

Abstract
Carboxylate-bridged diiron(II) centers activate dioxygen for the selective oxidation of hydrocarbon substrates in bacterial multicomponent monooxygenases. Synthetic analogues of these systems exist in which substrate fragments tethered to the diiron(II) core through attachment to an N-donor ligand are oxidized by transient species that arise following introduction of O2 into the system. The present study describes the results of experiments designed to probe mechanistic details of these oxidative N-dealkylation reactions. A series of diiron(II) complexes, having ligands N,N-(4-R-Bn)Bnen, where en is ethylenediamine, Bn is benzyl, and R-Bn is benzyl with a para-directing group R = Cl, F, CH3, t-Bu, or OCH3, were prepared. A Hammett plot of the oxygenation product distributions of these complexes, determined by gas chromatographic analysis, reveals a small positive slope, ρ = + 0.48. Kinetic isotope effect (KIEintra) values for oxygenation of [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(C6H5CDH)2en)2] and [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(C6H5CD2)(C6H5CH2)en)2] are 1.3 (1) and 2.2 (2) at 23 °C, respectively. The positive slope ρ and low KIEintra values are consistent with a mechanism involving one-electron transfer from the dangling nitrogen atom in N,N-Bn2en to a transient electrophilic diiron intermediate, followed by proton transfer and rearrangement to eliminate benzaldehyde.
Introduction
The activation of triplet ground state dioxygen and the hydroxylation of unactivated C–H bonds under ambient conditions by iron- and copper-containing metal-loenzymes have attracted considerable attention.1-4 Monooxygenase enzymes, which catalyze the hydroxylation of aliphatic hydrocarbons, are of particular interest because of their unique ability to activate the C–H bond in methane.5,6 The hydroxylase component of methane monooxygenase (MMOH) is one such enzyme that reacts with O2, generating reactive intermediate(s)7 for insertion of an oxygen atom into the C–H bond of a substrate inherently difficult to activate. Understanding such a remarkable process in molecular detail is an important objective that may lead to new insights into how the metalloproteins work and provide clues for the design of catalysts for O2 activation and C–H bond hydroxylation.
The active site of MMOH utilizes diiron sites, coordinated by four glutamate and two histidine ligands, as a working module to achieve its physiological function.8 Although a number of small model complexes duplicate the composition of the diiron(II) core in MMOH,9-14 none of these can yet mimic its chemistry.9,15-17 The carboxylate-rich composition of the diiron site has been reproduced with the use of sterically hindered terphenyl-derived carboxylate ligands.9-12,17-19 When the diiron(II) complex [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-Bn2en)2] (1), where ArTolCO2- is 2,6-di(p-tolyl)benzoate and N,N-Bn2en is N,N-dibenzylethylenediamine, was exposed to dioxygen, it converted to [Fe2(μ-OH)2(μ-O2CArTol)(O2CArTol)3(N,N-Bn2en)(N-Bnen)], a structural homologue of the active site of oxidized MMOH, with concomitant formation of benzaldehyde (Scheme 1).9,15 This oxidative N-dealkylation of a substrate tethered to a carboxylate-bridged diiron(II) complex by reaction with dioxygen has been a highlight in the pursuit of structural and functional MMOH model complexes.
Scheme 1.

The stoichiometric correspondence of 1 with the diiron active site of MMOH and early experimental results suggested that the N-dealkylation reaction might involve an oxidant similar to the di(μ-oxo)diiron(IV) intermediate in the MMOH reaction cycle.9,15 Three possible pathways were suggested for insertion of the oxygen atom from such a high-valent intermediate into the benzylic C–H bond of the N,N-Bn2en ligand. Concerted insertion of an oxygen atom into the C–H bond (Top, Scheme 1) and conventional recoil with hydrogen atom transfer (HAT) followed by rebound (Middle, Scheme 1) for oxygenation of 1 follow commonly proposed mechanisms for the MMOH catalytic cycle.20 Sequential single electron transfer (SET), deprotonation, and internal electron transfer (Bottom, Scheme 1) can equally well account for the observed chemistry. This possibility was suggested by the proximity of the lone pair electrons on the uncoordinated nitrogen atom of the N,N-Bn2en ligand to the proposed electrophilic high-valent intermediate and also by the precedence for such chemistry in heme iron oxidations.21
The work described here was performed to provide additional experimental detail about the hydroxylation reaction through the synthesis and characterization of diiron(II) complexes analogues having N,N-Bn2en ligands with a variety of para substituents on the phenyl ring or deuterium atoms at the benzylic carbon. A Hammett analysis and measurement of intramolecular deuterium kinetic isotope effects (KIEs), both based on product distributions, provided the desired information, as presented and discussed in this report.
Experimental Section
General Considerations
All reagents were obtained from commercial suppliers and used as received unless otherwise noted. Diethylether, CH2Cl2 and pentanes were saturated with argon and purified by passage through activated Al2O3 columns under argon.22 Dioxygen (99.994%, BOC Gases) was dried by passing the gas stream through a column of Drierite. The syntheses and characterization of N,N-Bn2en derivatives are reported in Supporting Information. The compounds α-d2-benzylamine,23 [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2],10 and [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-Bn2en)2]9,15 were prepared as described in the literature. Deuterated reagents were all > 95% pure, as judged by NMR spectroscopy. All syntheses and air-sensitive manipulations were carried out under nitrogen in an MBraun glovebox.
Physical Measurements
1H and 13C NMR spectra were recorded on a Bruker 400 spectrometer in the Massachusetts Institute of Technology Department of Chemistry Instrument Facility (DCIF); chemical shifts were referenced to residual solvent peaks. FT-IR spectra were recorded either with a Thermo Nicolet Avatar 360 or Perkin Elmer FTIR model 2000 spectrophotometer.
Synthesis of [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(4-R-Bn)Bnen)2] Complexes
To a rapidly stirred CH2Cl2 (3 mL) solution of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (37 mg, 0.026 mmol) were added 2 equiv of N,N-(4-R-Bn)Bnen or deuterated N,N-Bn2en. After 2 h, the solution was filtered through Celite and reduced to 3 mL under partial vacuum. Colorless block crystals were isolated upon the vapor diffusion of pentane into the solution and characterized by X-ray crystallography and elemental analysis.
[Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(4-Cl-Bn)Bnen)2] (2)
Yield 68 %. FT-IR (KBr, cm-1) 3321 (m), 3250 (w), 3056 (w), 3027 (m), 2920 (m), 2875 (w), 2835 (m), 1609 (s), 1549 (s), 1515 (s), 1489 (m), 1456 (s), 1408 (m), 1384 (s), 1346 (w), 1186 (w), 1098 (w), 1075 (m), 1018 (m), 856 (m), 837 (m), 818 (m), 801 (s), 784 (m), 764 (m), 735 (m), 704 (m), 610 (m), 584 (m), 546 (w), 522 (m), 456 (w). Anal. Calcd for 1·0.5(CH2Cl2) or C116.5Cl3H107Fe2N4O8: C, 73.29; H, 5.65; N, 2.93. Found: C, 73.21; H, 5.42; N, 2.67.
[Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(4-F-Bn)Bnen)2] (3)
Yield 46 %. FT-IR (KBr, cm-1) 3316 (m), 3268 (w), 3057 (w), 3024 (w), 2919 (s), 2861 (w), 1605 (s), 1547 (s), 1508 (s), 1454 (s), 1410 (m), 1383 (s), 1307 (w), 1263 (w), 1221 (s), 1187 (w), 1152 (w), 1110 (w), 1070 (w), 1041 (w), 1020 (w), 974 (w), 855 (w), 819 (s), 801 (s), 784 (s), 765 (s), 736 (s), 700 (s), 584 (m), 546 (m), 521 (m). Anal. Calcd for 3, C116F2H106Fe2N4O8: C, 75.98; H, 5.83; N, 3.06. Found: C, 75.45; H, 5.83; N, 3.50.
[Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(4-Me-Bn)Bnen)2] (4)
Yield 67 %. FT-IR (KBr, cm-1) 3319 (m), 3254 (w), 3057 (w), 3023 (w), 2917 (w), 2790 (w), 1604 (s), 1565 (s), 1514 (s), 1454 (s), 1410 (m), 1373 (s), 1307 (w), 1269 (w), 1242 (s), 1187 (w), 1142 (w), 1118 (w), 1082 (w), 1044 (w), 1017 (w), 974 (w), 852 (w), 819 (s), 801 (s), 787 (s), 765 (m), 739 (s), 698 (s), 584 (m), 543 (m), 521 (s). Anal. Calcd for 4·0.5(CH2Cl2), C118.5ClH113Fe2N4O8: C, 76.18; H, 6.10; N, 3.00. Found: C, 76.01; H, 6.37; N, 3.17.
[Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(4-tBu-Bn)Bnen)2] (5)
Yield 57 %. FT-IR (KBr, cm-1) 3321 (m), 3270 (w), 3057 (m), 3025 (m), 2961 (s), 2919 (m), 2866 (m), 2799 (m), 1608 (s), 1545 (s), 1515 (s), 1495 (m), 1455 (s), 1410 (s), 1384 (s), 1306 (m), 1269 (m), 1212 (w), 1187 (w), 1145 (w), 1109 (m), 1070 (m), 1041 (w), 1020 (m), 984 (m), 943 (w), 855 (m), 818 (m), 800 (s), 785 (m), 766 (m), 737 (m), 712 (m), 699 (m), 608 (w), 584 (m), 544 (m), 521 (m), 455 (w). Anal. Calcd for 5·0.5(CH2Cl2), C124.5H125ClFe2N4O8: C, 76.59; H, 6.45; N, 2.87. Found: C, 76.70; H, 6.71; N, 3.00.
[Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(4-OMe-Bn)Bnen)2] (6)
Yield 54 %. FT-IR (KBr, cm-1) 3318 (w), 3256 (w), 3057 (w), 3024 (m), 2918 (m), 2833 (w), 2795 (w), 1610 (s), 1584 (m), 1593 (m), 1513 (s), 1454 (s), 1407 (w), 1378 (m), 1329 (s), 1305 (m), 1246 (s), 1179 (w), 1143 (w), 1110 (m), 1070 (w), 1038 (m), 1021 (w), 977 (w), 820 (s), 802 (s), 786 (m), 766 (m), 739 (m), 716 (w), 699 (m), 609 (m), 546 (m), 524 (m), 467 (w). Anal. Calcd for 6·(CH2Cl2), C119Cl2H114Fe2N4O10: C, 73.57; H, 5.91; N, 2.88. Found: C, 73.61; H, 6.12; N, 3.26.
[Fe2(μ-O2CArTol)2(O2CArTol)2)((C6H5CDH)2en)2] (7)
Yield 26 %. FT-IR (KBr, cm-1) 3348 (m), 3293 (m), 3058 (w), 3025 (w), 2925 (m), 2881 (w), 1606 (s), 1576 (s), 1515 (s), 1495 (m), 1455 (s), 1413 (s), 1387 (s), 1306 (w), 1264 (m), 1186 (w), 1138 (w), 1110 (m), 1073 (m), 1020 (m), 820 (s), 801 (s), 786 (w), 766 (w), 736 (m), 701(s).
[Fe2(μ-O2CArTol)2(O2CArTol)(N,N-(C6H5CD2)(C6H5CH2)en)2] (8)
Yield 63 %. FT-IR (KBr, cm-1) 3315 (m), 3268 (m), 3058 (m), 3023 (m), 2919 (m), 2859 (w), 1612 (s), 1544 (s), 1515 (s), 1493 (m), 1454 (s), 1410 (m), 1381 (s), 1109 (w), 1038 (w), 1021 (w), 986(w), 913 (w), 857 (w), 820 (m), 800 (s), 783 (m), 765 (w), 736 (m), 714 (m), 699 (m), 584 (w), 546 (w), 520 (w), 452 (w).
X-ray Crystallographic Studies
Single crystals were mounted at room temperature on the tips of quartz fibers, coated with Paratone-N oil, and cooled under a stream of cold nitrogen. Intensity data were collected on a Bruker (formerly Siemens) APEX CCD diffractometer running the SMART software package, with Mo Kα radiation (λ = 0.71073 Å). Data collection and reduction protocols are described in detail elsewhere.24 The structures were solved by Patterson and difference Fourier methods and refined on F2 by using the SHELXTL software package.25 Empirical absorption corrections were applied with SADABS,26 part of the SHELXTL program package, and the structures were checked for higher symmetry by the program PLATON.27 All non-hydrogen atoms were refined anisotropically. In general, hydrogen atoms were assigned idealized positions and given thermal parameters equivalent to either 1.5 (methyl hydrogen atoms) or 1.2 (all other hydrogen atoms) times the thermal parameter of the carbon atoms to which they were attached. In the structures of 2 - 6, two CH2Cl2 solvent molecules were assigned in the lattice. In the structure of 7, one carbon atom connected to the primary amine in the (C6H5CDH)2en ligand is disordered over two position with occupancy factors assigned as 0.7 and 0.3; hydrogen atoms for the disordered carbon atom were omitted. One CH2Cl2 solvent molecule was identified in the lattice in the structure of 8. Data collection and experimental details for the complexes are summarized in Table S1 (Supporting Information) and relevant interatomic bond lengths and angles are listed in Table 1.
Table 1.
Selected Interatomic Distances (Å) and Angles (deg) for 2-8.a
| Compound | 2·2CH2Cl2 | 3·2CH2Cl2 | 4·2CH2Cl2 | 5·2CH2Cl2 | 6·2CH2Cl2 | 7 | 8·CH2Cl2 |
|---|---|---|---|---|---|---|---|
| Fe1···Fe1A | 3.9189(17) | 3.9260(7) | 3.9388(8) | 3.8680(8) | 3.8781(13) | 4.360(5) | 3.934(2) |
| Fe1–O1 | 1.897(3) | 1.896(2) | 1.895(2) | 1.921(2) | 1.899(3) | 1.966(4) | 1.952(4) |
| Fe1–O2 | 2.013(3) | 2.0149(19) | 2.002(2) | 2.025(2) | 2.021(3) | 2.011(4) | 1.949(5) |
| Fe1–O3 | 2.005(3) | 2.0083(17) | 1.9992(19) | 2.0747(18) | 2.024(3) | 2.111(4) | 2.064(4) |
| Fe1–O4 | 2.503(3) | 2.5142(18) | 2.5289(19) | 2.2571(17) | 2.473(3) | 2.213(4) | 2.267(4) |
| Fe1–N1 | 2.143(3) | 2.1391(19) | 2.129(2) | 2.142(2) | 2.131(4) | 2.136(6) | 2.141(5) |
| O1–Fe1–O2 | 124.79(19) | 124.15(15) | 124.31(15) | 127.56(17) | 128.4(2) | 114.86(18) | 128.0(2) |
| O1–Fe1–O3 | 126.29(16) | 126.95(12) | 125.86(13) | 128.25(14) | 122.8(2) | 123.50(16) | 110.94(19) |
| O1–Fe1–O4 | 97.76(11) | 97.73(8) | 97.56(9) | 96.75(9) | 96.18(15) | 105.27(16) | 94.12(16) |
| O1–Fe1–N1 | 97.69(14) | 98.23(10) | 97.58(10) | 93.04(10) | 95.98(16) | 92.7(2) | 98.76(18) |
| O2–Fe1–O3 | 105.66(11) | 105.47(10) | 109.70(7) | 101.51(11) | 106.07(17) | 119.75(16) | 118.1(2) |
| O2–Fe1–O4 | 95.16(11) | 95.28(7) | 95.38(8) | 93.83(8) | 97.72(13) | 92.09(16) | 95.36(17) |
| O2–Fe1–N1 | 102.11(11) | 102.37(8) | 102.18(9) | 92.95(9) | 101.97(14) | 92.3(2) | 90.10(19) |
| O3–Fe1–O4 | 57.29(9) | 57.03(6) | 56.70(7) | 60.77(6) | 57.38(11) | 60.65(15) | 60.35(14) |
| O3–Fe1–N1 | 87.69(11) | 87.40(7) | 88.58(8) | 100.57(7) | 87.64(13) | 98.4(2) | 98.91(16) |
| O4–Fe1–N1 | 144.17(10) | 143.57(7) | 144.48(7) | 161.10(7) | 143.89(12) | 157.6(2) | 158.61(15) |
Numbers in parentheses are estimated standard deviations of the last significant figures.
General Procedures for Oxidative N-Dealkylation Studies Using GC and GC/MS Analysis
Analyses were carried out on a Hewlett Packard HP-5970 gas chromatograph connected to a HP-5971 mass analyzer. An Alltech Econo-cap EC-WAX capillary column of dimensions (30 m × 0.25 mm × 0.25 μm) and HP-5 cross-linked 5% PhMe-silicone column (25 m × 0.32 mm × 0.5 μm) were used for GC/MS and GC studies, respectively. The following program was used to effect all separations: initial temperature = 50 °C; initial time = 6 min; temperature ramp = 50 – 200 °C at 20 deg/min. The products were identified by comparing their retention times and mass spectral patterns to those of authentic standards. The FID response was calibrated by running with authentic samples containing an internal standard of 1,2-dichlorobenzene. All samples were prepared in an anaerobic glove box. Analytical conditions were maintained to match those reported in the initial studies of compound 1. Solutions (12.0 mM in CH2Cl2) of the [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(4-R-Bn)Bnen)2] compounds were prepared under nitrogen. The solutions turned brownish-yellow immediately upon exposure to dioxygen at 23 °C. After 1 h of additional stirring, 1,2-dichlorobenzene was added as an internal standard, and the reaction mixture was filtered through a 1-cm silica column to separate the organic products from inorganic materials. The resulting clear solution was analyzed by GC. Standard deviations were obtained from the results of at least three independently prepared samples.
Kinetic Isotope Effect Measurements
The product distribution was initially monitored by using GC-MS. Because the peaks for PhCHO and deuterated benzaldehyde (PhCDO) were not well separated, 1H-NMR intensity ratios were ultimately employed. A 12.0 mM CH2Cl2 solution of 7 or 8 was gently purged with dioxygen gas for 1 h at 23 °C and filtered through a 1-cm dried silica column. The 1H NMR spectrum was recorded to determine the product distribution. Standard deviations were obtained from the results of three independently prepared samples.
Results
Ligand Syntheses
A drawback in planning a mechanistic study of the oxygenation pathway of 1 is the absence of unique optical spectroscopic features that can be used to follow the reaction. No intermediate electronic transition band develops over the course of the oxygenation, even at − 78 °C. Deeper insight into the nature of the C–H bond-breaking step, for which three pathways have been proposed (Scheme 1), were therefore sought by Hammett and KIE studies based on the distribution of oxygenation products of complexes [Fe2(μ-O2CArTol)2(O2CArTol)2(L)2], where L is N,N-(4-R-Bn)Bnen or (C6H5CDH)2en.
A series of asymmetric ligands N,N-(4-R-Bn)Bnen with para-R-groups Cl, F, Me, tBu, or OCH3 were prepared by a four-step route, as depicted in Scheme 2, starting from commercially available benzylamine and the respective benzaldehyde. Reaction of benzylamine with the para-substituted benzaldehyde (4-R-PhCHO) yielded (4-R-benzyl)benzylimines, which were converted to the (4-R-benzyl)benzylamines by treatment with NaBH4. Reaction of the (4-R-benzyl)benzylamines with chloroacetonitrile afforded the (4-R-benzyl)benzylaminoacetonitriles. Reduction of these products with LiAlH4 generated the targeted asymmetric (4-R-benzyl)benzylethylenediamines with overall good yields. The ligand (C6H5CDH)2en was synthesized from α-d-benzylamine, prepared by the reduction of benzaldehyde oxime with lithium aluminum deuteride. Reaction of α-d-benzylamine with benzaldehyde (PhCHO) yielded (α-d-benzyl)benzylimine, which was converted to α,α′-d2-dibenzylamine by treatment with NaBD4. With the resulting α,α′-d2-dibenzylamine, the compound (C6H5CDH)2en was prepared as described for the preparation of N,N-(4-R-Bn)Bnen. The asymmetric dideuterated N,N-Bn2en ligand, N,N-(C6H5CD2)(C6H5CH2)en, was obtained from α-d2-benzylamine, following a similar reaction route to that previously described for obtaining the N,N-(4-R-Bn)Bnen ligands.
Scheme 2.

Synthesis and Structural Characterization of Diiron(II) Complexes
The reaction of the precursor complex, [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2], with 2 equiv of the diamine ligands N,N-(4-R-Bn)Bnen (R = MeO, tBu, Me, F, Cl), N,N-(C6H5CDH)2en, and N,N-(C6H5CD2)(C6H5CH2)en afforded the neutral diiron(II) complexes 2 – 8. Their structures, shown in Figures 1 and S1 – S6 (Supporting Information), were determined by X-ray diffraction. All compounds contain a crystallographic inversion center. The pseudo-trigonal bipyramidal iron(II) centers in 5, 7, and 8 have an NO4 coordination environment with chelating terminal carboxylate ligands. In 2, 3, 4 and 6, however, the terminal carboxylates are monodentate, resulting in four-coordinate NO3 environments. The relatively long Fe1···O(terminal carboxylate) distance of ca 2.5 Å may be compared to the shorter distances of ∼2.25 Å in 5, 7, and 8 (Table 1). In all the compounds, the coordinatively unsaturated iron(II) centers are bridged by two carboxylates and the Fe···Fe distances vary from 3.8680(8) Å to 4.360(5) Å. The Fe···Fe distances in 7 and 8, which have similar chemical compositions, are 4.360(5) Å and 3.934(2) Å, respectively. These diverse Fe···Fe and Fe–O distances involving terminal carboxylates indicate the flexible character of the carboxylate-rich diiron(II) units. The 4-R groups in the N,N-(4-R-Bn)Bnen ligands of 2, 3, 4, and 6 are disordered over two positions, each with an 0.5 occupancy factor; however, the tert-butyl group in 5, which is larger relative to the other 4-R groups, is localized on just one of the phenyl rings of the N,N-Bn2en moiety.
Figure 1.
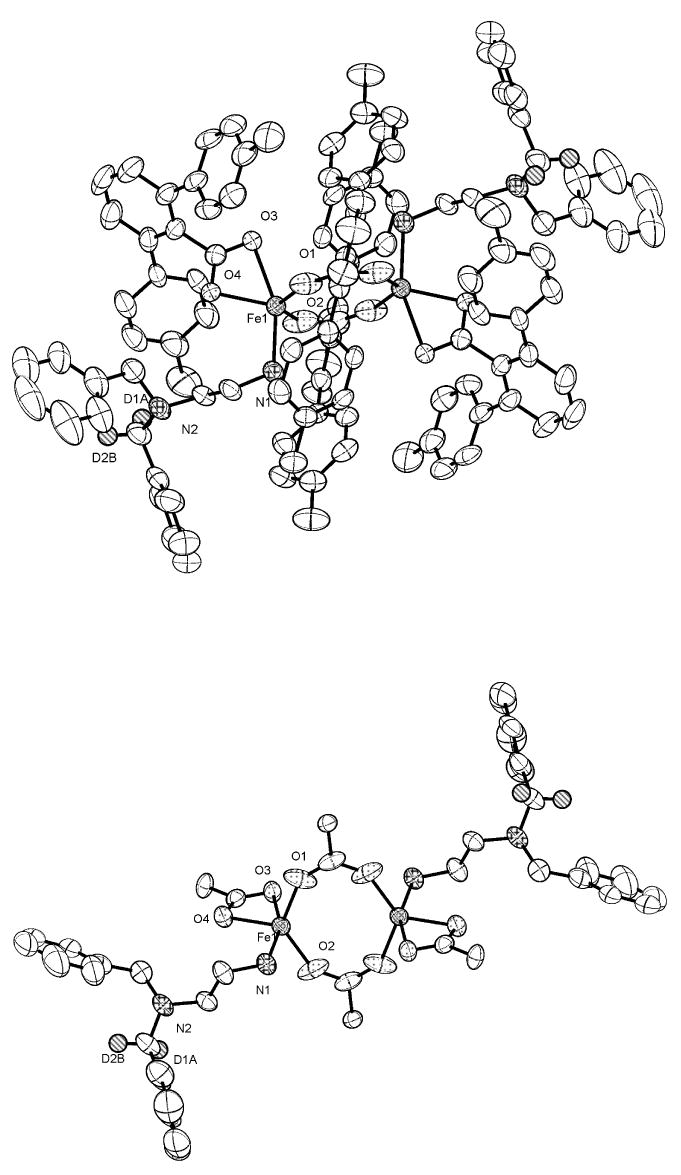
(Top) ORTEP drawing of [Fe2(μ-O2CArTol)2(O2CArTol)(N,N-(C6H5CD2)-(C6H5CH2)en)2] (8) showing 50 % probability thermal ellipsoids. The solvent molecules and hydrogen atoms are omitted for clarity. (Bottom) Structure in which all atoms of the ArTolCO2- ligands in 8, except for the carboxylate groups and the α–carbon atoms, were omitted for clarity.
Hammett Study of the Reaction of Diiron(II) Complexes 2 - 6 with Dioxygen
The series of diiron(II) complexes prepared with ligands (4-R)BnBnen (R = Cl, F, Me, t-Bu, or OCH3) were allowed to react with O2 at ambient temperature. Both 4-R-PhCHO and PhCHO were identified in the oxygenation product mixture by GC-MS (Scheme 2). The product distributions were analyzed by GC and the overall yields were ∼ 60 % based on the Fe2 unit (Table 2). In addition to GC analysis, the product ratios in the oxygenation reactions of 2 and 5 were confirmed by using 1H-NMR spectroscopy, comparing the integrated intensities of the aldehyde C–H protons of 4-R-PhCHO and PhCHO (Table 2, Figure 7S). The resulting ratios were the same within error to those determined by GC analysis. A plot of the product ratios against Hammett σ values28 (Figure 2) revealed a linear relationship, supporting the existence of similar transition states (TS) for the set of reactants. The positive slope, ρ = + 0.48 (R = 0.98), indicates that the TS is stabilized by electron withdrawing substituents. This result strongly suggests that C–H bond breaking occurs through generation of anionic character at the benzylic position in the transition state.
Table 2.
Yield and Product Distribution for Reactions of 2 – 6 with Dioxygen
| Complex | 4-R | σPa | YH (%)b PhCHO | YR (%)b 4-R-PhCHO | YR/YHc | YR/YHd | log (YR/YH) |
|---|---|---|---|---|---|---|---|
| 2 | Cl | 0.23 | 24(2) | 31(4) | 1.26 (7) | 0.10 (3) | |
| 3 | F | 0.06 | 28(2) | 32(1) | 1.16 (8) | 1.22 | 0.06 (3) |
| 4 | CH3 | -0.17 | 34(1) | 29(2) | 0.85 (6) | -0.07 (3) | |
| 5 | t-Bu | -0.20 | 33(1) | 24(3) | 0.78 (5) | 0.79 | -0.11 (3) |
| 6 | OCH3 | -0.27 | 34(4) | 25(2) | 0.74 (10) | -0.13 (6) |
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165-195.
YH and YR represent the product yields of PhCHO and 4-R-PhCHO, respectively.
Ratios were determined from GC analytical data.
Ratios were determined by using 1H NMR spectroscopy.
Figure 2.

Hammett plot for the oxygenation of [Fe2(μ-O2CArTol)2(O2CArTol)2((4-R)BnBnen)2] (R = Cl, F, H, Me, t-Bu, and OCH3) in CH2Cl2 at 23 °C. YH and YR represent product yields of PhCHO and 4-R-PhCHO, respectively. The line represents a least squares fit to the data with a slope ρ of 0.48 (R, 0.98).
Kinetic Isotope Effects
The determination and interpretation of intramolecular KIEs is one approach to delineate C–H bond breaking mechanisms in spectroscopically silent reactions.29-31 The method interrogates the effect on the product distribution of replacing an atom, from at least two equivalent atoms, by one of its isotopes. These types of intramolecular KIEs are not affected by steps that occur before and after C–H bond breaking. The observation of a KIEintra reflects the changes in the C–H(D) bonds from reactants to the TS in the C–H bond-breaking step.29,31
A KIEintra value of 1.3 (1) at 23 °C was obtained from the PhCDO/PhCHO ratio for oxygenation of [Fe2(μ-O2CArTol)2(O2CArTol)2((C6H5CDH)2en)2] (7). This value was determined by using product distribution analysis between PhCDO and PhCHO based on GC-MS and 1H NMR spectroscopy (Figure S8). Breaking of the C–H bond in 7 yields the amino alcohol, which eliminates PhCDO (see Scheme 3). The near-unity value of the KIE raises the question of whether it is a secondary instead of a primary effect. To address this issue, the compound [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-(C6H5CD2)(C6H5CH2)en)2] (8) having asymmetrically deuterated N,N-Bn2en ligands was prepared. If the KIE value for 7 were to originate from a secondary isotope effect, the KIE values for 8 should be the reciprocal of 1.3, whereas the primary KIE values for oxygenation of 7 and 8 are expected to be identical (Scheme 3). C–H bond breaking at the benzylic position of the (C6H5CDH)2en ligand and C–D bond breaking within the N,N-(C6H5CD2)(C6H5CH2)en ligand both afford a C–D bond at the benzylic position.
Scheme 3.

The KIEintra value of 8, measured by the same methods used for 7, is 2.2 (2) at 23 °C, obtained from the ratio of PhCHO/PhCDO. In the case of 8, C–H bond breaking generates PhCHO (Scheme 3). The KIEintra value, 2.2, is still small but larger than that obtained by oxygenation of 7. We interpret these results in the following manner. The observed KIE for 7 is a primary effect; although small, it is not the reciprocal of the KIE of 8. Evidently, the differences in two KIE values indicate that secondary effects contribute to the C–H bond-breaking step in addition to the primary KIEs. The combination of primary and secondary effects for the oxygenation of 7 and 8 is described by eqs 1 and 2, respectively. In the case of 7, the secondary KIE may detract from the
| (1) |
| (2) |
primary KIE, as shown in Scheme 3, whereas both primary and secondary effects are expected to contribute to the oxygenation of 8. The result is a larger KIE value for 8 than for 7. Based on these measured KIEs, primary and secondary KIE values were calculated to be 1.7 and 1.3, respectively.
Discussion
Oxidative N-dealkylation of the complex [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-Bn2en)2] (1) upon introduction of dioxygen9,15 parallels the activation of dioxygen and C–H bond hydroxylation by non-heme diiron enzyme active sites and is analogous to the dealkylation reactions catalyzed by cytochrome P450 enzymes.32 Reaction of compound 1 with dioxygen may afford a transient adduct [1-O2], which never builds up because the tethered substrates readily react with any species so generated. Formal possibilities for such a dioxygen adduct include Fe2II,III(O2-), Fe2III(O22-), and Fe2IV(O2-)2. We previously reported the formation and characterization of a high-valent Fe(III)Fe(IV) intermediate in the reaction between dioxygen and diiron(II) complexes of ArTolCO2−. We presented EPR and Mössbauer spectroscopic data consistent with this intermediate being generated through bimolecular electron transfer between a putative Fe2IV(O2-)2 species and the Fe2II,II starting material.10,11 The results suggested the involvement of a high-valent iron(IV)-oxo species as a potential oxidant in the N-dealkylation reaction of 1, analogous to well-studied N-dealkyations by cytochrome P450,32 Cu2(O)2,33-36 and Ni2(O)237 metal centers. Assuming the participation of such a high-valent iron oxo species, three reaction pathways were proposed for the oxidative N-dealkylation reaction of dioxygen with the diiron(II) complex [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-Bn2en)2] (Scheme 1).9 An electrophilic high-valent diiron oxo unit may perform 2-electron oxidative N-dealkylation by hydrogen atom abstraction followed by oxygen rebound or by concerted insertion of the oxygen atom into C–H bonds. Alternatively, sequential one-electron oxidation of dangling nitrogen atom by the proposed electrophilic high-valent iron(IV) oxo species, followed by α-proton abstraction and sequential oxygen rebound, could be the mechanism.
The present Hammett plot and KIEintra results indicate that anionic character develops in the transition state during the C–H bond-breaking step, thus ruling out Paths A and B in Scheme 4. Concerted oxygen atom insertion (Path A) by the electrophilic oxidant into the electron density of the C–H bond would generate partial positive character at the benzylic position in the transition state. Path B should not generate any charge, which would result in at most a small value for the Hammett parameter. Large KIE values (>20) have been reported for N-dealkylations, effected through HAT mechanisms by Cu35 and Ni36 complexes, in contrast to the small KIE values for oxygenation of 7 and 8. Single electron transfer from the nitrogen atom followed by α-proton abstraction (Path C in Scheme might proceed via a TS that has anionic character at the benzylic position. As shown in Path C/E/F, the KIEs would occur in the deprotonation step that follows electron transfer. The size of primary deuterium KIEs for proton transfer processes38-40 are generally considered as a measure of the symmetry of the transition state. The maximal primary KIE (∼ 7) occurs for the most symmetrical activated complex, usually one in which the proton is bound with equal strength to both the donor and acceptor. This situation occurs when the donor and acceptor have equal pKa values. The small primary KIE for 7 and 8 may originate from asymmetric character in the benzylic carbon donor and oxygen atom acceptor in a high-valent Fe–O intermediate. Tunneling, which is claimed to occur in catalytic systems including both enzymes20 and their small molecule mimics,35,36 is not involved in the C–H bond-breaking steps of 7 and 8.
Scheme 4.

N-Dealkylations of N,N-dimethylbenzylamines by iodosylbenzene catalyzed by a tetraphenylporphyrin metal complex, display small KIE and negative ρ ( ∼ -0.4 - -0.2) values.41 These results were interpreted in terms of a rate–limiting SET step, followed by deprotonation. The generation of positive charge on the nitrogen atom in the benzylamine substrates by rate-determining SET step leads to the small negative ρ values. Subsequent rapid proton transfer did not affect the Hammett parameters. Although we employ a benzylamine substrate in the Bn2en system described here, it is quite different. The nitrogen atom in the (R-Bn)Bnen ligands is adjacent to two different benzylic substituents. Electron transfer involving the N-atom of such a ligand should have a minimal impact on the Hammett parameter because generation of a positive charge on the nitrogen atom is stabilized by both R-Bn and Bn substituents. Only the subsequent deprotonation steps, which generate a negative charge on a benzylic carbon atom, will determine the Hammett parameter. Thus the ρ values in the (R-Bn)Bnen system will reflect relative charge buildup after the choice of one N-benzyl group over the other. In a somewhat related system, the reaction rates of para-substituted benzylamine analogues with purified monoamine oxidase A (MAO A) also resulted in a Hammett correlation with a positive ∼2.0 ρ value. This finding was explained in terms of a mechanism in which negative charge developed at the benzylic carbon atom following proton abstraction from the α-C–H bond, as in the present case.42
The small primary KIE in the present study may imply that the deprotonation has an early transition state and is non-linear. The observed secondary KIEs can be understood in the following manner. Species (E), generated after SET in Path C (Scheme 4), has sp3 hydridization at a benzylic carbon atom. On the other hand, the hydridization at the benzylic carbon in species F is sp2. The formation of the transition state in the Path C/E/F should involve an sp3 → sp2 conversion, resulting in a normal isotope effect (>1). The secondary KIE that occurs for oxygenation of the complexes is interpreted to reflect differing inductive and steric (a CD3 group is ca 10 % smaller than a CH3 group) effects for the isotopic forms.43 The steric effect cannot apply to the oxygenation of 7 and 8, however, since a secondary KIE <1 should have been obtained for the intramolecular competition, the CD2 group in 8 being less hindered than the CH2 group in 7. Therefore, the observed secondary KIE may originate from the fact that deuterium is slightly more electron-releasing than the proton.44 The generated anionic character at the benzylic position in the transition state (Hammett study) may be more destabilized by the electron-donating deuterium in the benzylic position, resulting in a secondary KIE reinforcing the primary effect in 8, but canceling it in the case of 7.
The reaction mechanism for N-dealkylation by high-valent iron-oxo porphyrinates has been continuously debated; both ET and HAT pathways have been claimed. In the case of cytochrome P450, it is possible that N-dealkylation by the enzyme may operate by the ET pathway for easily oxidized substrates, but by HAT for others. In the case of synthetic model systems, studies suggest that Cu2(O)2 complexes are capable of performing HAT reactions for benzylic amine substrates, which is in contrast to the present mechanistic studies on diiron complexes with tethered benzylamines. It should be noted, however, that the nitrogen atom in the copper complexes through which the substrate is tethered to the metal site does not have the free, non-bonding electron pair that occurs in a N,N-Bn2en ethylenediamine ligand. Consequently, it may be much more difficult for these systems to proceed by an ET pathway. Recently, a mechanistic study of dicopper complexes with substrates having a nitrogen atom with a free electron pair was reported.33,34 A shift in mechanism for oxidative N-dealkylation from ET to HAT was proposed, depending on the ease of substrate one-electron oxidation.
Although the observed results can be satisfactorily explained by a mechanism involving SET followed by proton transfer and rearrangement, such a pathway implies the participation of an electrophilic high-valent species as the oxidant.10,11 Recent observations, however, point to peroxo intermediates in oxygenation processes that occur at related carboxylate-rich diiron(II) complexes17,19 and proteins.45 It is therefore possible that the reactive species responsible for the observed KIE and Hammett plot results is an alternative, nucleophilic species, such as a diiron(III) peroxo complex. This possibility is depicted as Path D in Scheme 4. A nucleophilic peroxo intermediate might deprotonate the activated C–H bond of the tethered substrate, generating anionic character at the benzylic position, again resulting in the positive Hammett ρ parameter. The small KIE value could be rationalized if this route were only partially rate-determining. A bound peroxide (typically pKa in the 10-20 range) is not basic enough to deprotonate a benzylic C–H bond (typical pKa near 40), however, and to our knowledge there is not yet a reported precedent for the mechanism in Path D. From our present Hammett and KIEintra analyses for oxidative N-dealkylation of [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-Bn2en)2], therefore, we can rule out previously proposed concerted and hydrogen atom transfer mechanisms and favor the one involving SET from nitrogen atom, followed by proton transfer.
It would be desirable to have a system that can develop a spectroscopically observable intermediate with a tethered substrate, or lack a non-bonding electron pair in the tethered substrate. Such complexes would enhance our ability to refine the present working mechanism for oxidative N-dealkylation in carboxylate-rich diiron(II) system. Efforts are currently in progress to understand the oxidative N-dealkylation mechanism of substrates that lack a free non-bonding electron pair, but which can still be oxygenated by carboxylate-rich diiron(II) complexes.16-18 The reaction pathway may share common features with related intramolecular hydroxylation reactions mediated by carboxylate-bridged diiron complexes.46
Supplementary Material
Supporting Information Available: Table S1 presenting X-ray crystallographic information; Figures S1 – S6 showing ORTEP diagrams of 2 – 7; GC-MS traces and 1H NMR spectra of the oxidation product of 7 (Figure S8); 1H NMR spectra of the oxidation products of 3 and 6 (Figure S7) and 8 (Figure S9); and X-ray crystallographic (CIF) files. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by grant GM32134 from the National Institute of General Medical Sciences. NMR spectra were measured on a Bruker 400 spectrometer purchased with assistance from NIH Grant 1S10RR13886-01.
References
- 1.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 2.Holm RH, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 3.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 4.Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 5.Lindqvist Y, Huang W, Schneider G, Shanklin J. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 6.Tshuva EY, Lippard SJ. Chem Rev. 2004;104:987–1012. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- 7.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew Chem, Int Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 8.Whittington DA, Lippard SJ. J Am Chem Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 9.Lee D, Lippard SJ. Inorg Chem. 2002;41:827–837. doi: 10.1021/ic011008s. [DOI] [PubMed] [Google Scholar]
- 10.Lee D, Lippard SJ. Inorg Chem. 2002;41:2704–2719. doi: 10.1021/ic020186y. [DOI] [PubMed] [Google Scholar]
- 11.Lee D, Pierce B, Krebs C, Hendrich MP, Huynh BH, Lippard SJ. J Am Chem Soc. 2002;124:3993–4007. doi: 10.1021/ja012251t. [DOI] [PubMed] [Google Scholar]
- 12.Hagadorn JR, Que L, Jr, Tolman WB. J Am Chem Soc. 1998;120:13531–13532. doi: 10.1021/ja983333t. [DOI] [PubMed] [Google Scholar]
- 13.Hsu HF, Dong Y, Shu L, Young VG, Jr, Que L., Jr J Am Chem Soc. 1999;121:5230–5237. [Google Scholar]
- 14.Lee D, DuBois JL, Pierce B, Hedman B, Hodgson KO, Hendrich MP, Lippard SJ. Inorg Chem. 2002;41:3172–3182. doi: 10.1021/ic011050n. [DOI] [PubMed] [Google Scholar]
- 15.Lee D, Lippard SJ. J Am Chem Soc. 2001;123:4611–4612. doi: 10.1021/ja0100930. [DOI] [PubMed] [Google Scholar]
- 16.Carson EC, Lippard SJ. J Am Chem Soc. 2004;126:3412–3413. doi: 10.1021/ja031806c. [DOI] [PubMed] [Google Scholar]
- 17.Carson EC, Lippard SJ. Inorg Chem. 2006;45:828–836. 837–848. doi: 10.1021/ic051471v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon S, Lippard SJ. Inorg Chem. 2003;42:8606–8608. doi: 10.1021/ic0350133. [DOI] [PubMed] [Google Scholar]
- 19.Chavez FA, Ho RYN, Pink M, Young VG, Jr, Kryatov SV, Rybak-Akimova EV, Andres H, Münck E, Que L, Jr, Tolman WB. Angew Chem Int Ed. 2002;41:149–152. doi: 10.1002/1521-3773(20020104)41:1<149::aid-anie149>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Du Bois J, Mizoguchi TJ, Lippard SJ. Coord Chem Rev. 2000;200-202:443–485. [Google Scholar]
- 21.Sato H, Guengerich FP. J Am Chem Soc. 2000;122:8099–8100. [Google Scholar]
- 22.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 23.Wintner EA, Tsao B, Rebek J., Jr J Org Chem. 1995;60:7997–8001. [Google Scholar]
- 24.Kuzelka J, Mukhopadhyay S, Spingler B, Lippard SJ. Inorg Chem. 2004;43:1751–1761. doi: 10.1021/ic035009r. [DOI] [PubMed] [Google Scholar]
- 25.Sheldrick GM. Program Library for Structure Solution and Molecular Graphics, version 6.2. Bruker AXS; Madison, WI: 2000. [Google Scholar]
- 26.Sheldrick GM. SADABS: Area-Detector Absorption Correction. University of Göttingen; Göttingen, Germany: 2001. [Google Scholar]
- 27.Spek AL. PLATON, A Multipurpose Crystallographic Tool. Utrecht University; Utrecht, The Netherlands: 2000. [Google Scholar]
- 28.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 29.Adam W, Krebs O, Orfanopoulos M, Stratakis M, Vougioukalakis GC. J Org Chem. 2003;68:2420–2425. doi: 10.1021/jo0266240. [DOI] [PubMed] [Google Scholar]
- 30.Naróg D, Lechowicz U, Pietryga T, Sobkowiak A. J Mol Catal A Chem. 2004;212:25–33. [Google Scholar]
- 31.Frantom PA, Pongdee R, Sulikowski GA, Fitzpatrick PF. J Am Chem Soc. 2002;124:4202–4203. doi: 10.1021/ja025602s. [DOI] [PubMed] [Google Scholar]
- 32.Karki SB, Dinnocenzo JP, Jones JP, Korzekwa KR. J Am Chem Soc. 1995;117:3657–3664. [Google Scholar]
- 33.Shearer J, Zhang CX, Hatcher LQ, Karlin KD. J Am Chem Soc. 2003;125:12670–12671. doi: 10.1021/ja0359409. [DOI] [PubMed] [Google Scholar]
- 34.Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. J Am Chem Soc. 2005;127:5469–5483. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- 35.Mahapatra S, Halfen JA, Tolman WB. J Am Chem Soc. 1996;118:11575–11586. [Google Scholar]
- 36.Itoh S, Nakao H, Berreau LM, Kondo T, Komatsu M, Fukuzumi S. J Am Chem Soc. 1998;120:2890–2899. [Google Scholar]
- 37.Itoh S, Bandoh H, Nagatomo S, Kitagawa T, Fukuzumi S. J Am Chem Soc. 1999;121:8945–8946. doi: 10.1021/ja0104094. [DOI] [PubMed] [Google Scholar]
- 38.Ortiz de Montellano PR. Acc Chem Res. 1987;20:289–294. [Google Scholar]
- 39.Guengerich FP, Yun CH, Macdonald TL. J Biol Chem. 1996;271:27321–27329. doi: 10.1074/jbc.271.44.27321. [DOI] [PubMed] [Google Scholar]
- 40.Okazaki O, Guengerich FP. J Biol Chem. 1993;268:1546–1552. [PubMed] [Google Scholar]
- 41.Lindsay Smith JR, Mortimer DN. J Chem Soc, Chem Commun. 1985;2:64–65. [Google Scholar]
- 42.Miller RJ, Edmondson DE. Biochemistry. 1999;38:13670–13683. doi: 10.1021/bi990920y. [DOI] [PubMed] [Google Scholar]
- 43.Mislow K, Graeve R, Gordon AJ, Wahl GH., Jr J Am Chem Soc. 1963;85:1199–1200. [Google Scholar]
- 44.Laidler KJ. Chemical Kinetics. 3th. HarperCollins Publishers, Inc.; 1987. [Google Scholar]
- 45.Beauvais LG, Lippard SJ. J Am Chem Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- 46.Avenier F, Dubois L, Latour JM. New J Chem. 2004;28:782–784. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Table S1 presenting X-ray crystallographic information; Figures S1 – S6 showing ORTEP diagrams of 2 – 7; GC-MS traces and 1H NMR spectra of the oxidation product of 7 (Figure S8); 1H NMR spectra of the oxidation products of 3 and 6 (Figure S7) and 8 (Figure S9); and X-ray crystallographic (CIF) files. This material is available free of charge via the Internet at http://pubs.acs.org.