

Abstract
Background and Purpose
Multiple approaches are being studied to enhance the rate of thrombolysis for acute ischemic stroke. Treatment of myocardial infarction with a combination of a reduced-dose fibrinolytic agent and a glycoprotein (GP) IIb/IIIa receptor antagonist has been shown to improve the rate of recanalization versus fibrinolysis alone. The combined approach to lysis utilizing eptifibatide and recombinant tissue-type plasminogen activator (rt-PA) (CLEAR) stroke trial assessed the safety of treating acute ischemic stroke patients within 3 hours of symptom onset with this combination.
Methods
The CLEAR trial was a National Institutes of Health/National Institute of Neurological Disorders and Stroke–funded multicenter, double-blind, randomized, dose-escalation and safety study. Patients were randomized 3:1 to either low-dose rt-PA (tier 1=0.3 mg/kg, tier 2=0.45 mg/kg) plus eptifibatide (75 μg/kg bolus followed by 0.75 μg/kg per min infusion for 2 hours) or standard-dose rt-PA (0.9 mg/kg). The primary safety end point was the incidence of symptomatic intracerebral hemorrhage within 36 hours. Secondary analyses were performed regarding clinical efficacy.
Results
Ninety-four patients (40 in tier 1 and 54 in tier 2) were enrolled. The combination group of the 2 dose tiers (n=69) had a median age of 71 years and a median baseline National Institutes of Health Stroke Scale (NIHSS) score of 14, and the standard-dose rt-PA group (n=25) had a median age of 61 years and a median baseline NIHSS score of 10 (P=0.01 for NIHSS score). Fifty-two (75%) of the combination treatment group and 24 (96%) of the standard treatment group had a baseline modified Rankin scale score of 0 (P=0.04). There was 1 (1.4%; 95% CI, 0% to 4.3%) symptomatic intracranial hemorrhage in the combination group and 2 (8.0%; 95% CI, 0% to 19.2%) in the rt-PA–only arm (P=0.17). During randomization in tier 2, a review by the independent data safety monitoring board demonstrated that the safety profile of combination therapy at the tier 2 doses was such that further enrollment was statistically unlikely to indicate inadequate safety for the combination treatment group, the ultimate out-come of the study. Thus, the study was halted. There was a trend toward increased clinical efficacy of standard-dose rt-PA compared with the combination treatment group.
Conclusions
The safety of the combination of reduced-dose rt-PA plus eptifibatide justifies further dose-ranging trials in acute ischemic stroke.
Keywords: acute stroke, thrombolysis
Recombinant tissue-type plasminogen activator (rt-PA) is the only U.S. Food and Drug Administration–approved pharmacologic therapy for acute ischemic stroke.1 Despite the demonstrated efficacy of rt-PA in the National Institute of Neurological Disorders and Stroke (NINDS) rt-PA stroke trials, only 30% to 40% of occluded major vessels were recanalized in the first hour after treatment with rt-PA.2 In addition, recent studies have documented an appreciable risk of reocclusion.3 Methods that may enhance thrombolysis warrant clinical exploration. Glycoprotein (GP) IIb/IIIa inhibitors block the final common pathway of platelet aggregation and thus have considerable therapeutic potential in the treatment of thrombotic diseases, such as acute myocardial infarction (MI) and acute ischemic stroke. GPIIb/IIIa inhibitors are commonly used to treat acute MI and to prevent ischemic complications associated with percutaneous coronary interventions. For acute ischemic stroke treatment, experimental and clinical studies suggest that combination therapy (fibrinolytic agent plus GPIIB/IIIA inhibitor) could provide faster and more complete lysis than fibrinolytics alone.4
Eptifibatide is a cyclic heptapeptide GPIIb/IIIa inhibitor that is commercially available and is indicated for the treatment of patients with acute coronary syndrome, including patients who are to be managed medically and those undergoing percutaneous coronary intervention. Eptifibatide alone, without a fibrinolytic agent, has been found to decrease both the incidence of acute MI and mortality in patients with unstable angina.5 Eptifibatide alone has also been shown to reduce ischemic events and arterial reocclusions in patients undergoing percutaneous coronary interventions.6–8 Notably, the platelet aggregation inhibition that prevents the ischemic events after percutaneous transluminal coronary angioplasty has not been associated with higher rates of intracerebral hemorrhage (ICH).9 Though unavailable at the time of this study's design, data now show that full cardiac dose GPIIb/IIIa antagonists alone increase the rate of ICH versus placebo in stroke patients. Notably, the dosing of the GPIIb/IIIa antagonist in the CLEAR trial was significantly lower than full cardiac dosing.10 The eptifibatide dosing in the CLEAR trial was chosen to be slightly less than one half of the cardiac dose in this safety and dose-escalation trial.
On the basis of the potential to improve the efficacy of fibrinolysis for acute ischemic stroke, we hypothesized that combination therapy could be explored as an adjunct to standard thrombolytic therapy. Thus, we sought to execute a dose-escalation trial to assess the safety profile of combination therapy as the basis for potential further clinical exploration.
Subjects and Methods
The CLEAR trial was a multicenter, randomized, double-blind, sequential, dose-escalation and safety study of low-dose rt-PA in combination with eptifibatide versus standard-dose rt-PA alone given to patients diagnosed with acute ischemic stroke and treated within 3 hours of symptom onset. An investigational new drug application was approved by the U.S. Food and Drug Administration before trial initiation and is held by the principal investigator (A.M.P.). The institutional review board of each site approved the study protocol, and written, informed consent was obtained from each patient before study entry. The trial was sponsored by the NINDS as a Specialized Programs of Translational Research In Acute Stroke program project. Study drugs were supplied at no charge by the manufacturers. The trial design, execution, and data analysis were accomplished independent of the manufacturers. Eligible patients were 18 to 80 years of age with a clinical diagnosis of acute ischemic stroke and a National Institutes of Health Stroke Scale (NIHSS) score >5. The protocol required that the study drug be initiated within 3 hours from the time the patient was last seen normal. Inclusion and exclusion criteria are listed in Table 1. Note that these criteria differ from those used for the NINDS rt-PA stroke trials. The upper age limit was added because there was some concern for the potential for increased ICH in older patients treated with combination therapy in the cardiac literature.11 The NIHSS score >5 criterion was added because 81% of placebo-treated patients with an NIHSS score ≤5 in the NINDS rt-PA trials had a 90-day modified Rankin Scale (mRS) score of 0 to 1, making this group's potential benefit too small to include in a safety trial.12 The upper limit for creatinine was based on the renal excretion of eptifibatide.
Table 1. Inclusion and Exclusion Criteria.
| Inclusion Criteria |
|
| Exclusion Criteria |
| Clinical |
|
| CT scan exclusions |
|
INR indicates international normalized ratio.
From July 2003 to April 2007, 9 US centers (19 hospitals) enrolled 94 patients. Patients were enrolled into 2 sequential dosing tiers via a centralized telephone randomization at the clinical coordinating center at the University of Cincinnati. Patients were randomized in a 3-1 ratio to receive the combination of low-dose rt-PA and eptifibatide or standard rt-PA alone per the NINDS protocol. Dose tier 1 was designed to have 40 patients. After completion of dose tier 1 and a blinded review by the data safety monitoring board (DSMB), the study progressed. Dose tier 2 was designed to have a total of 60 patients. Drug dosing regimens in the 2 tiers were as follows. The dose of rt-PA was 0.3 mg/kg in tier 1 and 0.45 mg/kg in tier 2, with a maximum weight of 100 kg. The dose of eptifibatide was slightly less than one half of the cardiac dose and was the same in both tiers: a 75-μg/kg bolus followed by a 2-hour infusion of 0.75 μg/kg per minute.
To reach a therapeutic serum concentration of rt-PA with a lower total dose in the combination arm, the rate of infusion of rt-PA had to be greater than the standard rt-PA infusion rate. Blinding of the rt-PA dosing was maintained through a careful double-blind/double-dummy pattern. All patients received a bolus of rt-PA (either 15% of the low dose in the combination arm or 10% of 0.9 mg/kg in the control arm). These were prepared in the investigational pharmacies to a volume of 10 mL to avoid unblinding. All patients then received 2 sequential infusion bags that were either the remainder of the low-dose rt-PA over 30 minutes followed by placebo for 30 minutes or the remainder of the 0.9-mg/kg standard dose divided between 2 bags, each running for 30 minutes in the control group. The infusion bags were made to a volume of 50 mL to avoid unblinding. (See Figure 1 for method of drug delivery/blinding.) All study drugs were colorless and compatible in infusion lines, so there was no potential unblinding due to mixing of agents in the intravenous lines.
Figure 1.
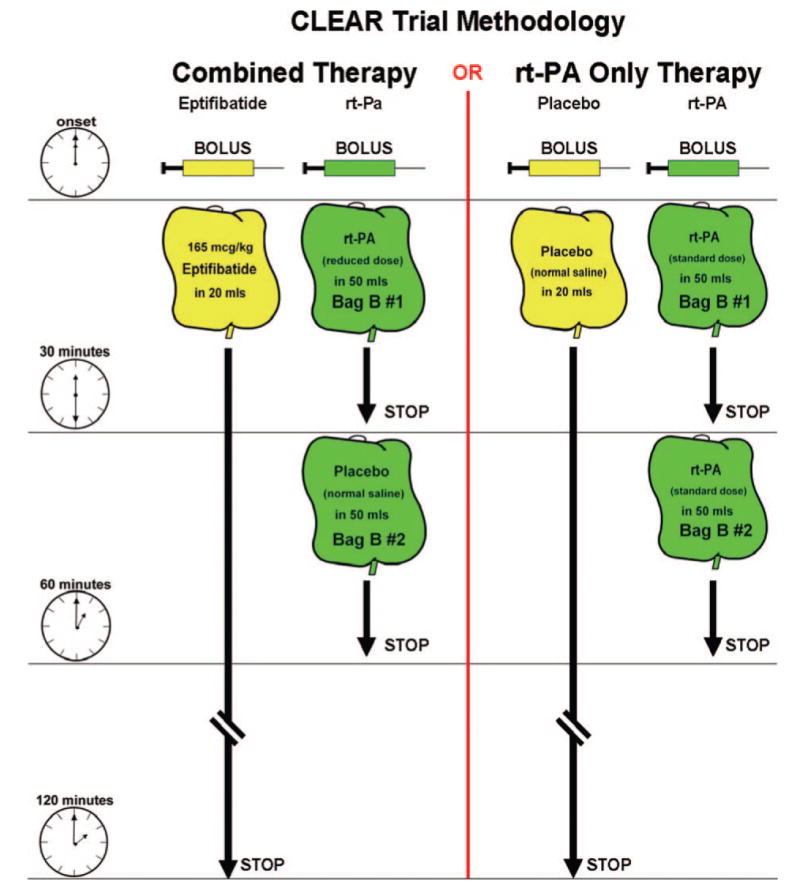
Bolus and infusion methodology for the 2 arms of the CLEAR trial. Eptifibatide or placebo was given as a bolus followed by a 2-hour infusion. The rt-PA was given as a bolus followed by 2 sequential infusion bags. Bag B#1 was rt-PA at either 85% of the lower-dose experimental regimens or 45% of the standard dose infused over 30 minutes. Bag B#2, started after bag B#1 was finished, was normal saline (lower-dose experimental regimens) or rt-PA (45% of the standard dose) infused during the remaining 30 minutes.
Patients were monitored clinically throughout the infusion by study personnel and then admitted to an intensive care unit for continued monitoring for at least 24 hours. Patients had standardized clinical evaluations at 2 hours, 24 hours, and 5 days or at discharge, followed by a telephone follow-up at 7 days and a final in-person standardized evaluation at 90 days. Outcomes measured were the NIHSS, Glasgow Outcome Scale, Barthel Index, EuroQOL, Stroke-Specific Quality of Life, and Stroke-Free Status questionnaire. Radiologic outcome measures included a 24-hour safety computed tomography (CT) scan to evaluate for ICH and a 24-hour magnetic resonance imaging (MRI) scan with magnetic resonance angiography to evaluate infarct volume and arterial patency.
The primary safety end point was the incidence of symptomatic ICH within 36 hours of onset. Secondary safety end points included asymptomatic ICH, death, and clinically significant extracranial bleeding. Although this was a safety trial, clinical outcomes were also collected to assess for potential efficacy. The primary outcome measures for this pilot study were evidence of early neurologic improvement, as measured by the incidence of an NIHSS score ≤2 at 24 hours, and late improvement, as measured by the incidence of an mRS score of 0 to 1 or a return to baseline at 3 months. Secondary functional end points at 90 days included a Barthel Index ≥95, Glasgow Outcome Scale of 1, and an NIHSS score of 0 or 1. In addition, the EuroQOL quality of life index and the Stroke-Specific Quality of Life Scale were administered at 3 months. Ninety-day outcome was assessed by study investigators not directly involved with acute treatment of the patients. All investigators were certified in the NIHSSS and received standardized training regarding the mRS, Barthel, Glasgow, and EuroQol assessments.
Data were managed and analyzed with SAS version 9.1 (SAS Institute Inc, Cary, NC). The case report forms were all double-entered into a custom SAS database. Univariate analysis included range checking, in particular against enrollment criteria. Bivariate analysis to compare baseline descriptors between the experimental and control groups consisted of Wilcoxon rank sum and Fisher's exact tests, as appropriate, based on the nature of the data being analyzed. Analysis of the efficacy outcome variables was done with multiple logistic regression to control for appropriate covariates. Variables considered for inclusion as potential covariates were age, baseline NIHSS score, history of prior stroke, history of diabetes, baseline mRS score, time to treatment, and the interaction of age and baseline NIHSS score. These variables were chosen on the basis of findings from the NINDS rt-PA trials.12 Dose tier was also considered as a potential covariate. Multivariable analyses were repeated for each dose tier.
Information was available for all subjects regarding the safety end point. However, even though every effort was made to obtain 90-day follow-ups, there were 4 subjects for whom information was unavailable. For these subjects, the worst-case scenario was assumed, as consistent with other stroke trials.
Results
The study enrolled 94 subjects, 40 in dose tier 1 and 54 in dose tier 2. With a combination of the 2 dose tiers, the experimental group (n=69) was older, had greater stroke severity at baseline, and was less likely to have a prestroke mRS score of 0 at baseline compared with the standard rt-PA group (n=25): a median age of 71 versus 61 (P=0.09) years, a median baseline NIHSS score of 14 versus 10 (P=0.04), and 52 (75%) with a baseline mRS score of 0 versus 24 (96%; P=0.04; Table 2). There were no other significant demographic or medical history differences between the 2 groups, and the median time to treatment was very similar, at 2.5 and 2.6 hours, respectively.
Table 2. CLEAR Trial Baseline Characteristics.
| Tier 1 | Tier 2 | Overall | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Combined | Control | P Value | Combined | Control | P Value | Combined | Control | P Value | Total | |
| n/N | 29 | 11 | 40 | 14 | 69 | 25 | 94 | |||
| Prior stroke | 4 (14%) | 1 (9%) | 1.0 | 9 (22%) | 3 (21%) | 1.0 | 13 (19%) | 4 (16%) | 1.0 | 17 (18%) |
| Hx diabetes | 8 (28%) | 1 (9%) | 0.40 | 9 (22%) | 3 (21%) | 1.0 | 17 (25%) | 4 (16%) | 0.58 | 21 (22%) |
| Hx hypertension | 23 (79%) | 6 (54%) | 0.12 | 26 (65%) | 9 (64%) | 0.96 | 49 (71%) | 15 (60%) | 0.31 | 64 (68%) |
| Baseline mRS score=0 | 19 (66%) | 11 (100%) | 0.04 | 33 (82%) | 13 (93%) | 0.66 | 52 (75%) | 24 (96%) | 0.04 | 76 (81%) |
| Base mRS score 0–1 | 24 (83%) | 11 (100%) | 0.30 | 37 (92%) | 14 (100%) | 0.56 | 61 (88%) | 25 (100%) | 0.10 | 86 (91%) |
| Age, y* | 72.7 (67, 77) | 60.1 (52, 74) | 0.03 | 68.0 (52, 77) | 61.5 (56, 77) | 0.79 | 71.4 (62, 77) | 61.2 (55, 74) | 0.09 | 68.8 (60, 77) |
| Baseline NIHSS score* | 14.0 (10, 20) | 14.0 (9, 16) | 0.23 | 13.5 (8, 17) | 8.5 (6, 12) | 0.02 | 14.0 (10, 20) | 10.0 (6, 14) | 0.01 | 13.0 (8, 17) |
| Time to treatment, min* | 2.5 (2.2, 2.8) | 2.6 (2.3, 2.8) | 0.61 | 2.5 (2.2, 2.8) | 2.6 (2.5, 2.9) | 0.27 | 2.5 (2.2, 2.8) | 2.6 (2.4, 2.8) | 0.28 | 2.6 (2.2, 2.8) |
| Glucose, mg/dL | 115 (101, 139) | 121 (99, 145) | 0.90 | 117 (99, 146) | 114 (96, 158) | 0.88 | 117 (100,141) | 116 (99, 146) | 0.90 | 117 (99, 146) |
| Systolic BP, mm Hg | 157 (141, 186) | 140 (122, 183) | 0.20 | 150 (138, 166) | 158 (137, 178) | 0.37 | 153 (139, 175) | 154 (137, 178) | 0.75 | 154 (137, 176) |
| Diastolic BP, mm Hg | 82 (71, 95) | 82 (74, 91) | 0.87 | 84 (74, 90) | 88 (77, 101) | 0.26 | 83 (74, 93) | 82 (77, 101) | 0.28 | 82 (74, 95) |
| MAP | 112 (96, 124) | 103 (90, 122) | 0.63 | 108 (94, 116) | 113 (97, 129) | 0.16 | 108 (95, 117) | 106 (97, 124) | 0.55 | 108 (96, 120) |
Hx indicates history; BP, blood pressure; and MAP, mean arterial pressure.
Data are expressed as n (%) or median and (25th, 75th percentiles).
Dose tier 1 consisted of 40 patients, including 29 randomized to combination therapy and 11 randomized to standard intravenous rt-PA. One symptomatic ICH did occur in the experimental group during the first dose tier, and there were no safety concerns after that tier was completed. Dose tier 2 was initiated after approval from the DSMB and the U.S. Food and Drug Administration. The trial was held briefly for DSMB review during dose tier 2 after release of the negative findings from the AbESTT trial because of concerns about the safety of GPIIb/IIIa blockade in acute ischemic stroke.10
The DSMB review found no safety concerns, and trial recruitment was continued. Dose tier 2 ultimately enrolled 54 of the planned 60 patients. At this point in the trial, an unblinded DSMB review demonstrated that the safety profile of the combination therapy at the current doses was such that further enrollment was statistically unlikely to indicate inadequate safety for the combination group, the ultimate outcome of the study. In fact, a futility analysis assuming the worst possible scenario for the 6 subjects remaining to be randomized resulted in a probability value equal to 1.0 for the safety outcome. Therefore, the study was halted.
The primary safety outcome of symptomatic ICH was found in 1 (1.4%; 95% CI, 0% to 4.3%) of 69 patients in the combination treatment group and 2 (8.0%; 95% CI, 0% to 19.2%) of 25 patients in the standard-treatment arm (P=0.17). Figure 2 shows the CT images of all ICHs, both symptomatic and asymptomatic, from the trial. Thus, despite the older age and higher baseline stroke severity in the combination treatment group, both factors previously associated with increased hemorrhagic risk, the symptomatic ICH rate for combination treatment was not higher. Similarly, asymptomatic ICH was less frequent in the experimental arm (n=7, 10.3%) than in the control arm (n=3, 12.0%).
Figure 2.

Occurrence of all hemorrhages in the CLEAR Trial.
*Symptomatic intracerebral hemorrhage.
Outcome data that compared standard intravenous rt-PA versus combination therapy in the total group, as well as by dose tier, are shown in Tables 3 and 4. Before a review of efficacy outcome data, it must be emphasized that the 2 groups in this safety study were very different with regard to variables that are known to predict outcome, including age, NIHSS score, and pretreatment mRS score. In the unadjusted analysis, there were no differences between the treatment groups with regard to primary measure of functional outcome, case fatality, symptomatic hemorrhage, or improvement in stroke severity. One secondary measure of functional outcome (the Barthel Index) was statistically significant in favor of standard therapy. After we controlled for covariates of age, baseline NIHSS score, prestroke baseline mRS score, time to treatment, history of diabetes, and age×NIHSS score interaction, there was no sign of efficacy compared with rt-PA for the combination therapy group.
Table 3. Clinical Outcomes at 3 Months for Tier 1, Tier 2, and the Overall Trial of Combination Treatment Versus rt-PA–Only Groups.
| Tier 1 | Tier 2 | Overall | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Combined | Control | P Value | Combined | Control | P Value | Combined | Control | P Value | |
| n | 29 | 11 | 40 | 14 | 69 | 25 | |||
| mRS score 0–1 or return to baseline* | 9 (31%) | 5 (45%) | 0.47 | 12 (30%) | 7 (50%) | 0.21 | 21 (30%) | 12 (48%) | 0.14 |
| Barthel index ≤95* | 12 (41%) | 8 (73%) | 0.16 | 20 (50%) | 10 (71%) | 0.22 | 32 (46%) | 18 (72%) | 0.04 |
| Glasgow outcome score 0* | 13 (45%) | 5 (45%) | 1.0 | 15 (38%) | 8 (57%) | 0.23 | 28 (41%) | 13 (52%) | 0.35 |
| Symptomatic ICH | 1 (3%) | 1 (9%) | 0.48 | 0 (0%) | 1 (7%) | 0.26 | 1 (1%) | 2 (8%) | 0.17 |
| Any ICH | 5 (17%) | 3 (27%) | 0.66 | 3 (8%) | 2 (14%) | 0.60 | 8 (12%) | 5 (20%) | 0.32 |
| Death at 90 days | 8 (28%) | 1 (9%) | 0.40 | 7 (18%) | 2 (14%) | 1.0 | 15 (22%) | 3 (12%) | 0.38 |
| Death at 7 days | 4 (14%) | 1 (9%) | 1.0 | 5 (12%) | 0 (0%) | 0.31 | 9 (13%) | 1 (4%) | 0.28 |
| NIHSS score decrease ≥4 in 24 hours | 14 (48%) | 6 (54%) | 1.0 | 15 (38%) | 5 (36%) | 1.0 | 29 (42%) | 11 (44%) | 1.0 |
| NIHSS score ≤2 at 24 hours | 2 (7%) | 1 (9%) | 1.0 | 7 (18%) | 4 (29%) | 0.45 | 9 (13%) | 5 (20%) | 0.51 |
Data are expressed as n (%); comparisons were made with Fisher's exact test owing to small numbers.
Deaths (n=18) and missing (n=4) patients were coded as “bad” outcome.
Table 4. Adjusted Odds Ratios (ORs) and Associated 95% CIs for Clinical Outcome at 3 Months*.
| Tier 1 | Tier 2 | Overall | ||||
|---|---|---|---|---|---|---|
| OR (95% CI) | P Value | OR (95% CI) | P Value | OR (95% CI) | P Value | |
| mRS score=0–1 or return to baseline† | 0.19 (0.02–1.84) | 0.15 | 0.64 (0.13–3.30) | 0.60 | 0.56 (0.19–1.63) | 0.29 |
| Barthel index ≤95† | 0.24 (0.01–4.60) | 0.34 | 0.28 (0.04–1.75) | 0.17 | 0.46 (0.14–1.53) | 0.20 |
| Glasgow outcome score=0† | 1.90 (0.25–14.76) | 0.54 | 0.55 (0.11–2.91) | 0.48 | 1.03 (0.36–3.00) | 0.95 |
| Survival at 90 days | 0.56 (0.01–33.3) | 0.78 | 0.35 (0.02–6.41) | 0.48 | 0.95 (0.19–4.81) | 0.95 |
| Survival at 7 days | Not estimable | Not estimable | 0.41 (0.04–4.05) | 0.45 | ||
| Any ICH | 0.18 (0.02–1.97) | 0.16 | 0.23 (0.01–5.30) | 0.36 | 0.28 (0.06–1.23) | 0.09 |
| NIHSS score decrease ≥4 in 24 hours | 0.51 (0.06–4.18) | 0.53 | 0.77 (0.16–3.69) | 0.74 | 0.61 (0.20–1.86) | 0.38 |
| NIHSS score ≤2 at 24 hours | 0.20 (0.01–20.8) | 0.50 | 0.34 (0.05–2.34) | 0.27 | 0.70 (0.17–2.87) | 0.62 |
Analysis controlled for covariates of age, baseline NIHSS score, baseline mRS score, time to treatment, history of diabetes, age×NIHSS interaction, baseline glucose, and baseline systolic and diastolic blood pressures.
Deaths (n=18) and missing (n=4) patients were coded as “bad” outcome.
Tables 5 and 6 offer a comparison of the combination treatment group with patients from the NINDS rt-PA trials that was similar to the CLEAR patients, in that they had a baseline NIHSS score >5 and an age range of 18 to 80 years. In addition, only the patients from the 91- to 180-minute group from the NINDS trials were selected, because the median time to treatment in the CLEAR trial was 2.55 hours, and patients treated at <90 minutes did not have an appropriate comparator. As shown, the only demographic difference was age, the NINDS control patients being younger, 66.7 versus 71.4 years (P=0.02). The only outcome variables that approached significance were the lower rate of symptomatic ICH in the combination treatment patients compared with rt-PA patients (1% versus 8%, P=0.08) and a trend toward a better Glasgow Outcome Score and greater likelihood of an NIHSS score ≤2 at 24 hours for combination treatment patients compared with NINDS control patients (P=0.06 for both after adjustment for baseline variables). Thus, the combination treatment patients had a trend toward a lower hemorrhage rate compared with rt-PA patients, no obvious difference in clinical outcome compared with rt-PA patients, and a signal of efficacy compared with controls in the NINDS study.
Table 5. Study Subject Characteristics: CLEAR Trial Combination Pts Versus NINDS Stroke Trial rt-PA and Control Pts.
| CLEAR | NINDS rt-PA | P Value | CLEAR | NINDS Control | P Value | |
|---|---|---|---|---|---|---|
| n or N | 69 | 115 | 69 | 151 | ||
| Prior stroke | 13 (19%) | 21 (18%) | 0.92 | 13 (19%) | 23 (15%) | 0.50 |
| Hx diabetes | 17 (25%) | 25 (22%) | 0.65 | 17 (25%) | 34 (22%) | 0.73 |
| Base mRS score=0 | 52 (75%) | 97 (84%) | 0.13 | 52 (75%) | 126 (83%) | 0.16 |
| Base mRS score 0–1 | 61 (88%) | 104 (90%) | 0.66 | 61 (88%) | 137 (91%) | 0.59 |
| Age, y* | 71.4 (62, 77) | 69.3 (59, 75) | 0.32 | 71.4 (62, 77) | 66.7 (56, 74) | 0.02 |
| Baseline NIHSS score* | 14.0 (10, 20) | 15.0 (9, 20) | 0.45 | 14.0 (10, 20) | 15.0 (10, 22) | 0.23 |
| Time to treatment, min* | 2.5 (2.2, 2.8) | 2.6 (2.2, 2.9) | 0.37 | 2.5 (2.2, 2.8) | 2.5 (2.1, 2.8) | 0.78 |
Hx indicates history.
Data are expressed as n (%) or median and (25th, 75th percentiles).
Table 6. Outcome Variables: CLEAR Trial Combination Pts Versus NINDS Stroke Trial rt-PA and Control Pts.
| CLEAR | NINDS rt-PA | P Value | CLEAR | NINDS Control | P Value | |
|---|---|---|---|---|---|---|
| n or N | 69 | 115 | 69 | 151 | ||
| mRS score 0–1 or return to baseline* | 21 (30%) | 46 (40%) | 0.19 | 21 (30%) | 36 (24%) | 0.30 |
| Barthel index ≤95* | 32 (46%) | 53 (46%) | 0.97 | 32 (46%) | 57 (38%) | 0.23 |
| Glasgow outcome score=0* | 28 (41%) | 47 (41%) | 0.97 | 28 (41%) | 44 (29%) | 0.09 |
| Symptomatic ICH | 1 (1%) | 9 (8%) | 0.08 | 1 (1%) | 2 (1%) | 1.0 |
| Any ICH | 8 (12%) | 13 (11%) | 0.95 | 8 (12%) | 8 (5%) | 0.10 |
| Death at 90 days | 15 (22%) | 21 (18%) | 0.56 | 15 (22%) | 31 (20%) | 0.84 |
| Death at 7 days | 9 (13%) | 9 (8%) | 0.25 | 9 (13%) | 14 (9%) | 0.40 |
| NIHSS score decrease ≥4 in 24 hours* | 29 (42%) | 47 (41%) | 0.88 | 29 (42%) | 57 (38%) | 0.55 |
| NIHSS score ≤2 at 24 hours* | 9 (13%) | 15 (13%) | 1.0 | 9 (13%) | 8 (5%) | 0.04 |
Data expressed as n (%).
Deaths and missing patients were coded as “bad” outcome.
The median time from onset to treatment for the combination treatment patients was 2.53 hours (interquartile range, 2.25 to 2.83), and for the rt-PA patients it was 2.62 hours (interquartile range, 2.41 to 2.83). The median time from symptom onset to Emergency Department arrival was 0.92 and 0.97 hour, respectively. The median times from Emergency Department arrival to CT were 0.23 hour for combination treatment patients and 0.30 hour for the rt-PA patients, and the median times from CT to treatment were 1.34 and 1.37 hours, respectively.
Discussion
The CLEAR trial was the first randomized, blinded, safety trial of the combination of low dose rt-PA and a GPIIb/IIIa antagonist for acute ischemic stroke. The primary goal was to assess the safety of combination therapy initiated within 3 hours of symptom onset. As a baseline for ICH rate for reference, we analyzed patients from the NINDS rt-PA trials who had an NIHSS score >5, were within the age range of 18 to 80 years, and were treated after 90 minutes. In that group, the rate of symptomatic ICH was 8.1%. This mirrors what was found in the 25 control patients treated with rt-PA in the CLEAR trial. Thus, the occurrence of only 1 symptomatic ICH in the 69 combination treatment patients (1.4%) was an encouraging safety datum for combination therapy. In addition to the primary safety outcome, other serious adverse events were carefully evaluated. Similar to ICH rates, no other serious adverse event rate was noted to be different between the 2 groups.
The CLEAR trial safety results must be considered in the context of the marked disparity in age and baseline NIHSS score between the combination treatment and control groups. It is well known that both older age and higher baseline NIHSS score are significant predictors of symptomatic ICH after rt-PA treatment of acute ischemic stroke.13 Despite the marked disparity favoring the standard intravenous rt-PA group (younger age and lower stroke severity and more zero baseline mRS scores), the combination therapy group did not show a higher rate of symptomatic or asymptomatic ICH. The fact that the study combination was safe despite these increased risk factors for ICH further suggests that the combination of eptifibitide and rt-PA for treatment of ischemic stroke within 3 hours of symptom onset is safe enough to consider further exploration.
From the first grant application submission, the plan of the CLEAR investigators was to proceed with a follow-up trial if the safety results of CLEAR were within prespecified limits. The safety stopping rules for each tier specified that if the observed symptomatic ICH rate exceeded the expected rate for comparable patients treated with rt-PA in the NINDS rt-PA stroke trials, the trial would have been placed on hold with a mandatory DSMB review. The trial was neither designed nor powered for efficacy. Notably, the trial design ultimately resulted in a large disparity in age and stroke severity between the 2 groups owing to the play of random chance and small numbers of patients in the randomly allocated groups. For future smaller phase II trials, adaptive or stratified randomization may be a necessary strategy to minimize group disparities.
These safety data allow for continued exploration of the combination of fibrinolytics and GPIIb/IIIa antagonists in acute stroke. Combination therapy of fibrinolytics with GPIIb/IIIa blockers has been an important focus of research in cardiac patients. The combination approach provides 2 simultaneous mechanisms for thrombolysis via disruption of the clot's fibrin meshwork, coupled with disaggregation of platelets. The result is improved arterial patency with the combination of GPIIb/IIIa blockade and fibrinolytic agents versus fibrinolytic agents alone.4 Of particular importance to acute ischemic stroke treatment, the rate of recanalization in acute MI that occurs with the combination of a fibrinolytic and GPIIb/IIIa inhibitor has consistently been faster and more effective than with a fibrinolytic alone. This has been demonstrated by continuous measurement of ST-segment elevation in patients with acute MI as a surrogate marker for recanalization.14
Animal research has demonstrated that significant micro-vascular platelet and fibrin accumulation occurs in the setting of middle cerebral artery occlusion in primates and rodents.15–18 Microvascular occlusions by platelet-fibrin aggregates have been shown to be preventable with the use of an experimental GPIIb/IIIa inhibitor given just before middle cerebral artery occlusion.19 In another study, infusion of an experimental GPIIb/IIIa inhibitor was associated with a significant reduction in microvascular platelet accumulation and was associated with a 70% reduction in cerebral infarct size.16 The data suggest an important role for postocclusive distal platelet deposition in the evolution of acute ischemic stroke. Further preclinical data from an embolic model in mice have demonstrated that the combination of a fibrinolytic agent and a GPIIb/IIIa inhibitor increases the efficiency of dissolving an embolus, decreases infarct volume, and improves microvascular patency compared with a fibrinolytic agent alone. In this research, treatment with rt-PA and a GPIIb/IIIa antagonist reduced perfusion deficits and significantly enhanced cortical perfusion.20
Thus, there is considerable cardiac and preclinical literature to support the hypothesis that the combination may, at appropriate dosages, prove efficacious in acute ischemic stroke. The CLEAR trial has demonstrated, despite significant imbalances in randomization, that there is adequate safety of the combination at the studied dosages to pursue a higher dose regimen.
Study Limitations
This was a pilot safety trial, and any efficacy conclusions beyond hypothesis generation are unwarranted. The disparities in baseline characteristics confound analysis as described earlier. This problem could be prevented with an adaptive randomization technique in a future trial.
There were also significant logistical limitations that became apparent during execution of the trial. The greatest limitation of the CLEAR trial was the cumbersome design of the double-blind drug dosing. To reach likely effective serum concentrations of rt-PA with lower dosing, the time interval for the low-dose infusion had to be shorter than the 1-hour infusion of standard-dose rt-PA. Thus, the rt-PA infusion was divided into 2 bags; for the low-dose arm, the entire rt-PA dose was in the first bag. The investigators were forced to wait while the investigational pharmacies prepared a total of 2 boluses and 3 infusion bags before initiation of drug administration. Although the investigators' practice was to inform the pharmacy as soon as a possible patient was identified, delays in treatment due to pharmacy preparation time did occur. As a surrogate marker for the delay, the median time from onset to treatment in these patients was 2.55 hours, but the median time from CT to initiation of the study drug was 1.35 hours, which is likely longer than the median time for open-label use at most experienced centers. As a comparison, in the NINDS rt-PA stroke trials, the median (and interquartile range) time from CT to initiation of drug therapy was 0.58 hour (0.38 to 0.83) for all patients in the trial. On a per-group basis, the median (and interquartile range) times from CT to treatment were 0.43 hour (0.27 to 0.58) for patients treated in <90 minutes and 0.75 hour (0.55 to 1.08) for patients treated between 91 and 180 minutes. Any future trial should consider starting open-label rt-PA as soon as clinically possible.
Conclusions
The CLEAR trial was a dose-escalation and safety study of the combination of low-dose rt-PA and a GPIIb/IIIa antagonist (eptifibatide) in acute ischemic stroke patients who were treated within 3 hours of symptom onset. The safety profile of the combination regimens was excellent. The investigational team believes that the combination of reduced-dose rt-PA and eptifibatide appears safe enough for consideration of further dose-ranging trials in acute ischemic stroke. The combined approach to lysis utilizing eptifibatide and rt-PA–enhanced regimen (CLEARER) trial will be a randomized trial of 0.6 mg/kg rt-PA plus eptifibatide versus standard-dose rt-PA. This trial is designed to evaluate the safety of this higher dosage combination as well as to determine whether the estimated signal of efficacy warrants proceeding to a phase III clinical trial.
Acknowledgments
The authors are grateful for the support of Genentech, Inc (rt-PA) and Schering Plough Corp (eptifibatide), who provided the indicated study drugs.
Sources of Funding: This study was supported by a grant from the NINDS (1 P50 NS44283-01).
Appendix: Participants in the CLEAR Study
The following investigators and institutions participated in the CLEAR study.
Principal Investigator
Arthur M. Pancioli, MD, University of Cincinnati.
Steering Committee
Joseph Broderick, MD, University of Cincinnati; Thomas Kwiatkowski, MD, and Richard Libman, MD, Long Island Jewish Medical Center; John Castaldo, MD, and Richard Mackenzie, MD, Lehigh Valley Hospital; Phillip Scott, MD, University of Michigan Medical Center; James Frey, MD, St. Joseph Hospital, Phoenix; Scott E. Kasner, MD, University of Pennsylvania Medical Center; R. Jason Thurman, MD, Vanderbilt University Medical Center; Sidney Starkman, MD, University of California at Los Angeles; James Gebel Jr, MD, Jewish Hospital and St. Mary Healthcare, Louisville; Scott Janis, PhD, NIH/NINDS; Thomas Brott, MD, Mayo Clinic, Jacksonville; Gregory del Zoppo, MD, University of Washington, Seattle; Thomas Tomsick, MD, University of Cincinnati; Chelsea Kidwell, MD, Georgetown University; Judy Bean, PhD, and Jane Khoury, PhD, Cincinnati Children's Hospital Medical Center.
Medical Safety Monitor
Steven Levine, MD, Mt. Sinai School of Medicine; Larry Brass, MD, Yale University.
DSMB
Claudia Moy, PhD, NIH/NINDS; Gretchen Tietjens, MD, Medical University of Toledo; K. Michael Welch, MD, Chicago Medical School; William R. Clarke, PhD, University of Iowa.
Study Coordinators
University of Cincinnati: Pamela Schmit, RN; Irene Ewing, RN; Judith Spilker, RN; Joyce Zeigler; Janice Carrozzella, RN; Long Island Jewish Medical Center: Rose Gonzaga-Camfield, RN; Marietta Manlulu, RN; Mary Schaefer, RN; Lehigh Valley Hospital: Maryjane Cerrone, RN; Carol Fox, RN; Donna Jenny, RN; Kathleen Knapp, RN; Claranne Mathiesen, RN; Joanne Rodgers, RN; University of Michigan: Shirley Frederiksen, RN; Kate Maddox, RN; Teresa Murrell, RN; Annette Sandretto, RN; St. Joseph Hospital, Phoenix: Susan Kurzer, RN; University of Pennsylvania: Michele Sellers, RN; Jean Luciano, RN; Vanderbilt University: Diane Brown, RN; University of California at Los Angeles: Judy Guzy, RN; Jewish Hospital and St. Mary Healthcare, Louisville: Pamela Adkisson, RN; Ramona Constant, RN.
Site Subinvestigators
University of Cincinnati: Daniel Woo, MD; Dawn Kleindorfer, MD; Pooja Khatri, MD; Opeolu Adeoye, MD; Matthew Flaherty, MD; Edward Jauch, MD; Daniel Kanter, MD; Brett Kissela, MD; Christopher Nichols, MD; Alexander Schneider, MD; Brian Stettler, MD; Long Island Jewish Hospital Medical Center: Laura Schoenberg, MD; Lehigh Valley Hospital: Peter Barbour, MD; Gary Clauser, MD; Alexander Rae-Grant, MD; Yevgenity Isayev, MD; Yuebing Li, MD; Glenn Mackin, MD; John Margraf, MD; Lorraine Spikol, MD; Jay Varrato, DO; University of Michigan: William Barsan, MD; Devin Brown, MD; Susan Hickenbottom, MD; Jennifer Majersik, MD; William Meurer, MD; Lewis Morgenstern, MD; Robert Silbergleit, MD; Rodney Smith, MD; Teresa Smith, MD; Michael Wang, MD, PhD; Darin Zahuranec, MD; University of Pennsylvania: Brett Cucchiara, MD; Steven Messe, MD; James Pacelli, MD; Lauren Sansing, MD; Qaisar Shah, MD; Vanderbilt University Medical Center: Kenneth Gaines, MD; Howard Kirshner, MD; Anne O'Duffy, MD; Adrian Jarquin-Valdivia, MD; University of California at Los Angeles: Latisha Ali, MD; Edwin Amos, MD; Brian Buck, MD; William Buxton, MD; Andre Fredieu, MD; Michael Gold, MD; Sheldon Jordan, MD: Doojin Kim, MD: David Liebeskind, MD; Chad Miller, MD; Edward O'Connor, MD; Andrew Oh, MD; Bruce Ovbiagele, MD; Verna Porter, MD; Venkatakrishna Rajajee, MD; Lucas Restrepo, MD; Nerses Sanossian, MD; Scott Selco, MD; Samir Shah, MD, Russ Shimizu, MD; Shuichi Suzuki, MD; Amytis Towfighi MD; Paul Vespa, MD Andrew Woo, MD.
Laboratory Specimen Coordinator
Laura Sauerbeck, RN, University of Cincinnati.
Data Entry Coordinators
Julie Brock and Bonnie Combs, University of Cincinnati.
Footnotes
Disclosures: J.B. was a consultant to Novo Nordisk (member of steering committee, Novo 7 trial), to Wyeth Pharmaceuticals (Stroke Advisory Board), to Johnson & Johnson (Stroke Advisory Board), and to Hoffman-La Roche, Ltd; and received honoraria for speaking from Boehringer Ingelheim. The Other authors have nothing to disclose.
References
- 1.Tissue plasminogen activator for acute ischemic stroke: the National Institute of Neurological Disorders and Stroke rt-PA stroke study group. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 2.Wolpert SM, Bruckmann H, Greenlee R, Wechsler L, Pessin MS, del Zoppo GJ. Neuroradiologic evaluation of patients with acute stroke treated with recombinant tissue plasminogen activator: the rt-PA acute stroke study group. AJNR Am J Neuroradiol. 1993;14:3–13. [PMC free article] [PubMed] [Google Scholar]
- 3.Alexandrov AV, Grotta JC. Arterial reocclusion in stroke patients treated with intravenous tissue plasminogen activator. Neurology. 2002;59:862–867. doi: 10.1212/wnl.59.6.862. [DOI] [PubMed] [Google Scholar]
- 4.Pancioli AM, Brott TG. Therapeutic potential of platelet glycoprotein IIB/IIIA receptor antagonists in acute ischaemic stroke: scientific rationale and available evidence. CNS Drugs. 2004;18:981–988. doi: 10.2165/00023210-200418140-00003. [DOI] [PubMed] [Google Scholar]
- 5.Inhibition of platelet glycoprotein IIB/IIIA with eptifibatide in patients with acute coronary syndromes: the PURSUIT trial investigators. Platelet glycoprotein IIB/IIIA in unstable angina: receptor suppression using integrilin therapy. N Engl J Med. 1998;339:436–443. doi: 10.1056/NEJM199808133390704. [DOI] [PubMed] [Google Scholar]
- 6.Randomised placebo-controlled trial of effect of eptifibatide on complications of percutaneous coronary intervention, IMPACT-II: integrilin to minimise platelet aggregation and coronary thrombosis-II. Lancet. 1997;349:1422–1428. [PubMed] [Google Scholar]
- 7.O'Shea JC, Hafley GE, Greenberg S, Hasselblad V, Lorenz TJ, Kitt MM, Strony J, Tcheng JE. Platelet glycoprotein IIB/IIIA integrin blockade with eptifibatide in coronary stent intervention: the ESPRIT trial: a randomized controlled trial. JAMA. 2001;285:2468–2473. doi: 10.1001/jama.285.19.2468. [DOI] [PubMed] [Google Scholar]
- 8.Tcheng JE, Harrington RA, Kottke-Marchant K, Kleiman NS, Ellis SG, Kereiakes DJ, Mick MJ, Navetta FI, Smith JE, Worley SJ, et al. Multicenter, randomized, double-blind, placebo-controlled trial of the platelet integrin glycoprotein IIB/IIIA blocker integrelin in elective coronary intervention. IMPACT investigators. Circulation. 1995;91:2151–2157. doi: 10.1161/01.cir.91.8.2151. [DOI] [PubMed] [Google Scholar]
- 9.Mahaffey KW, Harrington RA, Simoons ML, Granger CB, Graffagnino C, Alberts MJ, Laskowitz DT, Miller JM, Sloan MA, Berdan LG, MacAulay CM, Lincoff AM, Deckers J, Topol EJ, Califf RM. Stroke in patients with acute coronary syndromes: incidence and outcomes in the platelet glycoprotein IIB/IIIA in unstable angina. Receptor suppression using integrilin therapy (PURSUIT) trial. The PURSUIT investigators. Circulation. 1999;99:2371–2377. doi: 10.1161/01.cir.99.18.2371. [DOI] [PubMed] [Google Scholar]
- 10.Adams HP, Jr, Effron MB, Torner J, Davalos A, Frayne J, Teal P, Leclerc J, Oemar B, Padgett L, Barnathan ES, Hacke W. Emergency administration of abciximab for treatment of patients with acute ischemic stroke: results of an international phase III trial. Abciximab in emergency treatment of stroke trial (AbESTT-II) Stroke. 2008;39:87–99. doi: 10.1161/STROKEAHA.106.476648. [DOI] [PubMed] [Google Scholar]
- 11.Topol EJ. Reperfusion therapy for acute myocardial infarction with fibrinolytic therapy or combination reduced fibrinolytic therapy and platelet glycoprotein IIB/IIIA inhibition: The GUSTO V randomised trial. Lancet. 2001;357:1905–1914. doi: 10.1016/s0140-6736(00)05059-5. [DOI] [PubMed] [Google Scholar]
- 12.Ingall TJ, O'Fallon WM, Asplund K, Goldfrank LR, Hertzberg VS, Louis TA, Christianson TJ. Findings from the reanalysis of the NINDS tissue plasminogen activator for acute ischemic stroke treatment trial. Stroke. 2004;35:2418–2424. doi: 10.1161/01.STR.0000140891.70547.56. [DOI] [PubMed] [Google Scholar]
- 13.Khatri P, Wechsler LR, Broderick JP. Intracranial hemorrhage associated with revascularization therapies. Stroke. 2007;38:431–440. doi: 10.1161/01.STR.0000254524.23708.c9. [DOI] [PubMed] [Google Scholar]
- 14.Rebeiz AG, Johanson P, Green CL, Crater SW, Roe MT, Langer A, Giugliano RP, Lincoff AM, Newby LK, Harrington RA, Topol EJ, Califf RM, Wagner GS, Krucoff MW. Comparison of ST-segment resolution with combined fibrinolytic and glycoprotein IIB/IIIA inhibitor therapy versus fibrinolytic alone (data from four clinical trials) Am J Cardiol. 2005;95:611–614. doi: 10.1016/j.amjcard.2004.10.038. [DOI] [PubMed] [Google Scholar]
- 15.Del Zoppo GJ, Copeland BR, Harker LA, Waltz TA, Zyroff J, Hanson SR, Battenberg E. Experimental acute thrombotic stroke in baboons. Stroke. 1986;17:1254–1265. doi: 10.1161/01.str.17.6.1254. [DOI] [PubMed] [Google Scholar]
- 16.Choudhri TF, Hoh BL, Zerwes HG, Prestigiacomo CJ, Kim SC, Connolly ES, Jr, Kottirsch G, Pinsky DJ. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIB/IIIA receptor-mediated platelet aggregation. J Clin Invest. 1998;102:1301–1310. doi: 10.1172/JCI3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat) Am J Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang ZG, Zhang L, Tsang W, Goussev A, Powers C, Ho KL, Morris D, Smyth SS, Coller BS, Chopp M. Dynamic platelet accumulation at the site of the occluded middle cerebral artery and in downstream microvessels is associated with loss of microvascular integrity after embolic middle cerebral artery occlusion. Brain Res. 2001;912:181–194. doi: 10.1016/s0006-8993(01)02735-4. [DOI] [PubMed] [Google Scholar]
- 19.Abumiya T, Fitridge R, Mazur C, Copeland BR, Koziol JA, Tschopp JF, Pierschbacher MD, del Zoppo GJ. Integrin αIIBβ3 inhibitor preserves microvascular patency in experimental acute focal cerebral ischemia. Stroke. 2000;31:1402–1409. doi: 10.1161/01.str.31.6.1402. discussion 1409–1410. [DOI] [PubMed] [Google Scholar]
- 20.Ding G, Jiang Q, Zhang L, Zhang ZG, Li L, Knight RA, Ewing JR, Wang Y, Chopp M. Analysis of combined treatment of embolic stroke in rat with r-TPA and a GPIIb/IIIA inhibitor. J Cereb Blood Flow Metab. 2005;25:87–97. doi: 10.1038/sj.jcbfm.9600010. [DOI] [PubMed] [Google Scholar]