

Abstract
Mitochondrial outer-membrane permeabilization can lead to cell death even without activation of caspases. In this issue of Cell, Colell et al. (2007) identify the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase as a potent inhibitor of caspase-independent cell death that may allow metabolically active cells to survive mitochondrial insult.
Apoptosis is a tidy form of cell death. The dying cell quietly commits suicide without disturbing its neighbors and the corpse remains neatly packaged for clearance by phagocytes. This orderly dismantling of the apoptotic cell is the purview of caspases, enzymes that cleave cellular substrates, promoting the characteristic apoptotic morphology. Caspase activation often results from mitochondrial outer-membrane permeabilization, triggered by a variety of apoptotic stimuli that promote release of proapoptotic mitochondrial contents including cytochrome c. Although pathways linking mitochondrial permeabilization and caspase activation have received intense scrutiny, the oft-ignored truth is that mitochondrial permeabilization kills cells via a caspase-independent pathway if caspase activation is prevented. Nevertheless, some cells can evade caspase-independent cell death despite the blow to their mitochondria. In this issue, Colell et al. (2007) report the surprising discovery that the “housekeeping” gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) plays a key role in opposing caspase-independent death following release of mitochondrial cytochrome c.
Colell, Green, and their colleagues designed a clever screen for factors conferring survival only in cells that had undergone mitochondrial permeabilization in the absence of caspase activation, thereby avoiding repeated identification of established apoptotic inhibitors that prevent permeabilization of the mitochondrial outer membrane (such as Bcl-2 proteins). They introduced a retroviral cDNA library into Jurkat cells treated with staurosporine, a potent inducer of mitochondrial outer-membrane permeabilization, and sought cell clones able to proliferate in the continued presence of a pan-caspase inhibitor. Clones that also survived in the absence of caspase inhibitors were eliminated in a secondary screen (Figure 1). One can imagine the authors’ initial consternation when this screen yielded the ubiquitious glycolytic enzyme GAPDH. Nonetheless, they demonstrated that overexpression of GAPDH did not simply prevent mitochondrial permeabilization and did, indeed, confer protection against caspase-independent, but not caspase-dependent, cell death. Furthermore, GAPDH promoted survival in other scenarios of caspase-independent cell death, most notably in cells expressing proapoptotic oncogenes but lacking the cytochrome c-binding partner, Apaf-1.
Figure 1. GAPDH as a Regulator of caspase-Independent cell Death.
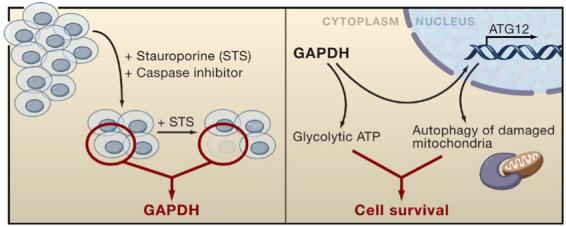
GAPDH was identified in a genetic screen in which cells were infected with a retroviral cDNA library and treated with caspase inhibitors and staurosporine (STS), which leads to mitochondrial outer-membrane permeabilization. Surviving cell clones were cultured with staurosporine alone to identify those that specifically resisted caspase-independent cell death but were susceptible to apoptosis. The proposed mechanism for GAPDH-mediated resistance to caspase-independent cell death occurs through dual cytoplasmic and nuclear functions. In the cytoplasm, GAPDH promotes glucose metabolism to sustain cellular ATP levels. In the nucleus, GAPDH leads to induction of ATG12 expression, which enhances degradation of damaged mitochondria through autophagy.
Given its glycolytic role, the isolation of GAPDH suggested that maintenance of ATP levels might play a role in the escape from caspase-independent death. Indeed, GAPDH expression conferred survival only in high-glucose media where GAPDH could increase intracellular ATP levels. Despite these observations, the authors may have suspected that ATP was not the whole story when other metabolic enzymes did not emerge from their screen. Although it has a cytoplasmic “day job” in glycolysis, GAPDH also has roles in the nucleus (Sirover, 2005). Colell et al. (2007) therefore produced mutant variants of GAPDH that either retained the ability to translocate to the nucleus but were unable to support ATP production, or variants that promoted ATP production but were unable to support nuclear functions. Remarkably, although neither of these mutant proteins could protect from caspase-independent death, coexpression of both mutants promoted survival following mitochondrial permeabilization/caspase inhibition.
As nuclear GAPDH has been implicated in transcriptional regulation, the authors performed microarray analysis to identify candidate genes induced by GAPDH expression. Interestingly, they identified ATG12, a known component of autophagic pathways (Tanida et al., 2004), dovetailing nicely with their observation that autophagy is increased in the cells expressing GAPDH. Moreover, ATG12 overexpression could substitute for nuclear GAPDH in the complementation assay described above, making it likely that increased ATP and enhanced ATG12 could cooperate to rescue cells from caspase-independent death via autophagy. These findings suggest that ATG12 can be rate limiting for the induction of autophagy, at least following mitochondrial permeabilization. It is unclear whether overexpression of ATG12 would also cooperate with other factors that elevate levels of ATP to rescue cells from caspase-independent death. If GAPDH was the only clone identified from their initial screen, it may be because it is the only protein endowed with the dual ability to elevate ATP and induce ATG12 (Figure 1). Under physiological circumstances, it may be that expression of multiple genes attains similar results.
It is attractive to speculate that autophagy protects against caspase-independent death through removal of damaged mitochondria. It is not clear whether the “recovery” of these cells (and the apparent ability of mitochondria to maintain membrane potential) is due to the proliferation of a small population of undamaged mitochondria or whether GAPDH expression also aids in the repair of damaged mitochondria. It will be of interest to monitor individual mitochondria within cells rescued from caspase-independent death to determine their fates.
It is important to consider how these findings apply to normal and disease physiology. Postmitotic cells, such as neurons and cardiomyocytes, are resistant to cell death even after cytochrome c release and have been shown to downregulate Apaf-1 expression, thus preventing the formation of the caspase 9/Apaf-1/cytochrome c apoptosome complex, precluding caspase activation. Induction of inhibitor of apoptosis (IAP) proteins can also limit or prevent caspase activity following cytochrome c release (Wright and Deshmukh, 2006). Therefore, cells with alterations in these proteins are candidates to survive even after mitochondrial permeabilization if caspase-independent death is inhibited. Nevertheless, mitochondria that have released cytochrome c are functionally compromised even if caspase activity is prevented. In contrast to its assumed stability as a housekeeping protein, GAPDH can be highly regulated and may, under specific conditions, be induced sufficiently to support viability of cells following mitochondrial permeabilization. Lymphocyte stimulation, for example, has been known to strongly upregulate glycolysis and GAPDH (Sabath et al., 1990). For a cell with damaged mitochondria, enhanced glycolysis would provide a bioenergetic source and also promote nonmetabolic functions of GAPDH, such as the induction of ATG12. Thus GAPDH may promote survival of lymphocytes that suffer partial or complete mitochondrial permeabilization during encounters with cytotoxic pathogens or in the normal execution of an immune response. Consistent with this, Colell et al. (2007) note that T cells resistant to caspase activation due to mutation in cytochrome c have a memory phenotype, suggesting they had been activated at some point in their lifetime (Hao et al., 2005).
The new work has demonstrated that a molecule considered by many to be among the most pedestrian of intracellular proteins can play a complex role in cell fate. There remain, however, a number of significant issues and questions. First, is there some particular metabolic feature of GAPDH that is required? This question is highly relevant to the survival of cancer cells, which Otto Warburg showed in the 1920s are more glycolytic than their normal counterparts. Since then, it has been shown that many different proglycolytic genes can be upregulated in cancer (Semenza et al., 2001). Although enhanced glycolysis can provide some initial protection from mitochondrial permeabilization and cytochrome c release (Gottlob et al., 2001; Zhao et al., 2007), it is not clear whether it affects the caspase-independent cell pathway after mitochondrial permeabilization. As many cancers carry mutations that impair cytochrome c-induced caspase activation, GAPDH overexpression may assist cancer cells in evading apoptosis, particularly when induced by chemotherapeutics that promote mitochondrial permeabilization.
How does GAPDH lead to ATG12 upregulation? There are a number of studies documenting nonmetabolic functions of GAPDH and Colell et al. (2007) provide fresh impetus to revisit these nontraditional roles. Autophagy can promote cell survival and function in tumor suppression. What allows autophagy to clear damaged mitochondria while preserving intact organelles thus aiding survival? Lastly, how frequently does the circumstance of inhibited caspase activity, elevated glycolysis, and autophagy occur in vivo? Clearly, there are instances when these events coincide, but is elevated GAPDH expression essential? This is an important and fertile ground for additional study as researchers consider how expression of this traditional gel-loading control may be regulated and how this may influence the fate of their favorite cells.
REFERENCES
- Colell A, Jean-Ehrland Ricci J-E, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse N-J, Li CW, et al. 2007 doi: 10.1016/j.cell.2007.03.045. Cell. this issue. [DOI] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z, Duncan GS, Chang CC, Elia A, Fang M, Wakeham A, Okada H, Calzascia T, Jang Y, You-Ten A, et al. Cell. 2005;121:579–591. doi: 10.1016/j.cell.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Sabath DE, Broome HE, Prystowsky MB. Gene. 1990;91:185–191. doi: 10.1016/0378-1119(90)90087-8. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P, Zhong H. Novartis Found. Symp. 2001;240:251–260. [PubMed] [Google Scholar]
- Sirover MA. J. Cell. Biochem. 2005;95:45–52. doi: 10.1002/jcb.20399. [DOI] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. Int. J. Biochem. Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KM, Deshmukh M. Cell Cycle. 2006;5:1616–1620. doi: 10.4161/cc.5.15.3129. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, Wofford JA, Dimascio LN, Ilkayeva O, Kelekar A, et al. Mol. Cell. Biol. 2007 doi: 10.1128/MCB.00153-07. in press Published online March 19, 2007. 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]