB (c) were prepared after the indicated durations of stimulation and analyzed by immunoblotting with antibodies against phosphorylated and total CREB proteins. Data are representative of four (b) or two (c) independent experiments.
(d) WT and p38αΔM BMDMs were treated with LPS for the indicated durations and analyzed by ChIP. The extent of promoter-bound phospho-CREB was determined by PCR analysis with primers specific to the indicated gene promoters. The chromatin preparations used in the immunoprecipitation (input) were separated by agarose gel electrophoresis and visualized by ethidium bromide. Data are representative of three independent experiments.
Among the transcription factors activated through direct phosphorylation by MSKs is the transcription factor CREB27. CREB-binding sites were found in the promoter sequences of all MSK-dependent p38α target genes identified (data not shown). A role for CREB in transcriptional activation of Il10, Dusp1, and Serpinb2 was previously demonstrated in the context of LPS-induced gene expression27,42,43. Induction of CREB phosphorylation in response to LPS and UVB was very robust in WT macrophages and keratinocytes, but much weaker or completely blocked in their p38α-deficient counterparts (Fig. 7b, c). The residual phosphorylation of CREB in p38αΔM macrophages (Fig. 7b) was suppressed by the ERK pathway inhibitor PD98059 (data not shown), showing a parallel contribution of ERK in LPS-induced CREB activation. The defect of CREB activation in p38αΔM macrophages was associated with reduced occupancy of the Il10 and Dusp1 promoters by phophorylated CREB (Fig. 7d). It is noteworthy that the genes induced via the p38α-MSK-CREB axis encode extracellular or intracellular regulators with known anti-inflammatory properties. Taken together, our gene expression data suggest a dual role of p38α in regulation of inflammatory gene expression (Supplementary Fig.11, online).
DISCUSSION
Here we analyzed the effect of cell type-specific ablation of p38α on inflammatory responses in mice. Our results illustrate that p38α can signal to induce inflammatory gene expression in both myeloid and epithelial cells, such as dermal macrophages and keratinocytes, and can thus provide an initiating cue for local inflammatory reactions from different cellular origins. However, p38α signaling in each cell type served distinct inflammatory functions, and varied depending on the nature of tissue injury. For instance, myeloid p38α was strictly required for inflammatory infiltration, epidermal hyperproliferation, and hyperkeratosis in our model of chronic SDS skin injury. On the other hand, acute inflammatory responses to UVB-induced epidermal injury were driven by p38α signaling in keratinocytes while myeloid p38α acted to attenuate UVB-induced vascular hyperpermeability. In all likelihood, stimulus-specific modes of p38α activation determine the cell type in which p38α target genes are first induced and exert their inflammatory function.
Our gene expression analysis identified KC and MIP-2 as induced in both macrophages and keratinocytes via a p38α-dependent mechanism. The impaired production of these neutrophil chemoattractants in p38αΔM macrophages and p38αΔK keratinocytes likely accounts for the reduced Gr-1+ infiltration in the respective mutant mice. Myeloid p38α also signaled to activate anti-inflammatory gene expression. In particular, the genes encoding IL-10 and Dusp1 may mediate the role of myeloid p38α in moderating acute vascular activation in epidermally injured skin. Intriguingly, p38α-dependent induction of these anti-inflammatory genes relied on the downstream MSK module. Serpinb2, another MSK-dependent p38α target gene, may also mediate the anti-inflammatory regulation exerted by myeloid p38α because plasminogen activator inhibitor-2 (PAI-2), the product of Serpinb2, was recently shown to inhibit IL-1β secretion in macrophages44. A critical role for MSK-dependent gene expression in negative regulation of inflammation is dramatically illustrated by the hyperinflammatory phenotype of Msk1, Msk2-DKO mice45. The divergence of pro- and anti-inflammatory signaling modules in the p38 MAPK pathway leaves room for selective targeting of the inflammatory processes governed by each signaling module.
Intracellular signaling pathways that are concurrently activated by the same stimulus often interact with one another via cross-regulatory feedback mechanisms. Signaling in such a molecular ‘network’ comprises the basis for coordinated cellular responses and, simultaneously, for unanticipated collateral effects of pharmacological signaling modifiers. We found that, in both macrophages and keratinocytes, p38α ablation led to enhanced activation of ERK and JNK. As has been discussed previously13, the p38α inhibition of ERK activation may involve RSKs suppressing a signaling step upstream of ERK although this idea remains to be experimentally verified. We expect that p38α-dependent Dusp1 induction in LPS- and UVB-exposed cells contributes to switching off both JNK and p38 MAPK signaling cascades. In addition, p38α has been shown to inactivate the protein kinase complex containing TAK1, a MAPK kinase kinase acting upstream of both JNK and p38 MAPK, through direct phosphorylation of its regulatory subunit, TAK1-binding protein 133. Hence, it appears that p38α imposes restraint on JNK signaling as well as its own activation at multiple levels.
Pharmacological inhibition of p38 MAPK has proved effective in treating or alleviating various inflammatory conditions5. However, the toxicity and undesired side-effects of p38 MAPK inhibitors have been recognized as well46. Adverse effects of anti-p38 MAPK therapy may arise from perturbed cross-regulatory signaling or self-limiting mechanisms that rely on p38α activity. In this respect, our findings have important implications for the clinical intervention of the p38 MAPK pathway; specific inhibition of a signaling module or a regulatory target that functions downstream of p38α may offer a more efficacious and selective anti-inflammatory strategy.
METHODS
Animals
Mapk14fl/fl and LysM-Cre mice were described previously22,23. p38αΔM mice were on the C57BL/6J background as were their parental strains. After derived from Mapk14fl/fl and K14-Cre mice24, p38αΔK mice were crossed into the C57BL/6J background for 6 generations. All animal studies were conducted under IACUC-approved protocols.
Primary cell culture
Primary macrophages47 and keratinocytes48 were prepared and cultured as described. The extent of Mapk14 deletion and p38α protein removal in each cell preparation were determined by quantitative PCR (qPCR) analysis of exon 2 (the floxed region of the gene) and immunoblotting, respectively. BMDM preparations from p38αΔM mice exhibited varying degrees of Mapk14 deletion, ranging from 85 to 97%, depending on the duration of differentiation and the cell density of culture batches. Cell preparations with gene deletion higher than 95% were further used for in vitro studies.
Skin inflammation models
To induce chronic disruption of skin barrier, 5% SDS solution in phosphate-buffered saline (PBS) was applied daily to the shaved back skin of mice for a period of 7 days. In the model of acute UVB injury, the shaved back skin was exposed to 160 mJ/cm2 UVB using UVB bulbs (Southern N.E. Ultraviolet Co.) and a Kodacel filter (Eastman Kodak Co.). The UV dose was monitored with a radiometer (International Light, Inc.). Skin samples were prepared from SDS- and UVB-challenged mice at indicated time points for histological and molecular analysis. For chemically induced skin irritation, 2 μg of TPA in 20 μl of acetone was applied to the right auricle and 20 μl of acetone to the left auricle.
Histology and immunofluorescence
Mouse skin samples were frozen in liquid nitrogen, and embedded in OCT medium. 5 μm sections mounted on slides were stained with H&E or with antibodies against the following markers: K14 (PRB-155P; Covance), loricrin (PRB-145P; Covance), Gr-1 (553125; BD Pharmingen), F4/80 (MCA497B; Serotec), TNF (557719; BD Pharmigen) or Ki67 (M7249; Dako). Secondary antibodies coupled to AlexaFluor488/594 (Molecular Probes) were used to visualize primary antibody-bound markers. Sections were counterstained with phalloidin-TRITC (Sigma) and with Hoescht (Molecular Probes) to visualize cell bodies and nuclei. TUNEL staining was performed using the In Situ Cell Death Detection kit (Roche).
MPO assay
To determine the extent of dermal neutrophil infiltration, skin samples were processed, and the MPO activity determined as described49.
Measurement of vascular permeability
100 μl of 1% Evans Blue in PBS was injected intravenously into the lateral tail vein of mice. The auricle samples were photographed 30 min after dye injection. To quantify extravascular dye content, auricle skin samples were minced, immersed in formamide at 0.5 ml per 100 mg tissue, and incubated for 16 h with vigorous shaking. After removing debris by centrifugation, the absorbance of the solution was measured at 620 nm.
Protein and RNA analysis
Whole-cell extracts for immunoblot analysis were prepared and analyzed as described29. Antibodies against the following proteins were used in immunoblotting: phospho-p38 (9211), phospho-JNK (9251), phospho-ERK (9101), ERK (9102) and CREB (9192; all from Cell Signaling Technology); p38α (sc-535), phospho-Stat3 (sc-8059), Stat3 (sc-482), RelA (sc-372), c-Rel (sc-71), Dusp1 (sc-1199) and TRAF1 (sc-874; all from Santa Cruz Biotechnology); JNK (554285, BD Pharmingen); phospho-CREB (AF2510, R&D Systems); COX-2 (160126, Cayman Chemical); actin (A4700; Sigma); PAI-2 (a gift from David Ginsburg); and MMP-13 (a gift from Stephen Krane). Secreted proteins in culture media were assayed by ELISA (eBioscience) and SearchLight protein array analysis (Pierce). Total RNA was isolated using Trizol (Invitrogen). RNA analysis using qPCR was performed as described29. Individual primer sequences are in the Supplementary Table online.
Chromatin immunoprecipitation
Cells were fixed with formaldehyde, and chromatin was isolated and subjected to mechanical sheering and immunoprecipitation as described50. Anti-phospho-CREB (R&D Systems) was used to immunoprecipitate specific chromatin DNA fragments. The sequences of the promoter-specific primers are in the Supplementary Table online.
Statistical Analysis
P-values between a pair of datasets were obtained from two-tailed Student’s t-test. Gene expression (qPCR) data are shown as the average ± standard deviation.
Supplementary Material
Acknowledgments
We thank S. Krane for the MMP-13-specific antibody and advice on its use; D. Ginsburg for the PAI-2-specific antibody; R. Bravo and C. Caelles for Dusp1-KO mice; and M. Karin for discussion about JNK activation mechanisms in p38α-deficient cells. Supported by the Cutaneous Biology Research Center through the Massachusetts General Hospital/Shiseido Co. Ltd. Agreement (J.M.P.), the National Institutes of Health (DK043351 to D. Podolsky) and the Center for the Study of Inflammatory Bowel Disease at Massachusetts General Hospital (J.M.P.).
References
- 1.Lee JC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 2.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–8. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 3.Rouse J, et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 4.Freshney NW, et al. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994;78:1039–1049. doi: 10.1016/0092-8674(94)90278-x. [DOI] [PubMed] [Google Scholar]
- 5.Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 6.Ashwell JD. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol. 2006;6:532–540. doi: 10.1038/nri1865. [DOI] [PubMed] [Google Scholar]
- 7.Goedert M, Cuenda A, Craxton M, Jakes R, Cohen P. Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6); comparison of its substrate specificity with that of other SAP kinases. EMBO J. 1997;16:3563–3571. doi: 10.1093/emboj/16.12.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, et al. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- 9.Jiang Y, et al. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38δ. J Biol Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- 10.Hale KK, Trollinger D, Rihanek M, Manthey CL. Differential expression and activation of p38 mitogen-activated protein kinase α, β, γ, and δ in inflammatory cell lineages. J Immunol. 1999;162:4246–4252. [PubMed] [Google Scholar]
- 11.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 12.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaru R, Ronkina N, Gaestel M, Arthur JS, Watts C. The MAPK-activated kinase Rsk controls an acute Toll-like receptor signaling response in dendritic cells and is activated through two distinct pathways. Nat Immunol. 2007;8:1227–1235. doi: 10.1038/ni1517. [DOI] [PubMed] [Google Scholar]
- 14.Karaman MW, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 15.Tamura K, et al. Requirement for p38α in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102:221–231. doi: 10.1016/s0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- 16.Adams RH, et al. Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6:109–116. [PubMed] [Google Scholar]
- 17.Allen M, et al. Deficiency of the stress kinase p38α results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J Exp Med. 2000;191:859–870. doi: 10.1084/jem.191.5.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mudgett JS, et al. Essential role for p38α mitogen-activated protein kinase in placental angiogenesis. Proc Natl Acad Sci USA. 2000;97:10454–10459. doi: 10.1073/pnas.180316397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hui L, et al. p38α suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39:741–749. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- 20.Ventura JJ, et al. p38α MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat Genet. 2007;39:750–758. doi: 10.1038/ng2037. [DOI] [PubMed] [Google Scholar]
- 21.Beardmore VA, et al. Generation and characterization of p38β (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454–10464. doi: 10.1128/MCB.25.23.10454-10464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishida K, et al. p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24:10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 24.Jonkers J. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 25.Thepen T, et al. Resolution of cutaneous inflammation after local elimination of macrophages. Nat Biotech. 2000;18:48–51. doi: 10.1038/71908. [DOI] [PubMed] [Google Scholar]
- 26.Cramer T, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JM, et al. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis--CREB and NF-κB as key regulators. Immunity. 2005;23:319–329. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 28.Carl VS, Gautam JK, Comeau LD, Smith MF., Jr Role of endogenous IL-10 in LPS-induced STAT3 activation and IL-1 receptor antagonist gene expression. J Leukoc Biol. 2004;76:735–742. doi: 10.1189/jlb.1003526. [DOI] [PubMed] [Google Scholar]
- 29.Berg DJ, et al. Interleukin 10 but not interleukin 4 is a natural suppressant of cutaneous inflammatory responses. J Exp Med. 1995;182:99–108. doi: 10.1084/jem.182.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol. 2007;8:1095–1104. doi: 10.1038/ni1503. [DOI] [PubMed] [Google Scholar]
- 31.Siewe L, et al. Interleukin-10 derived from macrophages and/or neutrophils regulates the inflammatory response to LPS but not the response to CpG DNA. Eur J Immunol. 2006;36:3248–3255. doi: 10.1002/eji.200636012. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Shi X, Hampong M, Blanis L, Pelech S. Stress-induced inhibition of ERK1 and ERK2 by direct interaction with p38 MAP kinase. J Biol Chem. 2001;276:6905–6908. doi: 10.1074/jbc.C000917200. [DOI] [PubMed] [Google Scholar]
- 33.Cheung PC, Campbell DG, Nebreda AR, Cohen P. Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J. 2003;22:5793–5805. doi: 10.1093/emboj/cdg552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathur RK, Awasthi A, Wadhone P, Ramanamurthy B, Saha B. Reciprocal CD40 signals through p38MAPK and ERK-1/2 induce counteracting immune responses. Nat Med. 2004;10:540–544. doi: 10.1038/nm1045. [DOI] [PubMed] [Google Scholar]
- 35.Perdiguero E, et al. Genetic analysis of p38 MAP kinases in myogenesis: fundamental role of p38α in abrogating myoblast proliferation. EMBO J. 2007;26:1245–1256. doi: 10.1038/sj.emboj.7601587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamata H, et al. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 37.Dolado I, et al. p38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 38.Salojin KV, et al. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol. 2006;176:1899–1907. doi: 10.4049/jimmunol.176.3.1899. [DOI] [PubMed] [Google Scholar]
- 39.Chi H, et al. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA. 2006;103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Q, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammer M, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. 2006;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu X, et al. IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 44.Greten FR, et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ananieva O, et al. MSKs act as negative regulators of TLR signaling. Co-submitted paper. [Google Scholar]
- 46.Dambach DM. Potential adverse effects associated with inhibition of p38α/β MAP kinases. Curr Top Med Chem. 2005;5:929–939. doi: 10.2174/1568026054985911. [DOI] [PubMed] [Google Scholar]
- 47.Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–2051. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- 48.Hu Y, et al. IKKα controls formation of the epidermis independently of NF-κB. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 49.Vakeva AP, et al. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: role of the terminal complement components and inhibition by anti-C5 therapy. Circulation. 1998;97:2259–2267. doi: 10.1161/01.cir.97.22.2259. [DOI] [PubMed] [Google Scholar]
- 50.Saccani S, Natoli G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002;16:2219–2224. doi: 10.1101/gad.232502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.